
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Porous ceria materials for efficient direct conversion of carbon dioxide and methanol to dimethyl carbonate†
Zhuxian
Yang
a,
Justin Tay
Zheng
b,
Xinhuan
Lu
c,
Monica Mengdie
Lin
b,
Dongming
Cai
d,
Yankun
Wang
a,
Wen-Yueh
Yu
 *b,
Yanqiu
Zhu
a and
Yongde
Xia
*a
*b,
Yanqiu
Zhu
a and
Yongde
Xia
*a
aDepartment of Engineering, Faculty of Environment, Science and Economy, University of Exeter, Exeter, EX4 4QF, UK. E-mail: y.xia@exeter.ac.uk
bDepartment of Chemical Engineering, National Taiwan University, No. 1, Sec. 4, Roosevelt Rd, Taipei, 106335, Taiwan. E-mail: wenyueh@ntu.edu.tw
cSchool of Chemical Engineering, Hubei University, 368 Youyi Dadao, Wuchang Qu, Wuhan, Hubei Province 430062, P. R. China
dHubei Key Laboratory of Energy Storage and Power Battery, School of Mathematics, Physics and Optoelectronics Engineering, Hubei University of Automotive Technology, Shiyan, 442002, P. R. China
First published on 15th July 2024
Abstract
Ceria (CeO2) is widely considered as a superior catalytic material for the direct conversion of carbon dioxide (CO2) and methanol into dimethyl carbonate (DMC). Developing porous structures is a versatile way to increase the surface area, to create defects, and to improve the mass transfer of the resulting materials, consequently enhancing their catalytic performance. However, most of the reported preparation methods of porous CeO2 involve complex hydrothermal (100–120 °C) or refluxing (95–140 °C) processes followed by calcination at temperatures of 500–650 °C. In this work, we report a simple and low temperature approach to prepare porous CeO2, which involves mixing the raw materials at room temperature, followed by drying and then calcining at 450 °C. A DMC formation rate of 14.8 mmol g−1 h−1 is achieved for one of the obtained porous CeO2, which is much higher than those of the most reported CeO2 (0.51–11 mmol g−1 h−1). Further studies show that the DMC formation rate has a positive link to the parameters following the order: the CO2 uptake amount at 25 °C, the amount of weak acidity, the Ce3+ concentration, the amount of weak basicity, and the BET surface area of the CeO2 catalysts in this study. In addition, there seems to be an optimum oxygen vacancy concentration of the CeO2 samples for the DMC formation rate. This study provides a simple strategy for the preparation of a porous CeO2 material as a highly efficient catalyst for the catalytic conversion of CO2, which can not only mitigate the greenhouse gas CO2, but also turn it into value-added and versatile chemical DMC.
1. Introduction
Dimethyl carbonate (DMC) exhibits a number of promising properties including low toxicity, high oxygen content (53%), low viscosity, good dissolving ability, and a high-octane number (105).1,2 DMC can be used as a green solvent and a fuel additive.3,4 Thanks to its multiple functionalities, DMC can also be used as a versatile reagent for methylation, carbonylation, and methoxycarbonylation.5 Recently the market demand for DMC has been increasing significantly because of its widespread applications.6There are a number of methods for DMC preparation, and some of them have been industrialised,6–8 while others are under study.9–12 There are, however, some issues with the industrialised processes, such as the involvement of hypertoxic phosgene, high cost, explosion risk, etc.6,13 Among the various methods that have been studied for DMC production, the direct synthesis of DMC from CO2 and methanol is attractive because it does not involve any toxicants, moreover it can turn the greenhouse gas CO2 into DMC,11,14–21 a value-added and versatile product. This method is based on the following equation:
| CO2 + 2CH3OH ⇌ (CH3O)2CO + H2O |
The major issues with this method are the high stability of CO2 and the low equilibrium constant of the reaction, leading to a low DMC yield. Catalysts, high CO2 pressure/temperature and dehydrating agents have been employed to address these issues.4,6,11,19,22–24 Applying dehydrating agents to remove the in situ produced water can shift the equilibrium to the product side and consequently increase the DMC yield.22,23,25–30 Regarding catalysts, a large number of catalysts have been investigated for direct DMC synthesis including, CeO2,17–19,31–33 ZrO2,34–37 V2O5,38 Y2O3,39etc. Among them, CeO2 based catalysts have shown superior catalytic performance and attracted considerable attention.31–33,40–42
Yoshida et al. first studied CeO2 prepared by calcining commercial CeO2 as the catalyst for direct synthesis of DMC from CO2 and methanol.11 Ever since this report, a number of CeO2 based catalysts prepared via various synthesis strategies have been evaluated for the direct synthesis of DMC, including the hydrothermal method,15,20,43–45 solvothermal method,46 precipitation method,23,32,40,41,47–49 templating method,50 refluxing method,19etc. These various strategies resulted in CeO2 based catalysts with different properties and accordingly significantly different catalytic performance. It has been found that the specific surface area,11,30,47 morphologies,15,44,51,52 acid–base properties,33,45,46,53–56 oxygen-vacancies,16,19,21,30,46,57,58 surface Ce3+ content19,21,58 or Ce4+ content32,59 of CeO2 based catalysts can affect their catalytic performance.
Developing porous catalysts is an efficient approach to increase the surface area, to create defects, and to improve the mass transfer of the resulting catalysts, and consequently enhance the catalytic performance.19,30,60–62 Porous CeO2 can be prepared by a polyol method,63 hydrothermal method,20,60,62 solvent evaporation-induced self-assembly method,30 sol method,64 hard template method,61 reflux process,19 soft-to-hard consecutive template method,65etc. Although these strategies can produce porous CeO2 with a surface area of ca. 100–220 m2 g−1, most of them are tedious and involve complex steps. It is therefore desirable to develop a simple synthesis approach to prepare highly porous CeO2 catalysts for the direct preparation of DMC from CO2 and methanol.
In this study, we report the preparation of porous CeO2 by a simple method at lower temperature, which involves mixing the raw materials at room temperature, followed by drying at 70 °C and then 100 °C, and finally annealing at 450 °C. The resulting CeO2 was characterised by N2 adsorption analysis, CO2 adsorption analysis, XRD, Raman spectroscopy, XPS, SEM, TEM, temperature-programmed desorption of NH3 (NH3-TPD) and CO2 (CO2-TPD), and further evaluated for the direct synthesis of DMC from CO2 and methanol without applying a dehydrating agent. A high DMC formation rate of 14.8 mmol g−1 h−1 has been achieved for the CeO2 sample calcined at 450 °C with a heating rate of 1.4 °C min−1, which is much higher than the majority of the reported CeO2 catalysts. This sample was further studied by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) for the DMC formation mechanism. The effect of the BET surface area (SBET), the CO2 uptake amount at 25 °C, the weak acidity, the weak basicity, the Raman peak intensity ratio of ID/IF2g, the XPS Ce3+ concentration (Ce3+%), and the XPS oxygen vacancy concentration (OV%) of the CeO2 samples prepared under different conditions on the DMC formation rate has been investigated.
2. Materials and methods
2.1. Materials
Pluronic F-127, phloroglucinol (C6H6O3), cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O), nitric acid (70%) (HNO3) and formaldehyde (37%) (HCHO) were purchased from Merck. Absolute ethanol (C2H5OH) was purchased from Fisher Scientific.2.2. Preparation of porous CeO2
The CeO2 precursor was prepared following a slightly modified procedure reported by Lee et al.65 In brief, 0.3 mmol Pluronic F-127 (C7H16O4), 10 mmol phloroglucinol (C6H6O3), and 10 mmol Ce(NO3)3·6H2O were dissolved in 25 mL of ethanol, followed by adding 1.1 mmol HNO3 (70%) under vigorous stirring for 30 min, and then 15 mmol HCHO was added under stirring for 2 h. The resulting mixture was dried in an oven at 70 °C for 24 h, and then at 100 °C for 24 h to produce the CeO2 precursor.The CeO2 precursor was calcined to generate porous CeO2via two different methods, i.e., two-step calcination and one-step calcination. In the two-step calcination process, the CeO2 precursor was heated in argon at 800 °C with a ramp rate of 5 °C min−1 for 3 h, followed by calcination in air at 450 °C with a ramp rate of 1.4 °C min−1 for 3 h to remove carbon, and the obtained CeO2 was named Ar800-5.0_Air450-1.4. In the one-step calcination process, the CeO2 precursor was directly calcined in air at 400 or 450 °C with a heating rate of 1.4 °C min−1 for 3 h to generate CeO2 named Air400-1.4 and Air450-1.4 respectively; while the CeO2 precursor underwent direct calcination in air at 450 °C with a heating rate of 5 °C min−1 for 3 h resulting in CeO2 named Air450-5.0.
2.3. Sample characterisation
N2 gas sorption analysis was measured with a Quantachrome Autosorb-iQ gas analyser using a conventional volumetric technique at −196 °C. Samples were outgassed under vacuum at 200 °C for 6 h prior to analysis. The surface area was calculated using the Brunauer–Emmett–Teller (BET) method using adsorption data in the partial pressure (P/P0) range of 0.05–0.2. The pore size distribution was calculated with the DFT model with adsorption data. CO2 gas sorption analysis was performed in the same way as N2 gas sorption analysis except that the analysis temperature was 25 °C. X-ray diffraction (XRD) patterns were obtained with a Bruker D8 Advance X-ray diffractometer with a Cu Kα X-ray source (λ = 0.15418 nm) at 40 kV and 40 mA. X-ray photoelectron spectroscopy (XPS) analysis was performed with a Kratos Axis Ultra system with a monochromated Al Kr X-ray source operated at an emission current of 10 mA and an anode potential of 15 kV. The XPS spectra were deconvoluted with CasaXPS software. Raman spectroscopy measurement was carried out with a Renishaw inVia Qontor with a 532 nm laser.In situ FT-IR spectra were recorded with an infrared spectrometer (Thermo Scientific Nicolet iS50) equipped with a mercury–cadmium–telluride (MCT) detector and a diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) chamber (Harrick Praying Mantis™ HVC-DRP-5).19,66–68 The measurements were carried out as follows:19 the chamber was pre-treated at 200 °C in Ar flow (40 mL min−1) for 20 min. After the chamber was cooled to 140 °C, the methanol in the saturator was introduced to the chamber by Ar flow (40 mL min−1), and the catalyst was purged with Ar flow (40 mL min−1) for 10 min, and then the flow was switched to high-purity CO2 gas (99.999+%, 40 mL min−1) for 10 min. The DRIFTS spectra were averaged from 16 scans with a resolution of 4 cm−1.
NH3-TPD was performed with a Quantachrome Chemstar TPX. 100 mg of sample was heated under a helium (He) flow of 30 mL min−1 at 300 °C for 40 min to remove surface impurities, followed by cooling. When the temperature was down to 35 °C, a mixture of 5% NH3/He with a flow rate of 30 mL min−1 was introduced into the sample for a period of 30 min for NH3 adsorption by the sample to take place. Then the sample was purged with nitrogen for 30 min to eliminate the physically adsorbed NH3 before the desorption of NH3 took place, which was measured under temperature programming with a heating rate of 10 °C min−1 in the range of 50–780 °C. CO2-TPD was also carried out with a Quantachrome Chemstar TPX. The above procedure used for NH3-TPD measurement was adopted for CO2-TPD measurement except that the probe gas NH3 was replaced by CO2, and TPD was measured in the range of 50–700 °C.
2.4. Evaluation of catalytic activity for CO2 conversion
The catalytic activity for CO2 conversion was measured with a 100-mL Parr reactor without applying a dehydration agent.19,21,69 370 mmol methanol and 10 mg of CeO2 were loaded in a reactor, followed by introducing 5.0 MPa CO2 at room temperature. The reactor was then heated to 140 °C under stirring and kept at 140 °C for 3 h. Afterwards, the product in the reactor was analysed by a gas chromatograph equipped with a flame ion detector (Agilent 6890N GC-FID) and a capillary column (Zebron ZB-WAX). A pre-determined amount of 1-propanol was applied to the system as the internal standard for quantitative analysis. The DMC formation rate was calculated by dividing the DMC amount obtained at a reaction time of 3 h by the mass of CeO2 used.3. Results and discussion
3.1. Structures, textural properties, and morphologies
Fig. 1a shows the XRD patterns of CeO2 samples prepared under various conditions. Diffraction peaks at 2θ values of 28.4, 32.9, 47.4, 56.3, 59.2, 69.3, 76.7 and 79.0°, corresponding to the (111), (200), (220), (311), (222), (400), (331), and (420) planes of cubic CeO2 are observed for all of them. The two-step calcined sample (Ar800-5.0_Air450-1.4) exhibits sharper peaks with a much higher intensity compared to those samples calcined by one-step, likely due to the high calcination temperature. The crystal sizes estimated with the Scherrer equation (using a Scherrer constant of 0.89 and a λ value of 0.15418 nm) from the (111) XRD peak of CeO2 are listed in Table 1. The crystal size of Ar800-5.0_Air450-1.4 was estimated to be 63.4 nm, which is much higher than that of the one-step calcined CeO2 samples (12.5–15.4 nm). This is reasonable given that Ar800-5.0_Air450-1.4 was first calcined at 800 °C in Ar for 3 h before further calcination at 450 °C in air, while the other three samples were only calcined at 450 °C in air for 3 h. | ||
| Fig. 1 XRD patterns (a), nitrogen adsorption isotherms (b), and pore size distributions (c) of CeO2 samples prepared under various calcination conditions which are indicated in their names. | ||
| Sample name | Preparation conditions | S BET (m2 g−1) | Crystal sizea (nm) | CO2 uptakeb (mmol g−1) | CO2 uptakec (μmol g−1) | NH3 uptaked (μmol g−1) | Raman ID/IF2g | XPS Ce3+% | XPS OV% | DMC formation ratee (mmol g−1 h−1) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Calculated with the Scherrer equation from the (111) XRD peak of CeO2. b Obtained from CO2 adsorption analysis with a Quantachrome Autosorb-iQ gas analyser at 25 °C. c Obtained from CO2-TPD (100–300 °C). d Obtained from NH3-TPD (100–300 °C). e Without applying a dehydration agent. | ||||||||||
| Ar800-5.0_Air450-1.4 | 800 °C in Ar + 450 °C in air | 89 | 63.4 | 0.60 | 110.6 | 276.3 | 0.0081 | 25.0 | 38.1 | 3.3 |
| Air450-1.4 | 450 °C in air 1.4 °C min−1 | 104 | 15.4 | 1.06 | 148.4 | 313.2 | 0.0080 | 25.6 | 31.6 | 14.8 |
| Air400-1.4 | 400 °C in air 1.4 °C min−1 | 127 | 12.5 | 0.95 | 168.2 | 276.9 | 0.0073 | 25.9 | 28.6 | 8.9 |
| Air450-5.0 | 450 °C in air 5 °C min−1 | 134 | 13.5 | 0.88 | 197.4 | 334.1 | 0.0086 | 24.6 | 28.2 | 8.4 |
| Commercial CeO2 (Strem Chemicals) | 60 | 6.7 | ||||||||
| CeO2 octahedra | 2 | 50 | 50 | 0.0615 | ||||||
Fig. 1b shows the nitrogen adsorption isotherms of all the CeO2 samples calcined under various conditions. A hysteresis loop at partial pressure (P/P0) above 0.4 appears in all the isotherms, suggesting the existence of mesopores in these samples. Fig. 1c reveals that all the samples have multi-mode mesopores, with most pore sizes following in the ranges of 2–4 nm, 4–6 nm and 6–8 nm, whereas samples Ar800-5.0_Air450-1.4 and Air450-5.0 exhibit two extra ranges of 8–10 nm and 10–12 nm. It seems that the heating rate plays a role in the pore size distribution of the resulting samples. The samples obtained from the heating rate of 5.0 °C min−1 (Ar800-5.0_Air450-1.4 and Air450-5.0) show one pore size distribution pattern, while the other two samples (Air450-1.4 and Air400-1.4) that resulted from the heating rate of 1.4 °C min−1 show another pattern. The pore sizes of these samples are much smaller than the reported values (e.g., 13.8 nm).19,32,33 This multi-mode pore structure of CeO2 samples is expected to improve the mass transfer when they are used as catalysts. The BET surface areas of all samples are summarised in Table 1. The one-step calcined samples show a BET surface area of 104–134 m2 g−1, which is higher than that of the two-step calcined sample (89 m2 g−1). It is likely that the higher calcination temperature for the two-step calcined sample leads to increased crystallinity of CeO2 (Fig. 1a) but a decreased surface area. The BET surface area values of the one-step calcined samples (104–134 m2 g−1) are comparable to the reported ones for CeO2 prepared by other methods,19,20,32,60,69 and higher than some reported values (74–88 m2 g−1),16,33 but lower than other reported values (170–220 m2 g−1).62–64
The CO2 isotherms at 25 °C of CeO2 samples prepared under various calcination conditions are presented in Fig. S1 (ESI†). They all show a hysteresis loop, indicating that the adsorbed CO2 is not fully desorbed, and sample Air450-1.4 exhibits the largest hysteresis loop, suggesting the strongest interaction between CO2 and the sample. The calculated physical adsorption capacities of CO2 are included in Table 1, and sample Air450-1.4 shows the highest CO2 uptake capacity of 1.06 mmol g−1.
The SEM images of the CeO2 samples calcined under various conditions are presented in Fig. 2. Fig. 2a–c and g show that all the CeO2 samples appear to be irregularly shaped lumps with a wide size range of up to around 4 μm × 8 μm, and these lumps are made of tiny particles (Fig. 2d–f and h) regardless of the different calcination conditions. So, calcination has no effect on the morphology of the resulting CeO2 samples.
 | ||
| Fig. 2 SEM images of CeO2 samples calcined under various conditions: Air450-1.4 (a) and (b), Air400-1.4 (c) and (d), Air450-5.0 (e) and (f), and Ar800-5.0_Air450-1.4 (g) and (h). | ||
The CeO2 samples obtained via one-step calcination (Air450-1.4) and two-step calcination (Ar800-5.0_Air450-1.4) were further observed by TEM as presented in Fig. 3. Fig. 3a and b reveal that both samples are made of irregular tiny particles, and most of which are with estimated sizes in the range of 5–10 nm. In addition, for both samples regardless of the calcination conditions, the (111) plane is the most observed one (Fig. 3c–h) while the (200) plane is occasionally observable (Fig. 3c and d), indicating that the (111) plane could be the active phase for DMC formation.11,69 These high-resolution TEM image results are in good agreement with the XRD results presented in Fig. 1a. These results indicate that the different calcination conditions do not affect the morphologies and the exposure of the (111) and (200) planes of the resulting CeO2 samples.
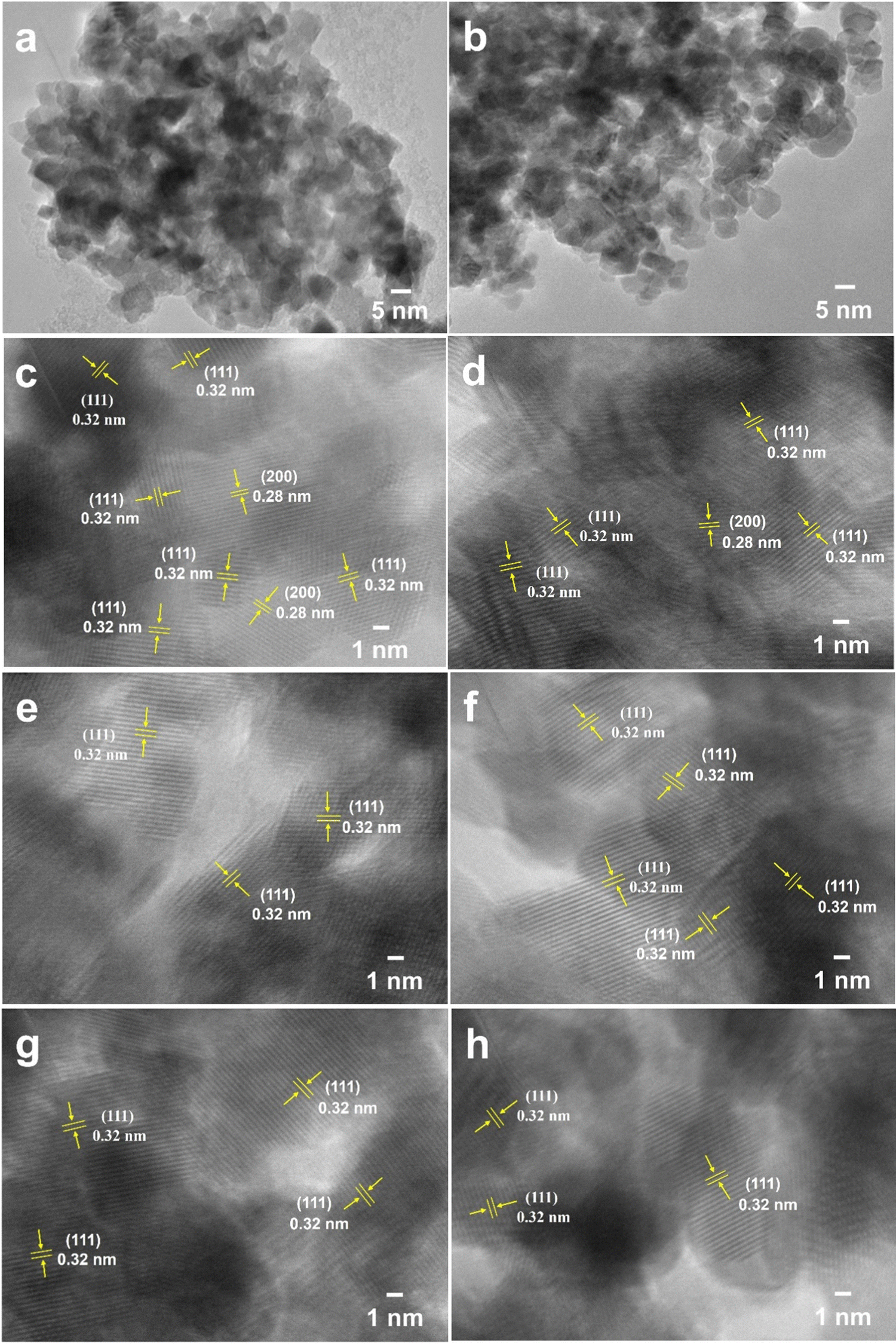 | ||
| Fig. 3 TEM images of CeO2 samples Air450-1.4 obtained via one-step calcination (a), (c), (e) and (g) and Ar800-5.0_Air450-1.4 obtained via two-step calcination (b), (d), (f) and (h). | ||
3.2. Acid–base properties
The surface acid–base properties of the samples were evaluated by NH3-TPD and CO2-TPD, and the corresponding profiles are shown in Fig. 4. A major broad desorption peak in the temperature range of 100–300 °C appears in the NH3-TPD profiles for all the CeO2 samples (Fig. 4a), indicating the presence of weak acidity. However, the intensity of this peak of the one-step calcined samples is higher than that of the two-step calcined sample (Ar800-5.0_Air450-1.4), suggesting that the one-step calcined samples exhibit stronger weak acidity. The amount of the weak acidity (the amount of NH3 adsorbed at 100–300 °C) of all the samples is listed in Table 1. The peak of sample Air450-5.0 is centred at ca. 166 °C while that of the other three samples is at ca.176 °C. In addition, a weak extra peak at ca. 357 °C is observable for the two-step calcined sample (Ar800-5.0_Air450-1.4), implying that two-step calcination results in medium acidity, in addition to weak acidity. Based on the above results, it can be concluded that one-step calcination is superior to two-step calcination in producing stronger weak acidity. This could be one of the reasons that all the one-step calcined samples show a higher DMC formation rate than the two-step calcined sample, as will be discussed later. | ||
| Fig. 4 NH3-TPD profiles (a), and CO2-TPD profiles (b) of CeO2 samples calcined under various conditions. | ||
Similar to the NH3-TPD profiles, a major broad CO2 desorption peak in the temperature range of 100–300 °C can be observed in the CO2-TPD profiles (Fig. 4b) for all the CeO2 samples, showing the presence of weak basicity, and the intensity of the CO2 desorption peak of the one-step calcined samples is also higher than that of the two-step calcined sample (Ar800-5.0_Air450-1.4), suggesting that the one-step calcined samples exhibit stronger weak basicity too. The peak of samples Air450-5.0 and Ar800-5.0_Air450-1.4 is centred at ca. 176 °C while that of samples Air450-1.4 and Air400-1.4 is at ca. 166 °C. In addition, a weak extra peak at ca. 354 °C is observable for the two-step calcined sample Ar800-5.0_Air450-1.4, showing the presence of medium basicity, in addition to weak basicity. According to the above results, it is evident that one-step calcination is superior to two-step calcination in enhancement of both weak acidity and weak basicity of the resulting CeO2 samples. This enhancement could be one of the reasons that all the one-step calcined samples show higher catalytic activity for the direct conversion of CO2 into DMC than the two-step calcined sample, as will be discussed later.
3.3. Defects, Ce3+% and OV%
Raman spectroscopy provides information on molecular vibration and rotation, which can be used to study the detects of samples. The Raman spectra (532 nm laser) of CeO2 samples calcined under various conditions are shown in Fig. 5. All of the samples display Raman peaks at 260.8, 462.8, and 601.6 cm−1, corresponding to the second-order 2TA peak, the first-order F2g peak, and the defect-induced (D) peak, respectively.70,71 The F2g peak corresponds to the symmetric contraction vibration of Ce–8O in the CeO2 lattice, which is sensitive to any disorder, doping, or particle-size-induced effects in the lattice caused by heating.72 It is generally accepted that the intensity ratio of the D peak to the F2g peak (ID/IF2g) is an indicator of the amount of defects of CeO2-based catalysts.16,19,73 The intensity ratio (ID/IF2g) values of all the CeO2 samples have been calculated and are listed in Table 1. The CeO2 samples calcined in air at 450 °C exhibit close ID/IF2g values of 0.0080–0.0086 regardless of one or two steps, while the sample calcined in air at 400 °C by one step shows a lower ID/IF2g value of 0.0073. These results suggest that a higher calcination temperature (in air) results in a larger amount of defects.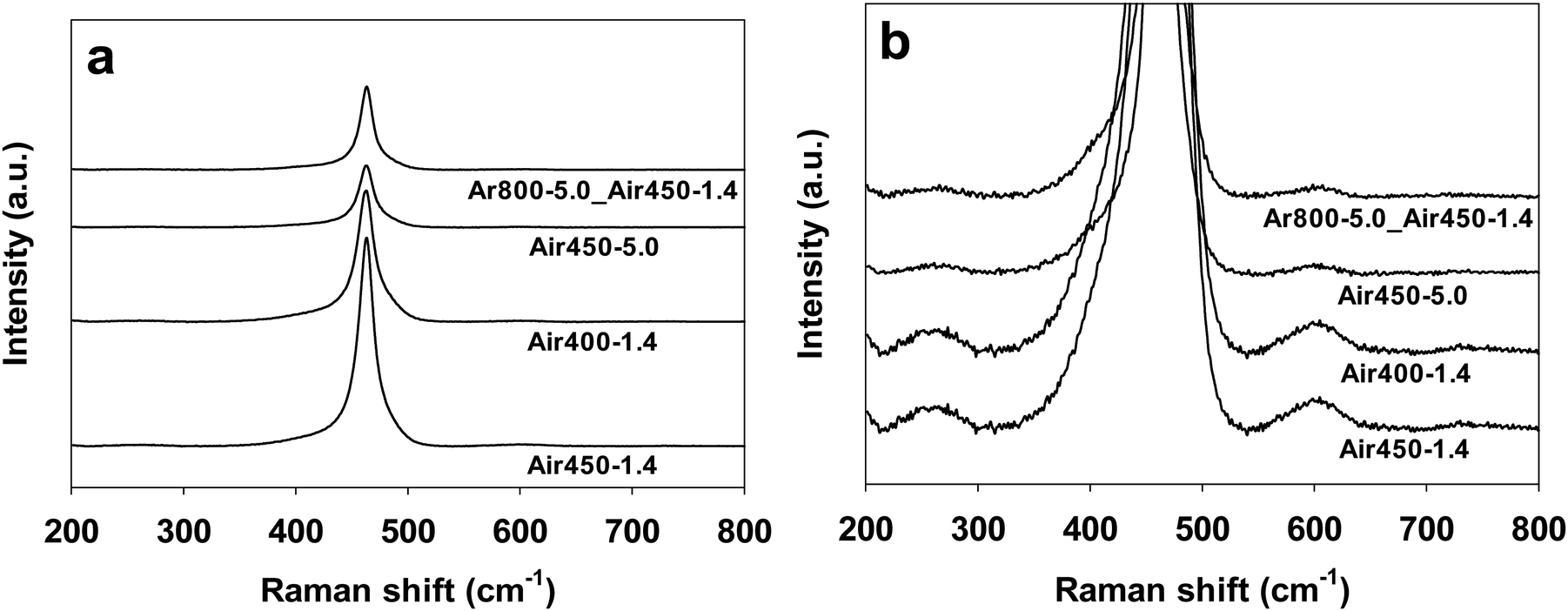 | ||
| Fig. 5 Raman spectra of CeO2 samples calcined under various conditions. (b) is the zoom-in image of (a). | ||
The XPS spectra of CeO2 samples calcined under various conditions are presented in Fig. 6. 8 Ten peaks can be deconvoluted for the Ce 3d spectra, i.e., μ0 (885.6 eV), μ1 (881.4 eV), μ0′ (903.5 eV), μ1′ (899.2 eV), ν0 (882.6 eV), ν1 (888.9 eV), ν2 (898.3 eV), ν0′ (900.9 eV), ν1′ (907.5 eV), and ν2′ (916.7 eV), corresponding to the spin–orbit splitting of Ce 3d5/2 (μ0, μ1, ν0, ν1, and ν2) and Ce 3d3/2 (μ0′, μ1′, ν0′, ν1′, and ν2′).59,73,74 The concentration of Ce3+ (Ce3+ %) is calculated from the ratio of peak areas of Ce3+ (μ0, μ1, μ0′, and μ1′) to the peak areas of Ce3+ and Ce4+ (ν0, ν1, ν2, ν0′, ν1′, and ν2′) based on the equation below:

 | ||
| Fig. 6 Ce 3d XPS spectra (a) and O 1s XPS spectra (b) of CeO2 samples calcined under various conditions. | ||
The calculated Ce3+% values are listed in Table 1. They are in the range of 24.6–25.9%, showing that the different calcination conditions have no significant effect on the Ce3+% values of the resulting CeO2 samples.
Regarding the O 1s XPS spectra, three peaks corresponding to the lattice oxygen (OL), the oxygen related to vacancy (OV), the chemisorbed oxygen (OC) can be deconvoluted at ca. 529.4 eV, 531.5 eV, and 533.5 eV, respectively.16,19 The concentration of surface oxygen vacancies is calculated from the ratio of peak areas of OV to peak areas of OL, OV and OC based on the equation below:
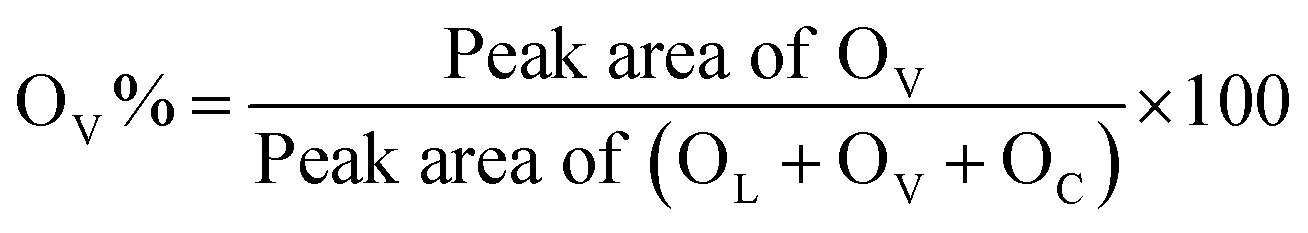
The calculated OV% values are listed in Table 1. The two-step calcined sample Ar800-5.0_Air-1.4 exhibits the highest OV% value of 38%, followed by Air450-1.4 (31.6%), and the samples Air400-1.4 and Air450-5.0 show OV% values of 28.6% and 28.2%, respectively. These results suggest that the first calcination step (at 800 °C in argon for 3 h) enhances the formation of oxygen vacancies. In addition, the XPS Ce3+% values do not correlate with the XPS OV% values of these CeO2 samples. A possible reason could be that not all the oxygen vacancies are correlated with the presence of Ce3+, because Frenkel-type oxygen vacancies can be formed due to the presence of interstitial oxygen ions.70,75 A study on CeO2 (obtained by calcination of cerium acetate at 600 °C) by pulsed neutron diffraction experiments has demonstrated the presence of Frenkel-type oxygen defects, which consist of interstitial oxygen ions and corresponding oxygen vacancies.75 Moreover, according to Table 1, the Raman peak density ratios, ID/IF2g values, do not correspond to the XPS Ce3+% values or the XPS OV% values. A possible reason could be that the defects measured by Raman do not solely originate from the presence of Ce3+ or oxygen vacancy, and as mentioned above, the F2g peak is sensitive to any disorder including oxygen vacancies, interstitial oxygen defects, dislocation, grain boundaries, etc.72,76,77
3.4. Catalytic activity
The catalytic activity of the CeO2 samples for the direct conversion of CO2 into DMC was evaluated by the DMC formation rate. For easy comparison, the data of a CeO2 sample without pores15 and a commercially available CeO2 sample (Strem Chemicals) with a lower BET surface area measured in this study are also included in Table 1. According to Table 1, all the one-step calcined CeO2 samples exhibit a much higher DMC formation rate (8.4–14.8 mmol g−1 h−1) than the two-step calcined CeO2 sample Ar800-5.0_Air-1.4 (3.3 mmol g−1 h−1), and higher than the nonporous CeO2 and the commercial CeO2. Given that all the samples exhibit a close Ce3+ concentration of 24.6–25.9%, the higher DMC formation rates of the one-step calcined samples could be due to the following reasons: the amounts of both the weak basicity and weak acidity, the CO2 uptake at room temperature, and the BET surface area of the one-step calcined catalysts are all higher than that of the two-step calcined sample, as these parameters have been reported to facilitate DMC formation.11,19,33,47,55,56,69 Regarding the oxygen vacancy concentration (OV%), although it is expected to improve DMC formation,16,19,21,30,46,57,58 there is a suggestion that areas with high oxygen vacancies where some of the carbonates can be trapped will activate the internal bonds in undesired manners, which would make the enrichment of more oxygen vacancies less efficient in DMC formation.78 Indeed, the two-step calcined sample has the highest OV% but does not show the highest DMC formation rate in this study, which is in good agreement with the previous report.69Among the three one-step calcined samples, the one (Air450-1.4) obtained with a heating rate of 1.4 °C min−1 at 450 °C shows the highest DMC formation rate of 14.8 mmol g−1 h−1. The data in Table 1 show that both the calcination temperature and the heating rate hugely affect the properties of the resulting CeO2, consequently affecting the catalytic performance, which is in good agreement with a previous report that the heating rate plays an important role in determining the properties of porous materials.79 The sample Air450-1.4 prepared by calcination at 450 °C with a ramp rate of 1.4 °C min−1 shows the highest DMC formation rate could be due to the reason that it exhibits the optimum combination of the parameters affecting the catalytic performance. As shown in Table 1, this sample shows the highest CO2 uptake at room temperature and the highest XPS OV% (yet lower than that of the two-step calcined sample) among the three samples, and moderate weak acidity and weak basicity. As all these parameters have been reported to play a role in the catalytic activity, it seems that the catalytic performance is a result of the superimposition of these factors. This is in good agreement with the claim made by Tomishige et al. that it is difficult to elucidate the crucial factors that can influence the catalytic activity of CeO2 catalysts towards DMC formation from CO2 and methanol.17
To further analyse the effect of the physicochemical properties of the CeO2 samples on the DMC formation rate of all the samples, the correlation between the DMC formation rates and various parameters is presented in Fig. 7. It can be seen that the DMC formation rate shows a positive correlation between the parameters following the order (based on the correlation coefficient R): the CO2 uptake amount at 25 °C (R = 0.96), the weak acidity (R = 0.49), the Ce3+ concentration (R = 0.45), the weak basicity (R = 0.39), and the BET surface area (R = 0.27) of the CeO2 samples. In addition, Fig. 7f suggests that there seems to be an optimum oxygen vacancy concentration for DMC formation. This is in agreement with previous reports that although the oxygen vacancy is thought to improve the DMC formation, areas with high oxygen vacancies where some of the carbonates can be trapped will activate the internal bonds in an undesired manner, which would make the enrichment of more oxygen vacancies less efficient in DMC formation69,78 In addition, Fig. 7g shows that there is no correlation between the DMC formation rate and the Raman peak intensity ratio ID/IF2g of the CeO2 catalysts in this study. This could be due to the fact that the Raman F2g peak is sensitive to any disorder including oxygen vacancies, interstitial oxygen defects, dislocation, grain boundaries, etc.72,76,77 while not all the defects contribute to the catalytic performance.
 | ||
| Fig. 7 Effect of various parameters of CeO2 samples on the DMC formation rate: (a) CO2 uptake at 25 °C, (b) amount of weak acidity (NH3 uptake at 100–300 °C), (c) XPS Ce3+%, (d) amount of weak basicity (CO2 uptake at 100–300 °C), (e) BET surface area (SBET), and (f) XPS OV%, and (g) Raman peak intensity ratio ID/IF2g. | ||
For easy comparison, Table 2 lists the CeO2 catalysts reported in the literature and those developed in this study, along with their preparation processes and their DMC formation rates measured under various conditions. Based on Table 2, the formation rate of 14.8 mmol g−1 h−1 is much higher than the values of most reported CeO2 samples that were prepared by a more complex hydrothermal process at 100–120 °C (5.46–8.03 mmol g−1 h−1)15,16,80 or reflux processes at 95 °C (5.97 mmol g−1 h−1)41 followed by calcination at 500–650 °C, although lower than the value of the samples (17.7–18.2 mmol g−1 h−1) that were prepared by the reflux process at 140 °C and calcination at 600 °C19 or by the reflux process at 100 °C and calcination at 400 °C followed by extra H2 heat treatment at 400 °C.69 These results demonstrate that the relatively simple preparation process at a lower temperature (450 °C) developed in this study is a promising approach to preparing highly active CeO2 catalysts for the direct conversion of CO2 and methanol to DMC.
| Catalyst | Preparation process | DMC formation rate (mmol g−1 h−1) | Reaction conditions | Ref. |
|---|---|---|---|---|
| CeO2 (Air450-1.4) | Mixing at room temperature + drying at 70 °C and 100 °C + calcination at 450 °C | 14.8 | 140 °C, 5 MPa, 3 h | This work |
| CeO2 | Calcination of commercial CeO2 at 600 °C | 11 | 130 °C, 0.2 mol CO2, 2 h | 11 |
| CeO2 spindles | Refluxing at 95 °C + calcination at 650 °C + chemical redox etching | 5.97 | 140 °C, 4.5 MPa, 3 h | 41 |
| CeO2 nanowires | Solvothermal at 140 °C + H2 heat treatment at 500 °C | 3.37 | 120 °C, 5 MPa, 5 h | 46 |
| CeO2 rods | Refluxing at 140 °C + calcination at 600 °C | 17.7 ± 2.3 | 140 °C, 5 MPa, 3 h | 19 |
| CeO2 | Refluxing at 100 °C + calcination at 400 °C + H2 heat treatment at 400 °C | 18.22 ± 0.64 | 140 °C, 5 MPa, 3 h | 69 |
| CeO2 | Hydrothermal at100 °C + calcination at 600 °C | 5.46 | 140 °C, 6.8 MPa, 2 h | 16 |
| CeO2 spindles | Hydrothermal at 120 °C + calcination at 600 °C | 8.03 | 140 °C, 5 MPa, 2 h | 15 |
| CeO2 quantum dots | Hydrothermal at 150 °C | 2.03 | 140 °C, 5 MPa, 2 h | 80 |
| CeO2 | Co-precipitation method + calcination at 600 °C | 0.51 | 120 °C, 15 MPa, 4 h | 33 |
| CeO2 | Precipitation + salcination at 550 °C | 2.26 | 140 °C, 3 MPa, 4 h | 32 |
The CeO2 sample with the highest DMC formation rate in this study, i.e., Air450-1.4, was further studied by in situ diffuse reflectance infrared Fourier transform spectroscopy. In situ DRIFTS measurement was carried out by pre-adsorption of methanol followed by purging CO2 at 140 °C, and the spectra are shown in Fig. 8. Bands at 1588, 1293 and 1016 cm−1 are ascribed to bidentate carbonate. Bands at 1040 and 1030 cm−1 are assigned to bridged methoxy groups and bands at 1100 cm−1 are on-top methoxy species. Bands at 1600 and 1349 cm−1 are due to monomethyl carbonate and bands at 1443, 1368 and 1016 cm−1 are corresponding to monodentate carbonate.19,81 According to the spectra in Fig. 8, it is expected that the methanol adsorption on the CeO2 catalyst leads to the formation of on-top methoxy species, while the CO2 adsorption on the CeO2 catalyst results in the formation of bidentate carbonate. The decrease of the on-top methoxy species is accompanied by the appearance of monomethyl carbonate, which suggests that monomethyl carbonate is formed due to the reaction between the on-top methoxy and the bidentate carbonate, and DMC is formed due to the reaction of the monomethyl carbonate and another on-top methoxy species. This is in good agreement with the reported DMC formation mechanism.19,82
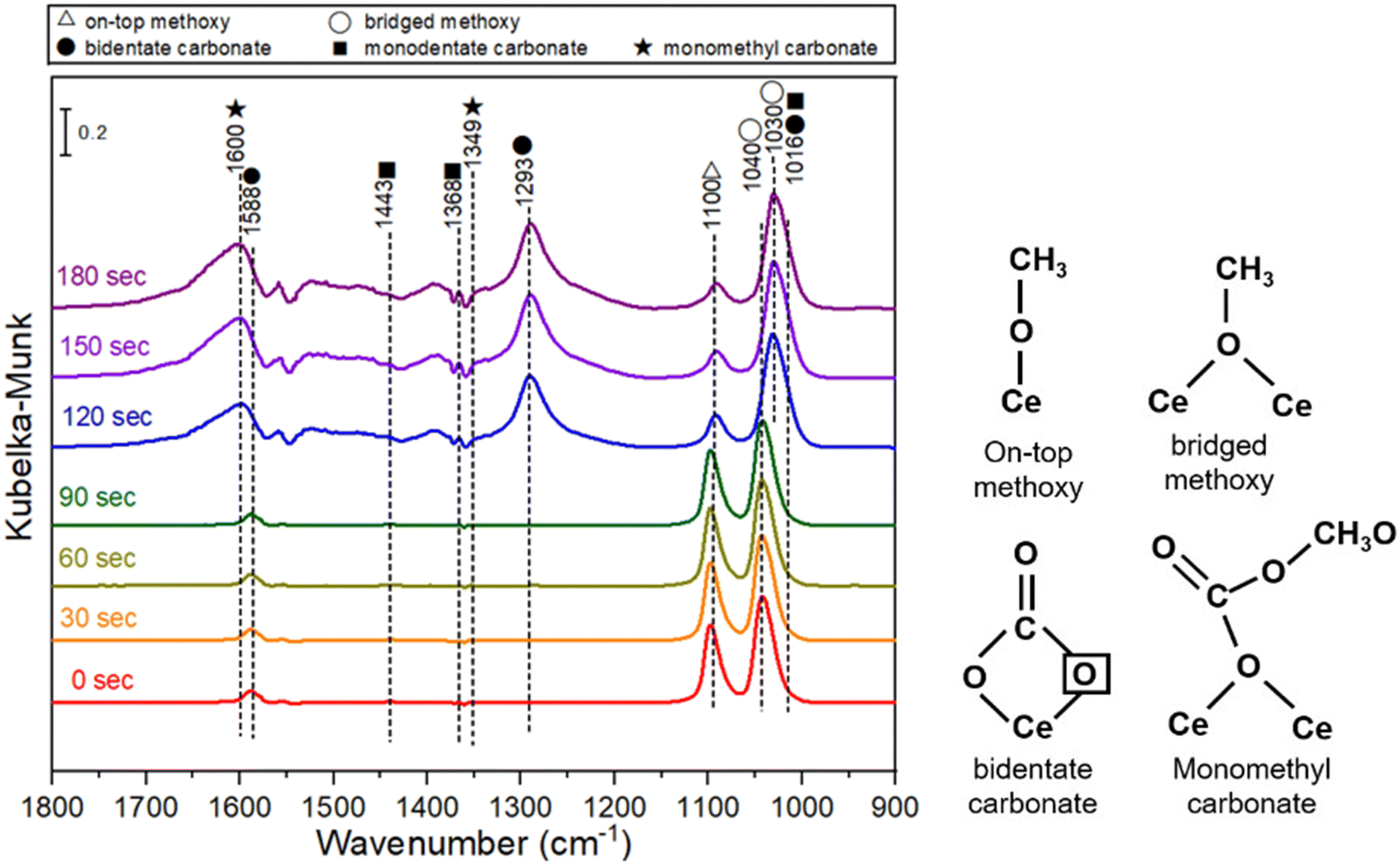 | ||
| Fig. 8 In situ DRIFTS spectra of CeO2 (Air450-1.4) with pre-adsorbed methanol followed by purging CO2 at 140 °C. | ||
4. Conclusions
Porous CeO2 was prepared by a simple method, in which the raw materials were mixed at room temperature and then dried at 70 °C and 100 °C consecutively, followed by calcination at 400 or 450 °C with a heating rate of 1.4 or 5.0 °C min−1. Calcination at 450 °C with a heating rate of 1.4 °C min−1 results in CeO2 showing the highest DMC formation rate of 14.8 mmol g−1 h−1, which is much higher than that of the majority of the reported CeO2 catalysts that were prepared by a more complex hydrothermal process at 100–120 °C (5.46–8.03 mmol g−1 h−1) or reflux processes at 95 °C (5.97 mmol g−1 h−1) followed by calcination at 500–650 °C. Further study shows that the DMC formation rate has a positive link to the parameters following the order (based on the correlation coefficient R): the CO2 uptake amount at 25 °C (R = 0.96), the weak acidity (R = 0.49), the Ce3+ concentration (R = 0.45), the weak basicity (R = 0.39), and the BET surface area (R = 0.27) of the CeO2 catalysts. In addition, there seems to be an optimum oxygen vacancy concentration for the DMC formation rate, while no correlation is found between the DMC formation rate and the Raman peak intensity ratio ID/IF2g of the CeO2 catalysts in this study. The in situ DRIFTS measurement suggests that monomethyl carbonate is formed due to the reaction between the on-top methoxy and the bidentate carbonate, and DMC is formed due to the reaction of the monomethyl carbonate and another on-top methoxy species. This study provides a simple and easy-to-scale-up strategy for the preparation of a porous CeO2 catalyst for the direct conversion of CO2 and methanol to DMC at a lower temperature (450 °C), which could be potentially applicable to other reactions (e.g., the synthesis of diethyl carbonate).Author contributions
Zhuxian Yang: investigation, methodology, data curation, and writing – original draft. Justin Tay Zheng: data curation, validation, and writing – review & editing. Xinhuan Lu: methodology, data curation, and validation. Monica Mengdie Lin: investigation, validation, and data curation. Dongming Cai: investigation, validation, and data curation. Yankun Wang: investigation and data curation. Wen-Yueh Yu: supervision, methodology, validation, and writing – review & editing. Yanqiu Zhu: supervision, and writing – review & editing. Yongde Xia: supervision, conceptualization, methodology, validation, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this manuscript.Acknowledgements
This work was supported by the Leverhulme Trust (RPG-2018–320), the Royal Society (IEC\NSFC\201121), and the National Science and Technology Council (NSTC), Taiwan (110-2221-E-002-012-MY3 and 112-2221-E-002-041-MY3).References
- G. Fiorani, A. Perosa and M. Selva, Green Chem., 2018, 20, 288–322 RSC.
- Y. Ono, Appl. Catal., A, 1997, 155, 133–166 CrossRef CAS.
- M. A. Pacheco and C. L. Marshall, Energy Fuels, 1997, 11, 2–29 CrossRef CAS.
- P. Kumar, V. C. Srivastava, U. L. Stangar, B. Music, I. M. Mishra and Y. Z. Meng, Catal. Rev., 2021, 63, 363–421 CrossRef CAS.
- F. Y. Tu, H. J. Lai, T. A. Chiu, J. C. Jiang and W. Y. Yu, Surf. Interfaces, 2023, 43, 103597 CrossRef CAS.
- D. C. Shi, S. Heyte, M. Capron and S. Paul, Green Chem., 2022, 24, 1067–1089 RSC.
- M. J. Schneider, M. Haumann, M. Stricker, J. Sundermeyer and P. Wasserscheid, J. Catal., 2014, 309, 71–78 CrossRef CAS.
- H. Z. Tan, Z. Q. Wang, Z. N. Xu, J. Sun, Z. N. Chen, Q. S. Chen, Y. M. Chen and G. C. Guo, Catal. Sci. Technol., 2017, 7, 3785–3790 RSC.
- L. G. Wang, Y. Wang, S. M. Liu, L. J. Lu, X. Y. Ma and Y. Q. Deng, Catal. Commun., 2011, 16, 45–49 CrossRef CAS.
- Z. Q. Hou, L. G. Luo, K. Liu, C. Z. Liu, Y. Y. Wang and L. Y. Dai, Chem. Eng. J., 2014, 236, 415–418 CrossRef CAS.
- Y. Yoshida, Y. Arai, S. Kado, K. Kunimori and K. Tomishige, Catal. Today, 2006, 115, 95–101 CrossRef CAS.
- S. S. Peng, X. B. Shao, M. X. Gu, G. S. Zhang, C. Gu, Y. Nian, Y. M. Jia, Y. Han, X. Q. Liu and L. B. Sun, Angew. Chem., Int. Ed., 2022, 61, e202215157 CrossRef CAS PubMed.
- H. Babad and A. G. Zeiler, Chem. Rev., 1973, 73, 75–91 CrossRef CAS.
- H. J. Lee, W. Joe and I. K. Song, Korean J. Chem. Eng., 2012, 29, 317–322 CrossRef CAS.
- S. P. Wang, L. F. Zhao, W. Wang, Y. J. Zhao, G. L. Zhang, X. B. Ma and J. L. Gong, Nanoscale, 2013, 5, 5582–5588 RSC.
- B. Liu, C. M. Li, G. Q. Zhang, X. S. Yao, S. S. C. Chuang and Z. Li, ACS Catal., 2018, 8, 10446–10456 CrossRef CAS.
- K. Tomishige, Y. Gu, T. Chang, M. Tamura and Y. Nakagawa, Mater. Today Sustain., 2020, 9, 100035 CrossRef.
- C. Daniel, Y. Schuurman and D. Farrusseng, J. Catal., 2021, 394, 486–494 CrossRef CAS.
- W. F. Kuan, W. Y. Yu, F. Y. Tu, C. H. Chung, Y. C. Chang, M. M. Lin, T. H. Yu and L. J. Chen, Chem. Eng. J., 2022, 430, 132941 CrossRef CAS.
- G. L. Yang, A. Z. Jia, J. D. Li, F. Li and Y. J. Wang, Mol. Catal., 2022, 528, 112471 CrossRef CAS.
- W. F. Kuan, C. H. Chung, M. M. Lin, F. Y. Tu, Y. H. Chen and W. Y. Yu, Mater. Today Sustain., 2023, 23, 100425 CrossRef.
- B. Peng, H. R. Dou, H. Shi, E. E. Ember and J. A. Lercher, Catal. Lett., 2018, 148, 1914–1919 CrossRef CAS.
- W. Donphai, O. Phichairatanaphong, R. Fujii, P. Li, T. Chang, M. Yabushita, Y. Nakagawa and K. Tomishige, Mater. Today Sustain., 2023, 24, 100549 CrossRef.
- D. Stoian, F. Medina and A. Urakawa, ACS Catal., 2018, 8, 3181–3193 CrossRef CAS.
- K. Tomishige and K. Kunimori, Appl. Catal., A, 2002, 237, 103–109 CrossRef CAS.
- D. J. Faria, L. M. dos Santos, F. L. Bernard, I. S. Pinto, M. Resende and S. Einloft, RSC Adv., 2020, 10, 34895–34902 RSC.
- J. C. Choi, L. N. He, H. Yasuda and T. Sakakura, Green Chem., 2002, 4, 230–234 RSC.
- Z. F. Zhang, Z. W. Liu, J. A. Lu and Z. T. Lie, Ind. Eng. Chem. Res., 2011, 50, 1981–1988 CrossRef CAS.
- N. Wang, Y. Liu, A. Huang and J. Caro, Microporous Mesoporous Mater., 2015, 207, 33–38 CrossRef CAS.
- G. L. Yang, H. B. Wang, A. Z. Jia, J. D. Li and Y. J. Wang, New J. Chem., 2023, 47, 12477–12486 RSC.
- W. Sun, P. L. Li, M. Yabushita, Y. Nakagawa, Y. Q. Wang, A. Nakayama and K. Tomishige, ChemSusChem, 2023, 16, e202300768 CrossRef CAS PubMed.
- G. Q. Zhang, Y. Zhou, Y. L. Yang, T. T. Kong, Y. Song, S. Zhang and H. Y. Zheng, Molecules, 2023, 28, 3785 CrossRef CAS PubMed.
- P. Kumar, P. With, V. C. Srivastava, R. Glaser and I. M. Mishra, J. Environ. Chem. Eng., 2015, 3, 2943–2947 CrossRef CAS.
- K. Tomishige, T. Sakaihori, Y. Ikeda and K. Fujimoto, Catal. Lett., 1999, 58, 225–229 CrossRef CAS.
- K. T. Jung and A. T. Bell, J. Catal., 2001, 204, 339–347 CrossRef CAS.
- C. J. Jiang, Y. H. Guo, C. G. Wang, C. W. Hu, Y. Wu and E. B. Wang, Appl. Catal., A, 2003, 256, 203–212 CrossRef CAS.
- Q. X. Zheng, R. Nishimura, Y. Sato, H. Inomata, M. Ota, M. Watanabe and S. Camy, Chem. Eng. J., 2022, 429, 132378 CrossRef CAS.
- X. L. Wu, M. Xiao, Y. Z. Meng and Y. X. Lu, J. Mol. Catal. A: Chem., 2005, 238, 158–162 CrossRef CAS.
- W. Sun, L. Zheng, Y. Q. Wang, D. D. Li, Z. R. Liu, L. Wu, T. Fang and J. Q. Wu, Ind. Eng. Chem. Res., 2020, 59, 4281–4290 CrossRef CAS.
- A. H. Tamboli, N. Suzuki, C. Terashima, S. Gosavi, H. Kim and A. Fujishima, Catalysts, 2021, 11, 223 CrossRef CAS.
- T. Kulthananat, P. Kim-Lohsoontorn and P. Seeharaj, Ultrason. Sonochem., 2022, 90, 106164 CrossRef CAS PubMed.
- Y. Z. Wang, S. Geng, F. Liu, M. Q. Yao, J. Ma, J. X. Cao and Z. W. Li, J. Colloid Interface Sci., 2023, 652, 1984–1993 CrossRef CAS PubMed.
- C. M. Marin, L. Li, A. Bhalkikar, J. E. Doyle, X. C. Zeng and C. L. Cheung, J. Catal., 2016, 340, 295–301 CrossRef CAS.
- S. Y. Zhao, S. P. Wang, Y. J. Zhao and X. B. Ma, Chin. Chem. Lett., 2017, 28, 65–69 CrossRef CAS.
- P. Kumar, L. Matoh, R. Kaur and U. L. Stangar, Fuel, 2021, 285, 119083 CrossRef CAS.
- Z. W. Fu, Y. H. Yu, Z. Li, D. M. Han, S. J. Wang, M. Xiao and Y. Z. Meng, Catalysts, 2018, 8, 164 CrossRef CAS.
- E. Leino, N. Kumar, P. Maki-Arvela, A. Aho, K. Kordas, A. R. Leino, A. Shchukarev, D. Y. Murzin and J. P. Mikkola, Mater. Chem. Phys., 2013, 143, 65–75 CrossRef CAS.
- M. Zhang, M. Xiao, S. J. Wang, D. M. Han, Y. X. Lu and Y. Z. Meng, J. Cleaner Prod., 2015, 103, 847–853 CrossRef CAS.
- J. Y. Zhang, S. Y. Zhao, Y. J. Zhao, X. B. Ma and S. P. Wang, Asia-Pac. J. Chem. Eng., 2021, 16, e2517 CrossRef CAS.
- P. Kumar, P. With, V. C. Srivastava, K. Shukla, R. Glaser and I. M. Mishra, RSC Adv., 2016, 6, 110235–110246 RSC.
- S. P. Wang, J. J. Zhou, S. Y. Zhao, Y. J. Zhao and X. B. Ma, Chin. Chem. Lett., 2015, 26, 1096–1100 CrossRef CAS.
- P. Unnikrishnan and S. Darbha, J. Chem. Sci., 2016, 128, 957–965 CrossRef.
- B. Liu, C. M. Li, G. Q. Zhang, L. F. Yan and Z. Li, New J. Chem., 2017, 41, 12231–12240 RSC.
- K. W. La, J. C. Jung, H. Kim, S. H. Baeck and I. K. Song, J. Mol. Catal. A: Chem., 2007, 269, 41–45 CrossRef CAS.
- H. J. Lee, S. Park, I. K. Song and J. C. Jung, Catal. Lett., 2011, 141, 531–537 CrossRef CAS.
- H. J. Lee, W. Joe, J. C. Jung and I. K. Song, Korean J. Chem. Eng., 2012, 29, 1019–1024 CrossRef CAS.
- S. Wada, K. Oka, K. Watanabe and Y. Izumi, Front. Chem., 2013, 1, 8 Search PubMed.
- C. H. Chung, F. Y. Tu, T. A. Chiu, T. T. Wu and W. Y. Yu, Chem. Lett., 2021, 50, 856–865 CrossRef CAS.
- H. Liu, W. J. Zou, X. L. Xu, X. L. Zhang, Y. Q. Yang, H. J. Yue, Y. Yu, G. Tian and S. H. Feng, J. CO2 Util., 2017, 17, 43–49 CrossRef CAS.
- J. Li, Z. Y. Zhang, Z. M. Tian, X. M. Zhou, Z. P. Zheng, Y. Y. Ma and Y. Q. Qu, J. Mater. Chem. A, 2014, 2, 16459–16466 RSC.
- Q. X. Zhou, S. L. Ma and S. H. Zhan, Appl. Catal., B, 2018, 224, 27–37 CrossRef CAS.
- S. W. Yang, H. D. Shen, F. Cheng, C. Wu, Y. L. Cao, S. F. Zhuo, Q. Y. Zhang and H. P. Zhang, J. Mater. Chem. A, 2020, 8, 14006–14014 RSC.
- C. M. Ho, J. C. Yu, T. Kwong, A. C. Mak and S. Y. Lai, Chem. Mater., 2005, 17, 4514–4522 CrossRef CAS.
- G. N. Li, S. Dissanayake, S. L. Suib and D. E. Resasco, Appl. Catal., B, 2020, 267, 118373 CrossRef CAS.
- S. K. Lee, C. Jo, J. Kim and R. Ryoo, Microporous Mesoporous Mater., 2020, 293, 109767 CrossRef CAS.
- Z. J. Gong, Y. R. Li, H. L. Wu, S. D. Lin and W. Y. Yu, Appl. Catal., B, 2020, 265, 118524 CrossRef CAS.
- Y. H. Wang, C. H. Chuang, T. A. Chiu, C. W. Kung and W. Y. Yu, J. Phys. Chem. C, 2020, 124, 12521–12530 CrossRef CAS.
- Z. J. Gong, C. C. Chien, S. Mudhulu, J. C. S. Wu, N. Daneu, M. M. Krzmanc and W. Y. Yu, J. Catal., 2022, 416, 222–232 CrossRef CAS.
- Z. Yang, M. M. Lin, X. Lu, J. T. Zheng, W.-Y. Yu, Y. Zhu, H. Chang and Y. Xia, Chem. Eng. J., 2024, 486, 150339 CrossRef CAS.
- Z. L. Wu, M. J. Li, J. Howe, H. M. Meyer and S. H. Overbury, Langmuir, 2010, 26, 16595–16606 CrossRef CAS PubMed.
- T. Taniguchi, T. Watanabe, N. Sugiyama, A. K. Subramani, H. Wagata, N. Matsushita and M. Yoshimura, J. Phys. Chem. C, 2009, 113, 19789–19793 CrossRef CAS.
- C. M. Yang, Y. X. Lu, L. Zhang, Z. J. Kong, T. Y. Yang, L. Tao, Y. Q. Zou and S. Y. Wang, Small Struct., 2021, 2, 2100058 CrossRef CAS.
- R. H. Gao, D. S. Zhang, P. Maitarad, L. Y. Shi, T. Rungrotmongkol, H. R. Li, J. P. Zhang and W. G. Cao, J. Phys. Chem. C, 2013, 117, 10502–10511 CrossRef CAS.
- F. Larachi, J. Pierre, A. Adnot and A. Bernis, Appl. Surf. Sci., 2002, 195, 236–250 CrossRef CAS.
- E. Mamontov and T. Egami, J. Phys. Chem. Solids, 2000, 61, 1345–1356 CrossRef CAS.
- B. Feng, H. Hojo, T. Mizoguchi, H. Ohta, S. D. Findlay, Y. Sato, N. Shibata, T. Yamamoto and Y. Ikuhara, Appl. Phys. Lett., 2012, 100, 073109 CrossRef.
- H. Hojo, E. Tochigi, T. Mizoguchi, H. Ohta, N. Shibata, B. Feng and Y. Ikuhara, Appl. Phys. Lett., 2011, 98, 153104 CrossRef.
- M. Capdevila-Cortada, G. Vilé, D. Teschner, J. Pérez-Ramírez and N. López, Appl. Catal., B, 2016, 197, 299–312 CrossRef CAS.
- Z. X. Yang and R. Mokaya, Microporous Mesoporous Mater., 2008, 113, 378–384 CrossRef CAS.
- Z. Q. Wang, M. J. Zhang, X. B. Hu, V. P. Dravid, Z. N. Xu and G. C. Guo, Chem. Commun., 2020, 56, 403–406 RSC.
- S. Rousseau, O. Marie, P. Bazin, M. Daturi, S. Verdier and V. Harle, J. Am. Chem. Soc., 2010, 132, 10832–10841 CrossRef CAS PubMed.
- M. Honda, M. Tamura, Y. Nakagawa, K. Nakao, K. Suzuki and K. Tomishige, J. Catal., 2014, 318, 95–107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00629a |
| This journal is © The Royal Society of Chemistry 2024 |