
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substituent-controlled regioselective arylation of carbazoles using dual catalysis†
M.
Shahid
,
Perumal
Muthuraja
and
Purushothaman
Gopinath
 *
*
Department of Chemistry, Indian Institute of Science Education and Research (IISER) Tirupati, Tirupati 517507, India. E-mail: gopi@iisertirupati.ac.in
First published on 20th December 2023
Abstract
Regioselective arylation of carbazoles is reported using dual palladium–photoredox catalysis. Controlled monoarylation and diarylation of symmetrical and unsymmetrical carbazoles were achieved under mild reaction conditions with a broad substrate scope and functional group tolerance. Steric and electronic control the regioselectivity of the arylation of unsymmetrical carbazoles. Late-stage functionalization of a caprofen drug derivative and large-scale synthesis of mono- and di-arylated carbazoles were demonstrated to showcase the synthetic versatility of the method. Finally, we also showcased the synthesis of hyellazole analogues (a marine alkaloid) in a short route using our strategy.
1. Introduction
Carbazoles represent a class of heteroaromatic nitrogen-containing molecules that have great significance in organic chemistry. They are encountered in a wide range of natural products1,2 such as hyellazole,3–6 chlorohyellazole,3 and in many drug and bio-active molecules such as rimcazole,7,8 carvedilol,9–11 carprofen,12–14 carazallol,15–17etc. Beyond their significance in natural products and pharmaceuticals, carbazole derivatives are also used as building blocks for LEDs,18,19 optoelectronic materials,20–22 and semiconductors.23–25 Similarly, biaryl moieties are vital molecular frameworks that are present in many drugs and bio-active compounds. Despite the importance of both carbazoles and biaryls, only a handful of reports are known for the direct arylation of carbazoles.26–31 Wu et al.30 and Nageswar et al.31 independently reported pyridine- and pyrimidine-directed ortho-arylation of carbazoles. These methods suffer from disadvantages such as the use of oxidants, additives, higher reaction temperature, etc. Another major challenge is achieving site-selective functionalization of unsymmetrical carbazoles and controlling the site-selectivity. Given that the majority of carbazole-based drugs and natural products are unsymmetrical in nature, a new method for site-selective functionalization is highly desirable.Recently, photo-mediated dual catalysis has emerged as an effective tool for functionalizing various sp2 and sp3 C–H bonds. In 2011, Sanford et al. reported the first dual Pd/Ru photoredox strategy32 for C–H arylation and in 2015, Toste and coworkers presented mechanistic insights into dual metallophotoredox catalysis.33 Following this, several other groups reported photomediated organic transformations involving a dual metallophotoredox strategy.34,35 In this direction our group reported a dual palladium-photoredox strategy for C–H arylation of phenylureas.36 Similarly, Itami and coworkers reported photo-induced arylation of carbazoles; however, it had limitations such as a higher reaction temperature (65 °C), lower yields, the use of near ultraviolet light and poor regioselecivity.37
Recently, Jain et al. reported C–H arylation/acylation of carbazoles using a dual palladium–photoredox approach.38 Unfortunately, this method suffers from limitations such as low to moderate yields, a narrow substrate scope (in terms of carbazoles) and the use of excess K2CO3 as an additive. More importantly, regioselective arylation of unsymmetrical carbazoles and diarylation of carbazoles were not well studied. In this context, we herein report a modular approach for site-selective and controllable ortho-arylation of carbazoles using a dual palladium–photoredox strategy under milder conditions without any additives. Furthermore, the regioselectivity of the reaction can be tuned by controlling the sterics and electronics of the substituents on the unsymmetrical carbazoles (Scheme 1).
 | ||
| Scheme 1 Previous reports and our work. | ||
2. Results and discussion
We began our investigation with 9-(pyrimidin-2-yl)-9H-carbazole (1) as a model substrate and an aryl diazonium salt as the arylating agent (Table 1). With Eosin Y (5 mol%) as a photocatalyst and Pd(OAc)2 (10 mol%) as a transition metal catalyst in 0.1 M methanol, 54% yield of the desired product was obtained (entry 1). When we changed the photocatalyst to Ru(bpy)3Cl2 (5 mol%), 74% yield of the desired product was obtained (entry 2). Next, we investigated different palladium catalysts such as Pd(TFA)2, Pd(MeCN)2Cl2, and Pd(PPh3)2Cl2, but unfortunately the yield didn't improve in all these cases (entries 3–5). Similarly, screening different solvents, such as toluene, DCE, dioxane, etc., didn't improve the yield further (entries 6–9). Reducing the catalyst loading of palladium acetate to 1 mol% and 5 mol%, the yield dropped (entries 11 & 12). Interestingly, when Ru(bpy)3Cl2 catalyst loading was decreased from 5 mol% to 1 mol%, a slight increase in the yield was observed (entry 13). Finally, when 2.5 mol% Ru(bpy)3Cl2 was used, the yield increased to 82% (entry 14). Changing the solvent concentration further didn't improve the reaction yield (entries 10, 15 & 16).|
|
||||
|---|---|---|---|---|
| S. no. | Photocatalyst | TM | Solvent | Yield (%) |
| a Conditions: carbazole 1 (1 equiv.), aryldiazonium salt 2a (4 equiv.), Pd(OAc)2 (10 mol%), Ru(bpy)3Cl2 (2.5 mol%), in methanol under argon at room temperature for 24 hours, 44 W blue LED (Kessil). | ||||
| 1 | Eosin Y (5 mol%) | Pd(OAc)2 (10 mol%) | MeOH (0.1 M) | 54% |
| 2 | Ru(bpy)3Cl2 (5 mol%) | Pd(OAc)2 (10 mol%) | MeOH (0.1 M) | 74% |
| 3 | Ru(bpy)3Cl2 (5 mol%) | Pd(TFA)2 (10 mol%) | MeOH (0.1 M) | 52% |
| 4 | Ru(bpy)3Cl2 (5 mol%) | Pd(MeCN)2 (10 mol%) | MeOH (0.1 M) | 30% |
| 5 | Ru(bpy)3Cl2 (5 mol%) | Pd(pph3)2Cl2 (10 mol%) | MeOH (0.1 M) | Traces |
| 6 | Ru(bpy)3Cl2 (5 mol%) | Pd(OAc)2 (10 mol%) | Toluene (0.1 M) | — |
| 7 | Ru(bpy)3Cl2 (5 mol%) | Pd(OAc)2 (10 mol%) | DCE (0.1 M) | 28% |
| 8 | Ru(bpy)3Cl2 (5 mol%) | Pd(OAc)2 (10 mol%) | Dioxane (0.1 M) | — |
| 9 | Ru(bpy)3Cl2 (5 mol%) | Pd(OAc)2 (10 mol%) | MeCN (0.1 M) | 41% |
| 10 | Ru(bpy)3Cl2 (2.5 mol%) | Pd(OAc)2 (10 mol%) | MeOH (0.05 M) | 60% |
| 11 | Ru(bpy)3Cl2 (5 mol%) | Pd(OAc)2 (1 mol%) | MeOH (0.1 M) | 62% |
| 12 | Ru(bpy)3Cl2 (5 mol%) | Pd(OAc)2 (5 mol%) | MeOH (0.1 M) | 68% |
| 13 | Ru(bpy)3Cl2 (1 mol%) | Pd(OAc)2 (10 mol%) | MeOH (0.1 M) | 76% |
| 14 | Ru(bpy) 3 Cl 2 (2.5 mol%) | Pd(OAc) 2 (10 mol%) | MeOH (0.1 M) | 82% |
| 15 | Ru(bpy)3Cl2 (5 mol%) | Pd(OAc)2 (5 mol%) | MeOH (0.05 M) | 68% |
| 16 | Ru(bpy)3Cl2 (2.5 mol%) | Pd(OAc)2 (10 mol%) | MeOH (0.2 M) | 75% |

With the optimized conditions in hand, we next investigated the scope of aryl diazonium salts (Scheme 2). Halo-substituted aryldiazoniums, such as meta-fluoro (80%), meta-bromo (78%), and ortho-bromo (69%) afforded good yields of the arylated products (3b–3d). Aryl diazonium salts bearing electron-withdrawing substituents such as p-nitro, p-cyano & m-trifluoromethyl were well tolerated and afforded the resultant biaryls (3e, 3f & 3g) in good yields. Similarly, electron-rich and disubstituted aryl diazonium salts also afforded good to excellent yields of the resultant arylated products (3h–3o). Surprisingly, the para-acetyl phenyl diazonium salt did not afford any arylated carbazole product (3p). Interestingly, the 2-amino dibenzo furan-based heteroaryl diazonium salt furnished the desired product 3q in 72% yield whereas the 8-aminoquinoline based diazonium salt did not furnish the expected product, 3r.
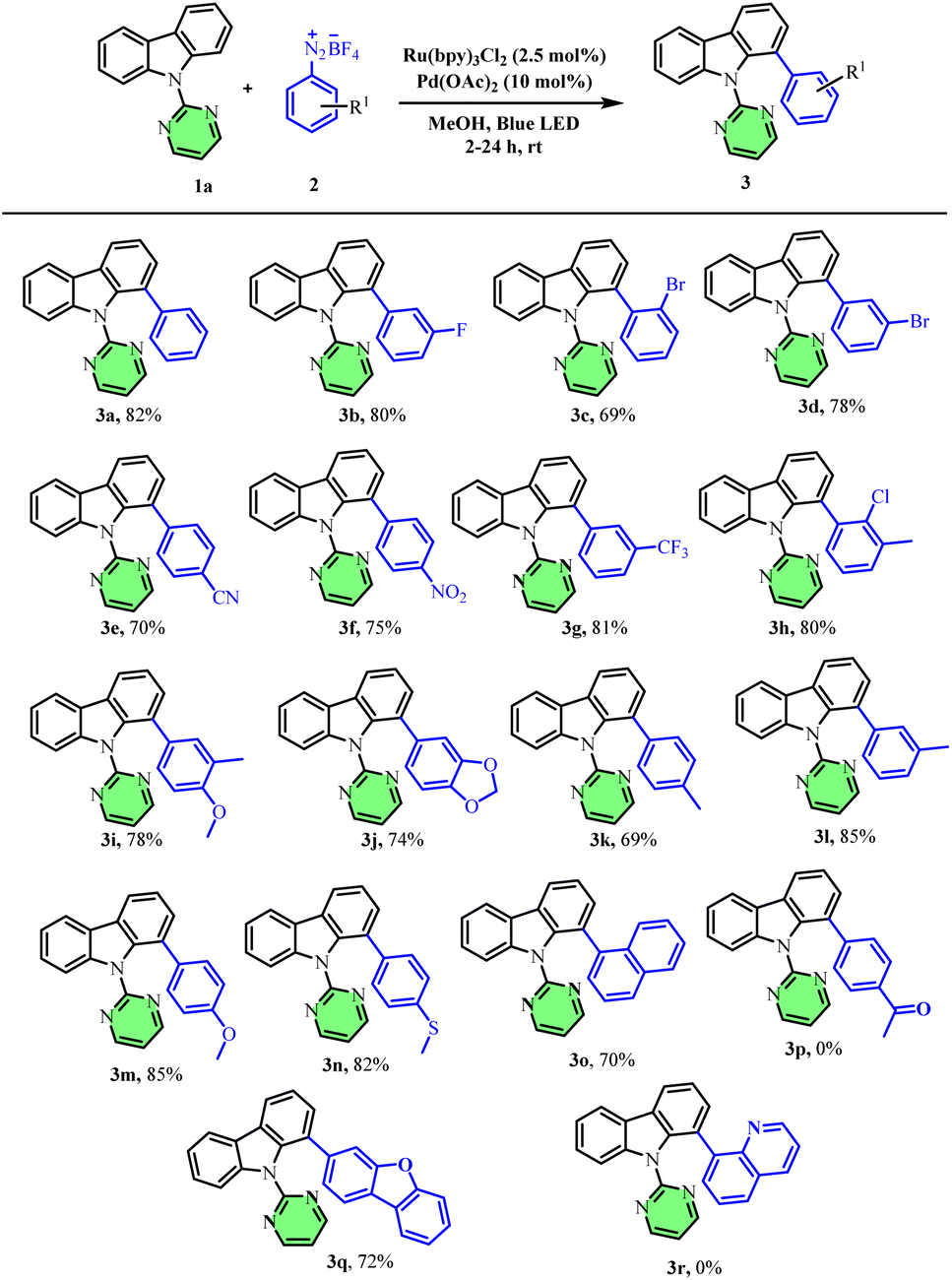 | ||
| Scheme 2 Scope of different aryl diazonium salts for carbazole arylation. Reaction conditions: carbazole 1 (1 equiv.), aryl diazonium salt 2 (4 equiv.), Ru(bpy)3Cl2 (2.5 mol%), Pd(OAc)2 (10 mol%), in methanol under argon at room temperature for 2–24 h, 44 W blue LED (Kessil). | ||
In general, most of the carbazole functionalizations are limited to simple and symmetrical carbazoles with very limited examples for unsymmetrical carbazoles as they generally give a mixture of regioisomers. Hence, we next focussed on the scope of less explored unsymmetrical carbazoles for regioselective arylation. Accordingly, we attempted arylation of various 2-aryl substituted carbazoles under standard conditions but using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of methanol and acetonitrile as the solvent. Interestingly, all these substrates afforded the corresponding arylated products, 4a–4d, (arylation on the less hindered side of carbazole aryl rings) with good yields and selectivity. Similarly, other C-2 substituted carbazoles such as 2-bromo, 2-chloro and 2-trifluoromethyl substituted carbazoles also afforded the corresponding C-8 arylated products (4e–4g) in good yields and high selectivity with a 12 W blue LED light. 2-Methyl substituted carbazole on the other hand furnished a mixture of regioisomers (4h & 4h′) in a 4:1 ratio, although it still afforded the C-8 regioisomer as the major product.
:1 mixture of methanol and acetonitrile as the solvent. Interestingly, all these substrates afforded the corresponding arylated products, 4a–4d, (arylation on the less hindered side of carbazole aryl rings) with good yields and selectivity. Similarly, other C-2 substituted carbazoles such as 2-bromo, 2-chloro and 2-trifluoromethyl substituted carbazoles also afforded the corresponding C-8 arylated products (4e–4g) in good yields and high selectivity with a 12 W blue LED light. 2-Methyl substituted carbazole on the other hand furnished a mixture of regioisomers (4h & 4h′) in a 4:1 ratio, although it still afforded the C-8 regioisomer as the major product.
Interestingly, electron-donating 2-methoxy and 2,3-methylenedioxy-substituted carbazoles afforded the corresponding C-1 arylated products (4i and 4j) regioselectively (arylation on the more substituted side) in good yields. In order to understand the mechanism and to know if the change in selectivity is due to mere electronic effects or the directing group ability of methoxy substituents, we attempted arylation of 4-methoxy-substituted carbazole. Interestingly, we again obtained the corresponding C-1 arylated product (4k) regioselectively. This shows that the methoxy substituent does not function as a directing group and an electrophilic palladation mechanism may be in operation.
We recently reported the regioselective acylation of carbazoles, wherein acylation selectively occurred on the less hindered carbazole aryl ring.39 Inspired by the current findings, we further extended the scope of carbazole acylation to electron-rich systems. Accordingly, 2-methoxy and 4-methoxy substituted carbazoles afforded the corresponding C-1 acylated products (4l and 4m) regioselectively in good yields. Single crystal XRD analysis of compounds 4g, 4i, 4k, and 4m further confirmed the structure of the correct regioisomer (Scheme 3).
 | ||
| Scheme 3 Scope of aryl diazonium salts for regioselective arylation of carbazoles. Reaction conditions: carbazole 1 (1 equiv.), aryl diazonium salt 2 (4 equiv.), Ru(bpy)3Cl2 (2.5 mol%), Pd (OAc)2 (10 mol%), in methanol under argon at room temperature for 2–24 h, 44 W blue LED (Kessil). a12 W blue LED, 2–6 h. bRegioisomeric ratio. | ||
We next demonstrated the diarylation of various symmetrical and unsymmetrical carbazoles since most of the existing methods suffer from controlling the mono vs. diarylation products. After some initial screening, with higher diazonium salt equivalents (6 equiv.), we obtained diarylated carbazoles as the major product. Accordingly, 3-methyl carbazole and 2,7-dimethoxy carbazole afforded the desired diarylated products in good yields (4n & 4o). Unsymmetrical carbazoles having substituents at the C-2 position also afforded the desired diarylated products in good yields (4p–4r). Furthermore, we investigated the scope of various aryl diazonium salts for the diarylation reaction using carbazole 1a as the substrate (Scheme 4). A simple aryl diazonium salt afforded the corresponding diarylated product, 3s, in 80% yield after 24 h, whereas other substituted aryl diazonium salts afforded the desired diarylated products with five equivalents only and in a shorter reaction time (12 h). Both para- and meta-substituted (3t & 3u) aryl diazonium salts were suitable substrates and afforded the desired products in good yields (3v and 3w).
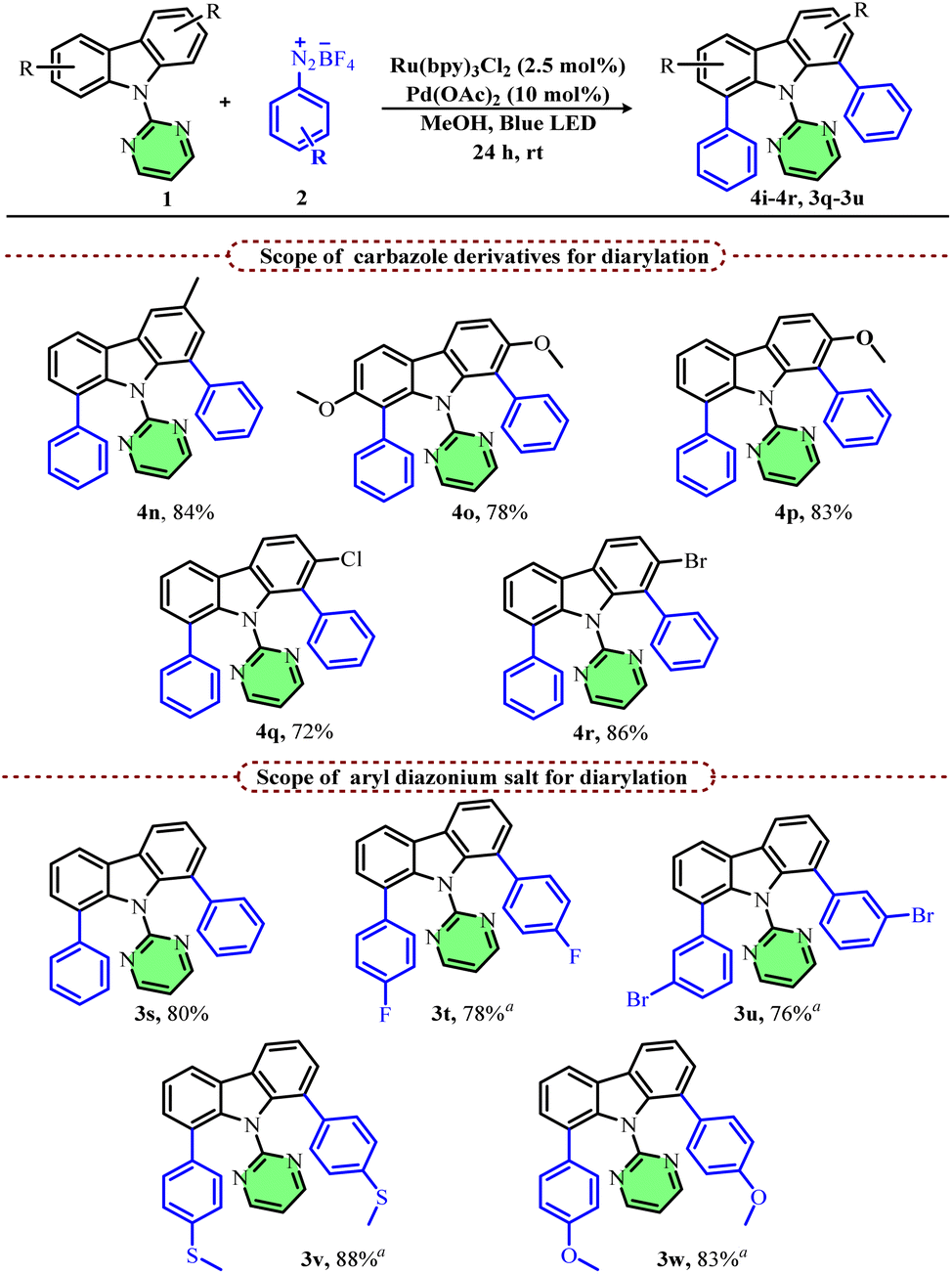 | ||
| Scheme 4 Scope of diarylation reaction with different diazoniums and substituted carbazoles. Reaction conditions: carbazole 1 (1 equiv.), aryl diazonium salt 2 (6 equiv.), Ru(bpy)3Cl2 (2.5 mol%), Pd(OAc)2 (10 mol%), in methanol under argon at room temperature for 24 h, 44 W blue LED (Kessil). aAryl diazonium salt 2 (5 equiv.). | ||
In order to demonstrate the synthetic utility of our methodology, we focused on the gram-scale synthesis of mono- and di-arylated carbazoles. At first, monoarylation of a simple carbazole was achieved on a gram scale in 72% yield using 3.5 equivalents of aryl diazonium salts under the standard conditions. Similarly, diarylation of a simple carbazole was performed on a gram scale under the standard conditions to afford the desired product in 80% yield (Scheme 5a). Caprofen, a class of NSAID (nonsteroidal anti-inflammatory drugs) commonly used for the treatment of inflammation, contains a carbazole core. Hence, we next attempted the site-selective late-stage functionalization of the caprofen drug derivative, 5, which afforded the desired product 5a in 72% yield (Scheme 5b). Next, we demonstrated an efficient route for the synthesis of hyellazole (a marine alkaloid obtained from blue green algae Hyella caespitosa) derivatives. Accordingly, we started the synthesis from 4-nitro-o-cresol, 6, which on simple methylation followed by reduction with iron and acetic acid gave 4-methoxy-3-methylaniline 8. Compound 8 on coupling with 1,2-dibromo benzene 9 using palladium acetate afforded the corresponding carbazole 10. Finally, the pyrimidine directing group was installed on compound 10 to form carbazole derivative 11. Compound 11 was first subjected to monoarylation conditions to afford carbazole 12, a regioisomer of hyellazole in 76% yield. Similarly, compound 11 under diarylation conditions afforded the corresponding aryl functionalized hyellazole derivative 13 in 82% yield (Scheme 5c). Finally, the removal of the pyrimidine directing group from 9-(pyrimidin-2-yl)-9H-carbazole was demonstrated by treating it with sodium methoxide (CH3ONa) in DMSO at 120 °C for 12 hours to showcase the synthetic versatility of our method (Scheme 5d).
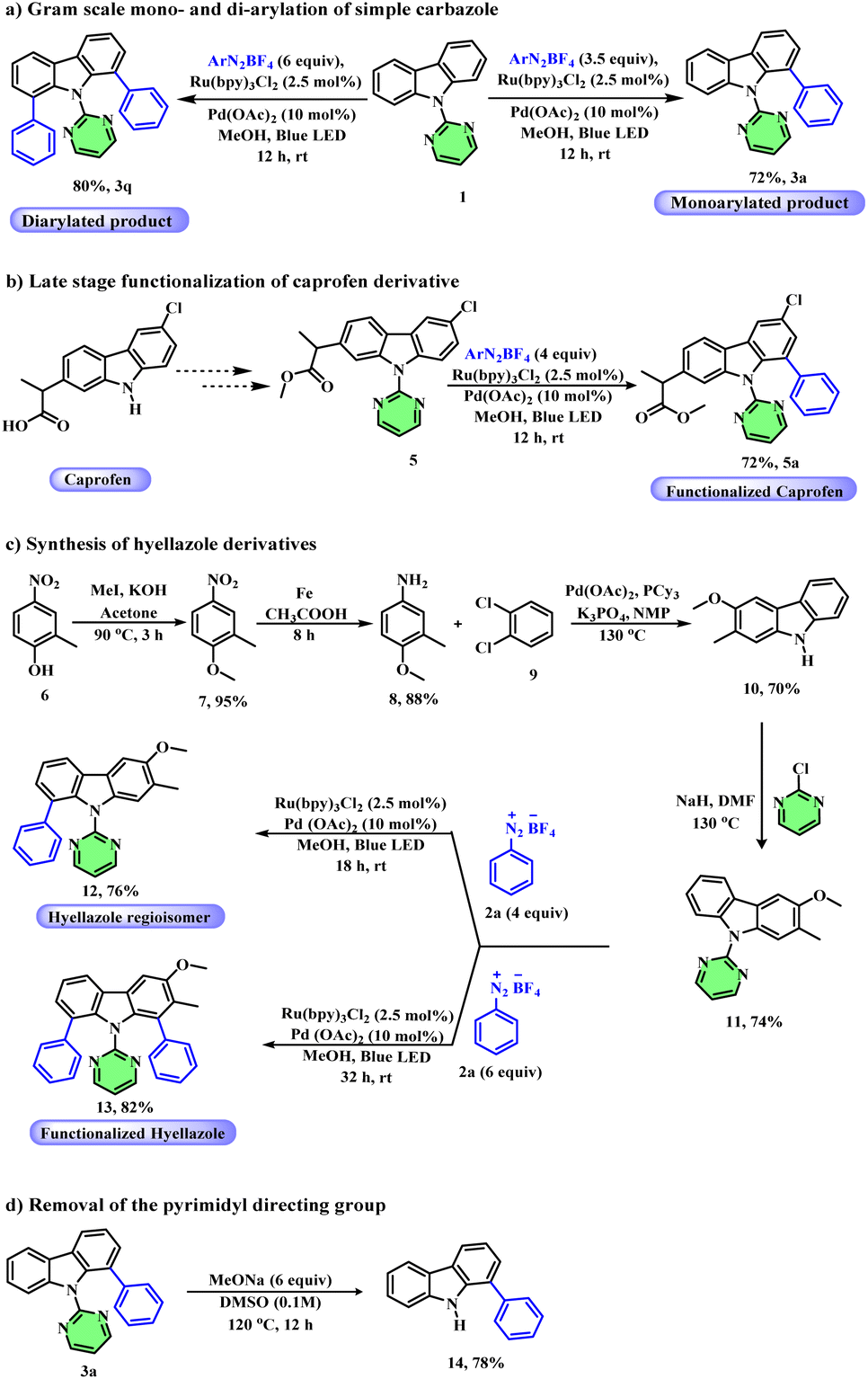 | ||
| Scheme 5 Application of the methodology. | ||
Finally, we carried out few mechanistic studies to understand the mechanism of this arylation reaction. Control experiments in the absence of blue LED irradiation led to no product formation. Similarly, in the absence of palladium acetate or the Ru(bpy)2Cl2 photocatalyst, the reaction didn't furnish any desired product (Table S1,† entries 9–11). Next, we conducted a radical trapping experiment using TEMPO as the radical scavenger, which didn't afford any desired product, and gave the corresponding aryl-TEMPO adduct (observed in HRMS), thereby confirming the radical pathway. Based on these observations, we propose the following mechanism for the transformation: coordination of palladium(II) acetate with substrate 1 leads to the formation of complex A. Next, the aryl radical generated from the photoredox cycle reacts with complex A to form the corresponding Pd(III) intermediate B. In the photoredox cycle, the Ru2+ catalyst upon irradiation with blue light forms the excited-state photocatalyst, which then undergoes SET with the aryl diazonium salt to form the corresponding aryl radical and Ru3+ complex. This Ru3+ complex then undergoes reduction by accepting an electron from complex B to form Ru2+ and Pd(IV) intermediate C. Finally, reductive elimination of intermediate C affords the desired arylated product 3 and regenerates the Pd(II) catalyst (Fig. 1).
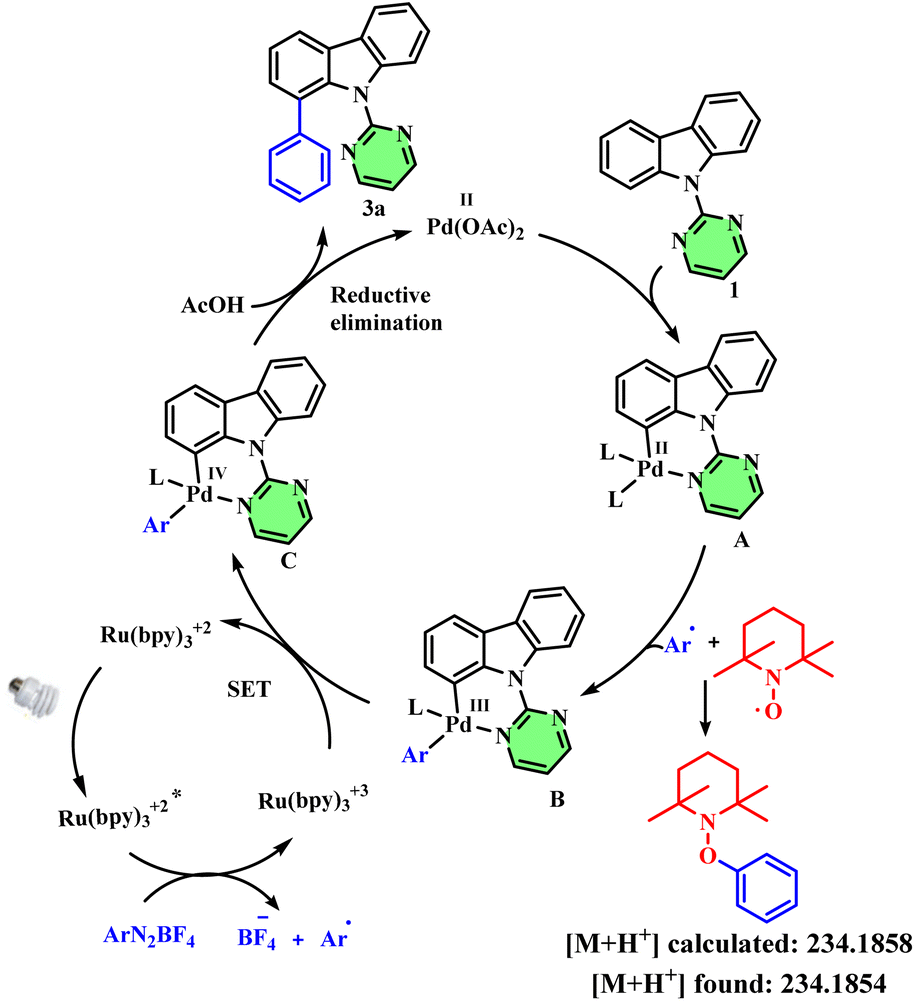 | ||
| Fig. 1 Proposed reaction mechanism. | ||
4. Conclusions
Herein, we have successfully developed a dual palladium–photoredox strategy for ortho-arylation of carbazoles and their derivatives using a pyrimidine directing group under mild conditions. We have achieved regioselective arylation of unsymmetrical carbazoles with good yields and selectivity. The regioselectivity of the carbazoles can be controlled by tuning the sterics and electronics of the substituents on the carbazole ring. Similarly, we also demonstrated controlled mono- vs. di-arylation of carbazoles by a slight modification of the reaction conditions. In general, both mono- and di-arylation reactions show a broad substrate scope with good functional group tolerance. Next, we showcased the synthetic utility of our method via the late-stage functionalization of the caprofen drug derivative and by the synthesis of hyellazole derivatives.Author contributions
The study was designed and conceptualized by PG and MS. MS and PM carried out all the experiments in consultation with PG. MS and PG wrote the manuscript. All authors agreed on the finalized version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the SERB (CRG/2022/008440) for the financial support. M. S. thanks IISER Tirupati for financial support. The authors thank Mr Sandip Das and Maria Francis from IISER Tirupati for their help and support in crystallographic experiments.References
- O. P. S. Patel, A. Mishra, R. Maurya, D. Saini, J. Pandey, I. Taneja, K. S. R. Raju, S. Kanojiya, S. K. Shukla, M. N. Srivastava, M. Wahajuddin, A. K. Tamrakar, A. K. Srivastava and P. P. Yadav, J. Nat. Prod., 2016, 79, 1276–1284 CrossRef CAS PubMed.
- J.-H. Yang, X.-Y. Wang, Y.-P. Zhou, R. Lu, C.-H. Chen, M.-H. Zhang, Y.-Y. Cheng, S. L. Morris-Natschke, K.-H. Lee and Y.-S. Wang, Molecules, 2019, 25, 99 CrossRef.
- S. Chakraborty and C. Saha, Eur. J. Org. Chem., 2018, 2018, 2013–2021 CrossRef CAS.
- T. Choshi, T. Sada, H. Fujimoto, C. Nagayama, E. Sugino and S. Hibino, J. Org. Chem., 1997, 62, 2535–2543 CrossRef CAS.
- E. M. Beccalli, A. Marchesini and T. Pilati, J. Chem. Soc., Perkin Trans. 1, 1994, 5, 579 RSC.
- S. Kano, E. Sugino and S. Hibino, J. Chem. Soc., Chem. Commun., 1980, 2, 1241 RSC.
- R. M. Ferris, H. L. White, F. L. M. Tang, A. Russell and M. Harfenist, Drug Dev. Res., 1986, 9, 171–188 CrossRef CAS.
- S. M. Husbands, S. Izenwasser, T. Kopajtic, W. D. Bowen, B. J. Vilner, J. L. Katz and A. H. Newman, J. Med. Chem., 1999, 42, 4446–4455 CrossRef CAS PubMed.
- M. O'Reilly, N. K. Kirkwood, E. J. Kenyon, R. Huckvale, D. M. Cantillon, S. J. Waddell, S. E. Ward, G. P. Richardson, C. J. Kros and M. Derudas, J. Med. Chem., 2019, 62, 5312–5329 CrossRef PubMed.
- M. E. Choi, H. Yoo, H.-R. Lee, I. J. Moon, W. J. Lee, Y. Song and S. E. Chang, Int. J. Mol. Sci., 2020, 21, 8796 CrossRef CAS PubMed.
- M. Packer, W. S. Colucci, J. D. Sackner-Bernstein, C. Liang, D. A. Goldscher, I. Freeman, M. L. Kukin, V. Kinhal, J. E. Udelson, M. Klapholz, S. S. Gottlieb, D. Pearle, R. J. Cody, J. J. Gregory, N. E. Kantrowitz, T. H. LeJemtel, S. T. Young, M. A. Lukas and N. H. Shusterman, Circulation, 1996, 94, 2793–2799 CrossRef CAS.
- M. Payne-Johnson, C. Becskei, Y. Chaudhry and M. R. Stegemann, Vet. Rec., 2015, 176, 284–284 CrossRef CAS.
- A. M. Gouda and F. A. Almalki, SN Appl. Sci., 2019, 1, 332 CrossRef CAS.
- J. V. Roughan and P. A. Flecknell, Pain, 2001, 90, 65–74 CrossRef CAS.
- A. S. Manalan, H. R. Besch and A. M. Watanabe, Circ. Res., 1981, 49, 326–336 CrossRef CAS PubMed.
- S. L. Heald, P. W. Jeffs, T. N. Lavin, P. Nambi, R. J. Lefkowitz and M. G. Caron, J. Med. Chem., 1983, 26, 832–838 CrossRef CAS.
- E. A. Dubois, J. C. van den Bos, T. Doornbos, P. A. P. M. van Doremalen, G. A. Somsen, J. A. J. M. Vekemans, A. G. M. Janssen, H. D. Batink, G. J. Boer, M. Pfaffendorf, E. A. van Royen and P. A. van Zwieten, J. Med. Chem., 1996, 39, 3256–3262 CrossRef CAS.
- Y. Liu, H.-Y. Wang, G. Chen, X.-P. Xu and S.-J. Ji, Aust. J. Chem., 2009, 62, 934 CrossRef CAS.
- A. van Dijken, J. J. A. M. Bastiaansen, N. M. M. Kiggen, B. M. W. Langeveld, C. Rothe, A. Monkman, I. Bach, P. Stössel and K. Brunner, J. Am. Chem. Soc., 2004, 126, 7718–7727 CrossRef CAS PubMed.
- J.-H. Tsai, C.-C. Chueh, M.-H. Lai, C.-F. Wang, W.-C. Chen, B.-T. Ko and C. Ting, Macromolecules, 2009, 42, 1897–1905 CrossRef CAS.
- B. Gieseking, B. Jäck, E. Preis, S. Jung, M. Forster, U. Scherf, C. Deibel and V. Dyakonov, Adv. Energy Mater., 2012, 2, 1477–1482 CrossRef CAS.
- M. A. Esteruelas, D. Gómez-Bautista, A. M. López, E. Oñate, J. Tsai and C. Xia, Chem. – Eur. J., 2017, 23, 15729–15737 CrossRef CAS PubMed.
- G. Bagdžiūnas and D. Palinauskas, Biosensors, 2020, 10, 104 CrossRef PubMed.
- S. Wakim, J. Bouchard, N. Blouin, A. Michaud and M. Leclerc, Org. Lett., 2004, 6, 3413–3416 CrossRef CAS PubMed.
- S. Suman, A. Siddiqui, M. L. Keshtov, G. D. Sharma and S. P. Singh, J. Mater. Chem. C, 2019, 7, 543–552 RSC.
- G. M. Reddy, N. S. S. Rao, P. Satyanarayana and H. Maheswaran, RSC Adv., 2015, 5, 105347–105352 RSC.
- V. P. Reddy, R. Qiu, T. Iwasaki and N. Kambe, Org. Lett., 2013, 15, 1290–1293 CrossRef CAS.
- R. Kaur, H. Singh and S. A. Babu, Synthesis, 2023, 55, 3535–3567 CrossRef CAS.
- Z.-C. Qi, Q.-X. Lou, Y. Niu and S.-D. Yang, Chem. Commun., 2021, 57, 2021–2024 RSC.
- J.-H. Chu, C.-C. Wu, D.-H. Chang, Y.-M. Lee and M.-J. Wu, Organometallics, 2013, 32, 272–282 CrossRef CAS.
- K. Harsha Vardhan Reddy, R. U. Kumar, V. P. Reddy, G. Satish, J. B. Nanubolu and Y. V. D. Nageswar, RSC Adv., 2016, 6, 54431–54434 RSC.
- D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef CAS PubMed.
- M. D. Levin, S. Kim and F. D. Toste, ACS Cent. Sci., 2016, 2, 293–301 CrossRef CAS PubMed.
- J. Jiang, W. M. Zhang, J. J. Dai, J. Xu and H. J. Xu, J. Org. Chem., 2017, 82, 3622–3630 CrossRef CAS.
- M. K. Sahoo, S. P. Midya, V. G. Landge and E. Balaraman, Green Chem., 2017, 19, 2111–2117 RSC.
- S. S. Babu, M. Shahid and P. Gopinath, Chem. Commun., 2020, 56, 5985–5988 RSC.
- B. Maeda, G. Mori, Y. Sakakibara, A. Yagi, K. Murakami and K. Itami, Asian J. Org. Chem., 2021, 10, 1428–1431 CrossRef CAS.
- N. Rajat and N. Jain, J. Org. Chem., 2023, 88, 8600–8608 CrossRef CAS.
- M. Shahid, A. J. Punnya, S. S. Babu, S. Sarkar and P. Gopinath, J. Org. Chem., 2023, 88, 13686–13698 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2267782–2267784 and 2292863. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01827j |
| This journal is © The Royal Society of Chemistry 2024 |