
Insights into the roles of superficial lattice oxygen in formaldehyde oxidation on birnessite†
Zhaoxia
Ma
 a,
Yongqi
Li
a,
Kongyuan
Sun
a,
Jahangeer
Ahmed
d,
Wei
Tian
*b and
Jinjia
Xu
*c
a,
Yongqi
Li
a,
Kongyuan
Sun
a,
Jahangeer
Ahmed
d,
Wei
Tian
*b and
Jinjia
Xu
*c
aCollege of Chemistry & Environment, Southwest Minzu University, Chengdu 610225, Sichuan, China
bSchool of Physical Science and Technology, Jiangsu Key Laboratory of Advanced Negative Carbon Technologies, Soochow University, Suzhou 215006, China
cDepartment of Chemistry and Biochemistry, University of Missouri-St. Louis, One University Blvd, St. Louis, 63121, MO, USA
dDepartment of Chemistry, College of Science, King Saud University, Riyadh-11451, Saudi Arabia
First published on 31st May 2024
Abstract
K+-modified birnessite materials were constructed to remove formaldehyde (HCHO) in this work. The introduction of K+ led to weakening of the Mn–O bonds and enhanced the migration of superficial lattice oxygen, resulting in improved redox properties and catalytic activity. MnO2-3K with the largest specific surface area and greatest abundance of superficial lattice oxygen showed the best catalytic performance at 30–130 °C. The operando analyses reveal that HCHO is primarily activated to dioxymethylene (DOM) and subsequently converted to formate species (*COOH). The accumulation of formate species caused a decline in catalytic performance during extended testing at 30 °C, a challenge that could be mitigated by raising the temperature. Theoretical studies disclose that the *COOH → *H2CO3 step with the largest energy barrier is the rate limiting step for HCHO deep decomposition. Molecular oxygen could be activated at oxygen vacancies to replenish the depleted lattice oxygen after decomposition of carbonate species (*H2CO3) and CO2 and H2O desorption. The adsorbed oxygen and water did not limit the deep oxidation of HCHO. This research presents a promising approach for designing highly efficient, non-noble metal catalysts for formaldehyde degradation.
 Jinjia Xu | Jinjia Xu is an assistant professor in the Department of Chemistry and Biochemistry at the University of Missouri-St. Louis, USA. She obtained her PhD in Materials Science and Engineering from the National Institute for Materials Science (NIMS), Japan, where she was awarded with the Junior Researcher Fellowship and JSPS Fellowship for Young Scientists. Her group's research interests lie in organic chemistry, polymer materials, nanotechnology, and biomedical engineering. The overall goal is to integrate fundamental research in novel soft matter nanotechnology and biomedical engineering with the creation of multifunctional polymers/macromolecules for advanced technologies to address problems related to energy, environment, and healthcare. |
1. Introduction
Formaldehyde (HCHO) primarily emanating from decorative furniture stands out as a prominent indoor pollutant.1–4 The pursuit of technological advancements for the enduring degradation of indoor HCHO at ambient temperature has become an important concern. Catalytic oxidation technology has shown promise in converting HCHO into carbon dioxide and water, devoid of harmful by-products.5–7 While noble-metal-based catalysts exhibit impressive efficiency in HCHO oxidation at room temperature, their widespread application is hindered by the high cost of noble metals and resource scarcity.8–13 Manganese oxides have recently emerged as promising materials for diverse applications in pollution abatement.14–22 However, it is still a great challenge to achieve high catalytic activity and long-term stability at ambient temperature for HCHO oxidation over MnOx-based catalysts.To address the challenge of catalyst inactivation at room temperature, numerous studies have explored modifications to the original MnO2 catalyst to enhance the decomposition of intermediates during catalysis. Recently, alkali metal doping has been identified as an effective method for promoting catalytic activity. Studies by Zhang et al. demonstrated that an increased concentration of K+ significantly enhanced the catalytic efficiency for HCHO oxidation by considerably boosting the lattice oxygen activity.23 Huang and co-authors found that a medium concentration of K+ resulted in large amounts of surface active oxygen species, including surface-adsorbed oxygen and surface hydroxyl groups, with a Langmuir–Hinshelwood mechanism being implicated during the HCHO oxidation process.24 Research by Shi's group indicated that surface-adsorbed oxygen on CoMn oxides could be generated by Na/K doping, which enhanced HCHO oxidation.25 Jia and co-authors reported that K+ enhanced the mobility of the lattice oxygen, which provided surface active oxygen to sustain HCHO oxidation.26 Liu et al. proposed that the addition of K+ over ε-MnO2 provided oxygen vacancies, inducing more active hydroxyl species through water activation.27 These studies collectively highlight the crucial role of potassium in generating surface reactive oxygen species (ROS), which mainly consists of surface lattice oxygen and chemisorbed active oxygen from molecular O2 activation.17,23,27–32 He's group proposed that intermediates are easily formed by the oxidation of surface lattice oxygen at ambient temperature.14 Guo et al. confirmed that surface lattice oxygen species of δ-MnO2 did not participate in HCHO oxidation.33 The above-discussed findings confirm the importance of ROS; however, the effects of ROS (especially surface lattice oxygen) and the detailed reaction route during HCHO oxidation remain to be determined.34
Herein, we present a strategy to construct a series of K+-modified birnessite materials. The weakened Mn–O bond and enhanced migration of superficial lattice oxygen promote the oxidation of catalysts and facilitate HCHO degradation. The adsorbed oxygen and water did not limit the deep oxidation of HCHO. We found that HCHO was first adsorbed on the surface of birnessite, then easily transformed into dioxymethylene (DOM), which was then transformed into formate species. The transformation from formate species to carbonate species is a rate limiting step, which could be accelerated by temperature increments. Finally, carbonate species mineralized to CO2 and H2O. Oxygen vacancies formed after superficial lattice oxygen was consumed and could be replenished by adsorbed oxygen molecules. This work might provide meaningful insights into designing the next generation of efficient MnOx-based catalysts.
2. Experimental section
2.1 Catalyst preparation
All samples were synthesized using a hydrothermal method. For an unmodified MnO2 sample, KMnO4 (8.40 mmol) was dissolved in 70 mL of deionized water by stirring. Then, the solution was transferred into a 100 mL Teflon-lined stainless autoclave and sustained at 140 °C for 8 h. The precursor was separated by filtration and washed with ethanol and H2O, and then dried at 60 °C in a vacuum oven for 12 h. The preparation of the other three samples could be tuned by changing the amount of KCl additive in the KMnO4 solution. Three KCl quantities, i.e., 0.84, 2.52, and 4.20 mmol, were selected in this study, and the prepared samples were labelled as MnO2-1K, MnO2-3K and MnO2-5K, respectively.2.2 Catalyst characterization
The samples were characterized by X-ray diffraction (XRD) patterns, Raman spectra, N2 adsorption and desorption isotherms, scanning electron microscopy (SEM), high-resolution transmission electron microscopy (HRTEM), X-ray photoelectron spectroscopy (XPS), oxygen-temperature-programmed desorption (O2-TPD), hydrogen-temperature-programmed desorption (H2-TPR), and in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). Detailed procedures for characterization and density functional theory (DFT) calculations are described in the ESI.†2.3 Activity test
The process of HCHO degradation was conducted on a fixed bed by filling 100 mg of the catalyst in a quartz tube (ϕin = 6 mm). HCHO was supplied by flowing Ar over paraformaldehyde in a glass bottle that was placed in a water bath. The total gas flow rate was set at 100 mL min−1, and the RH was controlled by bubbling O2 into the water. The concentration of HCHO was tested using the MBTH method, which is described in the ESI.† The produced CO2 was monitored online using a gas chromatograph (GC-7900, Tianmei, China). The HCHO conversion as well as CO2 yield (SCO2) are defined as follows: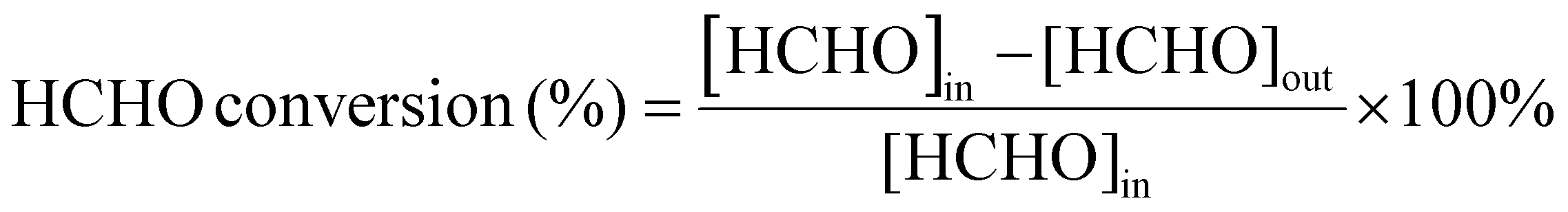 | (1) |
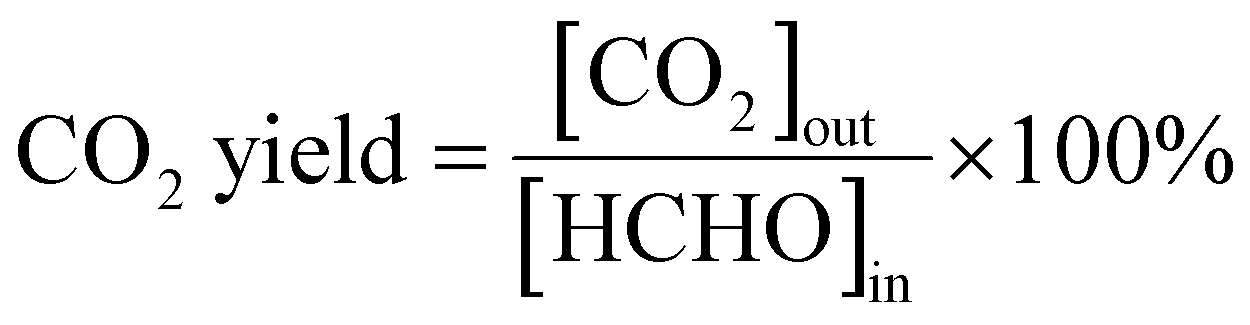 | (2) |
3. Results and discussion
3.1 Catalytic activity
The catalytic performance of MnO2, MnO2-1K, MnO2-3K, and MnO2-5K catalysts was evaluated for their removal efficiency of HCHO. Fig. 1a shows the HCHO conversion with increasing reaction temperature for all four catalysts. Notably, MnO2-3K exhibited the highest activity with 60% HCHO conversion at 30 °C and the lowest Ea at about 7.55 kJ mol−1. The durability of the catalyst is crucial for practical applications. As shown in Fig. 1b, the HCHO conversion of MnO2-3K remained as high as 50% for the initial 20 h, followed by a slight decrease to 40% over the subsequent 28 h at 30 °C. Remarkably, at 90 °C, the conversion remained constant at 100% without any decline within 48 h.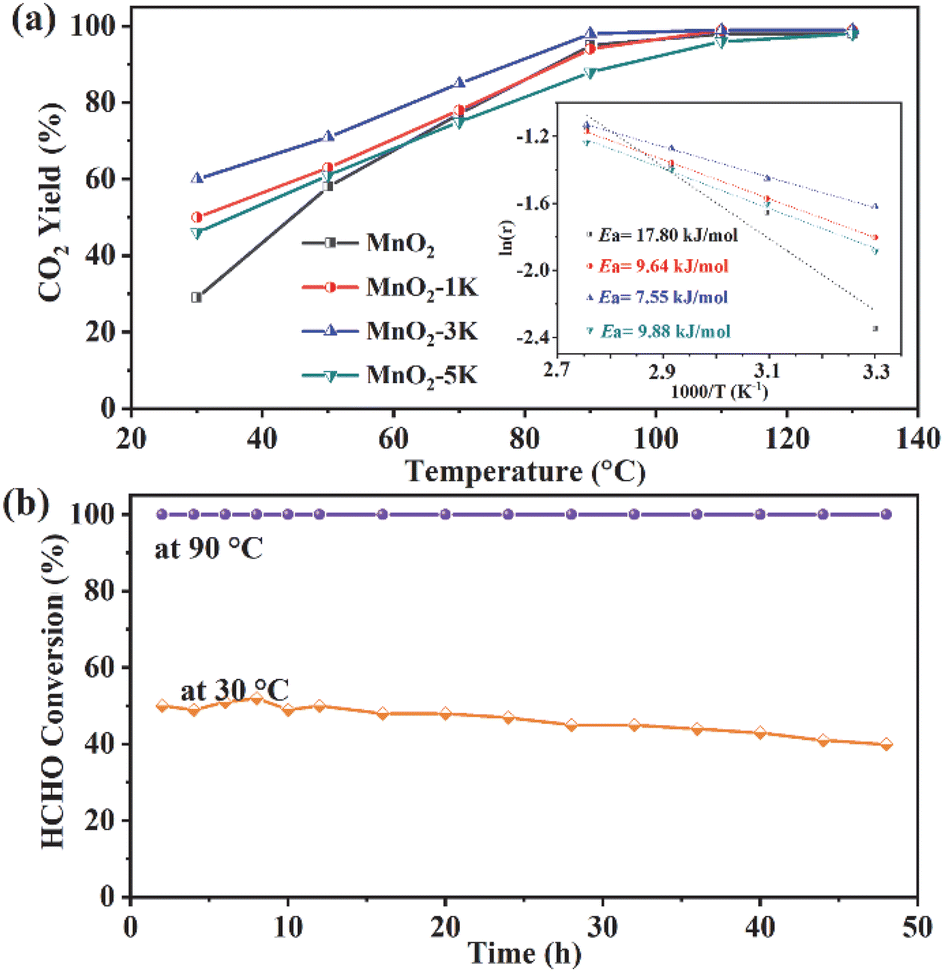 | ||
Fig. 1 (a) Removal efficiency of HCHO on different catalysts with varying temperatures; the inset shows Arrhenius plots for the samples. (b) Stability of HCHO oxidation on MnO2-3K for 48 h at 30 °C and 90 °C. Reaction conditions: 500 ppm HCHO, 20 vol% O2, RH = 70%, GHSV = 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL (g h)−1. 000 mL (g h)−1. | ||
3.2 Morphological properties
The crystal structure of the as-prepared catalysts was characterized by XRD. As shown in Fig. 2a, there are two weak peaks at 12.5° and 25.2° observed for the MnO2-1K sample, which can be assigned to the (001) and (002) planes of birnessite (JPCDS 80-1098). Two asymmetric broad peaks at 37.0° and 66.2° were identified as the (006) and (119) planes corresponding to birnessite-type manganese oxide (JCPDS 18-0802).24,35–38 However, for the other three samples, only the peaks at 37.0° and 66.2° appeared, and the (001) and (002) peaks were not visible, which could be attributed to random interstratification.39 The observation of broad and diffused diffraction peaks in the XRD analysis indicates the poor crystallinity of all samples. | ||
| Fig. 2 (a) XRD patterns and (b) Raman spectra of the prepared samples. (c) N2 adsorption–desorption isotherms measured for the samples; the inset shows the corresponding pore size distribution curves. | ||
Raman spectroscopy was used to further analyze the structural characteristics of the samples. As shown in Fig. 2b, all the samples showed specific peaks from 200 to 1000 cm−1 with two typical bands: the region from 300 to 400 cm−1 and the region from 630 to 640 cm−1, which suggested the assignment of a birnessite-type phase as the main dioxide component.40 The Raman band observed at 630–640 cm−1 corresponds to the stretching of the O–Mn–O bonds, which is perpendicular to the chains of O–Mn–O–Mn–O in the basal plane of the MnO6 octahedra.41–43 The peaks of this Raman mode over MnO2, MnO2-1K, MnO2-3K, and MnO2-5K are at 639, 637, 633, and 633 cm−1, respectively. The slight red shift of the Raman mode indicates the weakening of the Mn–O bond strength along the interlayer direction of the birnessite structure after K+ introduction, which improves oxygen mobility.27,44–46 The Raman and XRD results indicate that tuning the additive content of K+ could affect the crystal structure of birnessite.
N2 adsorption and desorption isotherms were recorded to analyze the porosity and specific surface area of the samples. As shown in Fig. 2c, all the isotherms correspond to a type IV curve. The appearance of H3-type hysteresis loops (P/P0 > 0.8) signified the mesoporous nature of the samples.16,43Table 1 presents the specific surface area calculated using the Brunauer–Emmett–Teller method and the corresponding pore volume data for the samples. Among the samples, MnO2-3K exhibited the highest specific surface area (101 m2 g−1) and pore volume (0.62 cm3 g−1), which may result in intensive adsorption capacity of HCHO, consequently promoting the removal efficiency of HCHO.
| Sample | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|
| MnO2 | 75 | 0.44 | 20 |
| MnO2-1K | 52 | 0.27 | 19 |
| MnO2-3K | 101 | 0.62 | 20 |
| MnO2-5K | 70 | 0.55 | 29 |
Fig. 3 shows the morphology and microstructure of the as-prepared catalysts, which were observed using the SEM and TEM techniques. The nanoparticles were first fabricated for all samples and ultrathin nanosheets grew along the particles. The nanosheets on MnO2-3K displayed the largest laminate size with a thickness of less than 2 nm, which is beneficial for exposing active sites. However, the growth of the nanosheets was inhibited over MnO2-1K and MnO2-5K, which indicated that the additive content of K+ greatly affected the microstructure of birnessite. In the HR-TEM images, lattice fringes with 0.24 nm d-spacing are indicative of the (006) plane of birnessite. Additionally, the HR-TEM images proved the poor crystallinity of all samples.
 | ||
| Fig. 3 SEM, TEM, and HR-TEM images (from left to right) of (a–c) MnO2, (d–f) MnO2-1K, (g–i) MnO2-3K, and (j–l) MnO2-5K. | ||
3.3 Electronic and redox properties
Fig. 4 shows the XPS spectra of all samples. The Mn 2p3/2 spectrum exhibits two distinct peaks at 642.5–643.3 and 641.1–641.3 eV, corresponding to Mn4+ and Mn3+, respectively (Fig. 4a).14,47,48 It is worth noting that the XPS peak for Mn4+ shifts from 642.5 eV (MnO2) to 643.3 eV (MnO2-xK (x = 1, 3, and 5)), which indicates Mn4+ over MnO2-xK (x = 1, 3, and 5) has stronger oxidation ability with lower negative charge density.49 The content of Mn3+ and Mn4+ can be estimated as ∼76% and ∼24% over all samples as shown in Fig. 4c based on their respective peak areas, which indicates the existence of oxygen vacancies. Additionally, Mn/O molar ratios in all samples remained at about 0.4, suggesting the existence of Mn defects, which could enhance the activity and mobility of adjacent oxygen atoms.32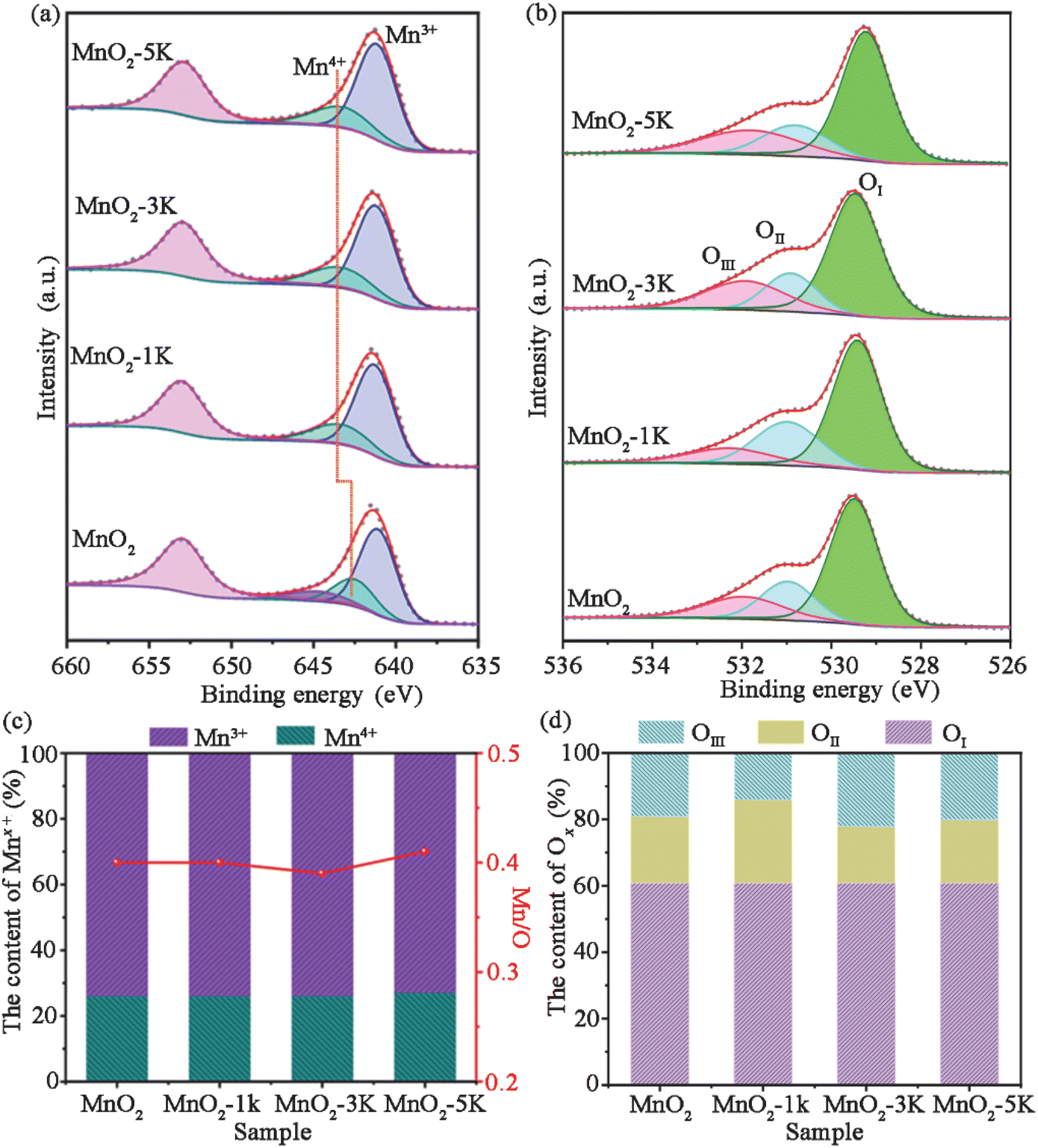 | ||
| Fig. 4 (a) Mn 2p and (b) O 1s regions of the XPS spectra of four samples. (c) The content of Mn3+ and Mn4+ and the ratio of Mn/O. (d) The content of oxygen species. | ||
O 1s spectra (Fig. 4b) were deconvoluted into three peaks at 532.3–532, 531–530.8, and 529.5–529.3 eV, assigned to adsorbed H2O (OIII), adsorbed oxygen and carbonates (OII), and lattice oxygen (OI), respectively.15,50,51 As shown in Fig. 4d, the atomic ratio of lattice oxygen was maintained at about 61% over all samples while MnO2-3K has a greater abundance of adsorbed H2O due to its larger specific surface area.
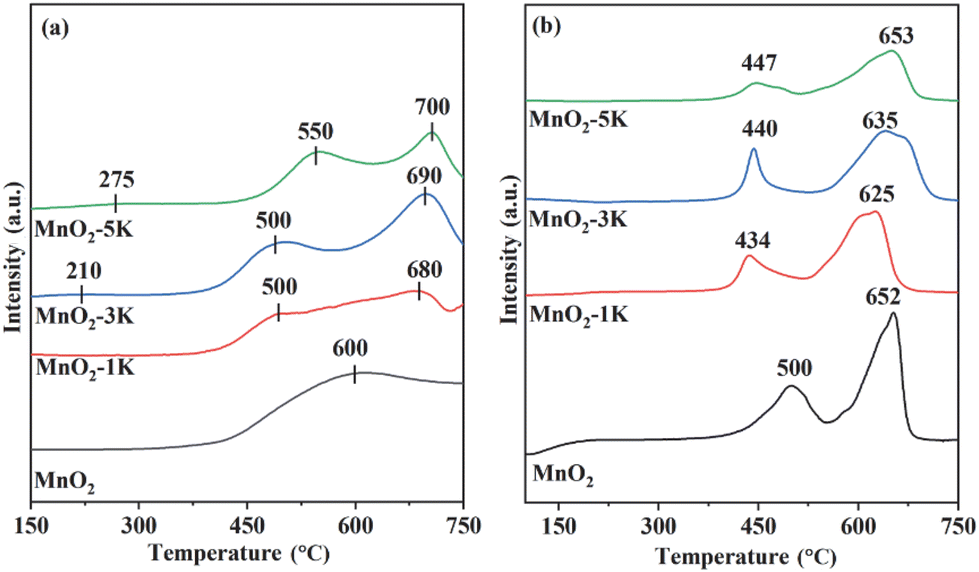
The O2-TPD technique was employed to determine desorption and mobility of oxygen species over the samples (Fig. 5a). TCD signals at 150–400 °C and above 400 °C belong to adsorbed oxygen and lattice oxygen, respectively.24,47,52,53 Chemically adsorbed oxygen was first detected at 300, 350, 210 and 275 °C over MnO2, MnO2-1K, MnO2-3K, and MnO2-5K with ultra-low peak intensity, respectively, and then a large amount of superficial lattice oxygen and bulk lattice oxygen was desorbed. This indicates that superficial lattice oxygen could provide the main active sites for HCHO oxidation over the samples and that adsorbed oxygen does not limit the oxidation of HCHO under the testing conditions with adequate O2, while modification by K+ could promote the migration of superficial lattice oxygen to construct more active centers.
 | ||
| Fig. 5 O2-TPD (a) and H2-TPR (b) profiles of the samples. | ||
As shown in Fig. 5b, H2-TPR profiles of all samples reveal hydrogen consumption in the range of 400–700 °C. MnO2 presents two reduction peaks located at around 500 and 652 °C, which can be assigned to Mn4+ → Mn3+ and Mn3+ → Mn2+, respectively.43 In the first reduction stage, MnO2-1K, MnO2-3K, and MnO2-5K show lower reduction temperatures, i.e., 434, 440, and 447 °C, respectively, which indicate that the migration of surface lattice oxygen was strengthened by K+ doping and induced greater redox properties. Generally, the interaction between oxygen species and hydrogen, and the metal reducibility at low temperatures, are closely associated with the catalytic performance.47 Furthermore, the area of the first reduction peak over K+-modified samples follows the order of MnO2-3K > MnO2-1K > MnO2-5K, suggesting that MnO2-3K displays the highest initial H2 consumption rate, thus leading to the best catalytic activity for HCHO oxidation, as shown in Fig. 1.
3.4 In situ DRIFTS and mechanism
To explore the reaction mechanism and the role of oxygen species during HCHO degradation, intermediate species on MnO2-3K under various reaction conditions were identified via in situ DRIFTS, as shown in Fig. 6. After surveying the literature, the main intermediates are dioxymethylene (CH2O2, DOM), formate species, and carbonate species (Table 2) found on the catalyst's surface.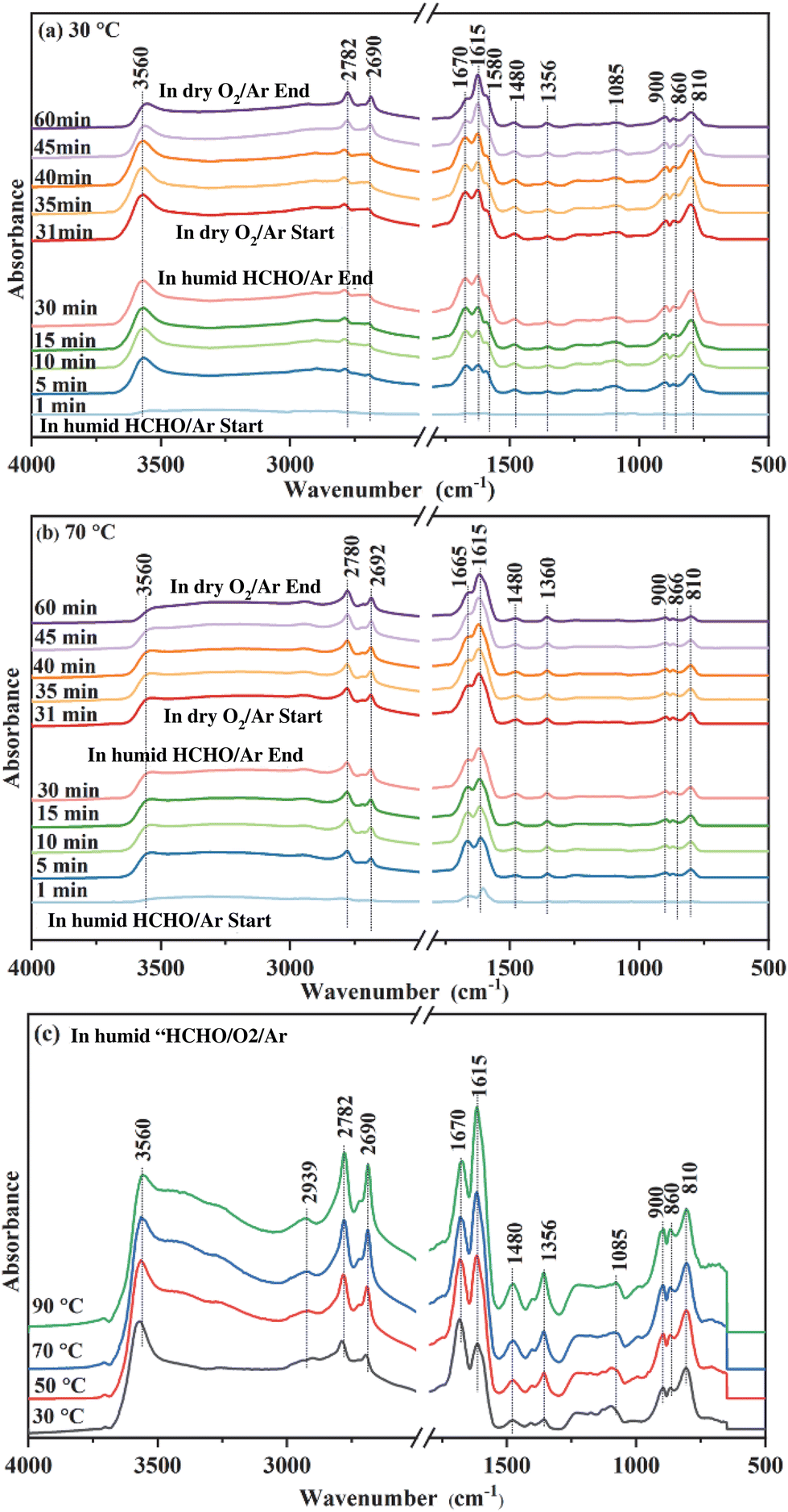 | ||
| Fig. 6 In situ DRIFTS of MnO2-3K, which was in situ heated in Ar at 400 °C for 30 min then cooled to 30 °C (a) and 70 °C (b). Humid HCHO/Ar was first introduced for 30 min, followed by dry O2/Ar for 30 min. (c) In situ DRIFTS of MnO2-3K at different temperatures under a flow of humid HCHO/O2/Ar. Each temperature was scanned for 10 min. | ||
| Peak (cm−1) | Assignment | Ref. |
|---|---|---|
| 810 | ν(CO) of DOM | 54 |
| 860 | ν(CO) of DOM | 54 |
| 900 | ν(CO) of DOM | 27 and 55 |
| 1085 | ν(OCO) of DOM | 56 |
| 1356 | ν s(OCO) symmetric stretch of formate | 55–57 |
| 1480 | Carbonate species | 56, 58–60 |
| 1580 | ν as(OCO) asymmetric stretch of formate | 56 and 57 |
| 1615 | Formate species | 6, 58–60 |
| 1670 | Carbonate species | 58 and 61 |
| 2690 | Formate species | 62 |
| 2780 | ν as(CH) of formate | 62 |
| 2939 | ν(CH) in DOM | 62 |
| 3560 | ν(OH) stretch of adsorbed water | 31 |
In situ DRIFT spectra of MnO2-3K exposed to humid HCHO/Ar at 30 °C over different time periods are shown in Fig. 6a. As the exposure time is prolonged, the intensity of the peaks for DOM (810, 860, 900, and 1085 cm−1), formate species (1356, 1580, 1615, 2690, and 2782 cm−1), carbonate species (1480 and 1670 cm−1), and adsorbed water (3560 cm−1) increased. These results indicate that HCHO could be easily transformed to DOM, which can be further oxidized to formate species by superficial lattice oxygen at 30 °C, leaving the surface oxygen vacancies.27 Then formate species transformed to carbonate species, which could finally decompose to CO2 and H2O. After 30 minutes, the flow of humid HCHO/Ar was switched off and dry O2/Ar was introduced. The peak intensity of DOM decreased significantly as the exposure time increased. As reported, the oxygen molecules could be activated on oxygen vacancies to replenish the lattice oxygen.27 Meanwhile, the peak intensity of the formate species strengthened, indicating that DOM is continuously transformed into formate species after O2 introduction and the peak intensity of the carbonate species was weakened. The above results indicate that formate species could continuously accumulate on the surface of MnO2-3K with adequate O2 in the testing process, which finally induced deactivation of catalytic activity as shown in Fig. 1b. Therefore, it can be concluded that adsorbed oxygen does not accelerate the deep oxidation of HCHO, and the transformation of formate species into carbonate species is the rate limiting step during the deep degradation of HCHO.
The in situ DRIFT spectra of MnO2-3K at 70 °C are displayed in Fig. 6b. DOM, formate species and carbonate species were detected after humid HCHO/Ar was introduced for 1 min. The peak intensity of DOM and formate species remained almost unchanged after 5 min. It can be found that the peak intensity of DOM in Fig. 6b is weaker than that in Fig. 6a. However, the peak intensity of formate species in Fig. 6b is stronger than that in Fig. 6a. These results indicate that the transformation of DOM to formate species was accelerated by the temperature increase, which could be attributed to more activated superficial lattice oxygen at a higher temperature. Furthermore, the peak intensity of carbonate species gradually weakened after 5 min, which indicates that the formed carbonate species decomposed to CO2 and H2O on the surface of MnO2-3K. Adsorbed water was detected as shown in both Fig. 6a and b, and the differing evolution trends of the peak intensity of formate species and carbonate species suggest that the adsorbed water could not significantly promote the transformation of DOM into formate species and the decomposition of carbonate species. Then the flow of humid HCHO/Ar was switched off and dry O2/Ar was introduced. The peak intensity of DOM, formate species and carbonate species decreased, which indicates that the consumed superficial lattice oxygen was replenished by oxygen molecules to continuously degrade HCHO to ensure stable and high catalytic activity at the higher temperature (Fig. 6b). The above results confirm that the transformation of formate species into carbonate species is highly dependent on the activity of superficial lattice oxygen but not on adsorbed oxygen and water.
Spectra from in situ DRIFTS of MnO2-3K exposed to humid HCHO/O2/Ar under different temperatures are shown in Fig. 6c. DOM, formate species, and carbonate species were detected while increasing the temperature. Meanwhile, no other intermediates were observed. This suggests that the coexistence of adsorbed oxygen and water did not affect the reaction route of the HCHO degradation over MnO2-3K.
Based on the results of in situ DRIFTS, the energetically favorable reaction pathways of HCHO oxidation by superficial lattice oxygen over the birnessite material were evaluated using DFT calculations. As shown in Fig. 7, the reaction proceeds in six elementary steps: HCHO adsorption (HCHO → *CH2O), adsorbed HCHO activation (*CH2O → *CHO + *OH), DOM formation (*CHO + *OH → *CHOOH), DOM dehydrogenation to form formate species (*CHOOH → *COOH + *OH), *H2CO3 formation (*COOH + *OH → *H2CO3), *H2CO3 decomposition and CO2 and H2O desorption. Obviously, DOM could be easily formed from adsorbed HCHO with an endothermic energy of 0.11 eV. Then, DOM is further dehydrogenated to form formate species with an exothermic energy of 0.76 eV, which indicates that the transformation of DOM into formate species can easily occur at a relatively low temperature. However, the step from formate species to H2CO3 is an endothermic process with the largest reaction heat of 0.86 eV, which is recognized as the rate determining step of HCHO degradation and in great agreement with the in situ DRIFTS results. The decomposition of *H2CO3 and desorption of CO2 and H2O could easily occur with an exothermic energy of 1.3 eV. Then, oxygen vacancies were formed and could be replenished by adsorbed oxygen molecules. According to the results of the in situ DRIFTS and DFT calculations, it is evident that the superficial lattice oxygen acts as an active center to break the C–H bond of HCHO.
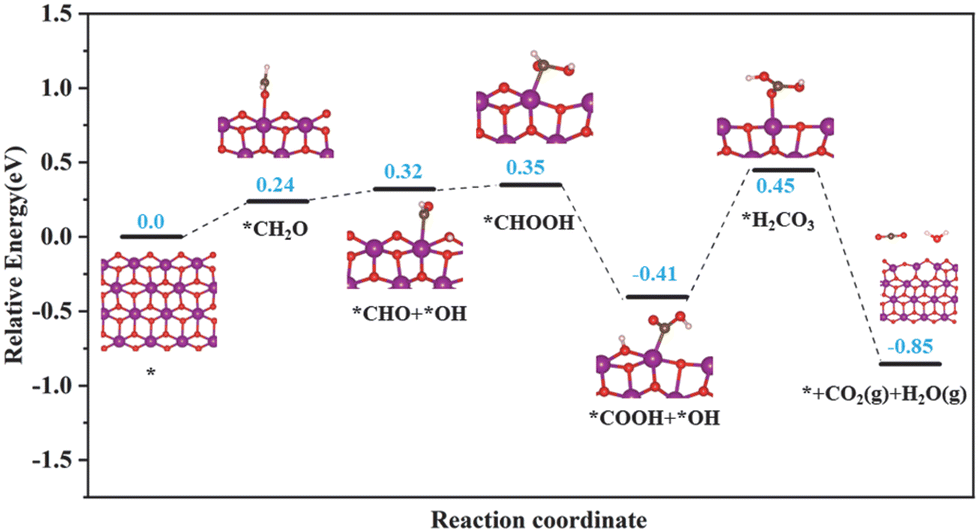 | ||
| Fig. 7 Free energy diagram of HCHO oxidation over the (100) plane of the birnessite material. O: red; Mn: purple; C: gray; and H: white. | ||
4. Conclusions
In summary, we developed a series of K+-modified birnessite materials enriched with reactive superficial lattice oxygen to promote HCHO degradation. Further characterization by various techniques provide evidence that K+ modification induced a weakened Mn–O bond and enhanced migration of superficial lattice oxygen to promote the oxidation of birnessite materials. Moreover, the content of the K+ additive regulated the crystal structure, micro-morphology, and specific surface area of the samples. As a result, MnO2-3K, with the largest specific surface area and the greater abundance of superficial lattice oxygen, exhibited the best catalytic activity and facilitated 60% HCHO conversion at 30 °C. We found that DOM, formate species, and carbonate species are the main intermediates during HCHO oxidation. The adsorbed oxygen and water did not limit the deep degradation of HCHO. Moreover, the accumulation of formate species resulted in the deactivation of MnO2-3K at the relatively low temperature of 30 °C. Elevated temperatures can accelerate the transformation of formate species to carbonate species then to CO2 and H2O. This work demonstrates that reactive superficial lattice oxygen on birnessite materials is the main active center for HCHO degradation.Author contributions
Zhaoxia Ma: methodology, investigation, writing – original draft, and funding acquisition. Yongqi Li: methodology. Kongyuan Sun: methodology. Jahangeer Ahmed: supervision. Wei Tian: writing – review & editing and supervision. Jinjia Xu: writing – review & editing and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of the Fundamental Research Funds for the Central Universities, Southwest Minzu University (Grant No. RQD2021085), and the Natural Science Foundation of Sichuan Province (Grant No. 24NSFSC1008). The authors also thank the Researchers Supporting Project (number RSP2024R391), King Saud University, Riyadh, Saudi Arabia. The University of Missouri-St. Louis Research Award to J. Xu is greatly acknowledged.References
- B. Robert and G. Nallathambi, Environ. Chem. Lett., 2021, 19, 2551–2579 CrossRef CAS.
- B. A. Welsh, M. E. Corrigan, E. Assaf, K. Nauta, P. Sebastianelli, M. J. T. Jordan, C. Fittschen and S. H. Kable, Nat. Chem., 2023, 15, 1350–1357 CrossRef CAS PubMed.
- J. Y. Zheng, H. Zhang, J. D. He, B. H. Tian, C. B. Han, Z. Cui and H. Yan, Nanoscale Adv., 2023, 5, 2027–2037 RSC.
- A. C. Lewis, D. Jenkins and C. J. M. Whitty, Nature, 2023, 614, 220–223 CrossRef CAS PubMed.
- J. Y. Zheng, W. K. Zhao, L. Song, H. Wang, H. Yan, G. Chen, C. B. Han and J. Zhang, Green Energy Environ., 2023, 8, 626–653 CrossRef CAS.
- E. Gao, Q. Jin, T. Zhang, L. Han, N. Li, J. Xu, S. Yao, Z. Wu, J. Li, J. Zhu and W. Wang, Chem. Eng. J., 2023, 474, 145618 CrossRef CAS.
- L. Zhao, Y. Yang, J. Liu and J. Ding, Chemosphere, 2023, 330, 138754 CrossRef CAS PubMed.
- X. Liu, C. Wang, Y. Chen, Q. Qin, Y. Li and H. He, J. Environ. Sci., 2023, 125, 811–822 CrossRef CAS PubMed.
- Y. Guo, Z. Di, X. Guo, Y. Wei, R. Zhang and J. Jia, J. Environ. Sci., 2023, 125, 135–147 CrossRef CAS PubMed.
- Z. Zhang, G. He, Y. Li, C. Zhang, J. Ma and H. He, Environ. Sci. Technol., 2022, 56, 10916–10924 CrossRef CAS PubMed.
- L. Zhang, Q. Bao, B. Zhang, Y. Zhang, S. Wan, S. Wang, J. Lin, H. Xiong, D. Mei and Y. Wang, JACS Au, 2022, 2, 1651–1660 CrossRef CAS PubMed.
- S. Sun, X. Wu, Z. Huang, H. Shen, H. Zhao and G. Jing, Chem. Eng. J., 2022, 435, 135035 CrossRef CAS.
- X. Chen, S. Hu, X. Qin, M. Chen, J. Zhang, X. Bao and C. Zhang, ACS Appl. Nano Mater., 2023, 6, 8763–8771 CrossRef CAS.
- C. Wang, J. Chen, Q. Li, S. Su, H. Jia and H. He, Environ. Sci. Technol., 2023, 57, 4598–4607 CrossRef CAS PubMed.
- L. Shi, X. Zhou, Y. Guo, Y. Li, C. Yan, Q. Han, L. Zhang and W. Zhang, J. Hazard. Mater., 2023, 441, 129836 CrossRef CAS PubMed.
- X. Liu, J. Wu, S. Zhang, Q. Li, Z. Wu and J. Zhang, Appl. Catal., B, 2023, 320, 121994 CrossRef CAS.
- R. Fang, X. Huang, X. A. Luo, Y. Sun, Z. Liu, L. Ao, F. Dong and H. Huang, J. Environ. Chem. Eng., 2023, 11, 109064 CrossRef CAS.
- G. Abdallah, J.-M. Giraudon, N. Nuns, A. Addad, R. Morent, N. De Geyter and J.-F. Lamonier, Appl. Surf. Sci., 2023, 618, 156559 CrossRef CAS.
- M.-S. Niu, H.-H. Yang, H. Zhou, X. Yi, X. Zhou, J. Zhan and Y. Liu, Chem. Eng. J., 2022, 431, 133928 CrossRef CAS.
- X. Li, G. Fang, X. Qian and Q. Tian, Chem. Eng. J., 2022, 428, 131052 CrossRef CAS.
- C. Ma, S. Sun, H. Lu, Z. Hao, C. Yang, B. Wang, C. Chen and M. Song, J. Hazard. Mater., 2021, 414, 125542 CrossRef CAS PubMed.
- R. Yang, Y. Fan, R. Ye, Y. Tang, X. Cao, Z. Yin and Z. Zeng, Adv. Mater., 2021, 33, 2004862 CrossRef CAS PubMed.
- S. Rong, K. Li, P. Zhang, F. Liu and J. Zhang, Catal. Sci. Technol., 2018, 8, 1799–1812 RSC.
- J. Ji, X. Lu, C. Chen, M. He and H. Huang, Appl. Catal., B, 2020, 260, 118210 CrossRef CAS.
- Z. Wang, B. Chen, M. Crocker, L. Yu and C. Shi, Appl. Catal., A, 2020, 596, 117512 CrossRef CAS.
- A. Yusuf, Y. Sun, C. Snape, J. He, C. Wang, Y. Ren and H. Jia, Mol. Catal., 2020, 497, 111204 CrossRef CAS.
- F. Wang, Y. Feng, Z. Wang, Y. Liu, H. Gu and X. Liu, Appl. Surf. Sci., 2023, 624, 157127 CrossRef CAS.
- J. Wang, J. Li, C. Jiang, P. Zhou, P. Zhang and J. Yu, Appl. Catal., B, 2017, 204, 147–155 CrossRef CAS.
- J. Wang, G. Zhang and P. Zhang, J. Mater. Chem. A, 2017, 5, 5719–5725 RSC.
- Z. Han, C. Wang, X. Zou, T. Chen, S. Dong, Y. Zhao, J. Xie and H. Liu, Appl. Surf. Sci., 2020, 502, 144201 CrossRef.
- S. Wang, J. Xie, Z. Deng, M. Wu and F. Wang, Fuel, 2023, 344, 128141 CrossRef CAS.
- T. He, Y. Zhou, D. Ding and S. Rong, ACS Appl. Mater. Interfaces, 2021, 13, 29664–29675 CrossRef CAS PubMed.
- Y. Wang, K. Liu, J. Wu, Z. Hu, L. Huang, J. Zhou, T. Ishihara and L. Guo, ACS Catal., 2020, 10, 10021–10031 CrossRef CAS.
- L. Miao, J. Wang and P. Zhang, Appl. Surf. Sci., 2019, 466, 441–453 CrossRef CAS.
- J. Zhang, Y. Hu, H. Zheng and P. Zhang, Catal. Sci. Technol., 2020, 10, 3603–3612 RSC.
- J. Yang, S. Ren, B. Su, Y. Zhou, G. Hu, L. Jiang, J. Cao, W. Liu, L. Yao, M. Kong, J. Yang and Q. Liu, Catal. Lett., 2021, 151, 2964–2971 CrossRef CAS.
- Y. Jian, X. Feng, M. Tian, Z. Jiang and C. He, Appl. Surf. Sci., 2021, 559, 149905 CrossRef CAS.
- N. Wang, W. Li, J. Liang, Y. Huang, Q. Cai, M. Hu, Y. Chen and Z. Shi, J. Alloys Compd., 2020, 846, 156396 CrossRef CAS.
- F. Polzer, D. A. Kunz, J. Breu and M. Ballauff, Chem. Mater., 2010, 22, 2916–2922 CrossRef CAS.
- H. Radinger, P. Connor, R. Stark, W. Jaegermann and B. Kaiser, ChemCatChem, 2020, 13, 1175–1185 CrossRef.
- Z. M. Chan, D. A. Kitchaev, J. N. Weker, C. Schnedermann, K. Lim, G. Ceder, W. Tumas, M. F. Toney and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5261–E5268 CAS.
- R. E. John, A. Chandran, J. George, A. Jose, G. Jose, J. Jose, N. V. Unnikrishnan, M. Thomas and K. C. George, Phys. Chem. Chem. Phys., 2017, 19, 28756–28771 RSC.
- J. Zhang, L. Zhang, Y. Cheng and Y. Liu, Fuel, 2023, 332, 126104 CrossRef CAS.
- A. M. Zhang, H. F. Cao, X. X. Pan, J. J. Zhu and X. S. Wu, Thin Solid Films, 2022, 750, 139186 CrossRef CAS.
- X. Duan, T. Zhao, B. Niu, Z. Wei, G. Li, Z. Zhang, J. Cheng and Z. Hao, Adv. Sci., 2022, 10, 2205054 CrossRef PubMed.
- C. Julien, M. Massot, R. Baddour-Hadjean, S. Franger, S. Bach and J. P. Pereira-Ramos, Solid State Ionics, 2003, 159, 345–356 CrossRef CAS.
- M. Zhang, G. Li, Q. Li, J. Chen, E. A. Elimian, H. Jia and H. He, Environ. Sci. Technol., 2023, 57, 4286–4297 CrossRef CAS PubMed.
- X. Zhai, L. Li, S. Song, J. Zhang, J. Ma, C. Xie and W. Chu, Fuel, 2023, 334, 126780 CrossRef CAS.
- G. Greczynski and L. Hultman, J. Appl. Phys., 2022, 132, 011101 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- J. Stoch and J. Gablankowska-Kukucz, Surf. Interface Anal., 1991, 17, 165–167 CrossRef CAS.
- S. Wu, H. Liu, Z. Huang, H. Xu and W. Shen, Appl. Catal., B, 2022, 312, 121387 CrossRef CAS.
- J. Mei, Y. Shen, Q. Wang, Y. Shen, W. Li, J. Zhao, J. Chen and S. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 35694–35703 CrossRef CAS PubMed.
- J. Xie, S. Wang, K. Zhao, M. Wu and F. Wang, Inorg. Chem., 2023, 62, 904–915 CrossRef CAS PubMed.
- L. Qin, S. Huang and H. Cheng, J. Colloid Interface Sci., 2023, 633, 453–467 CrossRef CAS PubMed.
- X. Wu, S. Sun, R. Wang, Z. Huang, H. Shen, H. Zhao and G. Jing, J. Hazard. Mater., 2023, 454, 131434 CrossRef CAS PubMed.
- M. Zhang, J. Chen, Z. Xu, Y. Ding, Z. Yan, L. Yue and L. Shi, Catal. Sci. Technol., 2023, 13, 3416–3426 RSC.
- K. Coenen, F. Gallucci, B. Mezari, E. Hensen and M. van Sint Annaland, J. CO2 Util., 2018, 24, 228–239 CrossRef CAS.
- F. Liu, S. Zhang, L. Wan, Y. Hao, J. Li, H. Wang, Z. Li, Q. Li and C. Cao, J. Hazard. Mater., 2023, 458, 131969 CrossRef CAS PubMed.
- J. Xu, T. Zhang, Y. Sun, S. Fang, Z. Wu, J. Zhu, L. Dai, W. Liu, B. Zhang, J. Zhang, S. Yao, E. Gao and J. Li, Appl. Catal., A, 2024, 670, 119519 CrossRef CAS.
- X.-H. Liu, T. Lu, X. Jiao, Z. Jiang, C. Chen, Y. Wang, Y. Jian and C. He, Environ. Sci. Technol., 2024, 58, 1752–1762 CrossRef CAS PubMed.
- J. Xie, S. Wang and F. Wang, Appl. Surf. Sci., 2024, 644, 158709 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01089b |
| This journal is © The Royal Society of Chemistry 2024 |