
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Switching the three-component Biginelli-like reaction conditions for the regioselective synthesis of new 2-amino[1,2,4]triazolo[1,5-a]pyrimidines†
Martina
Pacetti‡
 a,
Maria Chiara
Pismataro‡
a,
Tommaso
Felicetti
*a,
Federica
Giammarino
b,
Anna
Bonomini
c,
Matteo
Tiecco
d,
Chiara
Bertagnin
c,
Maria Letizia
Barreca
a,
Raimondo
Germani
e,
Violetta
Cecchetti
a,
Ilaria
Vicenti
b,
Oriana
Tabarrini
a,
Maurizio
Zazzi
b,
Arianna
Loregian
c and
Serena
Massari
a
a,
Maria Chiara
Pismataro‡
a,
Tommaso
Felicetti
*a,
Federica
Giammarino
b,
Anna
Bonomini
c,
Matteo
Tiecco
d,
Chiara
Bertagnin
c,
Maria Letizia
Barreca
a,
Raimondo
Germani
e,
Violetta
Cecchetti
a,
Ilaria
Vicenti
b,
Oriana
Tabarrini
a,
Maurizio
Zazzi
b,
Arianna
Loregian
c and
Serena
Massari
a
aDepartment of Pharmaceutical Sciences, University of Perugia, 06123 Perugia, Italy. E-mail: tommaso.felicetti@unipg.it; Tel: +39 075-5852185
bDepartment of Medical Biotechnologies, University of Siena, 53100 Siena, Italy
cDepartment of Molecular Medicine, University of Padua, 35121 Padua, Italy
dChemistry Interdisciplinary Project (ChIP), School of Pharmacy, University of Camerino, 62032 Camerino, MC, Italy
eDepartment of Chemistry, Biology and Biotechnology, University of Perugia, 06123 Perugia, Italy
First published on 19th December 2023
Abstract
Among the eight different triazolopyrimidine isomers existing in nature, 1,2,4-triazolo[1,5-a]pyrimidine (TZP) is one of the most studied and used isomers in medicinal chemistry. For some years, our group has been involved in developing regioselective one-pot procedures for the synthesis of 2-amino-7-aryl-5-methyl- and 2-amino-5-aryl-7-methyl-TZPs of interest in the preparation of antiviral agents. In this work, taking advantage of a Biginelli-like multicomponent reaction (MCR), we report the identification of finely tunable conditions to regioselectively synthesize C-6 ester-substituted amino-TZP analogues, both in dihydro and oxidized forms. Indeed, the use of mild acidic conditions is strongly directed toward the regioselective synthesis of 5-aryl-7-methyl C-6-substituted TZP analogues, while the use of neutral ionic liquids shifted the regioselectivity towards 7-aryl-5-methyl derivatives. In addition, the novel synthesized scaffolds were functionalized at the C-2 position and evaluated for their antiviral activity against RNA viruses (influenza virus, flaviviruses, and SARS-CoV-2). Compounds 25 and 26 emerged as promising anti-flavivirus agents, showing activity in the low micromolar range.
Introduction
Triazolopyrimidine represents a privileged structure in agrochemical and medicinal chemistry, with numerous derivatives that have found applications in several biological and medical fields.1–3 Among the eight different triazolopyrimidine isomers existing in nature, [1,2,4]triazolo[1,5-a]pyrimidine (hereafter abbreviated TZP for convenience) has been the most studied and used isomer in medicinal chemistry, especially to design and synthesize novel anti-infective agents.3–5 However, recent articles and reviews show the potential of this nucleus to develop compounds endowed with several biological activities, also taking advantage of its favourable pharmacokinetic properties.1,3,4,6For some years, our group has been involved in the synthesis of TZP-based compounds as inhibitors of RNA viruses, mainly as an anti-influenza virus (IV) agents able to inhibit RNA-dependent RNA polymerase (RdRP) PA–PB1 subunits interaction.7–10 Starting from the hit compound 1 (Scheme 1a), several compounds were designed and synthesized by exploring the role of the C-2 amide substituent and modifying the TZP nucleus by: (i) aromatization of the 4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine core, (ii) exchange of 5-methyl and 7-phenyl moieties, (iii) inversion of the C-2 amide link, (iv) decoration of the C-5/C-7 phenyl ring with different substituents, (v) removal of the methyl moiety while maintaining only a phenyl ring at the C-7, C-5, or C-6 position, and (vi) replacement of the C-7 methyl group by a hydroxyl group (Scheme 1a). These modifications led to the identification of compounds that exhibited an improved ability to inhibit PA–PB1 interaction with respect to 1 and, above all, acquired anti-IV activity at non-toxic concentrations.7,9
 | ||
| Scheme 1 (a) Structural modifications performed on the TZP core during the optimization of anti-IV hit compound 1. (b) Chemical procedures previously developed by us for the synthesis of 2-amino-TZP compounds 2–7. *Isolated yield. | ||
During the synthesis of anti-IV TZPs, efforts were directed towards the development of one-pot procedures to directly obtain oxidized TZPs. In particular, starting from 3,5-diaminotriazole, we reported facile and efficient one-step procedures for the regioselective synthesis of 2-amino-5-methyl-7-phenyl-[1,2,4]triazolo[1,5-a]pyrimidines (exemplified by compound 2),11 2-amino-7-methyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidines (exemplified by compound 3),11 2-amino-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4,8 and 7-phenyl-, 5-phenyl-, and 6-phenyl-2-amino-[1,2,4]triazolo[1,5-a]pyrimidines 5–7 (Scheme 1b).8
Keeping our interest on TZP-based compounds as antiviral agents, in this work, we focused on the synthesis of C-5, C-6, and C-7 trisubstituted TZPs taking advantage of a Biginelli-like multicomponent reaction (MCR) for their preparation. MCRs represent a powerful approach for achieving diversity and complexity of organic compounds, reducing the number of reaction steps and the environmental impact, thus combining molecular diversity, depending on the structures of the reagents, the solvent, and the catalyst, with eco-compatibility.
Some examples of Biginelli-like reactions for the synthesis of the TZP scaffold that combine aldehydes, compounds with an active methylene group, and polyfunctional aminotriazoles have been reported in the literature (readers are directed to reviews),3,12–16 mainly furnishing C-2 unsubstituted TZP derivatives, such as 4,5,6,7-tetrahydro-TZPs,17 4,7-dihydro-TZPs,18,19 and aromatic TZP analogues20 (Fig. 1 – general structures I–IV). Less attention has been paid to C-2-functionalized TZP derivatives, with only a few examples reporting 4,7-dihydro TZP analogues (general structure V in Fig. 1).21–23
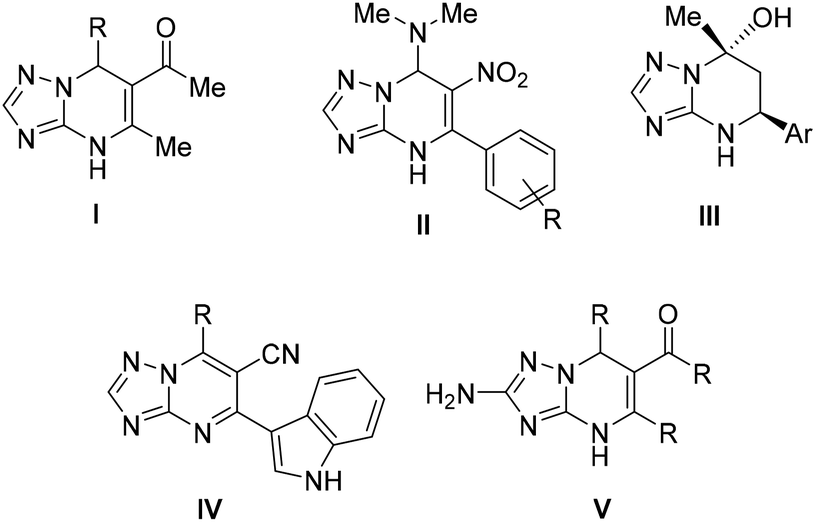 | ||
| Fig. 1 Examples of functionalized dihydro, tetrahydro, and aromatic TZP derivatives (general structures I–V) obtained by Biginelli-like MCRs. | ||
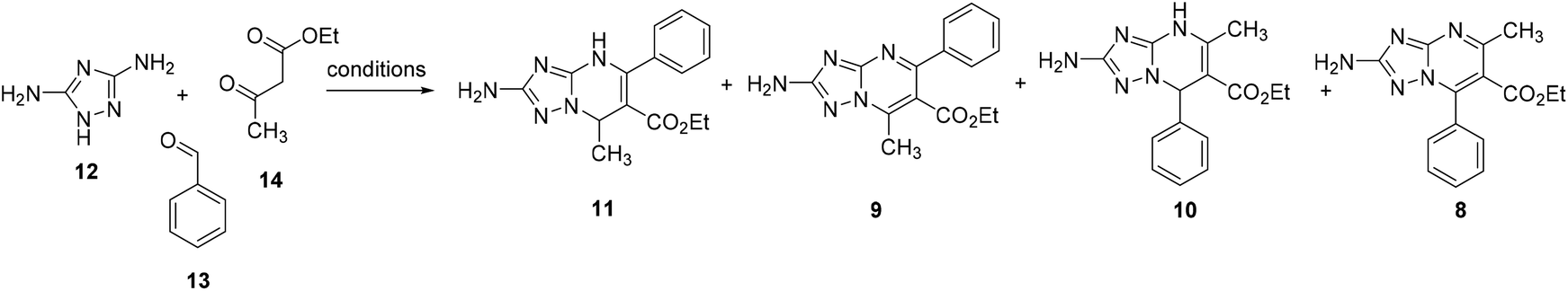
Herein, we reported for the first time two Biginelli-like MCRs to obtain directly and regioselectively aromatic 2-substituted TZPs. In particular, ethyl 2-amino-5-methyl-7-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate (8) and ethyl 2-amino-7-methyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate (9) (Scheme 2a) were regioselectively synthesized via the reaction of 3,5-diaminotriazole, benzaldehyde, and ethyl 3-oxobutanoate, under liquid ionic and acidic conditions, respectively. Moreover, the use of the same environments while changing the reaction conditions permitted us to regioselectively and efficiently obtain the dihydro analogues ethyl 2-amino-5-methyl-7-phenyl-4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate (10)21 and ethyl 2-amino-7-methyl-5-phenyl-4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate (11) (Scheme 2a).
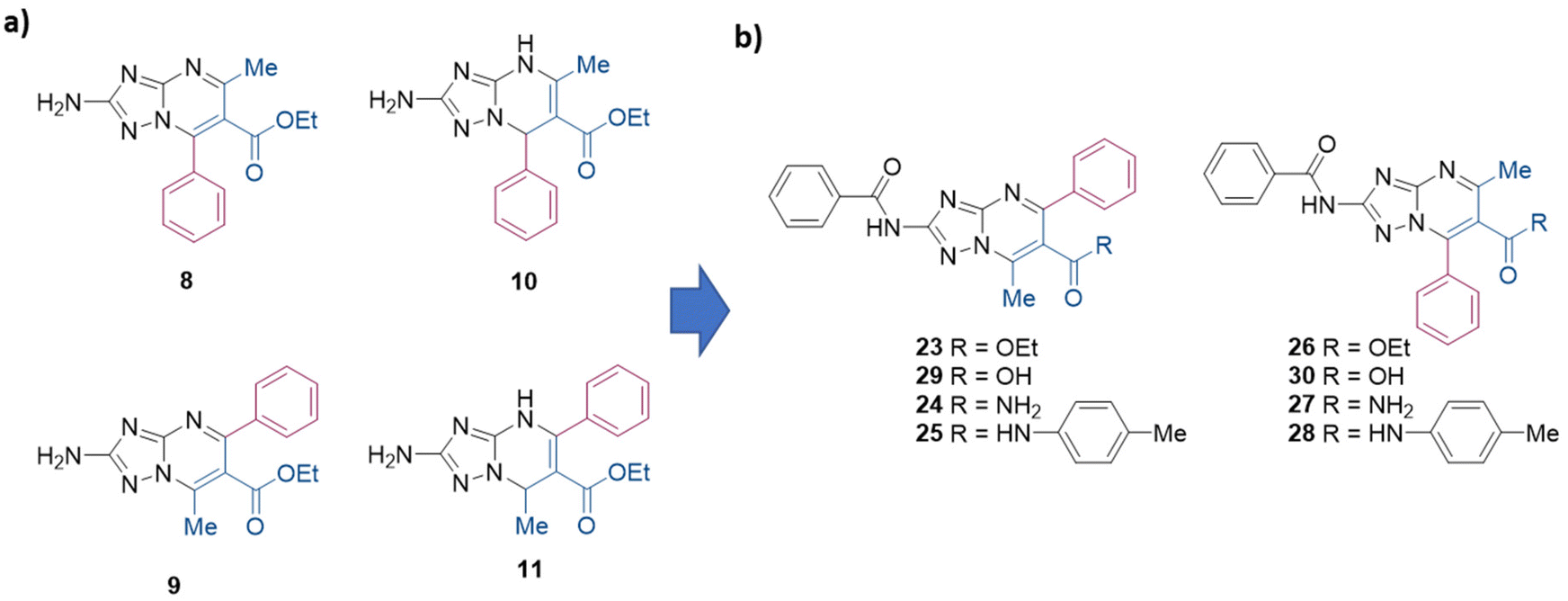 | ||
| Scheme 2 (a) Structures of TZP scaffolds 8–11 regioselectively synthesized in this work. (b) Successive target compounds synthesized and evaluated as antiviral agents. | ||
By taking advantage of the developed procedures, additional aromatic 2-amino-TZPs variously functionalized at the C-6 position were synthesized and used as key intermediates to be further functionalized at the C-2 position. Antiviral evaluation of the synthesized compounds (23–30, Scheme 2b) against IV, two flaviviruses (dengue and West Nile virus), and SARS-CoV-2 led to the identification of derivatives 25 and 26, which showed anti-flavivirus activity in the low micromolar range.
Results and discussion
Synthesis of ethyl 2-amino-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylates 8–11
Two procedures have been previously reported for the synthesis of non-oxidized 5-methyl-7-phenyl-TZP 10 by Chernyshev and coworkers (Schemes 3a and b).21,24 In the first procedure reported in 2007,21 3,5-diaminotriazole (12) (1 equiv.), benzaldehyde (13) (1 equiv.), and ethyl 3-oxobutanoate (14) (1 equiv.) were reacted in DMF at reflux for 30 min, furnishing a mixture of 10 and azomethine compound 15, which was treated with hydrazine hydrate to give 10 in 44% yield (Scheme 3a). In the successive procedure reported in 2017,24 the reaction of 13 and 14 in AcOH in the presence of piperidine furnished an intermediate bis-electrophile, which was then reacted with 12 yielding 10 in 71% yield (Scheme 3b). | ||
| Scheme 3 (a) and (b) Known procedures for the synthesis of 10. (b) Oxidation of 10 to aromatic TZP 8 performed in this work. (c) Synthesis of 11via the MCR of 3,5-diaminotriazole (12), benzaldehyde (13) and ethyl 3-oxobutanoate (14), and its oxidation to compound 9, respectively. *Isolated yield. | ||
Looking for a MCR allowing for the synthesis of compound 10 through a single step and a greener approach, we reacted 12 (1 equiv.), 13 (1 equiv.), and 14 (1 equiv.) in EtOH at reflux in the presence of citric acid (2.5 equiv.) (Scheme 3c) following a similar procedure previously reported for the synthesis of 1,4-dihydro-benzo[4,5]imidazo[1,2-a]pyrimidine analogues.25 After 5 h, the reaction furnished a mixture containing 10 only in traces, while, surprisingly, the main product (albeit obtained after purification in 16% yield) was isomer 11, as confirmed by 2D NMR (NOESY experiment), along with traces of another unknown compound (later characterized as the oxidized TZP analogue 9).
NMR signals and NOESY correlation for compounds 10 and 11 are shown in Fig. 2, and the superposition of NMR spectra and NOESY spectra is reported in Fig. S1–S4.† The values of the main 13C NMR signals are also reported in Table S1.† In the 1H NMR spectrum of compound 11, the signals of NH and H-7 are shifted upfield by 1.9 ppm and 0.5 ppm, respectively, with respect to those of isomer 10. Moreover, in the 13C NMR spectrum of compound 11, the signals of C-5 and C-6 are shifted downfield by 7.2 and 3.4 ppm, respectively, with respect to those of 10, while the signals of C-7 and CH3 carbon are shifted upfield by 6.6 and 3.2 ppm, respectively. In NOESY experiments, the singlet of NH of derivative 11 (8.55 ppm) correlated with multiplets (7.21–7.34 ppm) generated by the aromatic proton of the phenyl group at the C-5 position, while the singlet of NH (10.47 ppm) of 10 correlated with the singlet (2.37 ppm) of the methyl group at the C-5 position (violet arrows in Fig. 2a, and Fig. S3 and S4†). As a confirmation, the singlet of H-7 (5.94 ppm) of derivative 10 correlated with the multiplets (7.15–7.31 ppm) generated by the aromatic proton of the phenyl group at the C-7 position (violet arrow in Fig. 2a and S3†).
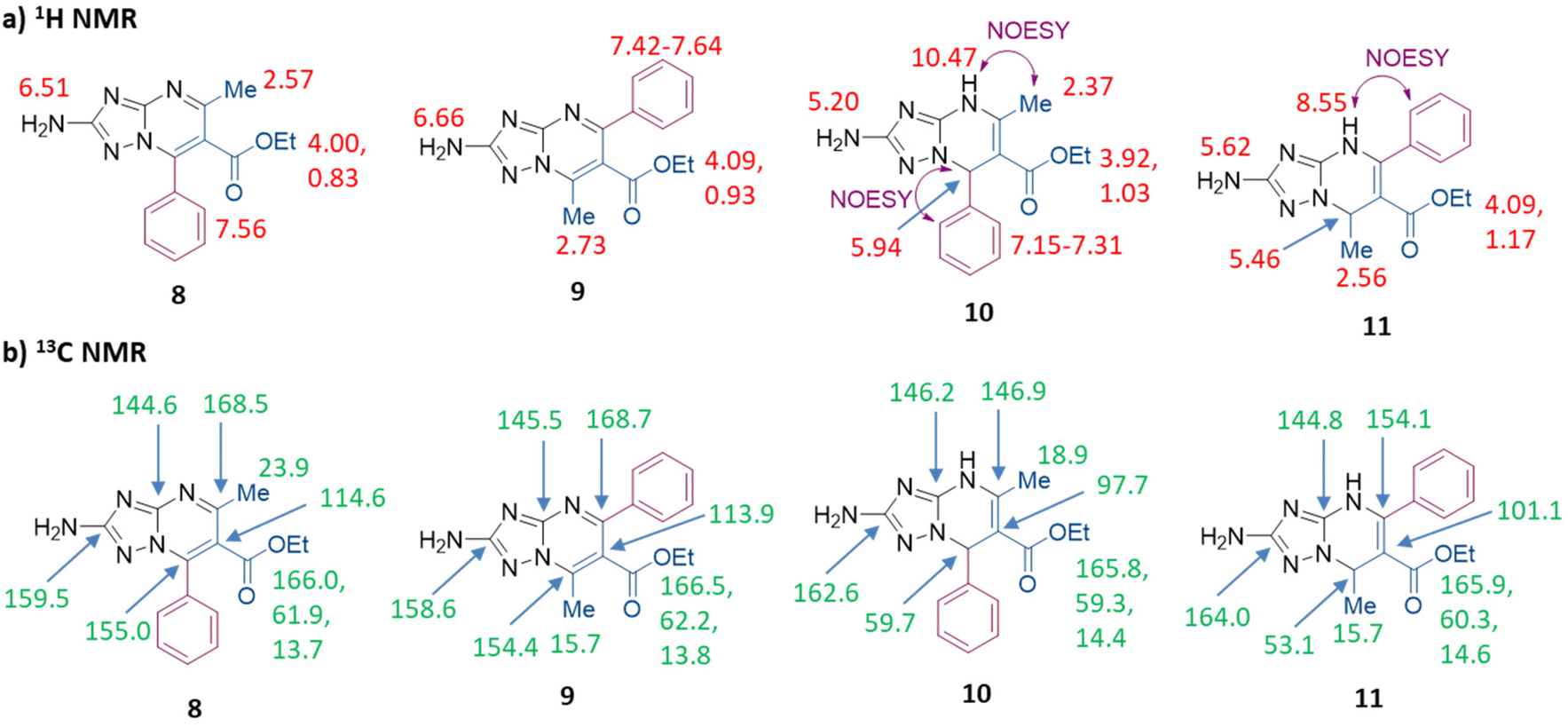 | ||
| Fig. 2 (a) 1H NMR and (b) 13C NMR spectral characteristics (chemical shifts δ, ppm) of compounds 8–11 in DMSO and key correlation in the NOESY spectra. | ||
At this point, compounds 10 (synthesized following the procedure reported in Scheme 3b) and 11 were oxidized by using NBS, furnishing compounds 8 and 9 in 48% and 26% yields, respectively (Scheme 3b and c). The NMR signals for compounds 8 and 9 are shown in Fig. 2 (the main 13C NMR signals are also reported in Table S1†) and the superposition of NMR spectra is reported in Fig. S5 and S6.† By comparing the 13C NMR spectrum of compound 9 to that of compound 8: (i) the signals of TZP carbons C-2, C-6 and C-7 of compound 9 are all slightly shifted upfield by 0.6–0.9 ppm (C-2 = 0.9 ppm, C-6 = 0.7 ppm, and C-7 = 0.6 ppm), (ii) the signals of C-3 and C-5 are slightly shifted downfield by 0.9 and 0.2 ppm, respectively, and (iii) the signal of CH3 carbon is shifted upfield by 8.2 ppm.
As mentioned previously, once the two oxidized isomers were obtained and characterized, we learned that the reaction reported in Scheme 3c furnished traces of oxidized compound 9, besides compounds 10 and 11. Based on the observation that these reaction conditions favoured the synthesis of 7-methyl-5-phenyl-TZP rather than 5-methyl-7-phenyl-TZP isomers, additional reaction conditions were further explored with the aim to regioselectively obtain the C-5 phenyl isomer derivatives 9 and 11. In this phase, efforts were also focused on the development of a HPLC method that, through the retention time (tR), allowed us to quickly detect isomers 8–11 and their ratios (Fig. 3).
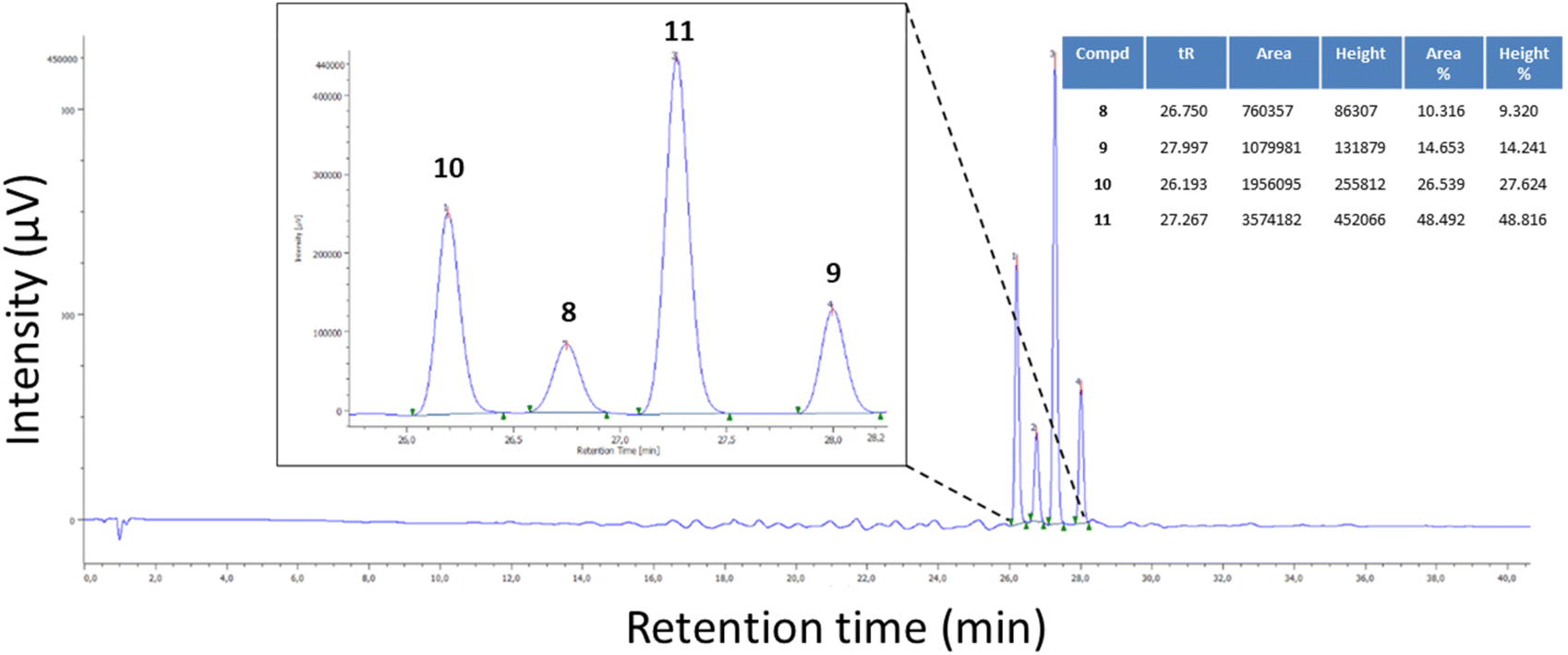 | ||
Fig. 3 Example of HPLC chromatogram of a mixture of compounds 8–11. HPLC method: RP-C18; flow: 0.4 mL min−1; 0.1% formic acid in H2O 100% to 0.1% formic acid in H2O 50%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3CN 50% in 40 min. :CH3CN 50% in 40 min. | ||
The reaction of 12 (1 equiv.), 13 (1 equiv.), and 14 (1 equiv.) in EtOH at reflux in the presence of citric acid (2.5 equiv.) (Table S2,† entry 1) was repeated by increasing the equiv. of citric acid (5 equiv., entry 2) or compound 13 (1.5 equiv., entry 3), but no significant changes in the outcome of the reaction were noted. Nevertheless, only by decreasing the reaction time from 5 h to 3.5 h (entry 4), compound 11 was regioselectively obtained in 83% yield (Table 1, entry a). Of note, the purification of compound 11 was performed by trituration of the solid in Et2O without the involvement of chromatography. Noteworthy is the propensity of compound 11, when solubilized in different organic solvents such as EtOAc, CHCl3, or CHCl3/MeOH, to spontaneously oxidize, furnishing derivative 9 in a few days at room temperature (rt), albeit not completely (about 50%). All attempts to completely convert derivative 11 into derivative 9 (solutions of 11 in different organic solvents were stirred overnight at rt, under light, or gently heated) failed. In contrast, it was stable when stored as a pure powder over time.
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Ratio 12:13:14 |
Catalyst (equiv.) | T° | Time (h) | % yield of 8–11 |
| a The reaction was performed on a 1.0 mmol scale of 12 in 3 mL of solvent. b Isolated yield. c H2O2 (1 mL) was added after 12 h. | ||||||
| a | EtOH | 1:1:1 |
Citric acid (2.5) | Reflux | 3.5 | 11 in 83%b |
| b | AcOH | 1:3:1 |
— | 60 °C | 9 | 9 in 55%b |
| c | TBMA MsO | 1:1:1 |
— | 120 °C | 24 | 10 in 75%b |
| d | BMIM MsO | 1:2:1 |
H2O2c |
120 °C | 24 | 8 in 40%b |
Searching for alternative procedures for the synthesis of compound 9 in higher yields, the reaction of 12 (1 equiv.), 13 (1 equiv.), and 14 (1 equiv.) was performed under microwave (μw) irradiation or in different solvents and/or by using different acid catalysts (Table S2,† entries 5–9). Overall, many spots were observed by TLC due to the formation of several side products. Of note, the reaction performed in THF in the presence of para-toluenesulfonic acid (PTSA) as the catalyst at reflux (entry 6) showed a certain degree of regioselectivity towards C-5 phenyl isomers, but after 24 h, not enough conversion from 11 to 9 was observed. Interestingly, the reaction performed in glacial acetic acid at reflux under nitrogen (entry 9) furnished compound 9 after 6 h with a certain degree of selectivity, even though several side products were formed. After purification, compound 9 was obtained in 21% yield, highlighting that the aromatic C-5 phenyl isomer can be directly achieved through a Biginelli-like MCR as a major compound.
Prompted by these results, the reaction was repeated in glacial acetic acid at reflux but in an open flask (Table S3†) in order to promote the oxidation reaction, analogously to procedures previously reported by us (Scheme 1b).8,11 In these reactions, the effects of the reaction time, the temperature, and the equiv. of each of the starting compounds were studied.
As expected, performing the reaction in an open flask favoured the oxidation of 11 into 9, accompanied by increased regioselectivity of the reaction. In particular, the best results were obtained by reacting 12 (1 equiv.), 13 (1 equiv.), and 14 (1 equiv.) in AcOH at 120 °C for 6 h (Table S3,† entry 2), with a percentage ratio of 89% for 9 and a yield of 37%. A shorter time (entry 1) did not permit a complete conversion of 11 into 9, while at longer times (entries 3 and 4), the formation of side products increased over time. Changing the equiv. of the starting materials (entries 7–11) was in general detrimental for the regioselectivity as well as for the efficiency of the reaction, owing to the excessive presence of side products. Very minor tarring was observed instead when performing the reaction at 60 °C (entries 5 and 6), although it was detrimental for the oxidation of 11 into 9.
Based on these results, we attempted again to perform the reaction under nitrogen (Table S4†). Thus, the reaction was repeated by evaluating the effect of the equiv. of the starting compounds, temperature, time, and/or the presence of a catalyst. The use of 2 equiv. of diaminotriazole 12 (entry 1) led to reaction tarring and a decrease in regioselectivity. On the other hand, the increase of benzaldehyde 13 (2 equiv., entries 2 and 3) furnished compound 9 as the major isomer. Of note, the best results were observed when performing the reaction at reflux (entry 2), with 9 being present with a percentage ratio of 77% (63% when the temperature was 60 °C, entry 3). According to the peak ratio, trituration using Et2O/EtOH gave compound 9 in 51% (entry 2) and 33% (entry 3) yields.
A further increase of benzaldehyde 13 (3 equiv., entries 4–6) led to good results only by performing the reaction at 60 °C, obtaining compound 9 after 9 h with a peak ratio of 78% and an isolated yield of 55% after purification by chromatography (Table 1, entry b); higher temperatures led to a decrease in regioselectivity. On the other hand, using 2 equiv. of 14 (entries 7–9), a higher peak ratio for 9 was obtained by performing the reaction at reflux, while a large amount of 11 was observed at lower temperatures. Although a good percentage peak ratio (83%) was obtained, the conditions used for entry 9 led to several side products, likely due to the reflux temperature, as demonstrated by the low reaction yield (23%). Analogous results were obtained by combining 3 equiv. of 13 and 2 equiv. of 14 at 60 °C (entry 10), which led to a good percentage peak ratio of compound 9 (85%), but a poor isolated yield was obtained after purification (36%) due to the presence of many side products.
At this point, the best reaction conditions identified in entry 4 (Table S4†) were used to perform the reaction under μw irradiation in order to decrease the reaction time and side products and improve the efficiency (entry 11). After 2.5 h under μw irradiation, many side products and the significant presence of the C-5 phenyl dihydro TZP derivative 11 were noticed. Thus, the reaction was repeated by adding, after 2 h at 60 °C under μw, H2O2 (1 mL) and continuing the reaction in an open flask at 110 °C (entry 12), in order to promote oxidation of 11 to 9. As expected, the desired oxidation occurred since the peak ratio of compound 9 was 72%, but the reaction showed a lot of side products leading to 9 in a very low yield (12%), after purification by chromatography. Analogously, no interesting results were achieved when performing the reaction in AcOH at 60 °C in the presence of I2 (entry 13), a catalyst used in the literature to promote cyclocondensation and oxidation in Biginelli MCRs.26,27
Thus, the best reaction conditions for the synthesis of compound 11 are reacting 12 (1 equiv.), 13 (1 equiv.), and 14 (1 equiv.) in the presence of citric acid (2.5 equiv.) in EtOH at reflux for 3.5 h (Table 1, entry a). Through this one-step procedure, compound 11 was regioselectively obtained in 83% yield. On the other hand, the best reaction conditions for the synthesis of 9 are reacting 12 (1 equiv.), 13 (3 equiv.) and 14 (1 equiv.) in acetic acid at 60 °C under nitrogen for 9 h (Table 1, entry b). Through this one-step procedure, compound 9 was regioselectively synthesized in 55% yield. Although the procedure for the synthesis of 9 showed a moderate yield, it was more efficient than the two-step procedure entailing the cyclocondensation reaction (83% yield) and the successive oxidation reaction (26% yield), showing a 22% overall yield. To the best of our knowledge, these two procedures are the first Biginelli-like MCRs in which the reaction of an aminotriazole, an aldehyde, and a β-ketoester leads to the formation of ethyl 2-amino-7-methyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate compounds.
Focusing our attention on the synthesis of ethyl 2-amino-5-methyl-7-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate (8) and searching for a procedure entailing a single step and without the use of oxidative agents, we evaluated some minor changes in the reaction conditions reported by Chernyshev and co-workers in Scheme 3a. Thus, DMF was first replaced with N-methyl-2-pyrrolidone (NMP) or pyridine, but only a complex mixture after 24 h was observed (data not shown). Then, the combination of the use of DMF with 1 equiv. of Et3N or 1,4-dioxane with 1 equiv. of K2CO3 led in both cases to the formation of the azomethine derivative 15 as the main product coupled with an incomplete consumption of the starting material 12 after 24 h (data not shown). The use of additional solvents was strongly affected by the limited solubility of the starting material 12 in most of the common solvents used for chemical reactions. At this point, also considering environmental sustainability, the use of ionic liquids (ILs) as the solvent was studied (Table S5†).
Initially, two ILs, i.e., 1-butyl-3-methylidazolium mesylate (BMIM-MsO) and N,N,N-tributylmethylammonium mesylate (TBMA-MsO)28 were used with 1 equiv. of each starting material (12, 13, and 14), and reactions were performed at 120 °C in an open flask up to the disappearance of 12 observed by TLC. Interestingly, on using BMIM-MsO (entry 1), after 48 h, the desired compound 8 was formed in a high percentage as monitored by HPLC (93%). However, after the aqueous reaction work-up and the filtration of the solid, compound 8 was obtained in 30% yield, even if no additional side products or remaining starting material was noticed. On the other hand, the same reaction performed with TBMA-MsO (entry 2) led to the disappearance of 12 after 24 h and the formation of the non-aromatic isomer 10, without showing any traces of compound 8. After the aqueous reaction work-up, compound 10 was obtained as a pure solid in 75% yield (Table 1, entry c). Therefore, the procedure to regioselectively obtain the dihydro-TZP analogue 10 with a high yield through a one-step, three-component reaction and without any purification step was identified.
Since BMIM-MsO allowed us to isolate the aromatic isomer 8 in a modest yield (30%), we considered the use of a similar IL having a different counterion. Thus, the reaction was repeated using the same conditions as entry 1 in BMIM-tetrafluoroborate (BMIM-TFB) (entry 3), but a loss of regioselectivity was observed, thus obtaining compound 8 in 25% yield. At this point, the effect of 2 equiv. of benzaldehyde 13 when using both BMIM-MsO (entry 4) and TBMA-MsO (entry 5) was evaluated. In both cases, after 12 h, 12 disappeared but the azomethine derivative 15 formed as the main product. Based on the observation that oxidation of compound 10 into 8 was accomplished in a low yield (48%, Scheme 3b) and that protection of the amino group led to improved efficiency of the oxidation in analogous TZP compounds,29 we attempted to oxidize derivative 15 and then deprotect the amino group in the same reaction to achieve compound 8. Thus, the reaction in three different ILs (entries 6–8) was carried out using 2 equiv. of benzaldehyde 13 and adding a mild and green oxidizing agent such as H2O2 (1 mL) after 12 h, at which time the disappearance of diamino-triazole 12 was observed. To our surprise, following the initial formation of the azomethine derivative 15, we did not observe its oxidation but the formation of the aromatic isomer 8 as the main product in all three reactions was observed. The highest regioselectivity was shown by the reaction performed in BMIM-MsO (entry 6), which, after the aqueous work-up, yielded compound 8 as a pure solid in 40% yield without any purification step (Table 1, entry d). On the other hand, the work-up of the other two reactions (entries 7 and 8) led to very dirty solids.
Based on these results, further studies were undertaken on the role of the equiv. of starting materials in the reaction of entry 6. Thus, the reaction in BMIM-MsO was repeated by using different ratios of the starting materials 12, 13 and 14 (i.e., 1:3:1; 1:2:2; 1:1:2; 2:1:1; 2:1:2) and adding H2O2 after the disappearance of the limiting starting material 12. Although the formation of compound 8 as the main product was observed, several side products characterized these reactions that led to the isolation of non-pure solids (not shown).
Finally, the role of the solvent was investigated by repeating the reaction of entry 6 in other ILs and also in deep eutectic solvents (DESs) (Table S6†). In particular, the reaction of 12 (1 equiv.), 13 (2 equiv.), and 14 (1 equiv.) was performed at 120 °C in six different ILs, i.e., TBMA tosylate (TsO), tributylmethylphosphonium (TBMP) MsO, 1,3-dimethylimidazolium (MMIM) TsO, tetrabutyl ammonium (TBA) TsO, TBA bromide, and TBA-MsO (entries 1–6), and in three different DESs, i.e., ethylene glycol/trimethylglycine (EG/TMG), glycolic acid/trimethylglycine (Gly/TMG), and urea/choline chloride (U/ChCl) (entries 7–9). In all the reactions, H2O2 (1 mL) was added after the disappearance of the starting material 12 (6, 20, or 24 h). With the exception of the reaction performed in MMIM-TsO (entry 3), which favoured the formation of compound 15 as the main product, all the other reactions performed in ILs furnished compound 8 as the main product. Nevertheless, all the reactions were characterized by extensive tarring and/or very low efficiency. Analogous results were obtained by the reactions in DESs, which were characterized by the formation of several side products, discouraging the successive work-up. Thus, the best reaction conditions for the synthesis of compound 8 are reacting 12 (1 equiv.), 13 (2 equiv.) and 14 (1 equiv.) in BMIM-MsO at 120 °C for 24 h and adding H2O2 (1 mL) after 12 h (Table 1, entry d). Through this one-step procedure, compound 8 was regioselectively obtained in 40% yield. Even though compound 8 was obtained in a moderate yield, this procedure is the first example of the Biginelli MCR in which the reaction between an aminotriazole, an aldehyde and a β-ketoester leads to the formation of an ethyl 2-amino-5-methyl-7-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate compound. Notably, this one-step MCR procedure having a 40% yield represents to date the best method to obtain oxidized 8, being more convenient in terms of yield, time, use of a mild oxidizing agent, and a green solvent, with respect to the two-step procedure (formation of 10 and subsequent oxidation, Scheme 3a) exhibiting a 34% overall yield. In addition, by changing some conditions, (i.e., IL, ratio, absence of H2O2, and time), for the first time, a novel method to regioselectively obtain the non-aromatic isomer 10 with an improved yield (75% vs. 71%) through a one-step MCR, and by using a greener solvent than DMF was also identified (Table 1, entry c).
Plausible pathways accounting for the formation of compounds 10 and 11 through the reaction of 12, 13, and 14 are speculatively reported and briefly described in Scheme S1.† In particular, analogously to the three plausible reaction mechanisms proposed for the classic Biginelli reaction,30 compounds 10 and 11 could be obtained through an imine route (Scheme S1a and d†), an enamine route (Scheme S1b and e†), or a Knoevenagel route (Scheme S1c and f†). While in the imine and enamine routes, the initial reaction plays a key role in the regioselectivity of the reaction, in the Knoevenagel route, the nucleophilic attack of 3,5-diaminotriazole on the adduct is crucial in driving the regioselective formation of 10 or 11.
Although a deep investigation of the mechanisms involved in this reaction is beyond the scope of this study, the imine and Knoevenagel routes, reported in Scheme 4, were hypothesized to be most likely to occur for the formation of compounds 11 and 10, respectively. The high rate at which compounds 10 and 11 are formed during the reaction impaired the isolation or TLC monitoring of intermediates. Nevertheless, previous studies provided important information. Indeed, we previously noticed a higher nucleophilicity of the C(3) amino group than the N(2) of 12 under both acidic and basic conditions,11 advising one to discard the enamine route for the synthesis of 11 (Scheme S1b†) and the imine route for the synthesis of 10 (Scheme S1d†). Regarding compound 11, we also discarded the Knoevenagel route (Scheme S1c†) based on previous studies suggesting that when reacting with α,β-enone such as Knoevenagel's adduct, the triazole C(3) amino group seems to undergo direct addition at the carbonyl carbon instead of conjugate addition at the β-carbon.8,11 Thus, in agreement with the most accredited mechanism proposed for the classic Biginelli reaction (which analogously occurs under acidic conditions),31 the imine route was hypothesized to be most likely to occur for the regioselective synthesis of 11 (Scheme 4).
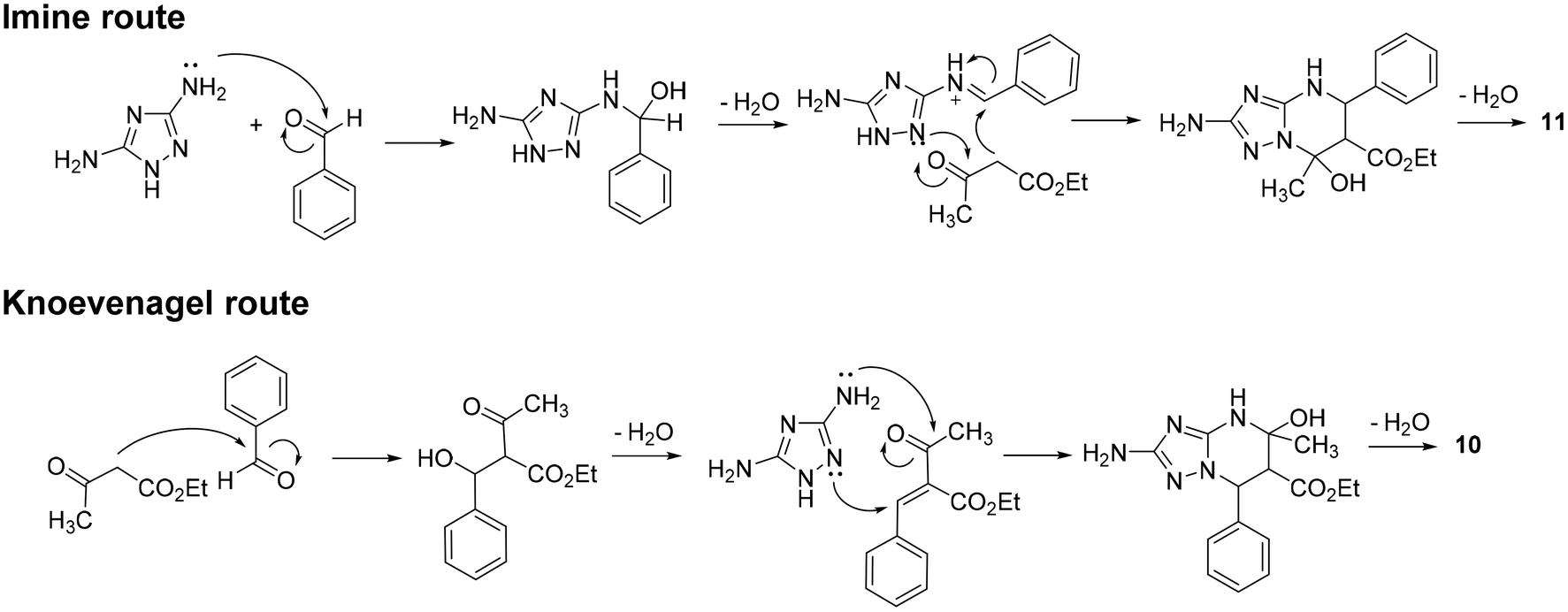 | ||
| Scheme 4 Plausible reaction mechanisms for the formation of 11 and 10. | ||
Focusing on compound 10, although both the enamine and Knoevenagel routes are possible (Scheme S1e and f†), the latter was the most plausible for us, based on two pieces of information. Firstly, as reported above, the synthesis of compound 10 was reported via a two-step procedure involving the initial formation of the Knoevenagel intermediate (Scheme 3b).24 Secondly, a variation of the classic Biginelli reaction, named Atwal modification,32,33 envisages the formation of a Knoevenagel intermediate in a neutral or slightly basic environment (analogously to our conditions in the IL) before the addition of urea to form the desired Biginelli compound. To investigate if, analogously, the formation of 10 occurs via the Knoevenagel route, we performed two parallel reactions by reacting ethyl 3-oxobutanoate 14 (1 equiv.) with benzaldehyde 13 (1 equiv.) or 3,5-diaminotriazole 12 (1 equiv.) in TBMA-MsO at 110 °C and, after 2 h, we added 12 or 13, respectively. Only the reaction allowing for the initial generation of Knoevenagel's adduct proceeds towards the formation of 10 after the addition of 12, thus supporting the hypothesis that the Knoevenagel route could be most likely to occur for the regioselective synthesis of compound 10 (Scheme 4). The regioselectivity of the reaction could be driven, as reported above, by the propensity of the more nucleophilic triazole C(3) amino group to undergo direct addition to the carbonyl carbon of the adduct instead of conjugate addition to the β-carbon.
Synthesis of target compounds 23–30
As mentioned above, for some years, we have been involved in the identification of TZP-based compounds as the inhibitors of RNA viruses, mainly as anti-IV agents by inhibiting the PA–PB1 subunits interaction of viral RdRP. In this study, in order to gain structure–activity relationship insights for this class of compounds, we aimed at studying the role of a substituent at the C-6 position. In particular, to prepare the designed compounds that need to be evaluated for antiviral activity, we exploited scaffolds 8 and 9, characterized by C-6 ethyl ester, and prepared an additional set of intermediates having carboxamide, p-tolyl-carboxamide, and carboxylic acid at the C-6 position.Firstly, we focused our attention on the synthesis of carboxamide C-6 functionalized scaffolds 16–19 (Schemes 5 and 6) by exploiting the reaction conditions identified for the synthesis of ester derivatives 8 and 9. Accordingly, the reactions were repeated by replacing ethyl 3-oxobutanoate (14) with 3-oxobutanamide (20) or N-acetoacetyl-p-toluidine (21). The isomers have been distinguished by NMR based on the chemical shifts of the pyrimidine methyl carbon of aromatic compounds, appearing at 23–24 ppm and 14–16 ppm for the 5-methyl-7-phenyl and 7-methyl-5-phenyl isomers, respectively (Table S1†).
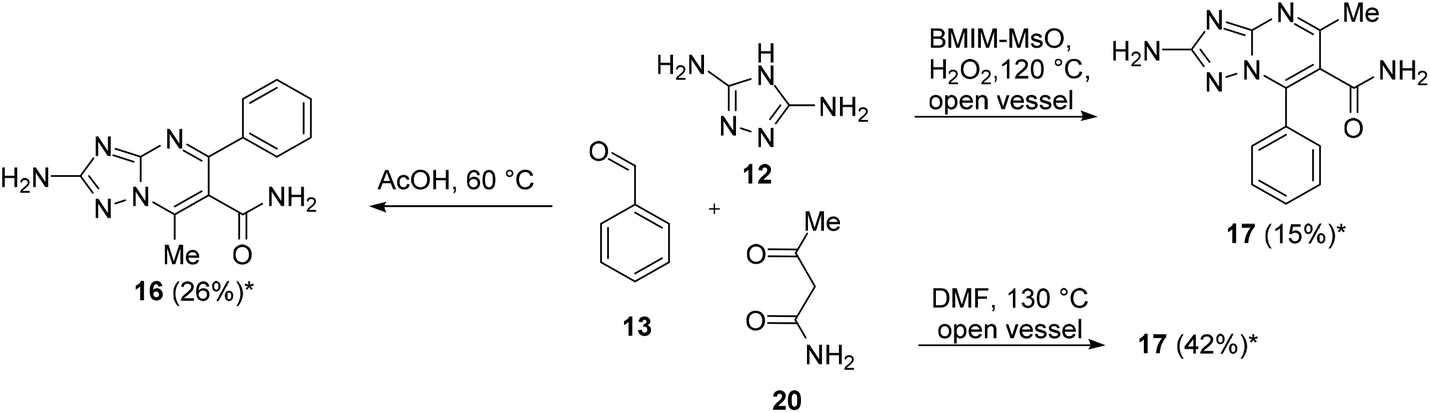 | ||
| Scheme 5 Chemical procedures for the synthesis of 2-amino-TZP compounds 16 and 17. *Isolated yield. | ||
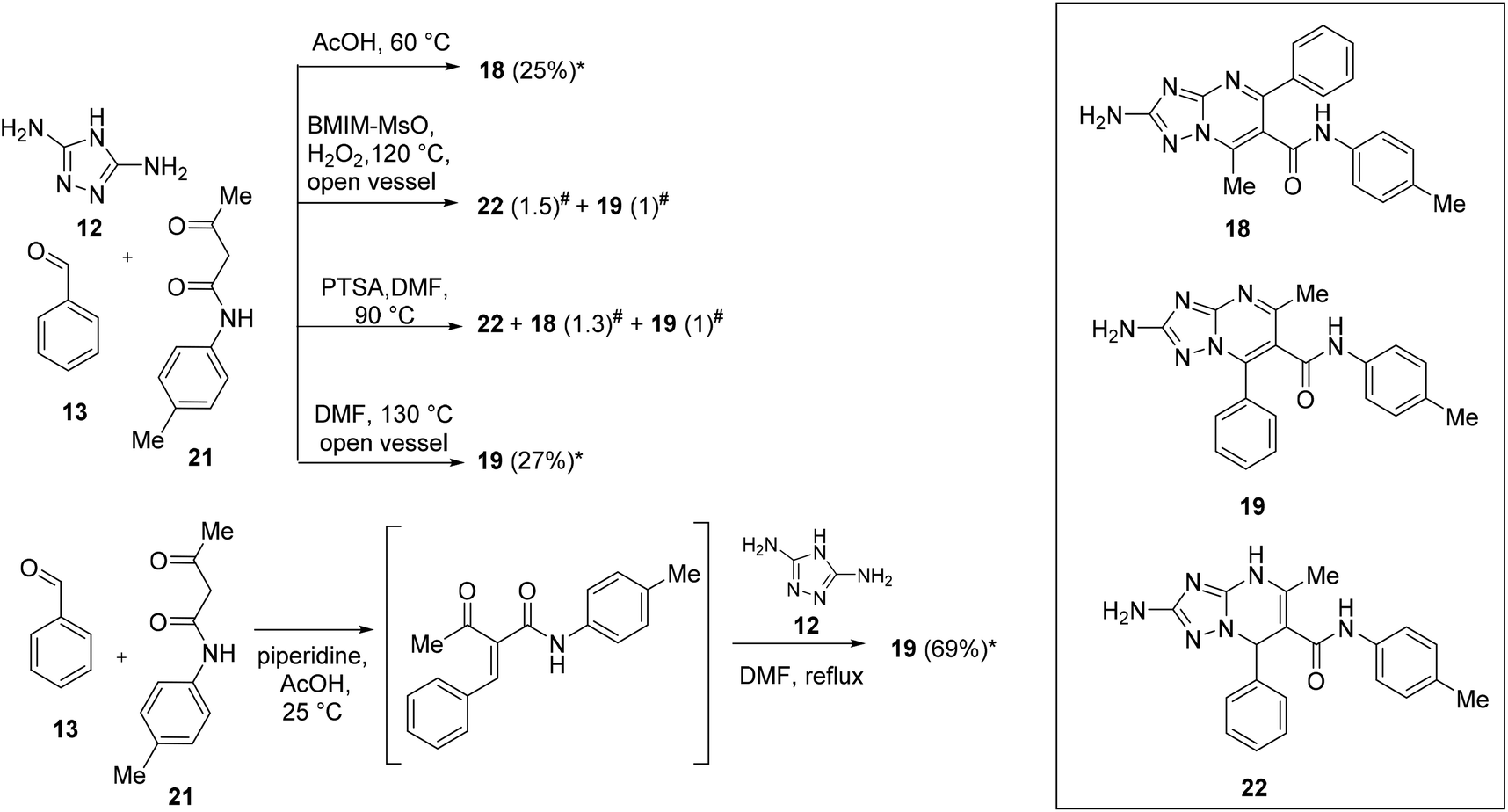 | ||
| Scheme 6 Chemical procedures for the synthesis of 2-amino-TZP compounds 18 and 19. *Isolated yield. # Ratios determined by NMR on the crude product. | ||
The reaction of 12 (1 equiv.), 13 (3 equiv.), and 20 (1 equiv.) in AcOH at 60 °C furnished compound 16 in 26% yield (Scheme 5). On the other hand, the reaction of 12 (1 equiv.), 13 (2 equiv.), and 20 (1 equiv.) in BMIM-MsO in an open flask at 120 °C with H2O2 after the disappearance of 12 furnished compound 17 only in 15% yield (Scheme 5). As an alternative reaction, we attempted to react 12 (1 equiv.), 13 (2 equiv.), and 20 (1 equiv.) in DMF at 130 °C in an open vessel to favour oxidation. Of note, compound 17 was obtained in 42% yield (Scheme 5).
Turning our attention to the synthesis of isomers 18 and 19 (Scheme 6), the reaction of 12, 13, and 21 in AcOH at 60 °C for 15 h yielded compound 18 in 25% yield. On the other hand, the reaction in BMIM-MsO furnished a mixture of 19 and non-oxidized analogue 22 in a ratio of 1:1.5. Thus, the synthesis of compound 19 was attempted by applying a synthetic procedure reported in the literature, in which the reaction of an equimolar mixture of 12, 13, and 21 in DMF in the presence of PTSA (0.05 equiv.) at 90 °C for 16 h furnished the target compound in 58% yield.34 However, when the reaction was performed under these conditions, a mixture containing several side products was obtained (i.e., the non-oxidized compound 22, and both the oxidized isomers 18 and 19 in a ratio of 1.3:1). Looking at the 13C NMR spectrum reported by the authors for the synthesized compound,34 the chemical shift of pyrimidine methyl carbon appears at 15.49 ppm, leading to a hypothesis that the compound obtained as the main product by the authors was the 7-methyl-5-phenyl isomer 18 and not the 5-methyl-7-phenyl isomer 19. Of note, in the manuscript, the authors also reported 22 compounds characterized by different substituents on the C-6 amide moiety, of which the chemical shift of the pyrimidine methyl carbon appears at ∼15.5 ppm for 13 compounds and ∼23 ppm for 9 compounds, suggesting that the 7-phenyl isomer was not always obtained as the major product as declared by the authors. At this point, the same conditions exploited to obtain compound 17 were used to synthesize compound 19, which was obtained in a low yield (27%). Thus, we attempted a one-pot two-step reaction entailing the reaction of 13 and 21 in AcOH and piperidine at room temperature followed by the addition of 12 and DMF leading the temperature at reflux. Under these conditions, oxidized compound 19 was directly obtained in 69% yield.
The synthesized scaffolds 8, 9, and 16–19 were reacted with benzoyl chloride in pyridine providing target derivatives 23–28 (Scheme 7). Then, to determine the effect of carboxylic acid at the C-6 position of the TZP core, compounds 23 and 26 were hydrolyzed in the presence of LiOH in dioxane, furnishing acid derivatives 29 and 30, respectively.
 | ||
| Scheme 7 Reagents and conditions: (i) benzoyl chloride, pyridine, from 0 °C to rt; (ii) LiOH, 1,4-dioxane, 45 or 60 °C. | ||
The synthesized compounds 23–30 were evaluated for their ability to inhibit the physical interaction between PA and PB1 subunits by an ELISA-based interaction assay (Table 2).
| Compd | R6 | ELISA PA-PB1 interaction assay | PRA in MDCK cells | Cytotoxicity (MTT assay) in MDCK cells |
|---|---|---|---|---|
| IC50,a μM | EC50,b μM | CC50,c μM | ||
|
a Activity of the compounds in ELISA-based PA–PB1 interaction assays. The IC50 value represents the compound concentration that reduces by 50% the interaction between PA and PB1 subunits.
b Activity of the compounds in plaque reduction assays with the IV A/PR/8/34 strain. The EC50 value represents the compound concentration that inhibits 50% of plaque formation.
c Cytotoxicity of the compounds in MTT assays. The CC50 value represents the compound concentration that causes a 50% decrease of cell viability. All the reported values represent the means ± SD of data derived from at least three independent experiments in duplicate. RBV=ribavirin.
|
||||
|
||||
| 23 | CO2Et | 132 ± 22 | >100 | >150 |
| 29 | CO2H | >200 | >100 | >250 |
| 24 | CONH2 | 109 ± 23 | >100 | >250 |
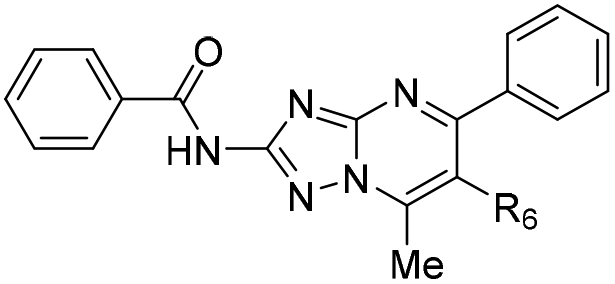
| 25 |

|
>200 | >20 | 123 ± 74 |
|
||||
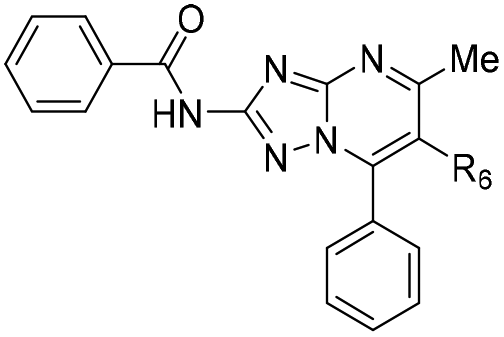
| 26 | CO2Et | >200 | >100 | >250 |
| 30 | CO2H | >200 | >100 | >250 |
| 27 | CONH2 | 5.5 ± 1.4 | >100 | >250 |
| 28 |
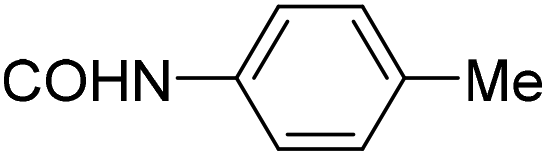
|
>200 | >100 | >250 |
| Tat-PB1 1–15 peptide | 35.2 ± 4.7 | 41.5 ± 5.2 | >100 | |
| RBV | 26.6 ± 5.2 | >250 | ||
In parallel, for all the synthesized compounds, the anti-IAV activity was tested by plaque reduction assays (PRA) in Madin–Darby canine kidney (MDCK) cells infected with a reference IV, the A/PR/8/34 strain. Ribavirin (RBV), a known broad-spectrum inhibitor of RNA virus polymerases, was also included as a positive control. To exclude that the observed antiviral activities could be due to cytotoxicity on the target cells, the compounds were also tested by MTT assays in MDCK cells.
As shown in Table 2, only compound 27 showed the ability to interfere with the PA–PB1 interaction with an IC50 value in the low μM range. However, unfortunately, all the tested compounds were devoid of anti-IV activity. In order to extend the potential antiviral activity of the newly synthesized compounds 23–30, cell-based antiviral assays were performed against additional RNA viruses, i.e., DENV, WNV, and SARS-CoV-2 (Tables 3 and S7†). Dengue virus (DENV), consisting of 4 serotypes (DENV-1–4), is a mosquito-borne flavivirus and is considered a major public health threat in many countries with no approved antiviral therapeutics available.35 DENV causes about 100–400 million infections each year, and led to 36100 deaths only in 2019.36,37 Similar to DENV, WNV is also a zoonotic, mosquito-borne flavivirus, containing a positive-sense, single-stranded RNA genome. Occurring in about 1% of human WNV infections, the neuroinvasive form of the infection represents the most severe form of the disease leading to meningitis, encephalitis, and acute flaccid paralysis/poliomyelitis.38 Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19, is an enveloped, positive-sense, single-stranded RNA virus belonging to the genus Betacoronavirus.39 Although we can consider the pandemic over, SARS-CoV-2 variants may continue to adapt in the human population for years or decades.40
| Compd | Anti-DENV-2 activity (Huh7 cells) EC50,a μM | Anti-WNV activity (Huh7 cells) EC50,a μM | Cytotoxicity (Huh7 cells) CC50,b μM | Anti-SARS-CoV-2 activity (A549 cells) EC50,c μM | Cytotoxicity (A549 cells) CC50,b μM |
|---|---|---|---|---|---|
| a Activity of the compounds as determined by the immunodetection assay. The EC50 value represents the compound concentration that reduces by 50% the expression of flavivirus envelope proteins in Huh7 cells infected with DENV or WNV. All the reported values represent the means ± SD of data derived from at least two independent experiments in duplicate. b Cytotoxicity of the compounds as determined by Cell Titer assay in A549 and Huh cell lines. The CC50 value represents the compound concentration that causes a 50% decrease of cell viability. c Activity of the compounds as determined by Cell Titer assay. The EC50 value represents the compound concentration that reduces by 50% the cytopathic effect in A549 cells infected with SARS-CoV-2. All the reported values represent the means ± SD of data derived from at least two independent experiments in duplicate. d NA = not active. NRM = nirmatrelvir, SOF = sofosbuvir. | |||||
| 25 | 4.3 ± 1.5 | 6.7 ± 3.7 | 18.4 ± 3.5 | NAd | 9.8 |
| 26 | 14.1 ± 4.1 | 19.3 ± 1.4 | 141 ± 1 | NA | 99.9 |
| NRM | — | — | — | 0.066 ± 0.007 | 36 |
| SOF | 8.1 ± 1.1 | 5.3 ± 2.5 | >243 | >243 | >243 |
The anti-DENV and anti-WNV activities were evaluated by a direct yield reduction assay in Huh7 cells infected with the New Guinea C DENV serotype 2 strain and WNV lineage 1 (Italy/2009) strain, respectively. Sofosbuvir (SOF) was used as a reference compound. Anti-SARS-CoV-2 activity was evaluated by a direct yield reduction assay in A549 cells infected with the SARS-CoV-2 strain belonging to lineage B.1. Nirmatrelvir (NRM) was used as a reference compound. In parallel, the cytotoxicity of compounds was determined by CellTiter-Glo 2.0 Luminescent Cell Viability Assay in Huh7 and A549 cells.
As shown in Table S7,† all the compounds were devoid of anti-SARS-CoV-2 activity and most of the compounds did not show anti-flavivirus activity. On the other hand, compounds 25 and 26 stood out as anti-flavivirus agents (Table 3). In particular, compound 25 showed EC50 values of 4.3 and 6.7 μM against DENV-2 and WNV, respectively, albeit it also showed certain cytotoxicity (CC50 = 18.4 μM). Instead, with EC50 values of 14.1 and 19.3 μM against DENV-2 and WNV, respectively, compound 26 was slightly less active but also markedly less toxic (CC50 = 141.1 μM), thus showing a promising selectivity index.
Conclusions
In this work, four facile one-step Biginelli-like MCRs for the regioselective synthesis of ethyl 2-amino-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylates were developed. The regioselectivity of the procedures was achieved by switching from an acidic environment, which allowed us to obtain ethyl 2-amino-7-methyl-5-phenyl-TZP-6-carboxylate 9 and its 4,7-dihydro analogue 11, to the use of ILs as a solvent, which resulted in ethyl 2-amino-5-methyl-7-phenyl-TZP-6-carboxylate 8 and its 4,7-dihydro analogue 10. In particular, aromatic derivative 9 was obtained in 55% yield by using AcOH as a solvent, while dihydro-TZP derivative 11 was synthesized in 83% yield under mild acidic conditions with citric acid in EtOH. On the other hand, different ILs were used to selectively obtain compound 10 in 75% yield and aromatic analogue 8 in 40% yield, with the in situ oxidation reaction prompted by performing the reaction in an open flask and adding H2O2.Although the two aromatic isomers 8 and 9 were not obtained in high yields, the two procedures represent the first examples of Biginelli-like MCRs in which aromatic TZP compounds have been regioselectively obtained; moreover, they were more efficient than the two-step procedure entailing the cyclocondensation reaction and the successive oxidation reaction.
The reaction in AcOH was also suitable to obtain aromatic 2-amino-7-methyl-5-phenyl-TZPs modified at the C-6 position, although not efficiently (about 25% yield). On the other hand, the reaction in the IL was not suitable to obtain C-6-modified aromatic 2-amino-5-methyl-7-phenyl-TZPs, which however, were obtained through two different chemical procedures in 42% and 69% yields.
In conclusion, it was evident that the electronic properties of the C-6 substituent strongly affect the reactivity of the reagent with an active methylene group needed to carry out the Biginelli-like MCR. Therefore, further efforts should be directed to the identification of reaction conditions compatible with the use of reactants as different as possible in order to extend the use of this MCR for the synthesis of several variously functionalized TZP compounds.
Regarding the biological activity of the new TZP-based compounds reported in this article, antiviral activities were evaluated against RNA viruses, i.e., IV, DENV-2, WNV, and SARS-CoV-2. Compounds 25 and 26 showed the ability to inhibit the viral replication of DENV-2 and WNV at concentrations in the low micromolar range, suggesting the use of the TZP scaffold in the discovery of anti-flavivirus agents. Indeed, these results are of particular interest since these compounds are the first TZP-based derivatives showing anti-flavivirus activity, thus paving the way for the design of new analogues.
Experimental
General chemistry
Commercially available starting materials, reagents, and solvents were used as supplied. All reactions were routinely monitored by TLC on silica gel 60F254 (Merck) and visualized using UV or iodine. Flash column chromatography was performed on Merck silica gel 60 (mesh 230–400). After extraction, organic solutions were dried over anhydrous Na2SO4, filtered, and concentrated with a Büchi rotary evaporator at reduced pressure. Melting points were determined in capillary tubes (Stuart SMP30) and were uncorrected. Yields are of purified products and were not optimized. 1H NMR and 13C NMR spectra were recorded on Bruker Avance DRX-400. Chemical shifts (δ) are reported in ppm relative to TMS and calibrated using residual undeuterated solvent as the internal reference. Coupling constants (J) are reported in Hz. The spectra were acquired at 298 K. Data processing was performed with Bruker TopSpin 4.2.0 software, and the spectral data are consistent with the assigned structures. The spin multiplicities are indicated using the symbols: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and bs (broad singlet). FT-IR measurements were carried out using a Shimadzu IR-8000 spectrophotometer (Kyoto, Japan). The spectral range measured was from 400 to 4000 cm−1, with a spectral resolution of 4 cm−1 acquiring 50 scans. The purity (>95%) of compounds was revealed at 254 nm and λmax of each compound by HPLC analysis using a Jasco LC-4000 instrument equipped with a UV-Visible Diode Array Jasco MD-4015 and an XTerra MS C18 Column, 5 μm, 4.6 mm × 150 mm. The methods and columns used have been specified for each compound. Chromatograms were analyzed using ChromNAV 2.0 Chromatography Data System software and the peak retention time is given in minutes. HRMS spectra were registered on an Agilent 6560 Ion Mobility Q-TOF LC/MS system.Ethyl 2-amino-5-methyl-7-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate (8)
Ethyl 2-amino-7-methyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate (9)
:3) to give 9 (0.04 g, 26%).
:10 for 10 min) to give 9 as a white solid (0.33 g, 55%); m. p. = 211.8–212.6 °C.1H NMR (400 MHz, DMSO-d6) δ: 0.93 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.73 (s, 3H, CH3), 4.09 (q, J = 7.1 Hz, 2H, OCH2CH3), 6.65 (bs, 2H, NH2), 7.42–7.64 (m, 5H, Ar–H). 13C NMR (101 MHz, DMSO-d6) δ: 13.84, 15.79, 62.20, 113.93, 128.44, 128.96, 130.04, 139.16, 145.51, 154.45, 158.67, 166.52, 168.71. FT-IR: 3382.25, 3309.38, 3175.06, 2983.58, 2937.86, 1713.27, 767.33 cm−1. HRMS (ESI) m/z [M + H]+ calcd for C15H15N5O2 298.1299 found 298.1315.
Ethyl 2-amino-5-methyl-7-phenyl-4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine-6 carboxylate (10)24
TBMA-MsO was heated at 120 °C and then, 12 (0.20 g, 2.02 mmol), 13 (0.20 mL, 2.02 mmol), and 14 (0.26 mL, 2.02 mmol) were added to an open flask. After 24 h, the reaction mixture was poured into ice/water obtaining a precipitate that was filtered to give 10 as a pale-yellow solid (0.45 g, 75%); m. p. = 230.2–231.0 °C. 1H NMR (400 MHz, DMSO-d6): δ 1.02 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.37 (s, 3H, CH3), 3.89–3.96 (m, 2H, OCH2CH3), 5.20 (s, 2H, NH2), 5.94 (s, 1H, H-7), 7.15–7.18 (m, 2H, Ar–H), 7.21–7.25 (m, 1H, Ar–H), 7.26–7.31 (m, 2H, Ar–H), 10.47 (s, 1H, NH). 13C NMR (101 MHz, DMSO-d6): δ 14.46, 18.98, 59.32, 59.73, 97.74, 127.53, 128.12, 128.71, 143.25, 146.28, 146.97, 162.68, 165.87. FT-IR: 3470.85, 3297.95, 3192.21, 2976.44, 2933.57, 1697.56, 750.18 cm−1. HRMS (ESI) m/z [M + H]+ calcd for C15H17N5O2 300.1455 found 300.1469.Ethyl 2-amino-7-methyl-5-phenyl-4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine-6 carboxylate (11)
Compounds 13 (0.21 mL, 2.02 mmol) and 14 (0.26 mL, 2.02 mmol), and citric acid (0.97 g, 5.05 mmol) were added to a suspension of 12 (0.2 g, 2.02 mmol) in EtOH (6 mL) under nitrogen and the reaction mixture was refluxed for 3.5 h. Then, it was poured into ice/water obtaining a precipitate that was filtered to give 11 as a yellow solid (0.50 g, 83%); m. p. = 218.4–218.9 °C. 1H NMR (400 MHz, DMSO-d6) δ: 1.17 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.56 (s, 3H, CH3), 4.09 (q, J = 7.1 Hz, 2H, OCH2CH3), 5.46 (d, J = 3.2 Hz, 1H, H-7), 5.62 (s, 2H, NH2), 7.21–7.25 (m, 2H, Ar–H), 7.25–7.30 (m, 1H, Ar–H), 7.32–7.34 (m, 2H, Ar–H), 8.55 (d, J = 3.2 Hz, 1H, NH). 13C NMR (101 MHz, DMSO-d6) δ: 14.64, 15.73, 53.15, 60.40, 101.12, 126.37, 128.18, 129.13, 143.80, 144.88, 154.11, 164.09, 165.92. FT-IR: 3420.83, 3312.24, 3179.35, 2979.30, 1683.27, 1633.26, 755.90 cm−1. HRMS (ESI) m/z [M + H]+ calcd for C15H17N5O2 300.1455 found 300.1458.2-Amino-7-methyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxamide (16)
The title compound was synthesized by following the same procedure as that used for the synthesis of 9 (method 2, 3 h) in 26% yield. 1H NMR (400 MHz, DMSO-d6) δ: 2.66 (s, 3H, CH3), 6.57 (s, 2H, NH2), 7.45–7.51 (m, 3H, Ar–H), 7.75–7.80 (m, 3H, CONH2 and Ar–H), 7.98 (s, 1H, CONH2). 13C NMR (101 MHz, DMSO-d6) δ: 14.47, 117.78, 127.55, 127.81, 128.84, 137.63, 142.02, 153.07, 155.73, 166.52, 167.29. HRMS (ESI) m/z [M + H]+ calcd for C13H12N6O 269.1146 found 269.1159.2-Amino-5-methyl-7-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxamide (17)
A mixture of 12 (0.2 g, 2.02 mmol), 13 (0.20 mL, 2.02 mmol), and 20 (0.20 g, 2.02 mmol) in DMF (6 mL) was heated at 130 °C for 2 h in an open flask. After cooling, the reaction mixture was poured into ice/water and filtered to give 17 as a pale-yellow solid (0.23 g, 42%). 1H NMR (400 MHz, DMSO-d6) δ: 2.54 (s, 3H, CH3), 6.38 (s, 2H, NH2), 7.49–7.70 (m, 6H, Ar–H and CONH), 7.84 (s, 1H, CONH). 13C NMR (101 MHz, DMSO-d6): δ 23.24, 119.80, 128.66, 129.89, 130.05, 130.82, 142.24, 154.69, 158.87, 167.16, 167.99. HRMS (ESI) m/z [M + H]+ calcd for C13H12N6O 269.1803 found 269.1160.2-Amino-7-methyl-5-phenyl-N-(p-tolyl)-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxamide (18)
The title compound was synthesized by following the same procedure as that used for the synthesis of 9 (method 2, 15 h). After purification by flash chromatography (Reveleris-X2) eluted with CHCl3/MeOH (from 99:1 to 97:3), compound 18 was obtained as a yellow solid in 25% yield. 1H NMR (400 MHz, DMSO-d6) δ: 2.24 (s, 3H, tolyl-CH3), 2.69 (s, 3H, CH3), 6.63 (s, 2H, NH2), 7.10 and 7.35 (d, J = 8.4 Hz, each 2H, Ar–H), 7.40–7.50 (m, 3H, Ar–H), 7.72–7.82 (m, 2H, Ar–H), 10.40 (s, 1H, NH). 13C NMR (101 MHz, DMSO-d6) δ: 15.68, 21.03, 118.61, 120.22, 128.83, 129.74, 130.13, 133.81, 136.42, 138.57, 143.86, 154.41, 157.24, 164.01, 168.61. HRMS (ESI) m/z [M + H]+ calcd for C20H18N6O 359.1615 found 359.1629.
2-Amino-5-methyl-7-phenyl-N-(p-tolyl)-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxamide (19)
A mixture of 13 (0.23 mL, 2.22 mmol), 21 (0.39 g, 2.02 mmol), and piperidine (0.1 mL, 1.01 mmol) in AcOH (0.06 mL, 1.01 mmol) was stirred at 25 °C for 2 h. Then, 12 (0.2 g, 2.02 mmol) and DMF (2 mL) were added and the reaction was stirred at reflux for 14 h. The mixture was poured into ice/water to give a precipitate that was filtered and purified by flash chromatography eluted with CHCl3/MeOH (95:5), furnishing 19 as a yellow solid (0.5 g, 69%). 1H NMR (400 MHz, DMSO-d6) δ: 2.22 (s, 3H, tolyl-CH3), 2.57 (s, 3H, CH3), 6.47 (s, 2H, NH2), 7.07 and 7.27 (d, J = 7.9 Hz, each 2H, Ar–H), 7.47–7.53 (m, 3H, Ar–H), 7.66–7.72 (m, 2H, Ar–H), 10.31 (s, 1H, NH). 13C NMR (101 MHz, DMSO-d6) δ: 21.00, 23.24, 119.41, 120.10, 128.74, 129.69, 129.74, 129.82, 130.99, 133.77, 136.31, 142.90, 154.90, 159.13, 163.53, 168.24. HRMS (ESI) m/z [M + H]+ calcd for C20H18N6O 359.1615 found 359.1627.
General procedure for C-2 amidation (Method A)
Benzoyl chloride (1.2 equiv.) was added dropwise at 0 °C to a solution of 2-amino-[1,2,4]triazolo[1,5-a]pyrimidine 8, 9, or 16–19 (1.0 equiv.) in dry pyridine and the reaction mixture was maintained at rt until the starting material was not detected by TLC (80 min–2 h). Then, the reaction mixture was poured into ice/water and 2 N HCl was added (pH ∼ 5–7), obtaining a precipitate that was filtered and purified as described.Ethyl 2-benzamido-7-methyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate (23)
The title compound was synthesized starting from compound 9 by Method A (80 min) and purified by flash chromatography eluted with CHCl3/MeOH (99:1) as a white solid in 60% yield; m. p. = 238.2–239.0 °C. 1H NMR (400 MHz, DMSO-d6) δ: 0.95 (t, J = 6.9 Hz, 3H, OCH2CH3), 2.88 (s, 3H, CH3), 4.14 (q, J = 6.9 Hz, 2H, OCH2CH3), 7.52–7.59 (m, 5H, Ar–H), 7.61–7.63 (m, 3H, Ar–H), 8.05 (d, J = 7.6 Hz, 2H, Ar–H), 11.57 (s, 1H, CONH). 13C NMR (101 MHz, DMSO-d6) δ: 13.86, 15.96, 62.58, 116.04, 128.57, 128.72, 192.02, 129.16, 130.56, 132.83, 134.08, 138.68, 148.17, 153.40, 160.84, 161.89, 165.35, 166.10. HPLC, 0.7 mL min−1, H2O 40%/CH3CN 60%, ret. time: 3.2133 min. HRMS (ESI) m/z [M + H]+ calcd for C22H19N5O3 402.1561 found 402.15619.
2-Benzamido-7-methyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxamide (24)
The title compound was synthesized starting from compound 16 by Method A (1 h) and purified by crystallization by a mixture of EtOH/DMF as a white solid in 64% yield; m. p. = 284.6–286.0 °C. 1H NMR (400 MHz, DMSO-d6) δ: 2.82 (s, 3H, CH3), 7.51–7.58 (m, 5H, Ar–H), 7.61–76.3 (m, 1H, Ar–H), 7.83–7.91 (m, 3H, Ar–H and CONH2), 8.04–8.09 (m, 3H, Ar–H and CONH2), 11.49 (s, 1H, CONH). 13C NMR (101 MHz, DMSO-d6) δ: 15.77, 120.90, 128.69, 128.89, 129.03, 129.12, 130.56, 132.80, 134.09, 138.35, 145.55, 153.17, 159.64, 161.54, 165.44, 167.14. HPLC, 0.5 mL min−1, H2O (0.1% FA) 80%/CH3CN 20% to CH3CN 100% in 10 min, ret. time: 6.8367 min. HRMS (ESI) m/z [M + H]+ calcd for C20H16N6O2 373.1408 found 373.14145.2-Benzamido-7-methyl-5-phenyl-N-(p-tolyl)-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxamide (25)
The title compound was synthesized starting from 18 by Method A (80 min) and purified by flash chromatography eluted with CHCl3/MeOH (99:1) as a pale pink solid in 55% yield; m. p. = 255.3–256.9 °C. 1H NMR (400 MHz, DMSO-d6) δ: 2.25 (s, 3H, tolyl-CH3), 2.86 (s, 3H, CH3), 7.12 and 7.36 (d, J = 8.0 Hz, each 2H, Ar–H), 7.45–7.51 (m, 3H, Ar–H), 7.55 (t, J = 7.4 Hz, 2H, Ar–H), and 7.64 (t, J = 7.4 Hz, 1H, Ar–H), 7.79–7.87 (m, 2H, Ar–H), 8.06 (d, J = 8.1 Hz, 2H, Ar–H), 10.51 and 11.54 (s, each 1H, CONH). 13C NMR (101 MHz, DMSO-d6) δ: 15.80, 21.04, 120.32, 120.49, 128.71, 129.00, 129.04, 129.81, 130.68, 132.83, 134.07, 136.21, 138.13, 146.35, 153.34, 159.93, 161.77, 163.43, 165.45. HPLC, 0.6 mL min−1, H2O (0.1% FA) 50%/CH3CN 50% to CH3CN 100% in 10 min, ret. time: 5.7000 min. HRMS (ESI) m/z [M + H]+ calcd for C27H22N6O2 463.1877 found 463.18877.
Ethyl 2-benzamido-5-methyl-7-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate (26)
The title compound was synthesized starting from compound 8 by Method A (2 h) and purified by flash chromatography eluted with CHCl3/MeOH (98:2) as a white/yellowish solid in 86% yield; m. p. = 250.1–251.7 °C. 1H NMR (400 MHz, DMSO-d6) δ: 0.88 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.68 (s, 3H, CH3), 4.07 (q, J = 7.1 Hz, 2H, OCH2CH3), 7.46–7.55 (m, 2H, Ar–H), 7.57–7.70 (m, 6H, Ar–H), 7.98 (d, J = 7.7 Hz, 2H, Ar–H), 11.42 (s, 1H, CONH). 13C NMR (101 MHz, DMSO-d6) δ: 13.84, 24.23, 62.34, 117.16, 128.66, 128.97, 129.04, 129.70, 131.47, 132.77, 133.98, 146.44, 154.09, 161.64, 161.91, 165.29, 165.71. HPLC, 0.7 mL min−1, H2O 40%/CH3CN 60%, ret. time: 3.0100 min. HRMS (ESI) m/z [M + H]+ calcd for C22H19N5O3 402.1561 found 402.15669.
2-Benzamido-5-methyl-7-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxamide (27)
The title compound was synthesized starting from compound 17 by Method A (1 h) and purified by flash chromatography eluted with CHCl3/MeOH (97:3) as a brown solid in 81% yield; m. p. = 248.2–248.9 °C. 1H NMR (400 MHz, DMSO-d6) δ: 2.66 (s, 3H, CH3), 7.50 (t, J = 7.3 Hz, 2H, Ar–H), 7.56–7.64 (m, 4H, Ar–H), 7.72–7.79 (m, 3H, CONH2 and Ar–H), 7.94–7.98 (m, 3H, CONH2 and Ar–H), 11.33 (s, 1H, CONH). 13C NMR (101 MHz, DMSO-d6) δ: 23.63, 121.94, 128.62, 128.82, 128.97, 129.30, 130.13, 131.33, 132.73, 134.00, 143.85, 153.76, 160.98, 161.93, 165.43, 166.62. HPLC, 0.5 mL min−1, H2O (0.1% FA) 80%/CH3CN 20% to CH3CN 100% in 10 min, ret. time: 5.5500 min. HRMS (ESI) m/z [M + H]+ calcd for C20H16N6O2 373.1408 found 373.14107.
2-Benzamido-5-methyl-7-phenyl-N-(p-tolyl)-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxamide (28)
The title compound was synthesized starting from 19 by Method A (1 h) and purified by crystallization by a mixture of EtOH/DMF as a white solid in 70% yield; m. p. = 240.3–240.9 °C. 1H NMR (400 MHz, DMSO-d6) δ: 2.23 (s, 3H, tolyl-CH3), 2.69 (s, 3H, CH3), 7.09 and 7.28 (d, J = 7.9 Hz, 2H, Ar–H), 7.47–7.58 (m, 5H, Ar–H), 7.59–7.63 (m, 1H, Ar–H), 7.72–7.81 (m, 2H, Ar–H), 7.99 (d, J = 7.4 Hz, 2H, Ar–H), 10.39 and 10.41 (s, each 1H, CONH).13C NMR (101 MHz, DMSO-d6) δ: 21.02, 23.64, 120.24, 121.48, 128.64, 128.90, 128.99, 129.09, 129.77, 129.99, 131.51, 132.76, 134.00, 136.10, 144.66, 153.94, 161.27, 162.12, 162.97, 165.43. HPLC, 0.5 mL min−1, H2O (0.1% FA) 35%/CH3CN 65%, ret. time: 4.3967 min. HRMS (ESI) m/z [M + H]+ calcd for C27H22N6O2 463.1877 found 463.18948.2-Benzamido-7-methyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylic acid (29)
1M solution of LiOH in H2O (16.80 mL, 16.80 mmol) was added to a solution of 23 (0.65 g, 1.68 mmol) in 1,4-dioxane (18 mL), and the reaction mixture was stirred at 60 °C for 24 h. Then, the reaction mixture was poured into ice/water and HCl was added until pH ≈ 3, obtaining a precipitate that was filtered. After purification by flash chromatography elution with CHCl3/MeOH (95:5), 29 was obtained as a white solid (0.207 g, 74%); m. p. > 300 °C. 1H NMR (400 MHz, DMSO-d6) δ: 2.50 (s, 3H, CH3), 7.36–7.42 (m, 5H, Ar–H), 7.50 (t, J = 7.1 Hz, 2H, Ar–H), 7.62 (t, J = 7.0 Hz, 1H, Ar–H), 7.98 (d, J = 7.2 Hz, 2H, Ar–H), 11.73 (bs, 1H, CONH). 13C NMR (101 MHz, DMSO) δ: 32.70, 110.53, 128.26, 128.56, 128.68, 128.95, 129.08, 133.17, 133.31, 141.93, 155.44, 157.33, 157.52, 164.67, 166.30, 200.24. HPLC, 0.8 mL min−1, MeOH (0.1% FA) 30%/H2O (0.1% FA) 30%/CH3CN (0.1% FA) 40%, ret. time: 2.3500 min. HRMS (ESI) m/z [M + H]+ calcd for C20H15N5O3 374.1248 found 374.12503.
2-Benzamido-5-methyl-7-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylic acid (30)
The title compound was synthesized starting from 26 using the same synthetic procedure as that used for compound 29 (45 °C for 1 h), and purified by crystallization using a mixture of EtOH/DMF in 78% yield; m. p. > 300 °C. 1H NMR (400 MHz, DMSO-d6) δ: 2.26 (s, 3H, CH3), 7.48–7.58 (m, 4H, Ar–H), 7.59–7.70 (m, 2H, Ar–H), 7.90–7.97 (m, 2H, Ar–H), 7.98–8.05 (m, 2H, Ar–H), 11.30 (s, 1H, CONH), 13.71 (bs, 1H, COOH). 13C NMR (101 MHz, DMSO-d6) δ: 18.05, 111.28, 128.64, 129.03, 129.28, 129.87, 132.78, 134.08, 134.28, 137.94, 150.00, 151.20, 154.66, 157.73, 165.45, 193.72. HPLC, 0.5 mL min−1, H2O (0.1% FA) 65%/CH3CN (0.1% FA) 35% to CH3CN (0.1% FA) 100% in 12 min, ret. time: 6.9467 min. HRMS (ESI) m/z [M + H]+ calcd for C20H15N5O3 374.1248 found 374.12512.Biological evaluation (IV)
:4000 in PBS supplemented with 2% FBS. Following the washings, substrate 3,3′,5,5′-tetramethylbenzidine (TMB, KPL) was added and the absorbance was measured at 450 nm using an ELISA plate reader (MultiSkan FC, Thermo Scientific). The values obtained from the samples treated with only DMSO were used to set 100% of PA–PB1 interaction.
Biological evaluation (Flaviviruses and SARS-CoV-2)
The adherent human cell lines Huh7 (kindly provided by Istituto Toscano Tumori, Core Research Laboratory, Siena, Italy) and A549 ACE2-TMPRSS2(101006) were used to determine the cytotoxicity and the antiviral activity of candidate compounds against flaviviruses and SARS-CoV-2, respectively. Adherent cell lines were propagated in high glucose Dulbecco's Modified Eagle's Medium with sodium pyruvate and L-glutamine (DMEM; Euroclone) with 10% Fetal Bovine Serum (FBS; Euroclone) and 1% Penicillin/Streptomycin (Pen/Strep; Euroclone) used for Huh-7 or Minimum Essential Medium Eagle (EMEM; Euroclone) used for A549 supplemented with 10% Fetal Bovine Serum (FBS; Euroclone), 1% Penicillin/Streptomycin (Pen/Strep, Euroclone), 2 mM L-glutamine, 2 mg ml−1 G418 and 200 μg ml−1 of hygromycin B. For viral propagation, cytotoxic and antiviral experiments, a propagation medium with a lower concentration of FBS (1%) and 1% of Pen/Strep was used. The cells were incubated at 37 °C in a humidified incubator supplemented with 5% CO2.
The absorbance (expressed in OD450) for IA and the luminescence for CPE (expressed as RLU) were measured using the absorbance and the luminescence Module of the GloMax® Discover Multimode Microplate Reader (Promega). In each plate, the corresponding reference compound, the mock control (uninfected cells), the virus control and the virus back titration, performed diluting 2-fold the initial viral inoculum, were included. All drug concentrations were tested in duplicate in two independent experiments. In each plate, sofosbuvir and remdesivir were used as reference compounds against flaviviruses and SARS-CoV-2, respectively. Infected and uninfected cells without drugs were used to calculate 100% and 0% of viral replication, respectively. The half-maximal efficacy concentration (EC50) was calculated through a non-linear regression analysis of the dose–response curves generated with GraphPad PRISM software version 8.0.
Author contributions
M. P., M. C. P., T. F., F. G., A. B., M. T. and C. B.: data curation, formal analysis, and investigation. M. L. B., R. G., V. C., I. V., O. T., M. Z. and A. L.: project administration and writing – review & editing. T. F. and S. M.: conceptualization, project administration, writing – original draft and writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by PRIN 2017 – cod. 2017BMK8JR (to T. Felicetti, I. Vicenti, M. Zazzi, V. Cecchetti, and S. Massari), PON “Ricerca e innovazione” 2014–2020, Azione IV.4 (tematiche dell'innovazione) – cod. J91B2100320006 (to T. Felicetti), Fondazione Umberto Veronesi – Post-doctoral Fellowships 2022 (to M. C. Pismataro), Associazione Italiana per la Ricerca sul Cancro, AIRC, grants IG 2016 – ID. 18855 and IG 2021 – ID. 25899 (to A. Loregian); Ministero dell'Istruzione, dell'Università e della Ricerca, PRIN 2017-cod. 2017KM79NN and PRIN 2022-cod. 20223RYYFC (to A. Loregian); Fondazione Cassa di Risparmio di Padova e Rovigo-Bando Ricerca Covid-2019 No. 55777 2020.0162-ARREST-COV: AntiviRal PROTAC-Enhanced Small-molecule Therapeutics against COronaViruses (to A. Loregian); EU funding within the NextGenerationEU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (Project No. PE00000007, INF-ACT) (to A. Loregian).References
- Q. Renyu, L. Yuchao, W. M. W. W. Kandegama, C. Qiong and Y. Guangfu, Mini-Rev. Med. Chem., 2018, 18, 781–793 CrossRef.
- S. Pinheiro, E. M. C. Pinheiro, E. M. F. Muri, J. C. Pessôa, M. A. Cadorini and S. J. Greco, Med. Chem. Res., 2020, 29, 1751–1776 CrossRef CAS.
- M. A. Salem, M. S. Behalo and R. E. Khidre, Mini-Rev. Org. Chem., 2021, 18, 1134–1149 CrossRef CAS.
- T. Felicetti, M. C. Pismataro, V. Cecchetti, O. Tabarrini and S. Massari, Curr. Med. Chem., 2021, 29, 1379–1407 CrossRef.
- R. Bivacqua, M. Barreca, V. Spanò, M. V. Raimondi, I. Romeo, S. Alcaro, G. Andrei, P. Barraja and A. Montalbano, Eur. J. Med. Chem., 2023, 249, 115136 CrossRef CAS.
- A. S. Abdelkhalek, M. Salah and M. A. Kamal, Curr. Med. Chem. DOI:10.2174/0929867330666230228120416.
- S. Massari, G. Nannetti, J. Desantis, G. Muratore, S. Sabatini, G. Manfroni, B. Mercorelli, V. Cecchetti, G. Palù, G. Cruciani, A. Loregian, L. Goracci and O. Tabarrini, J. Med. Chem., 2015, 58, 3830–3842 CrossRef CAS PubMed.
- M. C. Pismataro, T. Felicetti, C. Bertagnin, M. G. Nizi, A. Bonomini, M. L. Barreca, V. Cecchetti, D. Jochmans, S. De Jonghe, J. Neyts, A. Loregian, O. Tabarrini and S. Massari, Eur. J. Med. Chem., 2021, 221, 113494 CrossRef CAS.
- S. Massari, C. Bertagnin, M. C. Pismataro, A. Donnadio, G. Nannetti, T. Felicetti, S. Di Bona, M. G. Nizi, L. Tensi, G. Manfroni, M. I. Loza, S. Sabatini, V. Cecchetti, J. Brea, L. Goracci, A. Loregian and O. Tabarrini, Eur. J. Med. Chem., 2021, 209, 112944 CrossRef CAS.
- J. Desantis, S. Massari, A. Corona, A. Astolfi, S. Sabatini, G. Manfroni, D. Palazzotti, V. Cecchetti, C. Pannecouque, E. Tramontano and O. Tabarrini, Molecules, 2020, 25, 1183 CrossRef CAS.
- S. Massari, J. Desantis, G. Nannetti, S. Sabatini, S. Tortorella, L. Goracci, V. Cecchetti, A. Loregian and O. Tabarrini, Org. Biomol. Chem., 2017, 15, 7944–7955 RSC.
- S. Imtiaz, J. A. War, S. Banoo and S. khan, RSC Adv., 2021, 11, 11083–11165 RSC.
- Y. V. Sedash, N. Y. Gorobets, V. A. Chebanov, I. S. Konovalova, O. V. Shishkin and S. M. Desenko, RSC Adv., 2012, 2, 6719–6728 RSC.
- G. Fischer, Advances in Heterocyclic Chemistry, Academic Press, 2019, vol. 128, pp. 1–101 Search PubMed.
- P. K. Singh, S. Choudhary, A. Kashyap, H. Verma, S. Kapil, M. Kumar, M. Arora and O. Silakari, Bioorg. Chem., 2019, 88, 102919 CrossRef CAS.
- A. Hibot, A. Oumessaoud, A. Hafid, M. Khouili and M. D. Pujol, ChemistrySelect, 2023, 8, e202301654 CrossRef CAS.
- S. A. Komykhov, A. A. Bondarenko, V. I. Musatov, M. V. Diachkov, N. Y. Gorobets and S. M. Desenko, Chem. Heterocycl. Compd., 2017, 53, 378–380 CrossRef CAS.
- M. A. Kolosov, E. H. Shvets, D. A. Manuenkov, S. A. Vlasenko, I. V. Omelchenko, S. V. Shishkina and V. D. Orlov, Tetrahedron Lett., 2017, 58, 1207–1210 CrossRef CAS.
- S. Karami, M. Bayat, S. Nasri and F. Mirzaei, Mol. Divers., 2021, 25, 2053–2062 CrossRef CAS PubMed.
- M. A. A. Radwan, F. M. Alminderej, H. E. M. Tolan and H. M. Awad, J. Appl. Pharm. Sci., 2020, 10, 012–022 CAS.
- V. M. Chernyshev, A. N. Sokolov and V. A. Taranushich, Russ. J. Appl. Chem., 2007, 80, 1691–1694 CrossRef CAS.
- Z. Zhang, Y. Pang, Y. Wang, C. Cohen, Y. Zhao and C. Liu, Int. J. Antimicrob. Agents, 2015, 45, 491–495 CrossRef CAS.
- R. P. Brigance, W. Meng, A. Fura, T. Harrity, A. Wang, R. Zahler, M. S. Kirby and L. G. Hamann, Bioorg. Med. Chem. Lett., 2010, 20, 4395–4398 CrossRef CAS PubMed.
- D. A. Pyatakov, A. V. Astakhov, A. N. Sokolov, A. N. Fakhrutdinov, A. N. Fitch, V. B. Rybakov, V. M. V. V. Chernyshev and V. M. V. V. Chernyshev, Tetrahedron Lett., 2017, 58, 748–754 CrossRef CAS.
- P. P. Warekar, P. T. Patil, K. T. Patil, D. K. Jamale, G. B. Kolekar and P. V. Anbhule, Synth. Commun., 2016, 46, 2022–2030 CrossRef CAS.
- Y. M. Ren and C. Cai, Monatsh. Chem., 2009, 140, 49–52 CrossRef CAS.
- K. V. N. S. Srinivas and B. Das, Synthesis, 2004, 2004, 2091–2093 CrossRef.
- L. Brinchi, R. Germani, G. Savelli and D. Biondini, Lett. Org. Chem., 2006, 3, 207–211 CrossRef.
- V. M. Chernyshev, A. N. Sokolov and V. A. Taranushich, Russ. J. Appl. Chem., 2006, 79, 1134–1137 CrossRef.
- H. Nagarajaiah, A. Mukhopadhyay and J. N. Moorthy, Tetrahedron Lett., 2016, 57, 5135–5149 CrossRef CAS.
- C. O. Kappe, J. Org. Chem., 1997, 62, 7201–7204 CrossRef CAS PubMed.
- K. S. Atwal, G. C. Rovnyak, B. C. O'Reilly and J. Schwartz, J. Org. Chem., 1989, 54, 5898–5907 CrossRef CAS.
- C. O. Kappe, Acc. Chem. Res., 2000, 33, 879–888 CrossRef CAS PubMed.
- X. Huo, Y. Ma, Z. Chen, L. Yuan, X. Zheng, X. Li, F. Liang, W. You and P. Zhao, ChemistrySelect, 2021, 6, 4562–4565 CrossRef CAS.
- N. Khetarpal and I. Khanna, J. Immunol. Res., 2016, 2016, 6803098 Search PubMed.
- Dengue—Level 3 cause | Institute for Health Metrics and Evaluation, https://www.healthdata.org/results/gbd_summaries/2019/dengue-level-3-cause, (accessed 16 October 2021).
- X. Yang, M. B. M. Quam, T. Zhang and S. Sang, J. Travel Med., 2021, 28, 146 CrossRef PubMed.
- G. Habarugira, W. W. Suen, J. Hobson-Peters, R. A. Hall and H. Bielefeldt-Ohmann, Pathogens, 2020, 9, 1–51 CrossRef.
- R. Lu, X. Zhao, J. Li, P. Niu, B. Yang, H. Wu, W. Wang, H. Song, B. Huang, N. Zhu, Y. Bi, X. Ma, F. Zhan, L. Wang, T. Hu, H. Zhou, Z. Hu, W. Zhou, L. Zhao, J. Chen, Y. Meng, J. Wang, Y. Lin, J. Yuan, Z. Xie, J. Ma, W. J. Liu, D. Wang, W. Xu, E. C. Holmes, G. F. Gao, G. Wu, W. Chen, W. Shi and W. Tan, Lancet, 2020, 395, 565–574 CrossRef CAS.
- C. B. Jackson, M. Farzan, B. Chen and H. Choe, Nat. Rev. Mol. Cell Biol., 2022, 23, 3–20 CrossRef CAS PubMed.
- S. Fawell, J. Seery, Y. Daikh, C. Moore, L. L. Chen, B. Pepinsky and J. Barsoum, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 664–668 CrossRef CAS PubMed.
- G. Muratore, L. Goracci, B. Mercorelli, Á. Foeglein, P. Digard, G. Cruciani, G. Palù and A. Loregian, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6247–6252 CrossRef CAS.
- I. D'Agostino, I. Giacchello, G. Nannetti, A. L. Fallacara, D. Deodato, F. Musumeci, G. Grossi, G. Palù, Y. Cau, I. M. Trist, A. Loregian, S. Schenone and M. Botta, Eur. J. Med. Chem., 2018, 157, 743–758 CrossRef.
- A. Loregian, B. A. Appleton, J. M. Hogle and D. M. Coen, J. Virol., 2004, 78, 158–167 CrossRef CAS PubMed.
- A. Loregian and D. M. Coen, Chem. Biol., 2006, 13, 191–200 CrossRef CAS PubMed.
- G. Nannetti, S. Massari, B. Mercorelli, C. Bertagnin, J. Desantis, G. Palù, O. Tabarrini and A. Loregian, Antiviral Res., 2019, 165, 55–64 CrossRef CAS PubMed.
- I. Vicenti, M. G. Martina, A. Boccuto, M. De Angelis, G. Giavarini, F. Dragoni, S. Marchi, C. M. Trombetta, E. Crespan, G. Maga, C. Eydoux, E. Decroly, E. Montomoli, L. Nencioni, M. Zazzi and M. Radi, Eur. J. Med. Chem., 2021, 224, 113683 CrossRef CAS PubMed.
- M. G. Martina, I. Vicenti, L. Bauer, E. Crespan, E. Rango, A. Boccuto, N. Olivieri, M. Incerti, M. Zwaagstra, M. Allodi, S. Bertoni, E. Dreassi, M. Zazzi, F. J. M. van Kuppeveld, G. Maga and M. Radi, ChemMedChem, 2021, 16, 3548–3552 CrossRef.
- L. Fiaschi, F. Dragoni, E. Schiaroli, A. Bergna, B. Rossetti, F. Giammarino, C. Biba, A. Gidari, A. Lai, C. Nencioni, D. Francisci, M. Zazzi and I. Vicenti, Viruses, 2022, 14, 1374 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Superposition of 1H NMR and 13C NMR spectra of compounds 10 and 11, and 8 and 9. NOESY spectra of compounds 10 and 11. 13C NMR chemical shifts (δ, ppm) of compounds 8–11 and 16–19. Optimization of reaction conditions for compounds 8–11. Plausible reaction mechanisms for the formation of compounds 10 and 11. Anti-DENV-2, WNV, and SARS-CoV-2 activity, and cytotoxicity of TZP derivatives 23–30. 1H NMR and 13C NMR spectra for compounds 8–11, 16–19 and 23–30. HRMS analyses of compounds 8–11 and 23–30. HPLC chromatograms of compounds 23–30. FT-IR spectra for compounds 8–11. See DOI: https://doi.org/10.1039/d3ob01861j |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2024 |