
A robust single compartment peroxide fuel cell using mesoporous antimony doped tin oxide as the cathode material†
Anu Maria Chittilappilly
Devassy
,
Karuna Dagaji
Wankhede
,
Adithya
Kamalakshan
and
Sarthak
Mandal
 *
*
Department of Chemistry, National Institute of Technology, Tiruchirappalli, Tamil Nadu 620015, India. E-mail: smandal@nitt.edu; sarthakmandal@gmail.com
First published on 16th May 2024
Abstract
To date, metal oxide catalysts have not been explored as cathode materials for robust and high-performance single-compartment H2O2 fuel cells due to significant non-electrochemical disproportionation losses of H2O2 on many metal oxide surfaces. Here, for the first time, we demonstrate an acidic peroxide fuel cell with antimony doped tin oxide as the cathode and widely used Ni foam as the anode material. Our constructed peroxide fuel cell records a superior open circuit potential of nearly 0.82 V and a maximum power density of 0.32 mW cm−2 with high operational stability. The fuel cell performance is further improved by increasing the ionic strength of the electrolyte with the addition of 1 M NaCl, resulting in an increased maximum power density value of 1.1 mW cm−2.
 Sarthak Mandal | Dr. Sarthak Mandal is an Assistant Professor in the Department of Chemistry at the National Institute of Technology, Tiruchirappalli. Dr. Mandal received his Ph.D. from the Indian Institute of Technology, Kharagpur. Subsequently, he worked as a postdoctoral fellow at Columbia University, New York (2014–2015) and Arizona State University (2015–2018). His current research focuses on the design, synthesis, and characterizations of a wide variety of materials including inorganic nanomaterials, strongly coupled dye aggregates, one-dimensional donor-acceptor assemblies, inorganic-organic hybrid materials and nano-bio hybrids for energy-harvesting, energy conversion and biosensing applications. |
Introduction
Fuel cells are potential energy storage and conversion technologies that can nourish our energy demands to make a transition from fossil fuel-based energy to renewable and sustainable energy in terms of their zero (or less) emission of harmful chemicals, wide range fuel/feedstock utility, and easy portability. Hydrogen fuel cells are considered the most promising candidates with a maximum theoretical output potential of 1.23 V. This is because hydrogen fuel can be produced by water splitting using renewable solar energy in a photo(electro)catalytic system and can be utilized in a fuel cell to generate electricity with high efficiency. Besides, the high gravimetric energy density (33.3 kW h kg−1) of hydrogen and low or near-zero emission have led to an unprecedented growth of research on hydrogen fuel cells. However, the low efficiencies of solar-energy driven catalytic hydrogen fuel production, the difficulties in developing strategies for safe and efficient hydrogen fuel storage and transportation, and the need for costly proton exchange membrane separators are the common challenges in the successful utilization of hydrogen fuel cells.Hydrogen peroxide (H2O2) is an emerging alternative fuel because of its low cost, easy storage and transportation in the aqueous phase,1 and thermodynamically favourable synthesis from water and oxygen (abundant in air) by suitable photo(electro)catalytic systems either via two electron reduction of oxygen in acidic media (O2 + 2H+ + 2e− → H2O2 E° = 0.68 V) or two electron oxidation of water (2H2O → H2O2 + 2H+ + 2e− E° = 1.77 V).2–7 Moreover, an aqueous solution of H2O2 (60% V/V) has a similar energy density (3.5 MJ L−1 or 2.1 MJ kg−1) to that of compressed (at 35 MPa pressure) hydrogen (2.8 MJ L−1 or 3.5 MJ kg−1).8 A kind of conventional peroxide fuel cell which utilises H2O2 as a fuel (2H2O2 → 2O2 + 4H+ + 4e− E° = 0.68 V) and oxygen as an oxidant (O2 + 4H+ + 4e− → 2H2O E° = 1.23 V) allows for achieving a maximum theoretical voltage output of 0.55 V (ref. 9), which is too low compared to hydrogen fuel cells. To overcome this issue, researchers have introduced the concept of a single compartment (without any membrane separator) H2O2 fuel cell, in which the peroxide itself acts as the fuel and oxidizer. This eventually leads to the generation of water and oxygen (as environmentally benign by-products) with a theoretical voltage output of 1.09 V which is comparable to that of hydrogen fuel cells. The redox reactions involved in a single compartment H2O2 fuel cell are given by:
| Anodic: H2O2 → O2 + 2H+ + 2e− E° = 0.68 V | (1) |
| Cathodic: H2O2 + 2H+ + 2e− → 2H2O E° = 1.77 V | (2) |
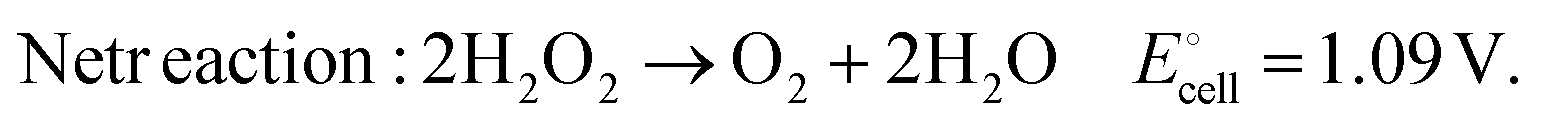 | (3) |
A schematic of the H2O2 fuel cell with the anodic and cathodic reactions in a single compartment generating electricity from H2O2 fuel is shown in Scheme 1.
 | ||
| Scheme 1 Schematic representation of the single-compartment H2O2 fuel cell along with the obtained current density vs. potential/power density plots. The fuel cell is developed with antimony doped tin oxide (ATO) coated on FTO as the cathode and Ni foam as the anode. | ||
Finding suitable catalysts or electrodes for selective oxidation and reduction reactions of H2O2 is a major challenge in developing robust and efficient H2O2 fuel cells. The decomposition of H2O2 by a disproportionation (2H2O2 → O2 + H2O) reaction on catalytic electrode interfaces is one of the major problems associated with this cell. In a seminal work, Yamazaki et al.10 introduced a single compartment peroxide fuel cell in alkaline medium using silver as the cathode and various other metals (Au, Pt, Ni, and Pd) as anodes. Although the platinum anode exhibited the highest current density, it caused the maximum extent of H2O2 decomposition. So far, nickel (Ni) electrodes have been extensively used as low-cost anodic materials for selective oxidation of H2O2 with the lowest degree of H2O2 decomposition. In recent years, there has been an increasing demand to design efficient electro-catalysts as cathode materials for selective reduction of H2O2 without causing much decomposition to achieve improved fuel cell performance in terms of higher open circuit voltages (OCV) and increased power density.
In alkaline medium, peroxide reduction is thermodynamically less favourable (lower positive cathode potential), and the decomposition rate is higher, resulting in lower performances of the previously reported alkaline H2O2 fuel cells. An acidic medium offers a higher cathodic potential to achieve a higher OCV for H2O2 fuel cells. By employing the iron–phthalocyanine [FeIII(Pc)Cl] complex as the cathode material, Yamada et al.11 earlier reported an OCV of 0.5 V and a maximum power density of 10 μW cm−2 at pH 3. They found a significant lowering of the fuel cell performance (an OCV of 0.125 V and a maximum power density of <0.03 μW cm−2) with the use of Fe(III)-porphyrin complexes as cathode catalysts. Pariyar et al.12 studied a single compartment peroxide fuel cell using the [FeIII(PLY)3] complex (PLY-H = 9-hydroxyphenalenone) as the cathode catalyst and obtained an OCV of 0.74 V and a maximum power density of 0.27 mW cm−2. An improved maximum power density of 1.43 mW cm−2 was achieved by the addition of 10 wt% carbon with the [FeIII(PLY)3] complex. Recently a few high performing single compartment peroxide fuel cells have been reported by Yamada and co-workers using Fe-containing bimetallic coordination complexes as cathode catalysts and Ni as the anode.9,13,14 The one with Prussian blue interfaced carbon as the cathode showed an OCV of 0.6 V and a maximum power density of 1.55 mW cm−2. Later, an improved cell performance with an OCV value of ∼0.75 V and a maximum power density of 4.2 mW cm−2 was achieved using pyrazine bridged iron-cyano complexes [Fe(Pyz)][Pt(CN)4] and [Fe(Pyz)][Pd(CN)4] as cathode materials in a single compartment fuel cell that contained 300 mM H2O2 fuel along with aqueous HClO4 (pH 1) and 1 M NaCl.9 A more significant enhancement of power density (up to ∼10 mW cm−2) has been reported by the addition of Sc3+ ions (to suppress the decomposition of H2O2 at the cathode) to the fuel cell that uses [FeII(H2O)2]3 [CoIII(CN)6]2 as the cathode and Ni mesh as the anode.15 So far, this is the highest reported power density of a single compartment H2O2 fuel cell.
Most of the previously reported peroxide fuel cells are based on the utilization of iron containing complexes as cathodic catalysts, where the Fe2+/Fe3+ redox shuttle plays a critical role in the selective reduction of H2O2.11,12 However, it is important to note that the Fe2+ ion-containing catalysts can cause significant decomposition of H2O2 through the well-known Haber–Weiss mechanism.15 This is why the previously reported high performing H2O2 fuel cells with Fe-based catalysts as cathodes have utilized Lewis acid metal cations (such as Sc3+ ions) and Brønsted acid (aqueous HClO4) as additives to reduce the decomposition of H2O2 and achieve high performances. Without these H2O2 stabilizing additives, the performances of the fuel cells were found to be significantly low. Besides, the complex preparation methods of metal-complex-based catalysts and costly ligands may pose a limit on their widespread application. It is therefore important to develop new alternative cathode materials which can conveniently switch their oxidation states along with better electron transfer properties and stability for the robust and efficient performance of a single compartment H2O2 fuel cell. Recently, Miglbauer et al.16 reported the first non-iron catalyst-based H2O2 fuel cell (having cell performances with OCV values of 0.5–0.6 V and power density values of 0.20–0.30 mW cm−2) by utilizing an organic conducting polymer, PEDOT-PSS, as the cathode and Ni as the anode.
Recently, metal oxide nanostructures (without or with doping) involving simple preparation strategies and important physicochemical characteristics (in terms of porous structures, active surface area, electronic conductivity, etc.) have attracted a lot of attention in various electrocatalytic applications as they provide many active sites for redox couples. However, so far, metal oxides have not been explored as cathode materials in a single compartment H2O2 fuel cell. Although many metal oxides17–21 have been demonstrated as nanozymes for peroxidase-like (reduction of H2O2 in solution) activities, their utilization in developing peroxide fuel cells has not been demonstrated presumably because of the high rate of H2O2 decomposition upon strong adsorption on the metal oxide surfaces. It has earlier been pointed out that the metal oxides with lower adsorption of H2O2 exhibit improved peroxidase activity than the metal oxides with higher adsorption of peroxide. Therefore, optimally weak adsorption of H2O2 is important for a metal oxide to be potentially utilized as a cathode material in a single compartment H2O2 fuel cell. Very high H2O2 adsorption can reduce the activity by increasing the rate of decomposition and by blocking the active sites with increased adsorption of intermediates and products. Keeping these in mind we aimed to search for metal–oxide based selective cathode materials for a robust and efficient peroxide fuel cell.
In this work, we have discovered a highly conducting mesoporous antimony-doped tin oxide (ATO) as a potential binder free cathodic catalyst (for the selective reduction of H2O2) for the development of a robust single-compartment H2O2 fuel cell, exhibiting high OCV and moderate power densities. ATO is a well-studied transparent conducting oxide, extensively utilized as an alternative to the expensive ITO (indium doped tin oxide) electrode support in many (photo)electrochemical, (photo)electronic and other applications.22–26 However, so far, its direct use as a (photo)catalyst or (photo)electrocatalyst in various catalytic reactions involved in the development of sustainable and renewable energy has remained largely unexplored.27 The doping of antimony (Sb) into semiconducting tin oxide (SnO2 – with a band gap of ∼3.6 eV) induces high electrical conductivity and optical transparency (in the visible region),28,29 and at the same time, it makes ATO a robust and active catalyst by providing highly dispersed Sb sites as catalytically active sites for a specific reaction. Very recently Naya et al.30 have reported two-electron oxygen reduction and one-electron water oxidation reactions (for the formation and consumption of H2O2) using their synthesised ATO nanoparticles (prepared via calcination or hydrothermal methods on FTO with precursors) as the catalyst.
The mesoporous ATO used in this work has been prepared following a reported one-pot sol–gel method using resorcinol–formaldehyde (R–F) forming a 3-D networked polymer gel as a template to obtain porous nanostructures of the ATO.31 In this method, the ATO composite gel is formed from the precursors (SnCl4·5H2O and 6 mol% SbCl3) concurrently with the polymerization of resorcinol and formaldehyde. A portion of the composite gel was coated (immediately after gelation) on conductive FTO glasses (0.8 × 0.9 cm2) using the ‘Doctor Blade Method’ and kept for subsequent drying and calcination to prepare highly conducting semi-transparent mesoporous ATO coated FTO slides for their utilization as cathodes in H2O2 fuel cells. The remaining portion of the gel was also processed under the same drying and annealing conditions to obtain ATO nano-powder for its utilization as the cathode material on interfacing with the glassy carbon electrode (GCE) without the requirement of costly Nafion as a binder. During the calcination process, as gel drying/shrinkage takes place, ATO is formed as porous structures integrated with the R–F polymer template. This highly conductive R–F template probably makes ATO a highly robust binder free cathodic electrocatalyst for H2O2 fuel cells. Furthermore, the lower extent of adsorptivity of H2O2 on the ATO surface than on the other commonly used metal oxide surfaces (as indicated by previous DFT calculations30) also plays a critical role in reducing H2O2 decomposition and achieving excellent H2O2 fuel cell performance.
2. Experimental methods
2.1. Synthesis of the ATO catalyst
We prepared ATO nanopowder (ATO NP) and an ATO coated FTO electrode (ATO/FTO) following a reported one-pot sol–gel method.31 In a typical procedure, 12 mg of SbCl3 (6 mol% Sb) and 280 mg of SnCl4·5H2O were dissolved sequentially in 0.52 and 1 mL of absolute ethanol, respectively, and then the precursor solutions were mixed together. Further, 151 mg of polyethylene glycol (PEG) was added to this solution (to achieve the required viscosity for ATO film preparation) followed by the addition of respective amounts of resorcinol (R) and formaldehyde (F) and the solution was kept stirring for about 10 min. Finally, 560 mg of epichlorohydrin was added to the solution and maintained at room temperature. Within 10 minutes, the solution turned into a gel by the acid-catalysed ring opening reaction of epichlorohydrin (this provides the suitable pH for ATO gel formation) along with the initialization of R–F polymerization. One portion of the gel was then immediately coated on conductive FTO glasses (0.8 × 0.9 cm2) using the ‘Doctor Blade Method’. The composite gel coated FTO slides were immediately placed inside a mineral oil bath to slow down solvent evaporation for about 20 hours. Here, the interpenetrating porous ATO is formed along with an in situ resorcinol–formaldehyde (R–F) polymeric framework, which provides more structural integrity to ATO pores. Now the oil bath containing these ATO/FTO slides was kept inside an air oven maintained at 70 °C for three more days and finally washed to remove oil and annealed at 500 °C for 10 hours to obtain ATO coated FTO electrodes (ATO/FTO). For reference the image of the ATO coated FTO electrode is given in Fig. S1 of the ESI.† The template SnO2 coated FTO electrode (SnO2/FTO) was also made in the same manner without the addition of the antimony precursor. The remaining portion of the gel was also processed under the same drying and annealing conditions as films to finally obtain ATO nanopowder (ATO NP).2.2. Electrochemical H2O2 reduction and fuel cell performance studies
To prepare the ATO/GCE electrode, 10 μL of 4 mg mL−1 ATO NP dispersion (in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol–water) was drop-cast on a clean GCE (3 mm inner diameter) in 5 μL steps and then dried at room temperature. The ATO coated FTO electrodes were prepared using doctor blade coating of the prepared ATO gel on rectangular FTO coated glass pieces as mentioned previously (Fig. S1†). For the ATO/FTO electrode, the electrolyte exposure area was 0.72 cm2 and the rest of the coated area was masked properly from electrolyte exposure. These prepared ATO/GCE and ATO/FTO electrodes were further used for electrochemical studies. All the electrochemical data were recorded using a CHI630E electrochemical workstation. The electrochemical H2O2 reduction at the ATO/GCE or ATO/FTO working electrode interfaces was carried out using a three-electrode set-up with Ag/AgCl and platinum electrodes as reference and counter electrodes, respectively. The fuel cell performance tests were performed in a two-electrode set-up using an ATO/GCE or ATO/FTO cathodic electrode and Ni-foam as a widely used anode in a single compartment that contained 0.05 M H2SO4 (pH 1) as the electrolyte and 300 mM H2O2 as the fuel. The distance of separation between the cathode (ATO coated electrode) and the anode (Ni foam) was maintained at 0.6 cm for these electrochemical measurements.
:1 ethanol–water) was drop-cast on a clean GCE (3 mm inner diameter) in 5 μL steps and then dried at room temperature. The ATO coated FTO electrodes were prepared using doctor blade coating of the prepared ATO gel on rectangular FTO coated glass pieces as mentioned previously (Fig. S1†). For the ATO/FTO electrode, the electrolyte exposure area was 0.72 cm2 and the rest of the coated area was masked properly from electrolyte exposure. These prepared ATO/GCE and ATO/FTO electrodes were further used for electrochemical studies. All the electrochemical data were recorded using a CHI630E electrochemical workstation. The electrochemical H2O2 reduction at the ATO/GCE or ATO/FTO working electrode interfaces was carried out using a three-electrode set-up with Ag/AgCl and platinum electrodes as reference and counter electrodes, respectively. The fuel cell performance tests were performed in a two-electrode set-up using an ATO/GCE or ATO/FTO cathodic electrode and Ni-foam as a widely used anode in a single compartment that contained 0.05 M H2SO4 (pH 1) as the electrolyte and 300 mM H2O2 as the fuel. The distance of separation between the cathode (ATO coated electrode) and the anode (Ni foam) was maintained at 0.6 cm for these electrochemical measurements.
3. Results and discussion
3.1. Characterization of the ATO catalyst
The microstructural characterization of the prepared ATO catalysts (for both the ATO nanopowder and the ATO film coated on the FTO slide) was carried out using field emission scanning electron microscopy (FESEM). FESEM images shown in Fig. 1a clearly indicate that the sizes of the prepared ATO particles are in the nano regime and they clearly showcase the extensively interconnected highly porous surface morphology. The higher magnification FESEM image (Fig. 1b) reveals that the surface of the particles is made up of uniformly distributed branches/networks of long and extensively interconnected pores. The ATO film coated on the FTO slide also exhibits a highly porous surface morphology (Fig. 1c and d). The high magnification image for the ATO/FTO film clearly indicates that the compactly packed arrays of pores are also in the nanoscale dimensional range (Fig. 1d). Hence the FESEM analysis confirms that both the prepared ATO catalysts (ATO nanopowder and the ATO coated on FTO) possess highly porous nanostructures. | ||
| Fig. 1 Field emission scanning electron microscopy images of ATO nanopowder (a and b) and ATO coated FTO slides (c and d). Scale bars are 1 μm, 200 nm, 2 μm and 500 nm in a, b, c, and d respectively. | ||
For further examining the porosity we performed the N2 adsorption–desorption isotherm. The adsorption–desorption isotherm and pore size distribution (BJH plot) curves of the ATO nanopowder catalyst are shown in Fig. S2.† The prepared catalyst displays good nitrogen adsorption/desorption ability in the relative pressure (ρ/ρ0) range of 0 to 1 and the isotherm (Fig. S2a†) is consistent with the representative type IV, which suggests the existence of mesopores.32,33 From the BJH pore distribution plot (Fig. S2b†) it is further evident that the material consists of pores in the pore diameter range of 2 to 30 nm, which confirms that the prepared ATO catalyst is mesoporous in nature. The calculated BET surface area, pore volume and average pore diameter for ATO nanoparticles are 106.37 m2 g−1, 0.084 cm3 g−1, and 2.76 nm respectively.
To probe the oxidation states of the constituting metal ions of our synthesized ATO catalyst, X-ray photoelectron spectroscopy (XPS) studies were performed (Fig. 2a and b). The deconvolution/peak fitting analysis of the Sb 3d3/2 peak (Fig. 2a) suggests that the Sb ions in ATO are mostly present in their higher oxidation state as Sb+5 (540.4 eV) with a small percentage in their lower oxidation state as Sb+3 (539.6 eV). The Sb 3d5/2 peak was not employed for analysis here because this peak generally overlaps with the O 1s peak. The XPS of the Sn 3d core level (Fig. 2b) shows two peaks at 487.31 and 495.75 eV corresponding to the binding energies of Sn 3d5/2 and Sn 3d3/2, respectively. These binding energy values confirm the +4-oxidation state of Sn in the prepared ATO sample. Overall, the XPS results confirm the formation of a Sb doped SnO2 n-type semiconductor material in which the active Sb+5/Sb+3 redox state plays a critical role in exhibiting the electrocatalytic activities of the ATO catalyst.34
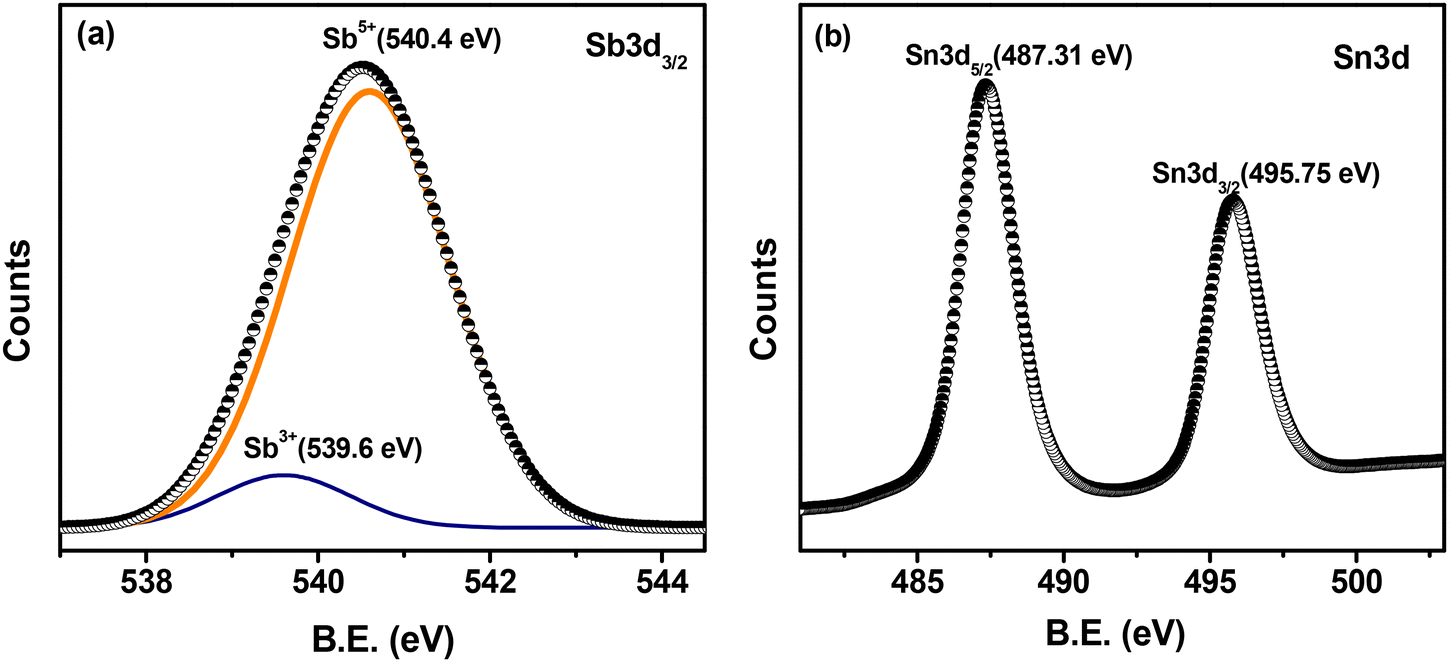 | ||
| Fig. 2 X-ray photoelectron spectra of ATO nanoparticles showing the peaks of (a) Sb 3d3/2 and (b) Sn 3d. | ||
Furthermore, the crystallinity of the prepared catalyst was confirmed by X-ray diffraction (XRD) analysis. We have performed XRD analysis (Fig. 3a) for both the undoped SnO2 and the Sb doped SnO2 (ATO) samples. The major peaks obtained at 26.8, 34, 38.1, 52, 54.8, 58, 62, 64.9, 66.2, 71.4, and 78.9 correspond to the (110), (101), (200), (211), (220), (002), (310), (112), (301), (202) and (321) planes for the as prepared SnO2 (Fig. 3a, lower panel). These peaks are suitably indexed to the tetragonal rutile phase of SnO2 (JCPDS card no. 88-0287), consistent with the literature reports.22,35,36 The XRD pattern for the synthesized ATO (6 mol% Sb doping) nanopowder catalyst (Fig. 3a, upper panel) was found to be similar to that of undoped SnO2 without the presence of any other additional peaks, implying that the Sb doping occurs by substituting Sn atoms in the SnO2 crystal lattice. Only minimal peak shifts were observed for the ATO sample with respect to undoped SnO2. This can be due to the relatively low level of Sb doping.36,37 However, the ATO sample shows a slightly increased XRD peak width than the undoped SnO2, suggesting a decrease in the SnO2 crystallite size with the introduction of the Sb dopant.38 Overall the XRD results suggest that the Sb ions are successfully doped into the crystal lattice of SnO2 replacing Sn+4 ions.
 | ||
| Fig. 3 (a) Powder XRD patterns of undoped SnO2 (red, lower panel) and 6 mol% Sb doped SnO2 (ATO) nanopowder (black, upper panel) samples and (b) the UV-visible absorption spectrum of the prepared ATO coated FTO electrode. | ||
The optical properties of the prepared ATO coated FTO electrode were also analysed by UV-visible absorption spectroscopy. As shown in Fig. 3b, the ATO coated FTO slide shows the onset of absorption at ∼450 nm, which is consistent with the literature reports for ATO.31 The result also indicates the transparency (in the visible range) of the prepared ATO coated FTO electrodes.
3.2. Electrochemical studies using the ATO nano-powder catalyst on GCE
 | ||
| Fig. 4 CV of ATO/GCE and bare GCE (a) without H2O2 and (b) with 300 mM H2O2 in 0.05 M H2SO4 solution at a scan rate of 10 mV s−1 using Ag/AgCl and platinum wire as reference and counter electrodes, respectively. | ||
Apart from the excellent electrocatalytic properties (providing a good onset potential for the selective reduction of H2O2), our prepared ATO catalyst exhibits strong adsorption onto the glassy carbon electrode without the use of any costly binders such as Nafion. The GCE usually requires binders to strongly hold catalysts. Almost all the previously reported cathodic electrocatalysts have used some kind of binder to interface the catalysts on the electrode support for demonstrating their applicability in single compartment peroxide fuel cells. Besides the cost, the use of a binder can lead to a reduction in electrocatalytic activities as it can block the active sites on the catalytic surface. Here, we have not used any such costly binder for the loading of the ATO catalyst on the electrode supports (GCE or FTO). This advantage originates from our organic R–F polymerization co-assisted ATO synthesis method, where the R–F polymer network forms in situ within metal oxide pore volumes and provides active support for binding ATO with GCE. The addition of Nafion during catalyst loading on the GCE was found to inhibit the electrocatalytic activity of ATO. Shiraishi et al.39 recently reported that the presence of Nafion creates a hydrophobic environment around the R–F particles that can eventually block the active sites of the catalyst from interacting with the reactive species.
As we have used Ni foam as the anodic counterpart in the ATO cathodic material based single compartment peroxide fuel cell, the CV using Ni foam (0.8 × 0.9 cm2 active area) as the working electrode was also monitored under the same electrolytic conditions (Fig. S4†). Ni foam shows H2O2 oxidation with an onset potential (∼0.43 V vs. RHE) lower than that of the H2O2 reduction potential achieved by our ATO catalyst. This is consistent with the previous literature report.11 Overall, the CV results suggest that a robust single compartment H2O2 fuel cell can be developed for electricity generation using R–F templated ATO as a potential cathodic material and the widely used Ni foam as the anode.
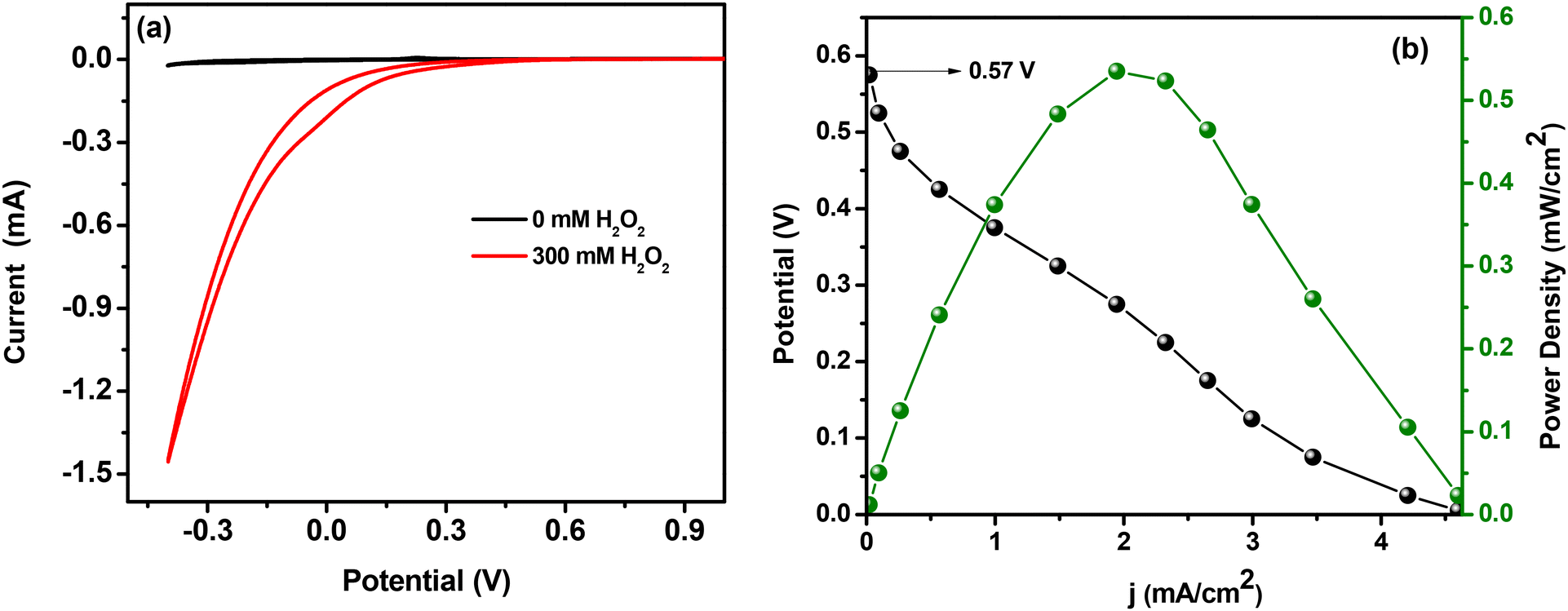 | ||
| Fig. 5 (a) Short circuit current vs. potential plots and (b) the corresponding current density vs. potential/power density plots (with 300 mM H2O2) for a two-electrode cell with ATO/GCE as the cathode and Ni foam as the anode in 0.05 M H2SO4 electrolyte solution at a scan rate of 10 mV s−1. Currents and powers were normalized to the geometric surface area of the working electrode. | ||
Although thermodynamics favoured the present molecular system to achieve an OCV value of 0.57 V, it is still lower than the values reported in a few iron-based systems (some of these systems utilized an external agent to suppress the decomposition of H2O2). Hence further efforts have been made to improve the performance of the single compartment H2O2 fuel cell. The non-electrochemical disproportionation of H2O2 is always a major cause behind the inefficiency of several peroxide fuel cells and the same is an obstacle to the deployment of many metal oxides as cathodes in this regard.40 Other than this, reduced conductivity and slow electron transfer kinetics can also significantly affect the efficiency of a fuel cell.12 Hence by considering these catalysis retardation factors, identical experiments were performed using an ATO/FTO thin film electrode (0.8 × 0.9 cm2) as the cathode, because improved conductivity, high capacitance and faster electron transfer properties are already reported as advantageous for an ATO coated FTO electrode. Extremely smaller and well defined ATO nanopores developed on thin films are said to be the reason behind its superior electrical properties (compared to bulk ATO nano-powder).31
3.3. Electrochemical performance using the ATO coated FTO electrode
 | ||
| Fig. 6 CV of the ATO/FTO electrode without and with 300 mM H2O2 in 0.05 M H2SO4 at a scan rate of 10 mV s−1 with Ag/AgCl and platinum wire as reference and counter electrodes, respectively. | ||
The respective CV measurements (with and without H2O2) using bare FTO and SnO2/FTO (with the same active area) as working electrodes were also examined in this regard. Bare FTO showed minute cathodic current generation upon the addition of peroxide (Fig. S5a†). Its cathodic current is almost two-hundred times less than that of the ATO/FTO electrode. Unlike only the FTO electrode, the SnO2/FTO electrode shows an appreciable amount of cathodic current (with a maximum current of 1 mA) which is still five times less than the cathodic current obtained for the ATO/FTO working electrode. This indicates that undoped SnO2 is also capable of electrocatalytically reducing H2O2, but with a very high onset potential of 0.5 V and a lower current density (Fig. S5b†). Here, the Sn(+4) centres can be catalytically active sites for peroxide reduction. But the superior onset potential and current generated by the ATO coated FTO implies that the doping of Sb into SnO2 imparts improved conductivity, high capacitance and faster electron transfer properties that significantly improve the electrocatalysis.
To study the effect of Sb doping on the catalytic activity of ATO, we have compared the electrocatalytic peroxide reduction of the above-mentioned ATO/FTO sample (6 mol% Sb) with two newly prepared ATO/FTO samples having 3 and 12 mol% Sb contents. From the CV plot (Fig. S6†) analysis, it is found that increasing the Sb content from 3 to 12 mol% induces an improvement in the peroxide reduction onset potential and cathodic current density. This indicates that Sb doping has an impact on the electrocatalytic H2O2 reduction activity of our synthesised ATO catalyst. However, the ATO film produced on the FTO substrate with a higher Sb content (12 mol%) did not provide a very smooth (in terms of surface roughness) and stable film, and hence, we chose to proceed with the 6 mol% Sb containing ATO/FTO film for further fuel cell performance study.
The enhanced electrocatalytic activity of the ATO can be attributed to several factors. The significantly higher electronic conductivity27,41 achieved through the doping of Sb ions in tin oxide (SnO2) can contribute toward its enhanced electrocatalytic activities. ATO has recently been demonstrated as a superior electrocatalyst to SnO2 towards a redox couple (VO2+/VO2+) in acidic solution due to improved electronic conductivity.41 As the electronic conductivity of ATO increases with increasing Sb content, the electrocatalytic activity also increases. In addition, the Sb dopants in SnO2 can act as active catalytic sites for a specific electrocatalytic reaction (peroxide reduction in this case). Recently Li et al.40 have demonstrated ATO nanoparticles as a moderately active peroxidase mimicking nanozyme for paper-based colorimetric detection of glucose. It has been proposed that the dynamic switching of Sb+5 ↔ Sb+3 arises from the potential electron transfer in the ATO material and this can be a possible reason behind its peroxidase like activity. Furthermore, the adsorption of H2O2 on the metal oxide surfaces also plays an important role in their catalytic activities. Strong H2O2 adsorption can reduce the surface catalytic activities by increasing the rate of H2O2 decomposition42 as well as by blocking the active sites with increased adsorption of intermediates and products. Yuan et al.17 have recently reported that, for two different metal oxides, the metal oxide with lower adsorption towards H2O2 exhibits improved peroxide reduction (peroxidase) activity compared to the metal oxide with higher adsorption of peroxide. Therefore, optimally weak adsorption of H2O2 is important for a metal oxide to be potentially utilized as a peroxide reduction cathode catalyst in a single compartment H2O2 fuel cell. From computational investigations, Tada et al.27,30 have recently reported that the strength of H2O2 adsorption is sensitive to the type of metal oxide used and it decreases with increasing Sb doping levels in SnO2. They also found an enhancement of the electrocatalytic activity of SnO2 by the introduction of the Sb dopant. Considering all these factors, it is reasonable to believe that Sb doping in SnO2 makes ATO an efficient peroxide reduction electrocatalyst.
 | ||
| Fig. 7 (a) Current vs. potential plots and (b) the corresponding current density vs. potential/power density plots (with 300 mM H2O2) for a single compartment peroxide fuel cell with ATO/FTO as the cathode and nickel foam as the anode in 0.05 M H2SO4 electrolyte solution at a scan rate of 10 mV s−1. | ||
To check the effect of electrolyte resistance on the output power density of the fuel cell, we performed experiments for an identical two-electrode single compartment fuel cell by increasing the ionic strength of the electrolyte (0.05 M H2SO4) through the addition of 1 M NaCl, which is known to decrease the electrolyte resistance of the cell. The result shows an impressive nearly 3-fold increase in the output current that enabled the maximum power density to reach 1.1 mW cm−2 (Fig. 8). This observation is consistent with the previous literature report.12
 | ||
| Fig. 8 Current density vs. potential/power density plot for a two-electrode system with an ATO/FTO cathode and a nickel foam anode in 0.05 M H2SO4 electrolyte containing 300 mM H2O2 and 1 M NaCl at a scan rate of 10 mV s−1. | ||
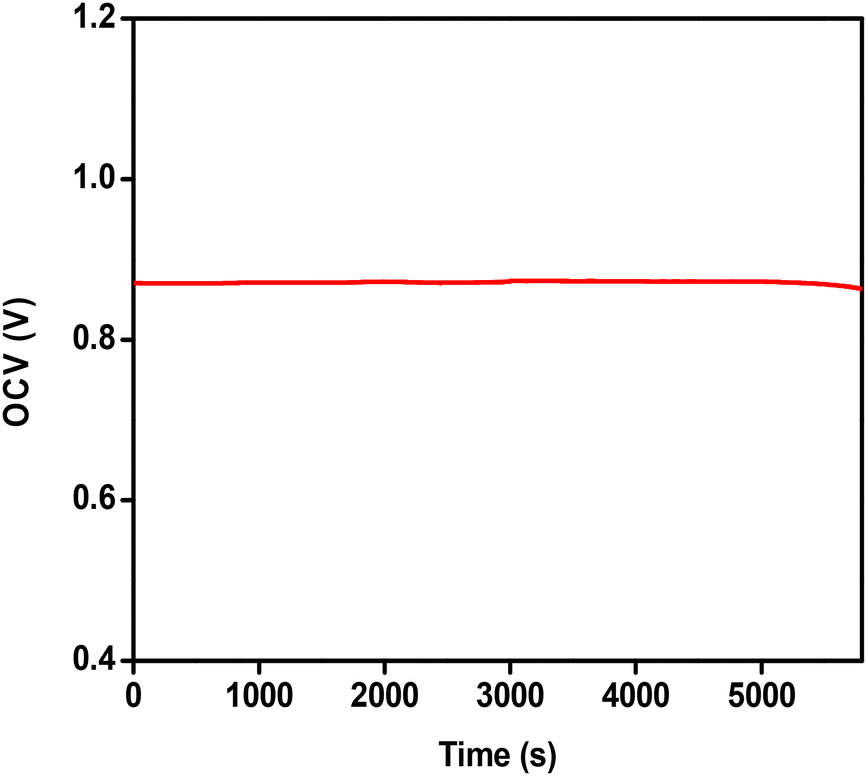 | ||
| Fig. 9 Open circuit voltage (OCV) recorded as a function of time for a two-electrode cell with ATO/FTO as the cathode and Ni foam as the anode in 0.05 M H2SO4 electrolyte solution at a scan rate of 10 mV s−1 with 300 mM H2O2. | ||
4. Conclusions
Here we have demonstrated, for the first time, an electrochemical single compartment acidic H2O2 fuel cell using highly conductive antimony doped tin oxide (ATO) as the cathode and Ni foam as the anode. The fuel cell performances were tested using an ATO coated GCE and ATO coated FTO electrodes as cathodes and widely used Ni foam as the anode. Our developed fuel cell with an ATO/FTO cathodic material achieved a superior OCV of 0.82 V and a maximum power density of 0.32 mW cm−2. The power density of the fuel cell was significantly improved by the addition of 1 M NaCl to the electrolyte, leading to a maximum power density value of 1.1 mW cm−2. The addition of salt decreases the electrolyte resistance and hence, improved performance of the fuel cell is observed. This is the first report on discovering a metal oxide as a potential cathode material for the formation of a robust peroxide fuel cell. Although by using R–F templated ATO we have achieved a superior OCV, there is still scope to further improve the power density. This can be achieved either by adding some external additives to the electrolyte or by small molecular functionalization on the ATO coated electrode surface.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. M. is thankful to the Department of Science and Technology (DST), Government of India for DST/TMD/IC-MAP/2K20/03(C) and DST/INSPIRE/04/2018/000869 grants. A. M. C. D. is thankful to the DST for a research fellowship. A. K. is thankful to the National Institute of Technology Tiruchirappalli for a research fellowship. The authors gratefully acknowledge the facilities provided by the SAIF of MG University, Kottayam for FESEM analysis, the SIF of NIT Trichy for XRD analysis, the CSIF of the University of Calicut for porosity measurements and the CIF of IISER Thiruvananthapuram for XPS measurements.References
- S. Fukuzumi and Y. Yamada, ChemElectroChem, 2016, 3, 1978–1989 CrossRef CAS.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- X. Sun, J. Chen, J. Zhai, H. Zhang and S. Dong, J. Am. Chem. Soc., 2022, 144, 23073–23080 CrossRef CAS PubMed.
- D. K. Perivoliotis, C. Stangel, Y. Sato, K. Suenaga and N. Tagmatarchis, Small, 2022, 18, 2203032 CrossRef CAS PubMed.
- W. Fan, B. Zhang, X. Wang, W. Ma, D. Li, Z. Wang, M. Dupuis, J. Shi, S. Liao and C. Li, Energy Environ. Sci., 2020, 13, 238–245 RSC.
- X. Hu, X. Zeng, Y. Liu, J. Lu and X. Zhang, Nanoscale, 2020, 12, 16008–16027 RSC.
- A. Mahata and B. Pathak, Nanoscale, 2017, 9, 9537–9547 RSC.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Inorg. Chem., 2014, 53, 1272–1274 CrossRef CAS PubMed.
- S.-I. Yamazaki, Z. Siroma, H. Senoh, T. Ioroi, N. Fujiwara and K. Yasuda, J. Power Sources, 2008, 178, 20–25 CrossRef CAS.
- Y. Yamada, S. Yoshida, T. Honda and S. Fukuzumi, Energy Environ. Sci., 2011, 4, 2822–2825 RSC.
- A. Pariyar, G. Vijaykumar, M. Bhunia, S. K. Dey, S. K. Singh, S. Kurungot and S. K. Mandal, J. Am. Chem. Soc., 2015, 137, 5955–5960 CrossRef CAS PubMed.
- Y. Yamada, Y. Fukunishi, S.-I. Yamazaki and S. Fukuzumi, Chem. Commun., 2010, 46, 7334–7336 RSC.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Chem. – Eur. J., 2013, 19, 11733–11741 CrossRef CAS PubMed.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Energy Environ. Sci., 2015, 8, 1698–1701 RSC.
- E. Miglbauer, P. J. Wójcik and E. D. Głowacki, Chem. Commun., 2018, 54, 11873–11876 RSC.
- B. Yuan, H.-L. Chou and Y.-K. Peng, ACS Appl. Mater. Interfaces, 2022, 14, 22728–22736 CrossRef CAS PubMed.
- H. Jia, D. Yang, X. Han, J. Cai, H. Liu and W. He, Nanoscale, 2016, 8, 5938–5945 RSC.
- W. Chen, S. Li, J. Wang, K. Sun and Y. Si, Nanoscale, 2019, 11, 15783–15793 RSC.
- M. Raineri, E. L. Winkler, T. E. Torres, M. V. Mansilla, M. S. Nadal, R. D. Zysler and E. Lima, Nanoscale, 2019, 11, 18393–18406 RSC.
- V. Baldim, F. Bedioui, N. Mignet, I. Margaill and J. F. Berret, Nanoscale, 2018, 10, 6971–6980 RSC.
- K. Suematsu, M. Sasaki, N. Ma, M. Yuasa and K. Shimanoe, ACS Sens., 2016, 1, 913–920 CrossRef CAS.
- Y. Sun, W. D. Chemelewski, S. P. Berglund, C. Li, H. He, G. Shi and C. B. Mullins, ACS Appl. Mater. Interfaces, 2014, 6, 5494–5499 CrossRef CAS PubMed.
- C. Liu, R. Félix, K. Forberich, X. Du, T. Heumüller, G. J. Matt, E. Gu, J. Wortmann, Y. Zhao, Y. Cao, Y. He, L. Ying, A. Hauser, M. F. Oszajca, B. Hartmeier, M. Rossier, N. A. Lüchinger, Y.-S. Liu, J. Guo, K. Nie, R. G. Wilks, J. Bachmann, M. Bär, N. Li and C. J. Brabec, Nano Energy, 2021, 89, 106373 CrossRef CAS.
- W. Chen, Z. Lei, T. Zeng, L. Wang, N. Cheng, Y. Tan and S. Mu, Nanoscale, 2019, 11, 19895–19902 RSC.
- X. Zhang, H. Liu, J. Wang, G. Ren, B. Xie, H. Liu, Y. Zhu and L. Jiang, Nanoscale, 2015, 7, 18763–18769 RSC.
- H. Tada and S.-I. Naya, J. Phys. Chem. C, 2022, 126, 13539–13547 CrossRef CAS.
- M. Karmaoui, A. B. Jorge, P. F. McMillan, A. E. Aliev, R. C. Pullar, J. A. Labrincha and D. M. Tobaldi, ACS Omega, 2018, 3, 13227–13238 CrossRef CAS PubMed.
- L. Liu, M. Ueda and T. Kawaharamura, RSC Adv., 2023, 13, 13456–13462 RSC.
- S.-I. Naya, H. Suzuki, H. Kobayashi and H. Tada, Langmuir, 2022, 38, 4785–4792 CrossRef CAS PubMed.
- S. Sharma, A. M. Volosin, D. Schmitt and D.-K. Seo, J. Mater. Chem. A, 2013, 1, 699–706 RSC.
- X. He, Y. Liu, Q. Wang, T. Wang, J. He, A. Peng and K. Qi, Sci. Rep., 2023, 13, 11107 CrossRef CAS PubMed.
- H. Luo, Z. Fang, N. Song, T. Garvey, R. Lopez and T. J. Meyer, ACS Appl. Mater. Interfaces, 2015, 7, 25121–25128 CrossRef CAS PubMed.
- S.-I. Naya, Y. Shite and H. Tada, Catal. Commun., 2020, 142, 106044 CrossRef CAS.
- S. M. Jassim, N. A. Bakr and F. I. Mustafa, J. Mater. Sci.: Mater. Electron., 2020, 31, 16199–16207 CrossRef CAS.
- J. M. Xu, L. Li, S. Wang, H. L. Ding, Y. X. Zhang and G. H. Li, CrystEngComm, 2013, 15, 3296–3300 RSC.
- R. Medhi, C.-H. Li, S. H. Lee, M. D. Marquez, A. J. Jacobson, T.-C. Lee and T. R. Lee, ACS Appl. Nano Mater., 2019, 2, 6554–6564 CrossRef CAS.
- A. M. Volosin, S. Sharma, C. Traverse, N. Newman and D.-K. Seo, J. Mater. Chem., 2011, 21, 13232–13240 RSC.
- Y. Shiraishi, M. Jio, K. Yoshida, Y. Nishiyama, S. Ichikawa, S. Tanaka and T. Hirai, JACS Au, 2023, 3, 2237–2246 CrossRef CAS PubMed.
- Y. Li, J. Sun, W. Mao, S. Tang, K. Liu, T. Qi, H. Deng, W. Shen, L. Chen and L. Peng, Microchim. Acta, 2019, 186, 403 CrossRef PubMed.
- Y. Lv, C. Yang, H. Wang, J. Zhang, Y. Xiang and S. Lu, Catal. Sci. Technol., 2020, 10, 2484–2490 RSC.
- C. M. Lousada, A. J. Johansson, T. Brinck and M. Jonsson, J. Phys. Chem. C, 2012, 116, 9533–9543 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01375a |
| This journal is © The Royal Society of Chemistry 2024 |