
Efficient reduction of CO2 and inhibition of hydrogen precipitation by polyoxometalate photocatalyst modified with the metal Mn†
Guifen
Li
a,
Yulan
Gu
a,
Rui
Ren
a,
Sitan
Li
*a,
Houen
Zhu
a,
Dongdong
Xue
a,
Xiangyi
Kong
a,
Ziyi
Zheng
a,
Nuo
Liu
a,
Bei
Li
*b and
Jiangwei
Zhang
 *ac
*ac
aCollege of Energy Material and Chemistry, Inner Mongolia University, Hohhot 010021, P. R. China. E-mail: sitanli@imu.edu.cn; zjw11@tsinghua.org.cn; jwz@imu.edu.cn
bCancer Centre and Institute of Translational Medicine, Faculty of Health Sciences, MoE Frontiers Science Center for Precision Oncology, University of Macau, Macau SAR 999078, China. E-mail: beili@um.edu.mo
cOrdos Laboratory, Ordos 017000, P. R. China
First published on 11th June 2024
Abstract
Photocatalytic reduction of CO2 to chemical fuels is attractive for solving both the greenhouse effect and the energy crisis, but the key challenge is to design and synthesize photocatalysts with remarkable performance under visible light irradiation. Efficient catalytic carbon dioxide reduction (CO2RR) with light is considered a promising sustainable and clean approach to solve environmental problems. Herein, we found a new photocatalyst ([Mn(en)2]6[V12B18O54(OH)6]) (abbreviated as Mn6V12) based on the modifiability of polyoxometalates, in which Mn acts as a modifying unit to efficiently reduce CO2 to CO and effectively inhibit the hydrogen precipitation reaction. This Mn modified polyoxometalate catalyst has a maximum CO generation rate of 4625.0 μmol g−1 h−1 and a maximum H2 generation rate of 499.6 μmol g−1 h−1, with a selectivity of 90.3% for CO generation and 9.7% for H2 generation. This polyoxometalate photocatalyst can effectively reduce CO and inhibit the hydrogen precipitation reaction. It provides a new idea for the efficient photocatalytic carbon dioxide reduction (CO2RR) with polyoxometalate catalysts.
 Jiangwei Zhang | Prof. Dr Jiangwei Zhang is currently a “Steed plan High level Talents” Professor, “Grassland Talents” of Inner Mongolia Autonomous Region, Principle Investigator from College of Energy Material and Chemistry under leadership of Dean Academician Dongyuan Zhao, Inner Mongolia University. He received his Ph.D. from the Department of Chemistry, Tsinghua University (THU) in 2016. He has published 184 innovative publications with H-index = 45. Currently, His research focuses on the common key scientific issues “materials structure and reaction mechanism dynamically and precisely by visual detection and determination” and “Advanced characterization methodology and energy catalytic materials – Interdisciplinary”. |
1. Introduction
With the burning of large quantities of carbon containing fossil fuels and the anthropogenic emission of CO2 into the atmosphere, the problem of global warming is worsening, threatening the safety of human society and the natural living environment.1 The environmental problems brought about by the greenhouse effect, energy shortages and fossil energy consumption have become major challenges for the common future of mankind.2–4 Therefore, Carbon Capture, Utilization and Storage (CCUS) is considered to be one of the most realistic ways to mitigate CO2 concentration and slow down global warming.5,6 Among various CO2 utilization technologies, the conversion of CO2 into chemicals and fuels through solar photocatalysis holds great promise for solving the global energy shortage and reducing the greenhouse effect.7,8 The reduction of CO2 to valuable hydrocarbon fuels also offers a promising strategy for a carbon neutrality energy cycle.9 The conversion of CO2 into commercial chemicals and useful fuels, similar to that of plant photosynthesis by modelling plant photosynthesis, has a wide range of applications in the organic industry and in oxidation fuel cells.10–17 This slows down energy consumption processes and reduces CO2 concentrations.18,19 The two-electron reduction products of syngas (from CO and H2) and formic acid are important raw materials and liquid fuels that are widely recognized as promising renewable fuels, which are also highly conducive to alleviating the energy crisis and climate change.20–22Polyoxometalates (POMs) are a class of clusters formed by the coordination of early transition metals such as Mo, W, V, etc. in their highest oxidation states with oxygen atoms.23–26 Which is an inorganic crystalline material with an oxygen-rich surface that has excellent physical and chemical properties.27 It has been extensively studied in the fields of catalysis, medicine and materials science.28,29 POMs can not only be structurally modified at the molecular level, but also have a good ability to carry and release electrons without changing their structural properties.30 Driven by light, electrons in POMs can be excited from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO), and the excited electrons are transferred from the valence band (VB) to the conduction band (CB) between the valence bands, resulting in the formation of electron–hole pairs in the valence and conduction bands, and the electron–hole pairs have a strong redox property and are used to catalyze the redox reactions.31,32 These advantages give POMs unique properties for photocatalytic CO2 reduction. In addition, the modifiability of the structure facilitates the introduction of functional groups and transition metals as active center to modulate the yield and selectivity of CO2 reduction.33 Transition metal modified polyoxometalates with redox pairs facilitate electron transfer and indicate structure–activity relationships in photochemical CO2 reduction, exhibiting good photocatalytic activity for CO2 reduction for more efficient solar-to-chemical energy conversion.34,35 As a result, more and more attention has been paid to the potential applications in various fields. However, currently developed catalysts for photocatalytic CO2 reduction still suffer from a number of limitations, including restricted use of visible light and poor selectivity.7 Therefore, it is urgent and challenging to explore new transition metal modified polyoxometalate photocatalysts with high activity and selectivity.
In this paper, we present a new vanadium borate cluster based on modification with the metal Mn,36–38 ([Mn(en)2]6[V12B18O54(OH)6]) (abbreviated as Mn6V12) can reduce CO2 to CO with high efficiency and inhibit the hydrogen precipitation reaction effectively. The Mn modified vanadium borate clusters showed a maximum CO production rate of 4625.0 μmol g−1 h−1 and a maximum H2 production rate of 499.6 μmol g−1 h−1, with a selectivity of 90.3% for CO production and 9.7% for H2 production. The high efficiency of CO reduction and the effective inhibition of hydrogen precipitation reaction were better than those of the same type of polyoxometalate catalysts. It provides a new way for the efficient photocatalytic carbon dioxide reduction (CO2RR ) of transition metal modified polyoxometalate photocatalysts.
2. Experimental component
2.1. Synthesis of [Mn(en)2]6[V12B18O54(OH)6]
The synthesis was based on known literature and replaced with Mn(CH3COO)2·4H2O (0.623 g, 2.5 mmol), other reaction conditions were not changed.39 Further details on the experimental methods and procedures are described in the ESI.†2.2. Photocatalytic reactions
Prior to the reduction, 2 mL of triethanolamine (TEOA) was used as a sacrificial reagent, [Ru(bpy)3] Cl2·6H2O 11.6 mg as a photosensitizer, 8 mL of acetonitrile (CH3CN) and 2 mL of deionized water as solvents, and they were transferred into a 60 mL quartz glass reactor with a dispersed catalyst (2 mg) with a lid to carry out the photocatalytic reduction of CO2 experiment. Afterwards, CO2 gas was bubbled into the mixed solution for 15 min until the CO2 concentration was saturated and oxygen was removed from the device to ensure that the system was carried out in a CO2 atmosphere. The reaction was kept at ∼20 °C by condensing circulating water and irradiated under a 300 W xenon lamp with a 420 nm cut-off filter. After 1 h of illumination under visible light, the gaseous products (CO and H2) were collected with a 1 mL syringe and then immediately tested and analysed with a GC-2014C.The EXAFS spectra were obtained by subtracting the post-edge background from the overall absorption and then normalizing with respect to the edge-jump step. Subsequently, the χ(k) data of were Fourier transformed to real (R) space using a hanging windows (dk = 1.0 Å−1) to separate the EXAFS contributions from different coordination shells. To obtain the quantitative structural parameters around central atoms, least-squares curve parameter fitting was performed using the ARTEMIS module of Demeter software packages
The following EXAFS equation was used:
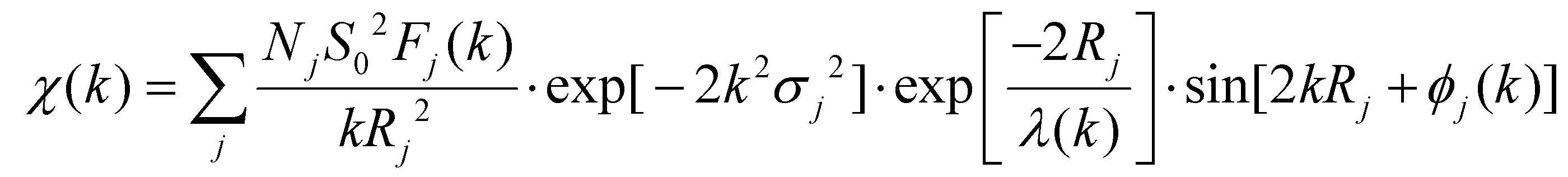
All fits were performed in the R space with k-weight of 2 while phase correction was also applied in the first coordination shell to make R value close to the physical interatomic distance between the absorber and shell scattered. The coordination numbers of model samples were fixed as the nominal values. While the S02, internal atomic distances R, Debye–Waller factor σ2, and the edge-energy shift Δ were allowed to run freely.
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (No. 148), a = b = 21.0608 Å, c = 21.1268 Å, α = β = 90°, γ = 120°, V = 8115.5 Å3, Z = 3, 3176 reflections measured, R1 = 0.0640, wR2 = 0.0826.
(No. 148), a = b = 21.0608 Å, c = 21.1268 Å, α = β = 90°, γ = 120°, V = 8115.5 Å3, Z = 3, 3176 reflections measured, R1 = 0.0640, wR2 = 0.0826.
CCDC 2324078 contains the supplementary crystallographic data for compound 1 respectively in this paper.†
3. Results and discussion
3.1. Catalyst structure
As shown in Fig. 1a and b, Mn6V12 has a B18O42 ring consisting of six B3O7 ternary rings sharing oxygen atoms. The top of the ring consists of six VO5 quadrangular cones conjoined by the common side of the lid, and the bottom is two segments consisting of three VO5 quadrangular cones conjoined by the common side.40 Overall there is a V12 cluster embedded in the B18 ring. In the [B3O7] unit, the B atom displays two different coordination geometries. Two B atoms are coordinated with four oxygen atoms to form a tetrahedral shape. One B atom is coordinated with three oxygen atoms to form a planar triangle. The oxygen atom on the periphery of the B18 ring is attached to the transition metal Mn and the ring is modified with six manganese ions. From the TEM image of Mn6V12 (Fig. 1c), it is observed that Mn6V12 is similar to spherical micron-sized plush clusters, and the plush edges are a lot of consisting of thin and long rod-like structures with a crystallographic spacing of d = 0.32 nm. The extensive rod-like structure is supposed to be the active center of the cluster and serves to increase the active site.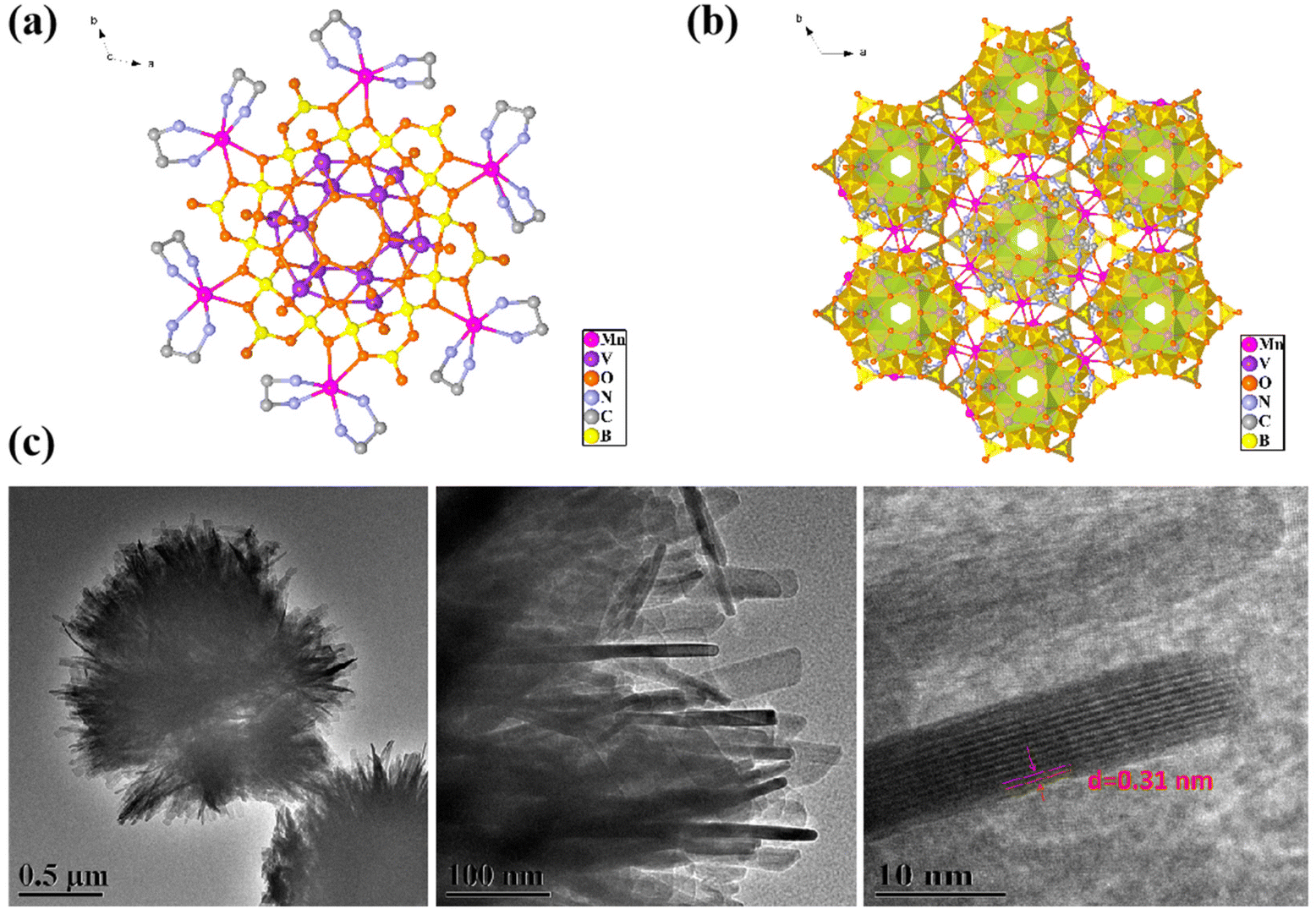 | ||
| Fig. 1 (a) Schematic crystal structure of Mn6V12; (b) crystal stacking diagram of Mn6V12; (c) TEM image of Mn6V12. | ||
In order to determine the valence and coordination structure of atoms at the atomic level, X-ray absorption fine structure (XAFS) measurements were performed.41,42 The electronic structures and coordination environments of V and Mn in Mn6V12 were analyzed using X-ray absorption spectroscopy (XAS), as well as Mn foil, MnO, MnO2, MnPc, V foil, and V2O5 as standard references. In Fig. 2a, X-ray measurements in terms of the K-edge of V in the absorption near edge structure (XANES) show that the absorption edge of Mn6V12 is located between the absorption edges of V foil and V2O5. Mn6V12 exhibits a V K-edge spectrum similar to that of V2O5, but the binding energy at the absorption edge of the V K-edge spectrum of Mn6V12 is lower than that of V2O5, thus determining that the valence state of V is close to +5 and lower than +5. The leading edge peak at ∼5470 eV in Fig. 2a is attributed to the dipole-prohibited 1s to 3d electron jumps,43the decrease in the intensity of the leading edge peaks compared to V2O5 indicates that the introduction of Mn atoms has resulted in an elevated local symmetry of Mn6V12. It is shown that Mn6V12 is structurally stable around the V atom. The k2-weighted Fourier transform EXAFS (FT-EXAFS) spectra (after phase correction) of standard V foil, V2O5 and Mn6V12 are shown in Fig. 2b. The corresponding specific fitting curves are shown in Fig. 2c and d, and the specific fitting parameters for Mn6V12 on the V k-side are shown in Table S1.† The absence of metal V–V scattering paths (2.55 Å) in the V k-edge EXAFS spectrum of Mn6V12 verifies the atomic dispersion of V (Fig. 2b). The first peak of Mn6V12 is located at 1.63 Å, which is 0.24 Å shorter compared to the representative V–O coordination in V2O5 (1.87 Å). The peak of Mn6V12 located at ∼3.30 Å is in the vicinity of the V–O–V of V2O5 (3.10 Å), which is broad and asymmetric, suggesting the presence of double coordination of V–O–V and V–O–B in Mn6V12, and in this way, further suggesting that due to the simultaneous presence of the two coordinating sites, there is a shortening of the V–O bond. The optimized model fitted well to the EXAFS spectra (Fig. 2c–d, and Table S1†), and the best-fit analysis clearly confirmed that the coordination numbers of V–O, V–O–B, and V–O–V were 5, 4, and 2, respectively. This means that the V center is connected to 5 O atoms, two of the V atoms are connected by 1 O atom and the other 2 oxygen atoms are each connected to a B atom, a result that is consistent with the structure of the material. The wavelet transform (WT) (Fig. 2e–g) was applied to the k2-weighted EXAFS data to further investigate the local environment of V on Mn6V12. As shown in Fig. 2g, two peaks exist for Mn6V12 at 1.5 Å–1.75 Å and 2.25 Å–2.5 Å, corresponding to V–O and V–O–V(B). Compared to the WT data for V2O5 (Fig. 2e), the V–O coordination in Mn6V12 is significantly shortened, consistent with the results in Fig. 2b. In addition, there is no V–V coordination in Mn6V12 compared to V foil (Fig. 2f), further illustrating the atomic dispersion of the V.
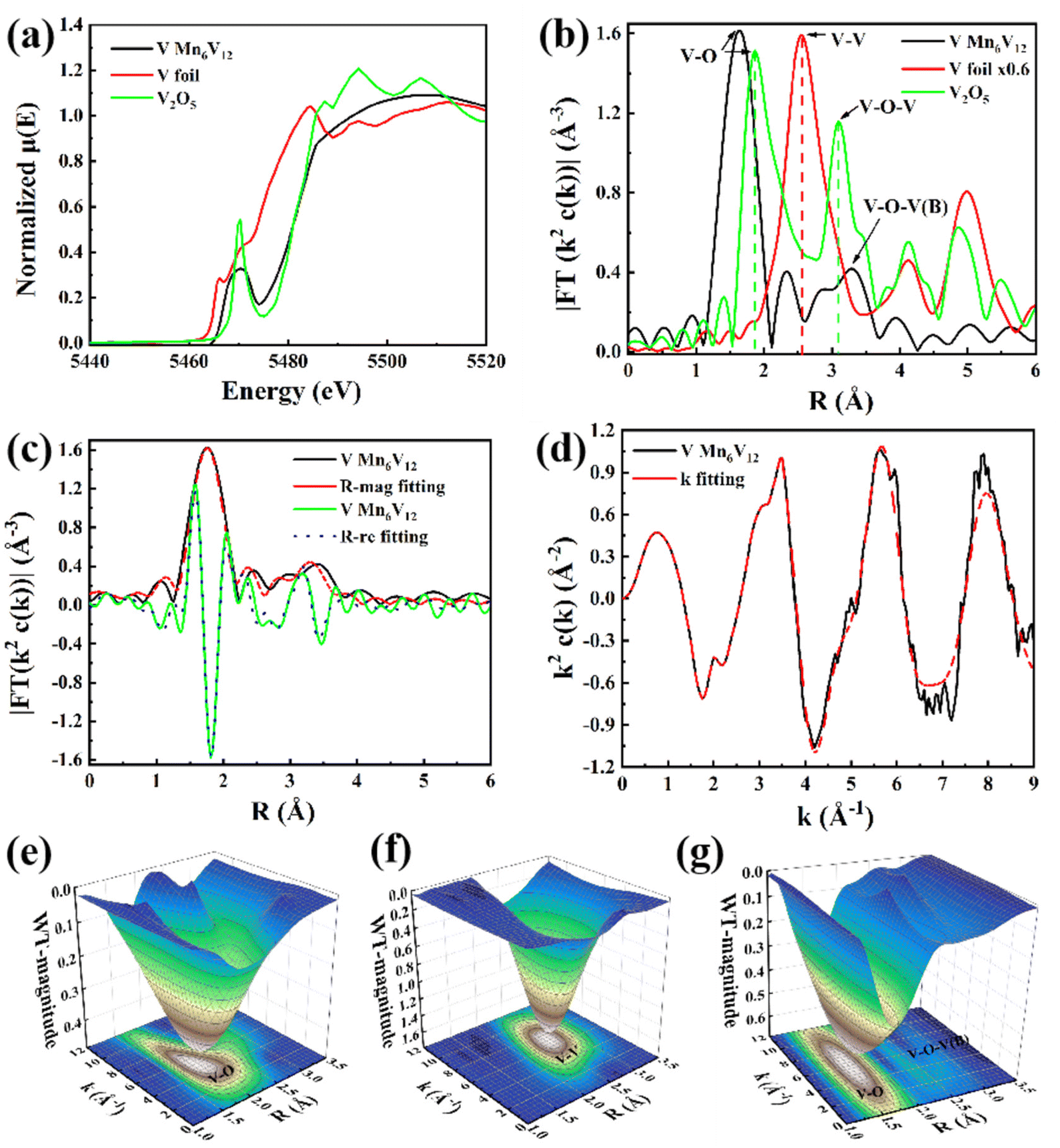 | ||
| Fig. 2 (a) V K-edge normalized XANES spectra, (b) FT curves of V K-edge EXAFS spectra, (c) V K-edge EXAFS curve fitting at R space and q space, (d) V K-edge EXAFS curve fitting at k space, (e) wavelet transform EXAFS of V2O5. (f) Wavelet transform EXAFS of V foil, (g) wavelet transform EXAFS of Mn6V12. | ||
From the Mn K-edge X-ray absorption near edge structure (XANES) in Fig. 3a, it can be seen that the absorption edge of Mn6V12 is intermediate between and close to Mn foil and MnO2, which indicates that the valence state of Mn in Mn6V12 is close to +4. The formation of Mn–N(O) and Mn–N(O)–C(B) coordination in Mn6V12 was directly confirmed by phase-corrected Fourier transform (FT) extended X-ray absorption fine structure (EXAFS) characterization (Fig. 3b). The first peak in the MnO standard sample is located at 2.0 Å and belongs to the Mn–O coordination. Therefore, the main peak at 2.02 Å in the FT-EXAFS spectrum of Mn6V12 can be attributed to the Mn–N44,45 or Mn–O double coordination environments. The second peak in the Mn6V12 material is located at 3.10 Å, which is 0.15 Å longer compared to Mn–O–Mn (2.95 Å) in MnO, and can be considered as the scattering path of Mn–N(O)–C(B) in the higher shell. It is noteworthy that the Mn–Mn scattering path (2.6 Å) does not exist in this material, verifying the atomic dispersion of Mn.
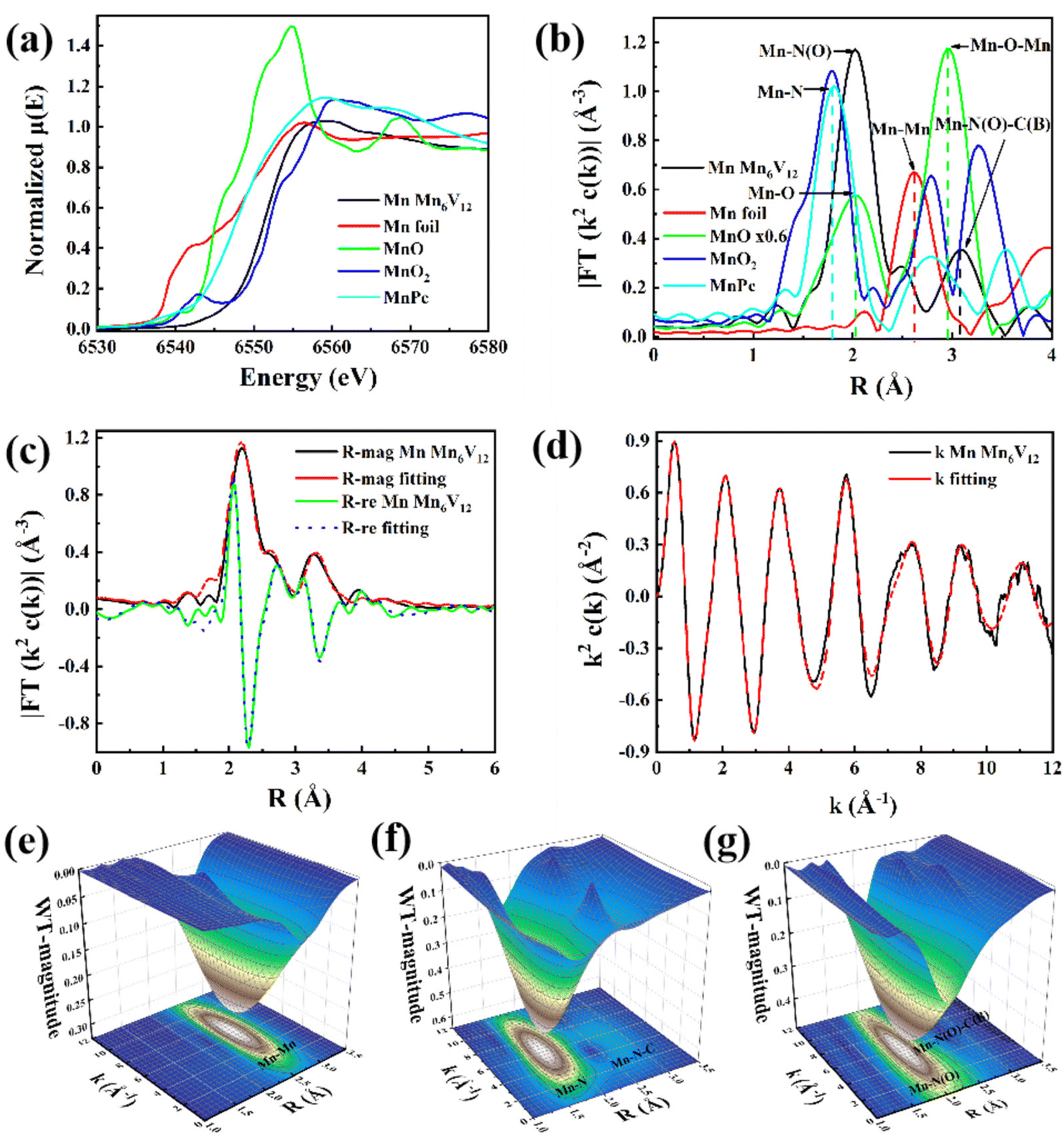 | ||
| Fig. 3 (a) Mn K-edge normalized XANES spectra, (b) FT curves of Mn K-edge EXAFS spectra, (c) Mn K-edge EXAFS curve fitting at R space and q space, (d) Mn K-edge EXAFS curve fitting at k space, (e) wavelet transform EXAFS of Mn foil, (f) wavelet transform EXAFS of MnPC, (g) wavelet transform EXAFS of Mn6V12. | ||
A quantitative EXAFS curve-fitting analysis of the structural parameters of Mn6V12 was carried out, and the best-fit analysis clearly confirms that the coordination numbers of Mn–N and Mn–N–C, and Mn–O–B are 4, 4, and 1, respectively, which implies that the Mn center is coordinated to four N atoms and one O atom, in accordance with the material structure (Fig. 3c and d; Table S1†). In contrast to Mn foil, MnPC, and MnO2 shown in Fig. 3e–g, the wavelet transform (WT) results indicate the absence of Mn–Mn bonds (with a maximum bond value (K) of 7 Å−1), as well as the presence of Mn–N(O) and Mn–N(O)–C(B) bonds (with a maximum bond value (K) of 5.94 Å−1, 6.0 Å−1) in this material. This result is consistent with the coordination environment exhibited in Fig. 3b and further illustrates the atomic dispersion of Mn.
Furthermore, the catalyst Mn6V12 was characterized by Fourier transform infrared (FTIR) spectroscopy in the range of 4000–500 cm−1. As shown in Fig. 4a, it is the characteristic peak of V–O bond in the range of 700–1000 cm−1. The strong peaks at 873, 811, and 701 cm−1 are due to the antisymmetric stretching vibration peaks of V–O–V (O is the bridging oxygen), and the peak at 945 cm−1 is the characteristic peak of V–O (O is the end-group oxygen). The peak at S3–1045 cm−1 belongs to the B–O antisymmetric stretching vibration peak belonging to the triangular coordination in the [B3O7] unit, and the peak at 1060–1104 cm−1 belongs to the B–O antisymmetric stretching vibration peak belonging to the tetrahedral coordination in the [B3O7] unit.46 The appearance of the characteristic peak at 2920 cm−1 is a result of symmetric and asymmetric stretching of the C–H bond. The stretching vibrations of N–H have distinct peaks at 1611–1635 cm−1 and 3394–3470 cm−1.47 The characteristic peaks in the infrared spectra of the catalyst before and after the reaction were basically unchanged, indicating that the catalyst structure was relatively stable.
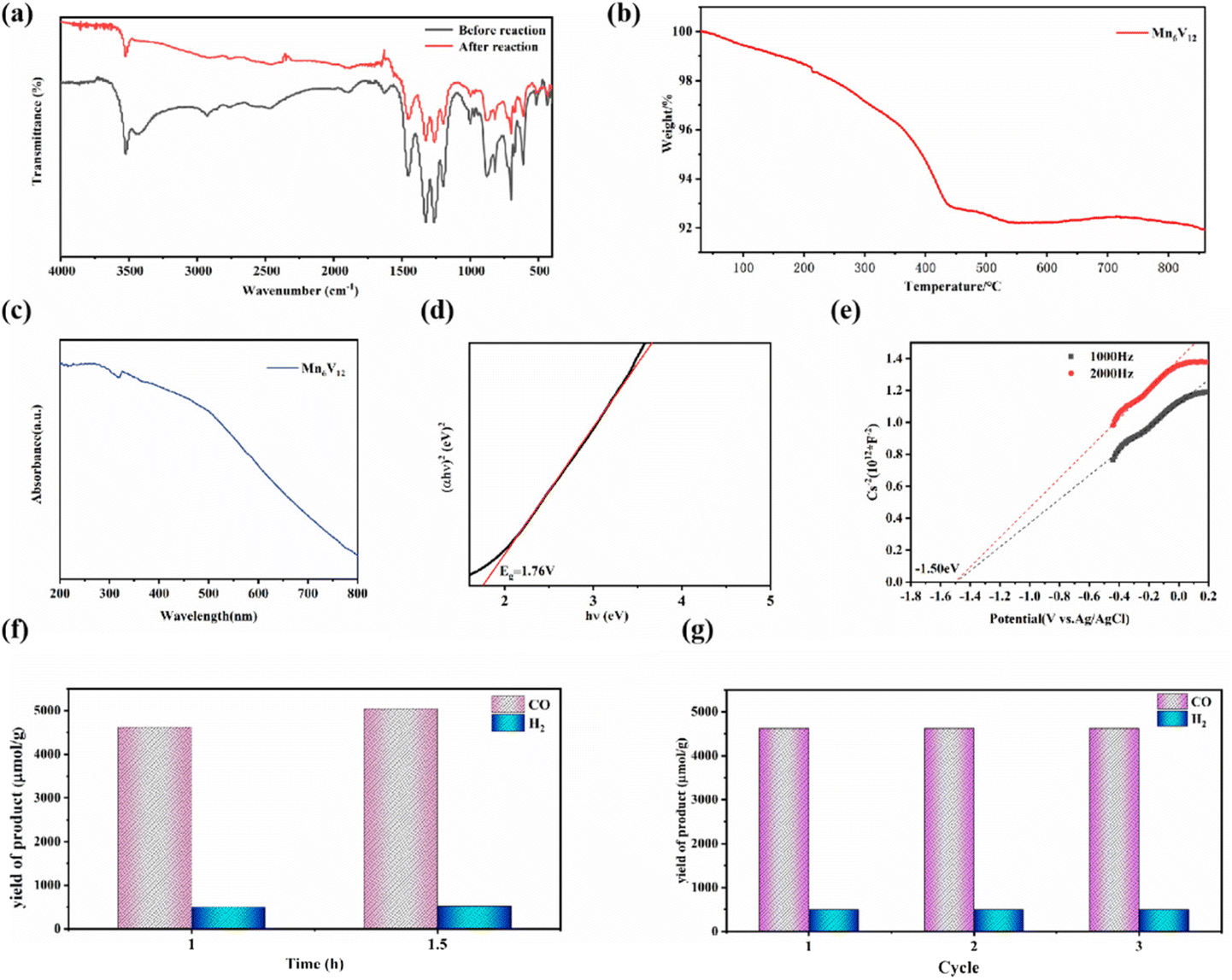 | ||
| Fig. 4 (a) IR spectra of Mn6V12; (b) TGA plot of Mn6V12; (c) UV-visible diffuse reflectance spectra of Mn6V12; (d) (αhν)2vs. hν curve of Mn6V12; (e) the Mott–Schottky plot of Mn6V12 in 0.2 M Na2SO4 aqueous solution; (f) gas yield diagram of Mn6V12 in photocatalytic system; (g) stability experiment of Mn6V12 in the same catalytic system. | ||
In order to further explore the thermal stability of Mn6V12, thermogravimetric analyses (TGA) were carried out in N2 atmosphere in the temperature range from room temperature to 800 °C. As shown in Fig. 4b, the TGA curve shows that the mass loss of Mn6V12 is about 8% at 0–800 °C, which is due to the decomposition of some free water molecules in the crystals, while the ligand does not collapse and decompose. Therefore, it can be assumed that Mn6V12 can be stabilized at least up to 800 °C. For the polyoxometalate photocatalysts [Co(en)2]6[V12B18O54(OH)6] and [Ni(en)2]6[V12B18O54(OH)6], which were also modified with metals, the thermal stability was classified as 200 °C and 300 °C.39 Therefore, the thermal stability of the polyoxometalate photocatalyst modified with metal Mn is better.
3.2. Photoreduction of CO2
The photocatalytic potential of Mn6V12 can be assessed based on the Mn6V12 UV-vis diffuse reflectance spectra (Fig. 4c). Probably due to the effective charge migration between the metal Mn and the ligand, the catalyst exhibited a very broad absorption band in the range of 200 nm to 700 nm, indicating that the catalyst possesses a good light absorption capacity. Meanwhile, the band gap (Eg) of the catalyst was estimated to be 1.76 eV by the Tauc plot method (Fig. 4d), which strongly suggests that Mn6V12 has the potential to be a photo catalyst.Mott–Schottky measurements under visible light irradiation show that the Mn6V12 flat band potential is −1.50 eV with respect to Ag/AgCl (Fig. 4e), suggesting that Mn6V12 is an n-type semiconductor. For n-type semiconductors, the position of the conduction band (CB) is 0.1 eV lower than the position of the flat band, which is −0.77 eV with respect to Ag/AgCl. According to the equation “(E(vs. RHE) = E(vs. Ag/AgCl) + 0.197 V + 0.0591 × pH)”, the conduction band (CB) of Mn6V12 is −0.77 eV vs. RHE. The position of the Mn6V12 conduction band (CB) is more negative than the potential for the reduction of CO2 to CO (−0.51 eV vs. RHE, pH = 7), suggesting that photo excited electrons can be transferred from the catalyst to the CO2 molecule. The valence band (VB) is 0.99 eV calculated by the equation Eg + ECB = EVB.
In the photocatalytic CO2 reduction system, Mn6V12 was used as a photocatalyst, 2 mL of triethanolamine (TEOA) was used as a sacrificial reagent, 11.6 mg of [Ru(bpy)3] Cl2·6H2O was used as a photosensitizer, and 8 mL of acetonitrile (CH3CN) and 2 mL of deionized water were used as solvents. Comparison test experiments were done to determine the source of the gaseous product CO. When photo catalysis was carried out for 1 h without passing CO2 gas but other catalytic reaction conditions were not changed, the yield of CO was found to be only 183 μmol g−1, whereas photo catalysis carried out with CO2 gas resulted in a higher yield of the product CO, which indicated that the carbon source of photo catalysis came from the passed CO2, rather than acetonitrile (CH3CN) and triethanolamine (TEOA) in the catalytic system. As shown in Fig. 4f, after 60 min of light exposure, a large amount of CO was detected, and the production of H2 was detected to be much smaller than that of CO. The production rates of CO and H2 were 4625.0 μmol g−1 and 499.6 μmol g−1 respectively, and the production rates of CO and H2 were 5048.1 μmol g−1 and 520.0 μmol g−1 respectively, after the light exposure was continued for 30 min. With increasing light time, the yields of CO and H2 increased successively. The yield of H2 was much lower than that of CO, implying that the adsorption energy of H2O on Mn ions and the high free energy of hydrogen precipitation could greatly inhibit the competing precipitation reaction. Comparing the product production rates at 1 h and 1.5 h, it was found that the rate of CO production was 3365.4 μmol g−1 h−1 and the rate of H2 production was 346.7 μmol g−1 h−1 for 1.5 h of light. The rate of product generation was higher for 1 h of light, with 4625.0 μmol g−1 h−1 for CO and 499.6 μmol g−1 h−1 for H2. The selectivity for CO generation was 90.3%, and that for H2 generation was 9.7%. Fig. S1† displays the CO and H2 yield in the long reaction time of the Mn6V12, showing a gradually increased trend. However, the output of the yield per 0.5 h is steadily decreased with time, which can be attributed to the consumption of the photosensitizer agent. In addition, the same type of photocatalysts [Co(en)2]6[V12B18O54(OH)6] and [Ni(en)2]6[V12B18O54(OH)6], after three hours of catalysis required in a similar catalytic system [Co(en)2]6[V12B18O54(OH)6] was catalyzed at a rate of 5700 μmol g−1 h−1 for CO and 3800 μmol g−1 h−1 for H2. [Ni(en)2]6[V12B18O54(OH)6] has a CO production rate of 3200 μmol g−1 h−1 and H2 production rate of 300 μmol g−1 h−1. By comparison, it was found that polyoxometalate catalyst modified with metal Mn could achieve efficient reduction of CO2 to CO in this photocatalytic system with only one hour of catalysis and could greatly inhibit the competitive hydrogen evolution reaction (HER). The performance of the Mn modified polyoxometalate photocatalyst was superior to that of the Co and Ni modified polyoxometalate photocatalysts. Moreover, a comparison with the previously reported photocatalysts for this purpose in similar systems (Table S2†) reveals that Mn6V12 exhibits comparatively better performance than other photocatalysts.
Control experiments were conducted to ensure the credibility of the experimental results (Fig. S2†). No observable products including CO and H2 were detected in the absence of either light or photocatalysts, indicating that both light and photocatalysts are crucial for photocatalytic CO2 reduction in this system.48 Almost no product was detected in the absence of the Ru photosensitizer, suggesting that the photogenerated electrons came from Ru photosensitization. Removing TEOA from the system leaded to a conspicuous decline of catalytic activity, because it consumes the photogenerated holes and lessens the recombination rate of photogenerated carriers.49 Furthermore, when pure CO2 was not introduced to the reactor, a notable drop of the amount of the products were detected, manifesting the carbon source of CO comes from CO2.
In addition, in order to test the stability of the photocatalyst, the catalyst was photo catalyzed for 1 h under the same conditions, and the results after repeating the test three times showed a small difference, with no significant change in yield observed (Fig. 4g). This indicates that the catalyst has stable catalytic activity and can efficiently reduce CO2 to CO and effectively inhibit hydrogen precipitation.
3.3. Photocatalytic mechanism
According to the experimental results, the energy levels of Mn6V12 at the highly occupied molecular orbital (HOMO) and the least occupied molecular orbital (LUMO) are −0.77 eV and 0.99 eV, respectively, with respect to RHE. The energy levels of [Ru(bpy)3] Cl2 at the high occupied molecular orbital (HOMO) and lowest occupied molecular orbital (LUMO) are 1.24 eV and −1.25 eV, respectively. Since the ECB value of Mn6V12 is −0.77 eV (vs. RHE) lower than the LUMO energy level of [Ru(bpy)3] Cl2, electrons on [Ru(bpy)3] Cl2 are preferentially transferred to Mn6V12. In addition, the SBET of Mn6V12 is higher than that of [Co(en)2]6[V12B18O54(OH)6]·17H2O (Fig. S3, S4 and Table S3†), suggesting Mn6V12 may promote CO2 reduction to CO and restrain the HER process, compared to [Co(en)2]6[V12B18O54(OH)6]·17H2O. Furthermore, the Mn6V12 exhibits higher photocurrent and lower interfacial charge transfer resistance than that of [Co(en)2]6[V12B18O54(OH)6]·17H2O (Fig. S5 and S6†), suggesting higher charge separation and transfer efficiency of Mn6V12, which directly influences the photocatalytic activity for CO2 reduction.The possible mechanism of photocatalytic CO2 reduction to CO and H2 is that there are two photoelectron transfers in the photocatalytic process. The photosensitizer reaches its excited state under the irradiation of UV-visible light and receives electrons from the triethanolamine electron donor to form the intermediate in the reduced state, while the intermediate in the reduced state then absorbs the visible light electrons to reach its excited state, and after that it transfers its electrons to the manganese metal ions on the POMs framework, which get the electrons to carry out a reduction reaction on the CO2 to achieve the photocatalytic reduction of CO2 to generate CO and H2. Moreover, according to the results of N2 adsorption–desorption test, a larger BET surface area of Mn6V12 may promote CO2 reduction to CO and restrain the HER process.50 Furthermore, combined with the measurements of photocurrent response and electrochemical impedance spectroscopy, a higher charge separation and transfer efficiency of Mn6V12, which directly influences the photocatalytic activity for CO2 reduction, thus leading to more CO production.51
4. Conclusions
Polyoxometalate photocatalyst ([Mn(en)2]6[V12B18O54(OH)6]) (Mn6V12) with Mn as the modifying unit were synthesized by hydrothermal method, and the highest production rate of CO for catalytic CO2 reduction was 4625.0 μmol g−1 h−1, and the highest production rate of H2 was 499.6 μmol g−1 h−1. The selectivity of CO production was 90.3% and that of H2 production was 9.7%. This polyoxometalate photocatalyst is good for efficient reduction of CO as well as effective inhibition of hydrogen precipitation reaction. The electronic structure and atomic coordination were elucidated using X-ray absorption near edge structure (XANES) spectroscopy and extended X-ray absorption fine structure (EXAFS) spectroscopy. The study in this work provides new ideas and possibilities for future transition metal modified polyoxometalate for photocatalytic carbon dioxide reduction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge BL17B beamline of National Facility for Protein Science (NFPS), Shanghai Synchrotron Radiation Facility (SSRF) Shanghai, China for providing the beam time. The financial support by “Grassland Talents” of Inner Mongolia Autonomous Region; Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (NJYT23030); “Steed plan High level Talents” of Inner Mongolia University; Carbon Neutralization Research Project (STZX202218); National Natural Science Foundation of China (U22A20107); Inner Mongolia Autonomous Region Natural Science Foundation (2023MS02002); Guangdong Provincial Key Laboratory of Materials and Technologies for Energy Conversion (MATEC2024KF011); National Key R&D Program of China (2022YFA1205201).References
- A. Rosas-Hernández, C. Steinlechner, H. Junge and M. Beller, Green Chem., 2017, 19, 2356–2360 RSC.
- S. You, S. Guo, X. Zhao, M. Sun, C. Sun, Z. Su and X. Wang, Dalton Trans., 2019, 48, 14115–14121 RSC.
- H. L. Zheng, S. L. Huang, M. B. Luo, Q. Wei, E. X. Chen, L. He and Q. Lin, Angew. Chem., 2020, 132, 23794–23798 CrossRef.
- A. A. Dubale, Y. Y. Zheng, H. L. Wang, R. Hübner, Y. Li, J. Yang, J. W. Zhang, N. K. Sethi, L. Q. He, Z. K. Zheng and W. Liu, Angew. Chem., Int. Ed., 2020, 59, 13891–13899 CrossRef CAS PubMed.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- L. R. Dou, L. D. Sun, W. Lyu, M. Y. Wang, F. Gao, M. Gao and H. Jiang, Pet. Explor. Dev., 2023, 50, 1246–1260 CrossRef.
- G. Huang, Q. Niu, Y. He, J. Tian, M. Gao, C. Li, N. An, J. Bi and J. Zhang, Nano Res., 2022, 15, 8001–8009 CrossRef CAS.
- L. Wang, L. Wang, S. Yuan, L. Song, H. Ren, Y. Xu, M. He, Y. Zhang, H. Wang and Y. Huang, Appl. Catal., B, 2023, 322, 122097 CrossRef CAS.
- Y. Benseghir, A. Lemarchand, M. Duguet, P. Mialane, M. Gomez-Mingot, C. Roch-Marchal, T. Pino, M.-H. Ha-Thi, M. Haouas and M. Fontecave, J. Am. Chem. Soc., 2020, 142, 9428–9438 CrossRef CAS PubMed.
- V. R. Calderone, N. R. Shiju, D. Curulla-Ferré, S. Chambrey, A. Khodakov, A. Rose, J. Thiessen, A. Jess and G. Rothenberg, Angew. Chem., Int. Ed., 2013, 52, 4397–4401 CrossRef CAS PubMed.
- H. X. Zhang, Q. L. Hong, J. Li, F. Wang, X. Huang, S. Chen, W. Tu, D. Yu, R. Xu and T. Zhou, Angew. Chem., Int. Ed., 2019, 58, 11752–11756 CrossRef CAS PubMed.
- Y.-C. Qiu, S. Yuan, X.-X. Li, D.-Y. Du, C. Wang, J.-S. Qin, H. F. Drake, Y.-Q. Lan, L. Jiang and H.-C. Zhou, J. Am. Chem. Soc., 2019, 141, 13841–13848 CrossRef CAS PubMed.
- J. Zhou, W. Chen, C. Sun, L. Han, C. Qin, M. Chen, X. Wang, E. Wang and Z. Su, ACS Appl. Mater. Interfaces, 2017, 9, 11689–11695 CrossRef CAS PubMed.
- M. Jiang, Y. Gao, Z. Wang and Z. Ding, Appl. Catal., B, 2016, 198, 180–188 CrossRef CAS.
- X. Lin, Y. Gao, M. Jiang, Y. Zhang, Y. Hou, W. Dai, S. Wang and Z. Ding, Appl. Catal., B, 2018, 224, 1009–1016 CrossRef CAS.
- B. Su, M. Zheng, W. Lin, X. F. Lu, D. Luan, S. Wang and X. W. Lou, Adv. Energy Mater., 2023, 13, 2203290 CrossRef CAS.
- G. Chen, Z. Zhou, B. Li, X. Lin, C. Yang, Y. Fang, W. Lin, Y. Hou, G. Zhang and S. Wang, J. Environ. Sci., 2024, 140, 103–112 CrossRef PubMed.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed.
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- Y. Richardson, J. Blin and A. Julbe, Prog. Energy Combust. Sci., 2012, 38, 765–781 CrossRef CAS.
- N. Scarlat, J.-F. Dallemand and F. Fahl, Renewable Energy, 2018, 129, 457–472 CrossRef.
- J. Zhou, H. Wu, C.-Y. Sun, C.-Y. Hu, X.-L. Wang, Z.-H. Kang and Z.-M. Su, J. Mater. Chem. A, 2018, 6, 21596–21604 RSC.
- J. Zhang, Y. Huang, G. Li and Y. Wei, Coord. Chem. Rev., 2019, 378, 395–414 CrossRef CAS.
- Z.-J. Liu, X.-L. Wang, C. Qin, Z.-M. Zhang, Y.-G. Li, W.-L. Chen and E.-B. Wang, Coord. Chem. Rev., 2016, 313, 94–110 CrossRef CAS.
- J. J. Walsh, A. M. Bond, R. J. Forster and T. E. Keyes, Coord. Chem. Rev., 2016, 306, 217–234 CrossRef CAS.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- Z.-Y. Du, Z. Chen, R.-K. Kang, Y.-M. Han, J. Ding, J.-P. Cao, W. Jiang, M. Fang, H. Mei and Y. Xu, Inorg. Chem., 2020, 59, 12876–12883 CrossRef CAS PubMed.
- H. Zhang, W. Liu, A. Li, D. Zhang, X. Li, F. Zhai, L. Chen, L. Chen, Y. Wang and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 16110–16114 CrossRef CAS PubMed.
- M. A. Moussawi, M. Haouas, S. Floquet, W. E. Shepard, P. A. Abramov, M. N. Sokolov, V. P. Fedin, S. Cordier, A. Ponchel and E. Monflier, J. Am. Chem. Soc., 2017, 139, 14376–14379 CrossRef CAS PubMed.
- Z. Wu, Y. Zhai, W. Zhao, Z. Wei, H. Yu, S. Han and Y. Wei, Green Chem., 2020, 22, 737–741 RSC.
- B. N. Li, X. J. Yu, H. J. Pang, Q. B. Shen, Y. Hou, J. Ma and H. Y. Xin, Chem. Commun., 2020, 56, 7199–7202 RSC.
- S. Li, S. Liu, S. Liu, Y. Liu, Q. Tang, Z. Shi, S. Ouyang and J. Ye, J. Am. Chem. Soc., 2012, 134, 19716–19721 CrossRef CAS PubMed.
- W. Zhang, K. Xie, Y. H. Tang, C. Qin, S. Cheng and Y. Ma, Prog. Chem., 2022, 34, 2638–2650 CAS.
- N. Lu, Y. Wang, S. Ning, W. Zhao, M. Qian, Y. Ma, J. Wang, L. Fan, J. Guan and X. Yuan, Sci. Rep., 2017, 7, 17298 CrossRef PubMed.
- Y. Qi, J. Zhang, Y. Kong, Y. Zhao, S. Chen, D. Li, W. Liu, Y. Chen, T. Xie, J. Cui, C. Li, K. Domen and F. Zhang, Nat. Commun., 2022, 13, 484 CrossRef CAS PubMed.
- X. Liu, J. Zhou, T. R. Amarante, F. A. Almeida Paz and L. Fu, Dalton Trans., 2021, 50, 1550–1568 RSC.
- H. Chen, Z. B. Yu, Z. Bacsik, H. Zhao, Q. Yao and J. Sun, Angew. Chem., Int. Ed., 2014, 53, 3608–3611 CrossRef CAS PubMed.
- J. T. Rijssenbeek, D. J. Rose, R. C. Haushalter and J. Zubieta, Angew. Chem., Int. Ed., 2003, 36, 1008–1010 CrossRef.
- X. Yu, C.-C. Zhao, J.-X. Gu, C.-Y. Sun, H.-Y. Zheng, L.-K. Yan, M. Sun, X.-L. Wang and Z.-M. Su, Inorg. Chem., 2021, 60, 7364–7371 CrossRef CAS PubMed.
- P. Hermosilla-Ibáñez, K. Wrighton-Araneda, L. Scarpetta-Pizo, W. Cañón-Mancisidor, M. Gutierrez-Cutiño, E. Le Fur, V. Paredes-García and D. Venegas-Yazigi, New J. Chem., 2019, 43, 17538–17547 RSC.
- C. Liu, Y. Wu, K. Sun, J. Fang, A. Huang, Y. Pan, W.-C. Cheong, Z. Zhuang, Z. Zhuang and Q. Yuan, Chem. Commun., 2021, 7, 1297–1307 CAS.
- Z. Cai, P. Wang, J. Zhang, A. Chen, J. Zhang, Y. Yan and X. Wang, Adv. Mater., 2022, 34, 2110696 CrossRef CAS PubMed.
- K. Zhu, S. Wei, H. Shou, F. Shen, S. Chen, P. Zhang, C. Wang, Y. Cao, X. Guo, M. Luo, H. Zhang, B. Ye, X. Wu, L. He and L. Song, Nat. Commun., 2021, 12, 6878 CrossRef CAS PubMed.
- L. Lai, H. Ji, H. Zhang, R. Liu, C. Zhou, W. Liu, Z. Ao, N. Li, C. Liu, G. Yao and B. Lai, Appl. Catal., B, 2021, 282, 119559 CrossRef CAS.
- D. Zhao, X. Wang, W. Zhang, Y. Zhang, Y. Lei, X. Huang, Q. Zhu and J. Liu, Adv. Funct. Mater., 2023, 2211412 CrossRef CAS.
- Z. H. Lin, H. H. Zhang, C. C. Huang, R. Q. Sun, Y. P. Chen and X. Y. Wu, Acta Chim. Sin., 2004, 62, 391–398 CAS.
- S. Li, B. Feng, X. Zhang, J. Tian, D. Wang, Y. Pei, M. Qiao, Y. Li and B. Zong, Appl. Catal., B, 2023, 335, 122879 CrossRef CAS.
- S. Liu, F. Chen, S. Li, X. Peng and Y. Xiong, Appl. Catal., B, 2017, 211, 1–10 CrossRef CAS.
- Z. Liu, Z. Chen, M. Li, J. Li, W. Zhuang, X. Yang, S. Wu and J. Zhang, ACS Catal., 2023, 13, 6630–6640 CrossRef CAS.
- W.-J. Wang, K.-H. Chen, Z.-W. Yang, B.-W. Peng and L.-N. He, J. Mater. Chem. A, 2021, 9, 16699–16705 RSC.
- S. Li, S. Shi, G. Huang, Y. Xiong and S. Liu, Appl. Surf. Sci., 2018, 455, 1137–1149 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2324078. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nr00097h |
| This journal is © The Royal Society of Chemistry 2024 |