
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkali metal-assisted nucleation and growth of stable 1T/2H MoS2 for the hydrogen evolution reaction
Avala
Ramesh
a,
Manoj
Goswami
b,
Surender
Kumar
 b and
Sukanti
Behera
*a
b and
Sukanti
Behera
*a
aMaulana Azad National Institute of Technology, Bhopal 462003, India. E-mail: sukantib@manit.ac.in
bCSIR - Advanced Materials and Processes Research Institute (AMPRI), Bhopal 462026, India
First published on 5th June 2024
Abstract
The present work investigates the extent of mixed phase 1T/2H MoS2 formation as the same precursor solution was maintained for 0 to 8 days in an alkaline medium. To confirm the mixed phase of MoS2, powder X-ray diffraction (XRD), Raman spectroscopy, scanning electron microscopy, and transmission spectroscopy measurements were performed. The major XRD peak of the 1T phase for the 2θ value was at 13.8° and was indexed as the (002) plane along with the 2H phase, and Raman spectra E12g = 381 cm−1 and A1g = 405 cm−1, including extra peaks J1, J2, E1g, and J3. The high-resolution transmission electron spectroscopy interplanar spacing of 1T-MoS2 and 2H-MoS2 was 0.246 nm and 0.688 nm, respectively. Our studies indicate that the alkali metal Na+ plays a crucial role via intercalation, which expanded the inter-layer spacing and increased the catalytic surface area and conductivity, hence affecting the hydrogen evolution reaction (HER) efficiency. The HER showed an overpotential @10 mA cm−2, and Tafel slope of 260 mV, 65 mV dec−1 for the instantly prepared 0-MoS2 sample compared to longer duration samples of 2 to 8 days.
1. Introduction
The widespread reliance on fossil fuels has severely polluted the atmosphere, driving an urgent need for cleaner, renewable sources of energy. One promising alternative is hydrogen fuel cell technology.1 These cells generate electrical energy through an electrochemical reaction between hydrogen (H2) and oxygen (O2) gases, and the amount of energy produced directly correlates to the quantity of these gases used.2 Unfortunately, the naturally occurring concentration of hydrogen gas in the environment is insufficient to power large-scale hydrogen fuel cells, but electrochemical cells can be employed to produce the necessary H2. In a typical setup, these cells comprise three electrodes: a reference electrode, a counter electrode, and a working electrode (where hydrogen generation occurs). Highly effective platinum (Pt) is traditionally used as the working electrode (cathode) to catalyze the hydrogen evolution reaction (HER), where protons are reduced to hydrogen.The high cost of platinum, motivates the search for more affordable yet efficient alternatives,3,4 such as non-noble metals (viz., metal-carbides, phosphides, chalcogenides),5,6 metal oxides (viz., spinel oxide, perovskite oxide),7 and composite hybrid materials.8,9 Two-dimensional transition metal dichalcogenides (2D-TMDs) are widely explored materials in this field because they can be used in the HER, and they are also commonly applied in sensors,10 electronic devices,11 optoelectronics, and energy storage.12,13 2D-TMDs (viz., molybdenum disulfide (MoS2), tungsten disulfide (WS2), and niobium disulfide (NbS2)14) are created by sandwiching a single atomic layer of transition metal atoms between two layers of sulphur atoms.15
In recent years, 2D-MoS2-based materials have shown remarkable promise in lowering the cost of hydrogen production while demonstrating excellent efficiency and stability in the HER.16 Efficient catalysts are crucial for enhancing the kinetics of this reaction and enabling applications in energy conversion and storage systems. MoS2 possesses several unique properties that make it attractive as an HER catalyst. Its layered structure provides a large surface area, which increases the availability of active sites for the reaction. The catalytic activity of MoS2 stems from its electronic structure, which enables efficient charge transfer during the HER in comparison to traditional catalysts, viz., platinum.
2D-MoS2 exists in three distinct phases (2H, 1T, and 3R),17–19 as well as a mixed phase (1T/2H). The 2H and 3R phases of MoS2 are thermodynamically more stable at high temperatures, which can be achieved by physical and chemical methods,20–22 but higher HER efficiency was achieved only in a limited synthesis process. Compared to the pure semiconductor 2H-MoS2, whose active sites are primarily on its edges, the metallic 1T phase increases active sites on both edges and basal surfaces. Zhang et al. (2020) reported that an increase in the percentage of the 1T phase leads to a corresponding increase in the efficiency of the HER. This enhancement is attributed to the higher basal plane catalytic activity of the sulfur sites in the 1T phase for hydrogen binding.23 The 1T phase exhibits several advantages, but its unstability, makes attaining an octahedral structure challenging.
Researchers have rigorously worked to prepare the 1T phase using hydrothermal and traditional intercalation/exfoliation ((CH3)4N+, Li) techniques. Hongqiang et al. created exfoliated 1T-MoS2 nanosheets using sonication and (CH3)4N+ ion intercalation. The intercalated ions expanded the interlayer spacing and decreased the van der Waals force between the layers, which exfoliated it into the 1T phase.24 Mengyao et al. synthesized 1T-MoS2 with superior ambient stability using a hydrothermal method via hydrazine intercalation.25 In the mixed phase, the interaction with the surrounding 2H phase stabilized the 1T phase, allowing it to contribute its beneficial properties without compromising the structure. Browne et al. developed a partially converted 1T phase from 2H using a two-step chemical vapor deposition (CVD) method, followed by a five-day tert-butyllithium immersion treatment.26 The presence of the metallic 1T phase within the mixed phase contributed to its overall high electrical conductivity compared to pure semiconducting 2H-MoS2. This improves conductivity, which exhibits wide applications for acquiring efficient charge transfer in electrocatalytic hydrogen evolution.
A wet chemical approach is one of the synthesis techniques studied to determine greater insight on the nucleation and development of 2D-MoS2. It also provides an alternative and adaptable method to produce a mixed phase (1T/2H) of bulk and atomically thin layers of molybdenum disulphide.27 This is a facile method that can be used to manufacture 2D-MoS2 (1T/2H), in comparison with vapour deposition techniques, and thus, it is suited for extensive applications and large-scale production.28,29 In 2D-MoS2 nucleation and growth by wet chemical methods, MoS2 nuclei or seeds are formed in a solution. This is accomplished by combining precursors such as molybdenum salts (e.g., ammonium molybdate) with sulphur sources (e.g., thiourea) in a solvent, or by employing alternative techniques such as hydrothermal or solvothermal reactions.30
These precursors undergo a reaction to generate MoS2 nuclei, which serve as a basis for the growth of MoS2 particles under optimized experimental parameters for bulk samples, as previously reported.31 The MoS2 nuclei serve as seeds for the deposition of additional MoS2 layers from solution, and these layers laterally expand on the nuclei as the reaction advances, resulting in a continuous 2D-MoS2 film.32 Temperature, reaction time, and precursor concentrations can be optimized to influence the growth kinetics and quality of the final MoS2 layers.33,34 Also, with thin films, a thin seeding layer (Na+) is deposited prior to MoS2 layer deposition. This Na layer affects the growth of thin films, and is advantageous and disadvantageous for MoS2 samples.
Obtaining a mixture of 1T and 2H phases (1T/2H) of MoS2 using chemical vapor deposition (CVD) is challenging due to the high-temperature requirements for CVD (typically 600–800 °C), which destabilizes the desired 1T phase. Browne et al. (2019) achieved the synthesis of 1T/2H MoS2 films through a dual CVD method followed by a lengthy (five-day) immersion treatment with tert-butyllithium, but this approach resulted in a low yield and extended processing time.26 A more efficient method for 1T/2H MoS2 synthesis was developed by Hongmei et al., who employed a two-step hydrothermal approach. This involved a 20-hour reaction at 200 °C, followed by chemical reduction with sodium borohydride (NaBH4) under an argon atmosphere.35 Another approach, reported by Huanran Li et al. (2022), involved treating as-synthesized MoS2 with sodium hydroxide (NaOH) to induce the formation of the mixed 1T/2H phase.36 The hydrothermal method is conducted at a temperature of 130–250 °C in aqueous solution with significant vapor pressure (0.3–4 MPa).37 This approach promotes improved crystallinity, which is required for catalytic activity.
Herein, the present work focused on determining (i) how variation of the synthesis parameters, viz., time, temperature, and pressure regulates the HER activity, whereby the wet chemical synthesis approach was adopted for experimentation, and (ii) how alkali metal ion insertion affects the catalytic activity involved in the HER. Hence, we report a wet-chemical synthesis method at 90 °C for 10 min, followed by filtration and annealing under atmospheric pressure for the stable 1T/2H-MoS2 phase. Mixed phase 1T/2H MoS2 was instantly formed from the precursor solution under normal atmospheric pressure. For a qualitative understanding of the nucleation seeding and growth of the MoS2 phase formation, the precursor was maintained in the same solution and then collected after regular intervals of 0, 2, 4, 6, and 8 days. Then, the solution was filtered, and the residue powder was annealed at 450 °C for 1 h in a sulphur atmosphere to form crystalline MoS2.
MoS2 phase samples were characterized by different structural and morphological techniques, and electrocatalytic HER applications were studied. We observed that crystallite size varied (increasing from 0-MoS2 to 4-MoS2, and then decreasing for 8-MoS2) over time, thereby regulating the HER activity of the samples. Among all samples, the highest HER efficiency in terms of current density (2.07 mA cm−2), onset potential (96 mV), and Tafel slope (65 mV dec−1) was for 0-MoS2. Furthermore, Na+ ion insertion widened the interlayer spacing of metallic 1T/2H MoS2, and thus enhanced the HER performance of the samples.
2. Experimental section
2.1 Synthesis of MoS2 powder
The 2D-MoS2 nanosheets were prepared using the wet chemical method38,39 and Mo and S as precursors, with reaction time intervals of 0, 2, 4, 6 and 8 days. The precursors were 30 mM of ammonium heptamolybdate ((NH4)6Mo7O24·H2O; 99% purity, AR, SRL) for the Mo source, and 0.5 M of thiourea (N2H4CS; 99% purity, AR, SRL) for the sulphur source, and these were added to 1 M of hydrazine hydrate (N2H4; 80% extrapure) in basic NaOH medium (pH 10). Initially, the precursor solution was mixed at room temperature and heated to 90 °C (10 min) on a hot plate for the nucleation of MoS2.The prepared solution was filtered after varied reaction times for controlling the growth of MoS2 (0, 2, 4, 6, and 8 days), with transfer from one filter to another filter solution. The instant filtrate, i.e., the first powder residue product, was named 0-MoS2 (0-day). The filtered solution was undisturbed and settled so that the next batch of MoS2 powder could be created for different durations. After two days, the first solution was filtered, and the collected residue powder sample was named 2-MoS2 (2-days). The same procedure was repeated after 4 days, 6 days, and 8 days, and the solutions were named 4-MoS2, 6-MoS2, and 8-MoS2respectively. All the residue powder samples (0-MoS2, 2-MoS2, 4-MoS2, 6-MoS2, and 8-MoS2) were annealed under a sulphur atmosphere at 450 °C for 1 h in a tubular furnace with the flow rate of carrier Argon (Ar) gas at 100 standard cubic centimeters per minute (sccm). For annealing, the residue powder samples (in a ceramic boat), and 2 g sulphur (in a brass holder) were maintained 10 cm apart in a quartz tube, as illustrated in Fig. 1. The formation of MoS2 can be written by chemical reaction as:
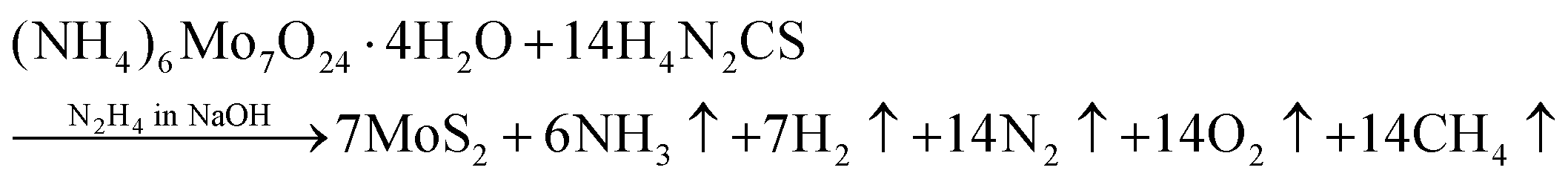
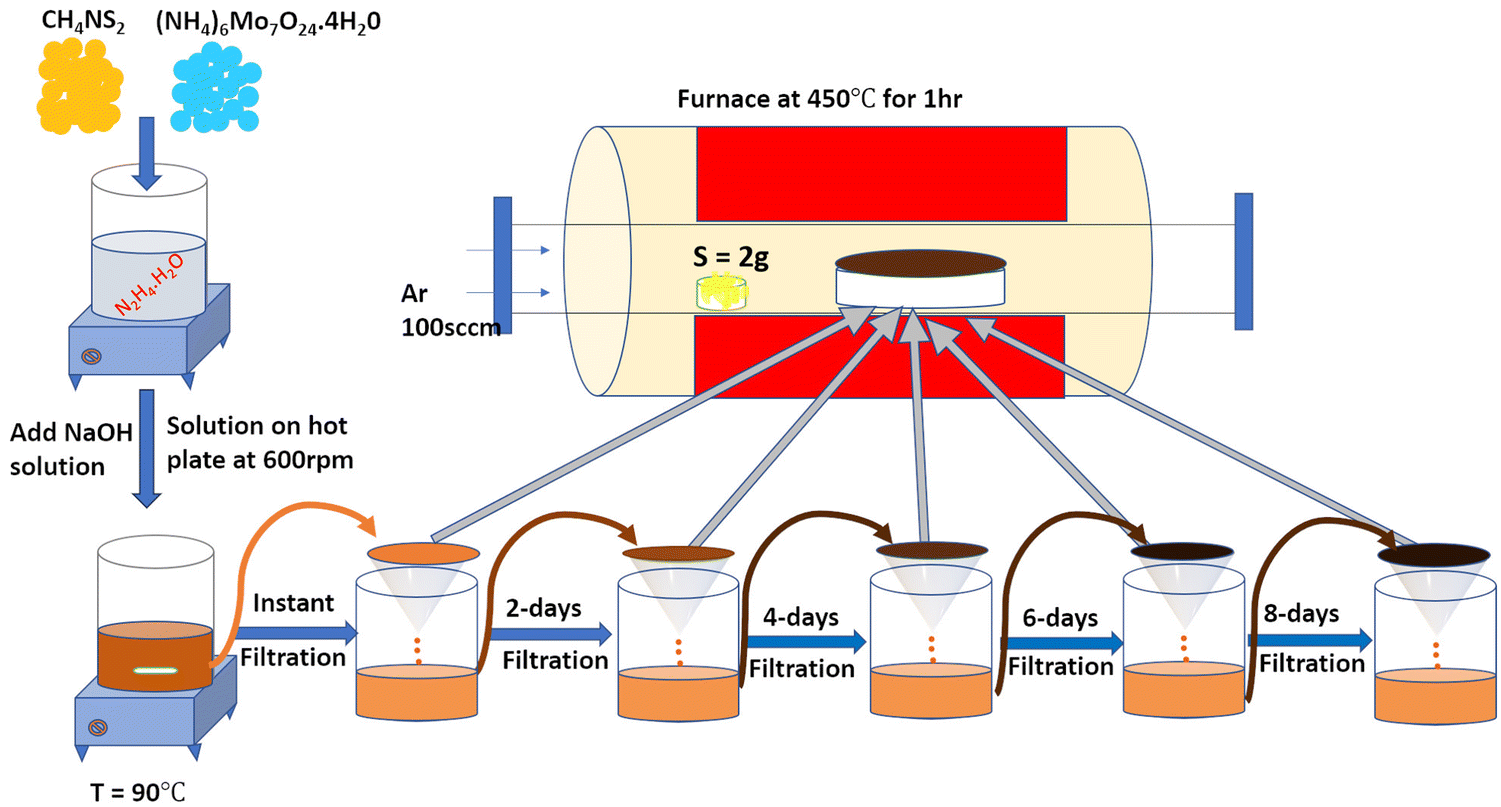 | ||
| Fig. 1 Schematic representation of the experimental setup: A wet chemical method was used to prepare MoS2 material by varying the duration time. Further annealing was conducted under a sulfur atmosphere at 450 °C for 1 hour. | ||
2.2 Characterization of MoS2 powder
Powder X-ray diffraction (PXRD) measurement of MoS2 powder samples was performed using a D8-Bruker (CSR-Indore) instrument with Cu-Kα (1.54178 Å) radiation as the X-ray source in the range of 5–80° (two theta values). Raman spectra were recorded using a LabRam excited with a 473 nm laser source. The morphology of the sample was examined using field emission scanning electron microscopy (FESEM) with energy dispersive X-ray spectroscopy (EDS) mapping, carried out on a Nova Nano 450 with 1 μm resolution, and acceleration voltage of 20 kV. The atomic structure of MoS2 was examined by high-resolution transmission electron microscopy (HRTEM, JEM F-200).2.3 Electrochemical measurements
For HER characterization, the electrochemical measurements were studied using linear sweep voltammetry (LSV), Tafel slope, and electrochemical impedance spectroscopy (EIS) at ambient conditions in 0.5 M H2SO4 electrolyte solution using a CHI electrochemical workstation with a three-electrode cell system.40 The MoS2 catalyst was deposited on a glassy carbon electrode (GCE) and acted as the working electrode (WE), while platinum wire (Pt) and a calomel electrode were the counter electrode and reference electrode, respectively. The WE preparation was carried out using a 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 ratio of catalyst, graphene, and polyvinylidene difluoride (PVDF) binder dissolved in N-methyl pyrrolidone (NMP) to form a homogenous ink solution. Then, the black ink was deposited on a 3-mm-diameter GCE (loading 0.004 g cm−2) using a drop-cast method. At room temperature, the properties of the HER were evaluated using linear sweep voltammetry (LSV) from 0 to −0.8 V (V vs. RHE) at a scan rate of 5 mV s−1. The following equation was applied to assign all potential contours to the reversible hydrogen electrode (RHE) according to the Nernst equation:
:10:10 ratio of catalyst, graphene, and polyvinylidene difluoride (PVDF) binder dissolved in N-methyl pyrrolidone (NMP) to form a homogenous ink solution. Then, the black ink was deposited on a 3-mm-diameter GCE (loading 0.004 g cm−2) using a drop-cast method. At room temperature, the properties of the HER were evaluated using linear sweep voltammetry (LSV) from 0 to −0.8 V (V vs. RHE) at a scan rate of 5 mV s−1. The following equation was applied to assign all potential contours to the reversible hydrogen electrode (RHE) according to the Nernst equation:| E(V) versus RHE = E(V) versus Hg/HgCl + 0.242 V + 0.059 pH |
3. Results and discussion
Analysis of the powder X-ray diffraction (PXRD) pattern of mixed-phase 1T/2H-MoS2 samples (as shown in Fig. 2(a)) reveals a key distinction compared to the pattern of bulk 2H-MoS2. This difference lies in the behavior of the characteristic (002) peak at 14.41° (2θ) for pristine 2H-MoS2. Also from the XRD data, the peak shifted to a lower 2θ angle (002) peak at 13.86°,18 and this signified a crucial structural change within the material. The (002) peak corresponds to the interlayer spacing of 0.61 nm between the MoS2 sheets. When the peak shifts, this indicates that the interlayer spacing is no longer uniform.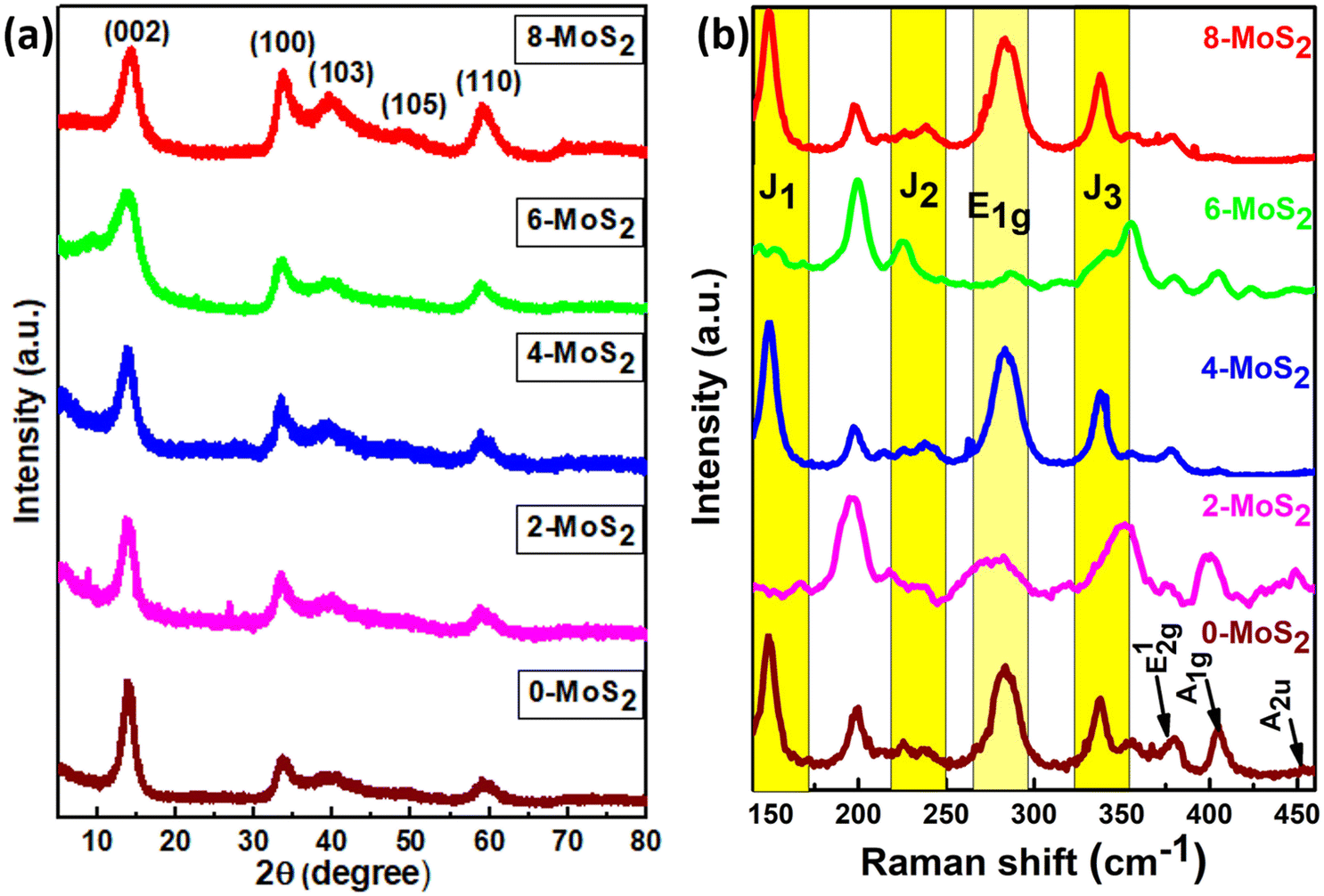 | ||
| Fig. 2 (a) Corresponds to the X-ray diffraction pattern and (b) Raman spectra of the prepared MoS2 samples with duration time of 0, 2, 4, 6, and 8 days. | ||
In the current study, the presence of a lower angle peak shift suggests an expansion of the interlayer spacing, which probably resulted from the Na+ intercalation between the layers (discussed further in the HRTEM analysis) compared to pure 2H-MoS2. The Na+ intercalated between the layers, as reported by Nuwan et al. (2017), expanded the gap between the layers to 1.2 nm, and as a result, the (002) peaks shifted to a lower angle at low-temperature synthesis, suggesting the 1T phase.41 Also, Huanran et al. (2022) reported that the concentration of Na+ decreased when the hydrothermal temperature at 180 °C increased. Hence, the gap was decreased to 0.65 nm causing the (002) peaks to shift to a higher angle, suggesting the emergence of a mixed phase (1T/2H).36
In this work, the sample was annealed at the high temperature of 450 °C, and the gap between the layers was 0.688 nm. This expansion is a well-established signature of the presence of the 1T phase within the MoS2 lattice.42 The overall XRD pattern exhibited the characteristic peaks associated with the hexagonal and trigonal crystalline phases of MoS2 at 33.36° (100), 39.9° (103), 49° (105), and 59° (110) 2θ values consistent with JCPDS No. 37-1492. The lower angle shift of the (002) peaks provides crucial evidence for the successful incorporation of the 1T phase within the semiconductor 2H phase MoS2 lattice,39,43 and no additional peaks were observed for all MoS2 samples. The increased interlayer spacing and phase transformation due to the alkali metals increased the catalytically active surface area and movement of electrons within the MoS2 lattice. This led to increased conductivity and greater catalytic activity. The crystallite size of the prepared MoS2 nanoparticles was calculated using Scherrer's formula:44
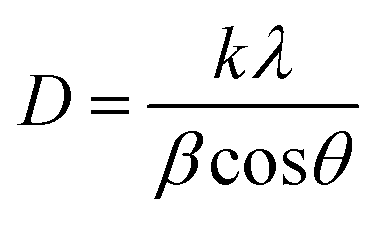
| Sample | Average D (nm) value |
|---|---|
| 0-MoS2 | 17.7 |
| 2-MoS2 | 20.3 |
| 4-MoS2 | 37.97 |
| 6-MoS2 | 23.45 |
| 8-MoS2 | 20.07 |
The structural phase of all synthesized MoS2 material was further analyzed by Raman spectra, and Fig. 2b shows Raman modes of the 1T/2H mixed phase of MoS2. From the figure, Raman spectra were found in seven peaks at 148, 238, 283, 337, 381, 405, and 452 cm−1 representing the J1, J2, E1g, J3, E12g, A1g, and A2u phonon modes of all synthesized MoS2, respectively. Out of seven phonon modes, two modes are the E12g in-plane vibration of Mo–S, and A1g is an out-of-plane vibration of S–S at 381 cm−1 and 405 cm−1 of the 2H phase with the A2u phonon mode at 452 cm−1 (0, 4, 6, 8-MoS2). However, for 2-MoS2, the modes slightly shifted to the higher energy side (375 cm−1 and 399 cm−1). The Raman mode (A1g) intensity of MoS2 samples decreased at 405 cm−1 as the number of days passed from 0 to 6 days and disappeared for 8-MoS2. This is because of the absence of Raman phonon mode (A1g), which is a relatively out-of-plane (S–S) bond vibration47 for 8-MoS2. The trigonal (1T) phase of all as-synthesized MoS2 was significant because it exhibited additional peaks at 148, 238, 283, and 337 cm−1, which corresponded to J1, J2, E1g, and J3, respectively.25,43 However, the 2- and 6-MoS2 sample exhibited J1, J2, and J3 peaks, which are broad with very weak intensity,48,49 while the J3 peak of 2-MoS2 slightly shifted to the lower energy side of 350 cm−1. The E1g band was attributed to the octahedral coordination of Mo in 1T MoS2.
The surface morphologies and elemental distribution of MoS2 samples were examined using FESEM and EDS mapping, respectively. In Fig. 3(a)–(c), a FESEM image shows all the prepared MoS2 samples, and the inset depicts the change in solution colour from 0 days to 4 days. There was a wine-red colour to the initial solution (after the first filtration, which changed to brown after two days, and darkened further), and with an increase in the number of days (4, 6, and 8 days), the solution changed to dark brown. A spherical nanoparticle shape was found for 0-MoS2, 2-MoS2, and 4-MoS2, as shown in Fig. 4. The dimension compared to 0-MoS2 increased with increasing reaction time duration (2 and 4 days), and can be said that the MoS2 particles were involved in nucleation. This nucleation further increased while the reaction duration proceeded, and thus additional agglomeration occurred, which led to an increase in crystallite size.
 | ||
| Fig. 3 FESEM images of (a) 0-MoS2, (b) after 2 days, and (c) after 4 days. The inset shows solution colour changes as time increased from 0 to 4 days. | ||
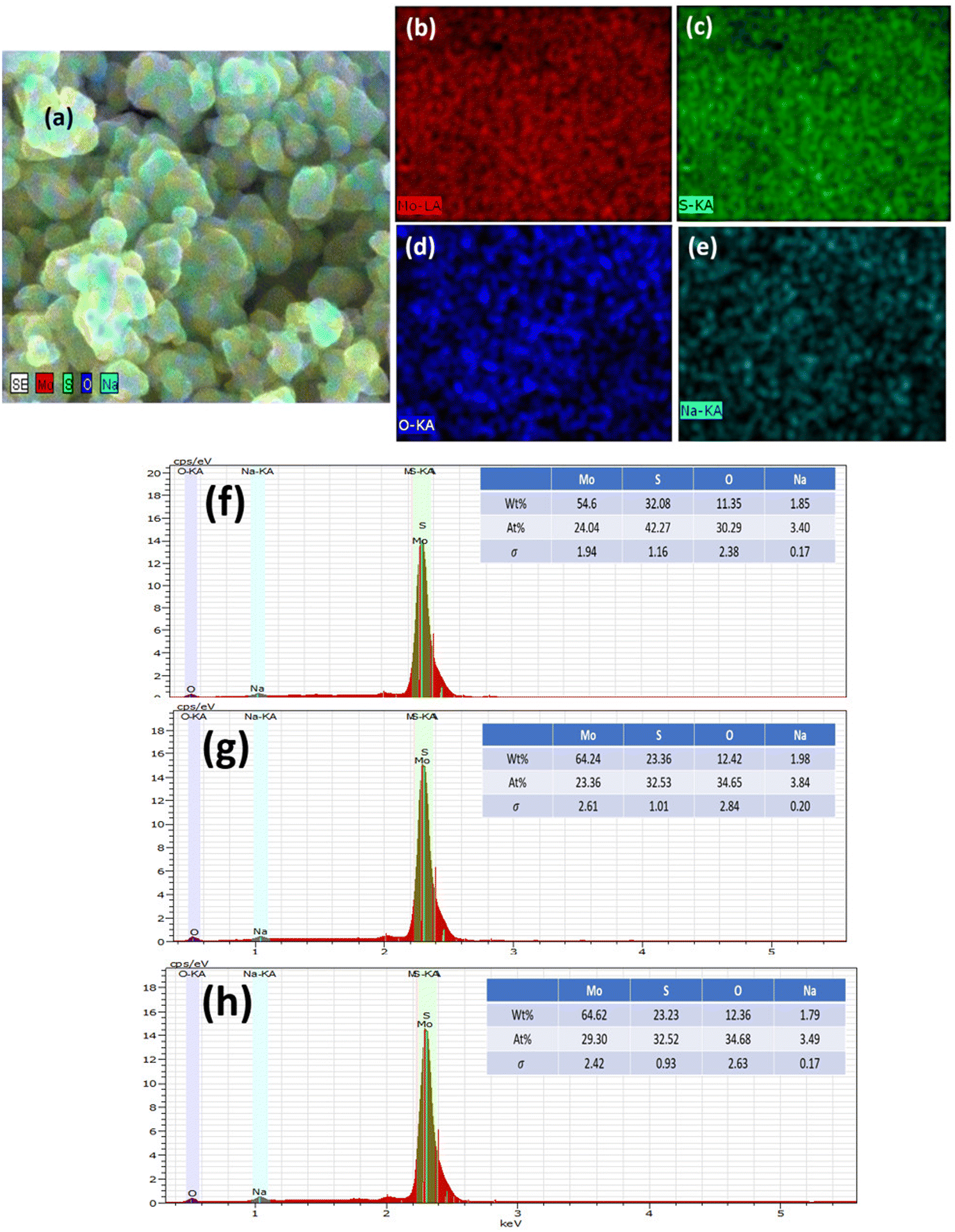 | ||
| Fig. 4 Elemental mapping. (a) FESEM image of the 0-MoS2 sample, (b) molybdenum element in red, (c) sulphur element in green, (d) oxygen in blue, and (e) sodium in light blue. Energy dispersive X-ray spectroscopy (EDS) study of (f) 0-MoS2, (g) 2-MoS2, and (h) 4-MoS2 samples. | ||
The elemental composition of MoS2 was determined using EDS, as demonstrated in Fig. 4(a)–(h). Extracted images show strong peaks, confirming the presence of Mo, S, and weak peaks of Na and O. However, the elemental mapping of 0-MoS2 shown in Fig. 4(a)–(e) indicates that Mo and S were uniformly dispersed, as shown in Fig. 4(b) in red, and Fig. 4(c) in green, respectively. Nevertheless, some O and Na were detected (shown in Fig. 4(d) and (e)), where Na+ was observed due to the addition of NaOH to the precursor solution to maintain the pH value, while O comes from the atmosphere.50 Each Mo and S element was evenly distributed in the 0-MoS2, 2-MoS2, and 4-MoS2 compounds (0, 2, and 4 days). However, when the reaction duration time advanced, the elemental mapping of Na wt% increased. The Na elements were dense in the case of the larger crystallite size for 4-MoS2. As a result of this, Na supports enhanced the crystallinity of the samples.51 However, as the particle growth proceeded, the saturation of the Na bonds decreased, which lowered the energy barrier and increased the growth rate.52,53 The increase in the number of days provided the solution sufficient time to create a greater yield with large particle sizes.
HRTEM analysis was carried out on 0-MoS2, to examine phase crystallinity and dimensions, as shown in Fig. 5(a). The nanosheets were formed by MoS2 layer by layer, as shown in Fig. 5(b), and the d-spacing between the interplane was 0.688 nm and corresponds to the (002) plane of 0-MoS2. Generally, bulk 2H-MoS2 has an inter-layer distance of 0.61 nm.38,39 However, the interplanar spacing of the prepared 0-MoS2 sample was slightly higher (0.688 nm) than that of the bulk 2H-MoS2, which is consistent with our XRD data, as displayed in Fig. 2(a). The 1T/2H-MoS2 crystal surface was studied to determine discrimination between the two phases using the high magnification of HRTEM, as shown in Fig. 5(c).
 | ||
| Fig. 5 (a) Transmission electron microscopy image, (b) high-resolution transmission electron microscopy (HRTEM) image of 0-MoS2, (c) low magnification HRTEM of 1T/2H-MoS2, (d) HRTEM image of region magnified by the yellow ellipse in (a) and (e) white region magnified in (a) and (f) selected area electron diffraction pattern of 0-MoS2. | ||
The enlarged image of the MoS2 2H phase shown in Fig. 5(d) (represented by the yellow ellipses in Fig. 5(c)) displays lattice fringes as a honeycomb structure (trigonal prismatic structure). Also in Fig. 5(e), the magnified white ellipse region in Fig. 5(c) shows the 1T phase of the lattice fringes with a trigonal structure (octahedral structure).25,35 The interplanar spacing of the 1T phase is 0.246 nm, as shown in Fig. 5(c), and indexed as the (102) plane. Selected area electron diffraction (SAED) was performed to evaluate the material's crystallinity, and also support the matching crystallinity plane,6 as shown in Fig. 5(f). It consisted of four blurred diffraction rings with multiple bright spots that were indexed as (100), (103), (110), and (108) accordingly (JCPDS No. 37-1492).54 The SAED image confirmed the polycrystalline behaviour of the 0-MoS2 sample.55
3.1 Electrocatalytic application
The sample polarization curves were examined using linear sweep voltammetry (LSV) with a scan rate of 5 mV s−1 in 0.5 M of H2SO4 electrolyte solution under ambient conditions, as shown in Fig. 6(a). The Pt electrode (0.5 mm in diameter and 32 mm in length) acted as a reference electrode, and all MoS2 samples showed catalytic activity towards the HER with a high current density up to 110 mA cm−2 and low overpotential. For the performance of samples in the HER, onset potentials were between −90 to −270 mV, which can be attributed to the nanoparticle structure assembled by the nanosheets. It is widely accepted that the HER active sites are located on the edge S-sites, and hence, changing the microstructure of MoS2 to obtain additional edge planes enhances the HER activity. Among the prepared set of catalyst samples of the present work, the 0-MoS2 shows the best catalytic activity due to the smaller crystallite size towards the HER, with positive onset potential at −96 mV and a current density of 10 mA cm−2 at −260 mV overpotential. This indicates that Na+ intercalation can promote the stabilization of the 1T phase, and this phase enhancement is crucial for the HER. However, it also increases the overall electrical conductivity and facilitates electron transport during the HER process.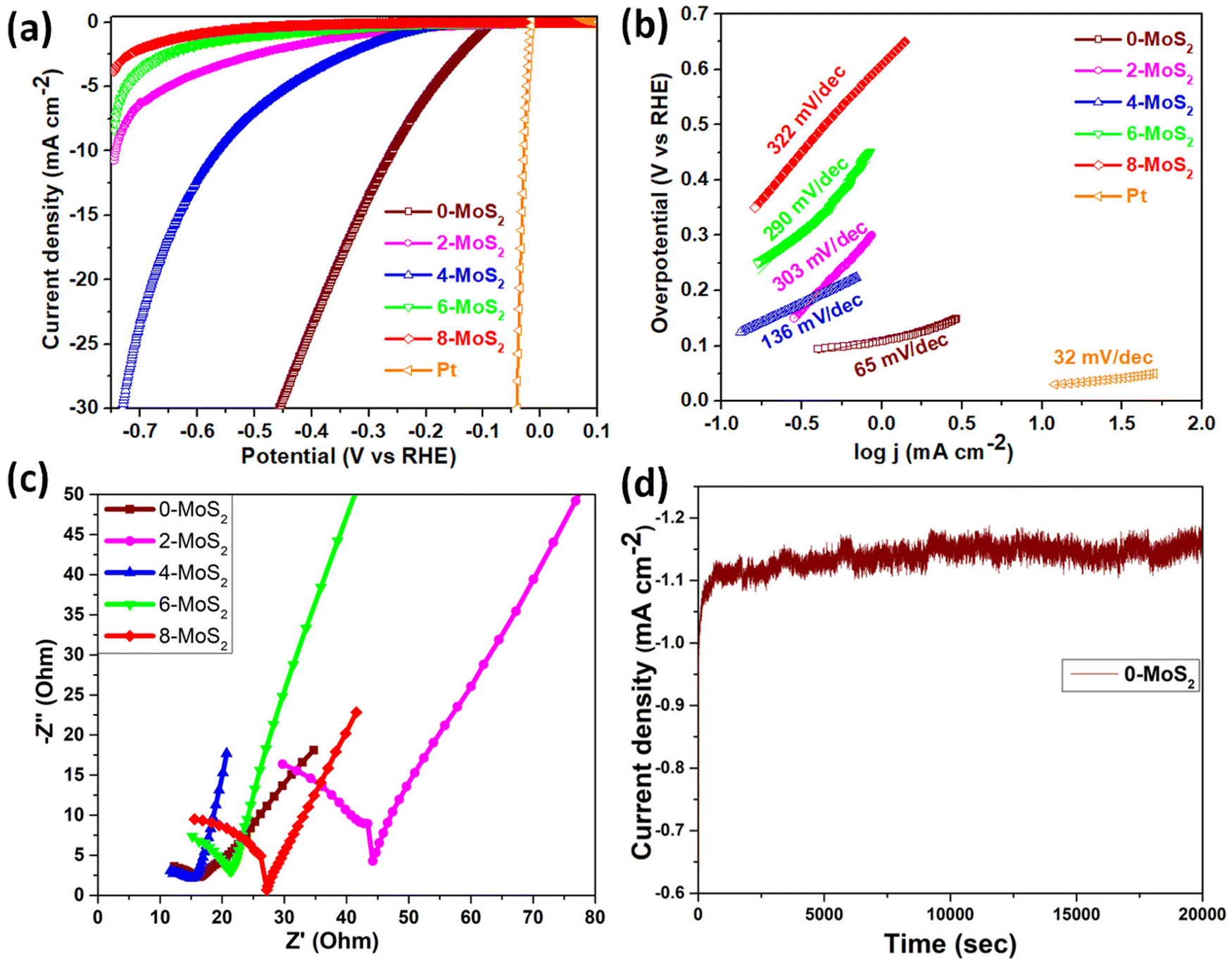 | ||
| Fig. 6 Electrocatalytic performance of Pt- and MoS2-based material. (a) Linear sweep voltammetry (LSV) curve, (b) Tafel slope, (c) electrochemical impedance spectroscopy (EIS) of 0-MoS2, 2-MoS2, 4-MoS2, 6-MoS2, and 8-MoS2, and (d) stability test of 0-MoS2. | ||
The LSV value of the characterized 0-MoS2 sample is equal to earlier reported values for MoS2-based catalytic materials determined using CVD, intercalation, and hydrothermal techniques, as listed in Table 2. However, materials with different reaction duration times, i.e., 2-MoS2, 4-MoS2, 6-MoS2, and 8-MoS2, exhibited different catalytic activity due to their intrinsic structure and the synthesized experimental parameters. Fig. 6(a) shows all MoS2 samples producing HER activity. The overpotential values @10 mA cm−2 current density were −260 mV, −744 mV, and −560 mV for 0-MoS2, 2-MoS2, and 4-MoS2, respectively. Although there was an increase in the number of days the current density decreased to −8.3 mA cm−2 and −3.8 mA cm−2, corresponding to 6-MoS2 and 8-MoS2 at −746 mV overpotential, respectively, and 8-MoS2 sample exhibited the least catalytic activity towards the HER.
| Name of the catalyst | Synthesis method | Overpotential (mV) η@10 mA cm−2 | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|
| 1T/2H-MoS2 | CVD, immersion treatment | 320 | 70 | 26 |
| 1T-MoS2 (Na–MoS2 at 160 °C) | Hydrothermal | 255 | 44 | 36 |
| 1T/2H-MoS2 (Na MoS2 at 180 °C) | Hydrothermal | 271 | 49 | 36 |
| 1T/2H-MoS2 (HF treated) | Hydrothermal | 317 | 90 | 36 |
| 1T@2H-MoS2 | Exfoliation, and dispersion in PVP | 330 | 70 | 56 |
| (1T/2H) MoS2 scrolle@pt | Exfoliation, rolling up | 251 | 57 | 57 |
| (1T/2H) MoS2@pt scrolle | Exfoliation, rolling up | 125 | 39 | 57 |
| 1T@2H-MoS2 nanosheet 8 h C−1 | Hydrothermal | 64 | 49 | 27 |
| 1T@2H-MoS2-4 h C−1 | Hydrothermal | 99 | 57 | 27 |
| 1T/2H-MoS2 | Hydrothermal | 296 | 59.6 | 58 |
| 1T-2H-MoS2 | Solid vapour reaction | 280 | 65 | 39 |
| 1T@2H-MoS2 | Hydrothermal | 270 | 88 | 30 |
| 1T-MoS2 (MoS2-4HZ) | Hydrothermal | 230 | 64 | 25 |
| 1T/2H-MoS2 (MoS2-1HZ) | Hydrothermal | 284 | 74 | 25 |
| 1T/2H-MoS2 | Wet chemical method | 260 | 65 | Current work |
The Tafel plots of all samples were derived from the polarization curves mentioned in Fig. 6(b). The Tafel slope is determined by fitting the linear portion of the Tafel plot to the Tafel equation η = blog|j| + a, where η denotes overpotential, j denotes current density, b denotes the Tafel slope, and a denotes the intercept. The lower value of the Tafel slope indicates a more optimal chemical kinetic reaction for HER applications with low potential. In Fig. 6b, the smallest Tafel slope of 65 mV dec−1 was obtained for the 0-MoS2 sample, and the Tafel slopes of 2-MoS2, 4-MoS2, 6-MoS2, and 8-MoS2 were 303 mV dec−1, 136 mV dec−1, 290 mV dec−1, and 322 mV dec−1, respectively. From this analysis, the smaller Tafel slope of 0-MoS2 as compared to the others indicates that it performs the HER with greater efficiency at a lower overpotential. The HER reaction occurred on the surface of the MoS2 electrode in acidic (0.5 M H2SO4) medium, and can be explained by the following mechanism of the Volmer–Heyrovsky reaction, as shown in Fig. 7:
| H3O+ + e− → Hads + H2O (Volmer reaction) | (1) |
| Hads + H3O+ + e− → H2 + H2O (Heyrovsky reaction) | (2) |
| Hads + Hads → H2 (Tafel reaction) | (3) |
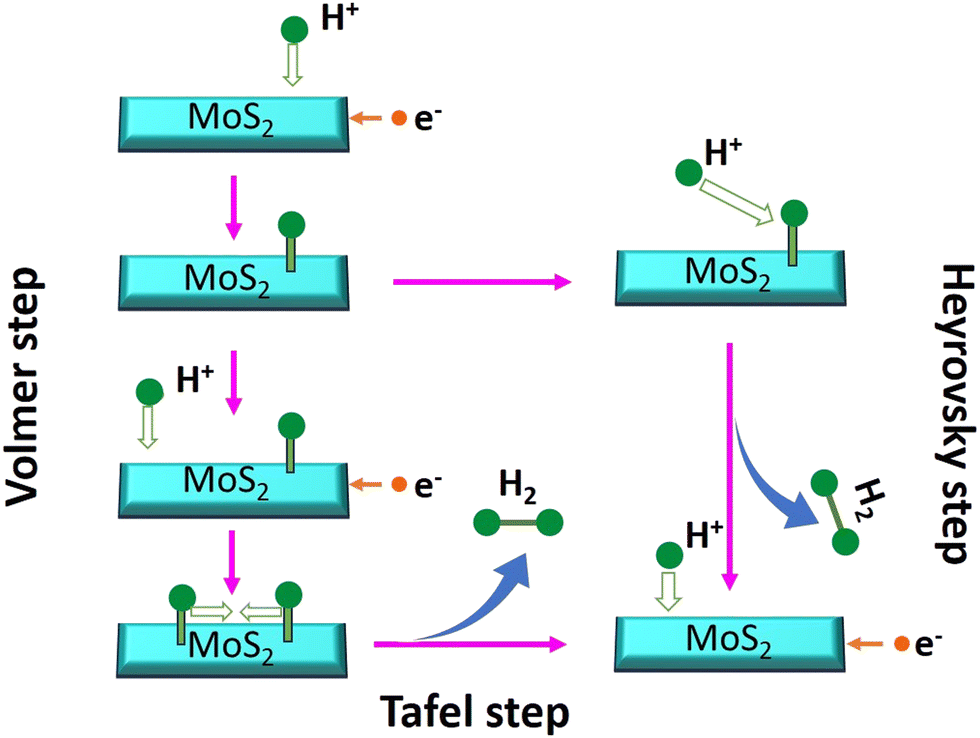 | ||
| Fig. 7 Schematic representation of the reaction pathway diagram for a catalytic process. | ||
Furthermore, EIS was used to investigate the interface reactions and electrode kinetics during the catalytic HER reaction. Fig. 6c depicts the Nyquist plots of the prepared MoS2 samples. The Nyquist plot shown in the figure is divided into two sections: a semicircle and a straight line, and these portions relate to the high and low frequency bands, respectively.59 The point at which the figure intersects the x-axis at higher frequencies denotes the electrolyte resistance (Rs) that is potentially independent. The semi-circular arc at the lower frequency end is potentially dependent and reflects the resistance-to-charge transfer (Rct) across the electrode–electrolyte interface.60 This resistance is connected to the ease with which charges may be separated during the electrochemical process. The charge-transfer resistance for an electrocatalyst with a smaller semicircle diameter is decreased, which indicates increased electrocatalytic activity.61 It was found that the lowest semicircle was for 4-MoS2, in comparison to the other samples (0-, 2-, 6-, and 8-MoS2). The calculated (Rct) values of prepared samples 0, 2, 4, 6, and 8-MoS2 were 6.1, 35.4, 5.6, 14.2, and 18.4 Ω, respectively. 4-MoS2 conveyed unusually low values due to its exceptional intrinsic conductivity, although the second-lowest Rct value was for 0-MoS2. The most inherent measure of activity for the HER is the exchange current density, Iex, which is determined from the EIS:62
| Exchange current density (Iex) = RT/nFΘ |
| Sample | Onset potential mV | Overpotential (mV) @10 mA cm−2 | Tafel (mV dec−1) | R s (Ω) | R ct (Ω) | I ex (mA cm−2) |
|---|---|---|---|---|---|---|
| 0-MoS2 | 96 | 260 | 65 | 9.543 | 6.175 | 2.07 |
| 2-MoS2 | 133 | 744 | 303 | 3.124 | 35.36 | 0.36 |
| 4-MoS2 | 270 | 560 | 136 | 7.917 | 5.631 | 2.28 |
| 6-MoS2 | 251 | — | 290 | 6.144 | 14.29 | 0.8 |
| 8-MoS2 | 251 | — | 322 | 5.94 | 18.47 | 0.6 |
Besides HER activity, stability is a crucial parameter for evaluating an advanced electrocatalyst's longevity. A stability test in an acidic medium was performed to assess durability. The time-dependent curve, which was obtained by applying a constant overpotential of 96 mV for 20000 s of data, as displayed in Fig. 6d, revealed that a continuous HER process occurred to create molecular hydrogen without any current deterioration.
4. Conclusion
In summary, this work successfully synthesized mixed phase 1T/2H-MoS2via a wet chemical synthesis method. To study nucleation seeding and growth, the reaction was carried out at various durations of time, i.e., 0, 2, 4, 6, and 8 days. It was observed that the crystallite size of MoS2 increased from 17.7 to 37.9 over a duration of 0 to 4 days, and subsequently decreased to 23.45 and 20 nm after 6 and 8 days, respectively. This observation may be attributed to the growth of MoS2 particles with a precursor concentration, which gradually decreased with time. This observation was further validated by XRD data using Scherrer's formula. Among the materials studied, 1T/2H 0-MoS2 exhibited the smallest crystallite size of 17.7 nm and the highest catalytic activity with an onset potential of −96 mV, an exchange current density of 2.07 mA cm−2, a low Tafel slope of 65 mV dec−1, and a charge transfer resistivity of 6.175 Ω. Moreover, the presence of Na+ ions expanded the space between the layers, creating a larger catalytically active surface area and increasing the material's electrical conductivity. Both improved conductivity and increased surface area were beneficial for enhanced electrocatalytic activity. The developed mixed 1T/2H phase 0-MoS2 exhibited superior performance as an instantly prepared catalyst and can be used in the scalable synthesis of MoS2 nanocrystals, with promising use in acid electrolytes. Furthermore, the intercalated sodium ions (Na+) hold promise for future research in sodium-ion batteries and hydrogen cells. Our data could significantly contribute to the advancement of renewable energy technologies.Conflicts of interest
There are no conflicts to declareAcknowledgements
The authors would like to thanks UGC-DAE CSR-Indore for providing the PXRD facility, and assistance with the Raman spectroscopy analysis studies. Also, Avala Ramesh thanks Mr Himanshu Shekhar for his help and support with the FESEM analysis.References
- H. G. Shiraz, X. Crispin and M. Berggren, Int. J. Hydrogen Energy, 2021, 46, 24060–24077 CrossRef CAS.
- K. Rajashekara, IEEE Trans. Ind. Appl., 2005, 41, 682–689 CrossRef.
- A. A. Feidenhans’l, Y. N. Regmi, C. Wei, D. Xia, J. Kibsgaard and L. A. King, Chem. Rev., 2023, 124, 5617–5667 CrossRef.
- Y. Qiu, X. Dai, Y. Wang, X. Ji, Z. Ma and S. Liu, J. Colloid Interface Sci., 2023, 629, 297–309 CrossRef CAS.
- A. D. Marinov, L. Bravo Priegue, A. R. Shah, T. S. Miller, C. A. Howard, G. Hinds, P. R. Shearing, P. L. Cullen and D. J. L. Brett, ACS Nano, 2023, 17(6), 5163–5186 CrossRef CAS.
- K. Song, H. Zhang, Z. Lin, Z. Wang, L. Zhang, X. Shi, S. Shen, S. Chen and W. Zhong, Adv. Funct. Mater., 2024, 34, 1–9 Search PubMed.
- Y. Zhu, Q. Lin, Y. Zhong, H. A. Tahini, Z. Shao and H. Wang, Energy Environ. Sci., 2020, 13, 3361–3392 RSC.
- Z. Wang, Z. Lin, Y. Wang, S. Shen, Q. Zhang, J. Wang and W. Zhong, Adv. Mater., 2023, 35(25), 2302007 CrossRef CAS.
- S. Shen, H. Zhang, K. Song, Z. Wang, T. Shang, A. Gao, Q. Zhang, L. Gu and W. Zhong, Angew. Chem., Int. Ed., 2024, 63(1), e202315340 CrossRef CAS.
- S. Y. Cho, S. J. Kim, Y. Lee, J. S. Kim, W. Bin Jung, H. W. Yoo, J. Kim and H. T. Jung, ACS Nano, 2015, 9, 9314–9321 CrossRef.
- K. K. Liu, W. Zhang, Y. H. Lee, Y. C. Lin, M. T. Chang, C. Y. Su, C. S. Chang, H. Li, Y. Shi, H. Zhang, C. S. Lai and L. J. Li, Nano Lett., 2012, 12, 1538–1544 CrossRef CAS.
- Y. Arora, A. P. Shah, S. Battu, C. B. Maliakkal, S. Haram, A. Bhattacharya and D. Khushalani, Sci. Rep., 2016, 6, 1–7 CrossRef.
- D. Wang, Y. Xiao, X. Luo, Z. Wu, Y. J. Wang and B. Fang, ACS Sustainable Chem. Eng., 2017, 5, 2509–2515 CrossRef CAS.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef.
- G. Habibi Jetani and M. B. Rahmani, Opt. Mater., 2022, 124, 111974 CrossRef CAS.
- M. He, F. Kong, G. Yin, Z. Lv, X. Sun, H. Shi and B. Gao, RSC Adv., 2018, 8, 14369–14376 RSC.
- E. Benavente, M. A. Santa Ana, F. Mendizábal and G. González, Coord. Chem. Rev., 2002, 224, 87–109 CrossRef CAS.
- S. Shi, Z. Sun and Y. H. Hu, J. Mater. Chem. A, 2018, 6, 23932–23977 RSC.
- D. Wang, X. Zhang, S. Bao, Z. Zhang, H. Fei and Z. Wu, J. Mater. Chem. A, 2017, 5, 2681–2688 RSC.
- J. Cheng, C. Shen, Y. He, H. Wei, S. Liu, P. Qiu, Y. Song, S. Wei, Z. Wang, X. Zheng and M. Peng, J. Alloys Compd., 2021, 853, 157374 CrossRef CAS.
- P. Yang, X. Zou, Z. Zhang, M. Hong, J. Shi, S. Chen, J. Shu, L. Zhao, S. Jiang, X. Zhou, Y. Huan, C. Xie, P. Gao, Q. Chen, Q. Zhang, Z. Liu and Y. Zhang, Nat. Commun., 2018, 9, 1–10 CrossRef CAS.
- U. Krishnan, M. Kaur, K. Singh, M. Kumar and A. Kumar, Superlattices Microstruct., 2019, 128, 274–297 CrossRef CAS.
- Y. Zhang, Y. Kuwahara, K. Mori, C. Louis and H. Yamashita, Nanoscale, 2020, 12, 11908–11915 RSC.
- H. Jin, Y. Yu, Q. Shen, P. Li, J. Yu, W. Chen, X. Wang, Z. Kang, L. Zhu, R. Zhao, L. Zheng, W. Song and C. Cao, J. Mater. Chem. A, 2021, 9, 13996–14003 RSC.
- M. Li, Z. Zhou, L. Hu, S. Wang, Y. Zhou, R. Zhu, X. Chu, A. Vinu, T. Wan, C. Cazorla, J. Yi and D. Chu, ACS Appl. Mater. Interfaces, 2022, 14, 16338–16347 CrossRef CAS.
- M. P. Browne, F. Novotný, C. L. Manzanares Palenzuela, J. Šturala, Z. Sofer and M. Pumera, ACS Sustainable Chem. Eng., 2019, 7, 16440–16449 CrossRef CAS.
- S. Shi, D. Gao, B. Xia, P. Liu and D. Xue, J. Mater. Chem. A, 2015, 3, 24414–24421 RSC.
- Q. Liu, X. Li, Q. He, A. Khalil, D. Liu, T. Xiang, X. Wu and L. Song, Small, 2015, 11, 5556–5564 CrossRef CAS.
- S. Das, G. Swain, B. P. Mishra and K. Parida, New J. Chem., 2022, 46, 14922–14932 RSC.
- Y. Yao, K. Ao, P. Lv and Q. Wei, Nanomaterials, 2019, 9, 844 CrossRef CAS.
- Y. Xi, M. I. Serna, L. Cheng, Y. Gao, M. Baniasadi, R. Rodriguez-Davila, J. Kim, M. A. Quevedo-Lopez and M. Minary-Jolandan, J. Mater. Chem. C, 2015, 3, 3842–3847 RSC.
- B. Song, K. He, Y. Yuan, S. Sharifi-Asl, M. Cheng, J. Lu, W. A. Saidi and R. Shahbazian-Yassar, Nanoscale, 2018, 10, 15809–15818 RSC.
- J. Mun, H. Park, J. Park, D. Joung, S. K. Lee, J. Leem, J. M. Myoung, J. Park, S. H. Jeong, W. Chegal, S. Nam and S. W. Kang, ACS Appl. Electron. Mater., 2019, 1, 608–616 CrossRef CAS.
- A. Singh, M. Moun, M. Sharma, A. Barman, A. Kumar Kapoor and R. Singh, Appl. Surf. Sci., 2021, 538, 148201 CrossRef CAS.
- H. Fan, R. Wu, H. Liu, X. Yang, Y. Sun and C. Chen, J. Mater. Sci., 2018, 53, 10302–10312 CrossRef CAS.
- H. Li, X. Han, S. Jiang, L. Zhang, W. Ma, R. Ma and Z. Zhou, Green Energy Environ., 2022, 7, 314–323 CrossRef CAS.
- A. K. da Silva, T. G. Ricci, A. L. de Toffoli, E. V. S. Maciel, C. E. D. Nazario and F. M. Lanças, in Handbook on Miniaturization in Analytical Chemistry, Elsevier, 2020, pp. 77–98 Search PubMed.
- D. Vikraman, K. Akbar, S. Hussain, G. Yoo, J. Y. Jang, S. H. Chun, J. Jung and H. J. Park, Nano Energy, 2017, 35, 101–114 CrossRef CAS.
- S. Wang, D. Zhang, B. Li, C. Zhang, Z. Du, H. Yin, X. Bi and S. Yang, Adv. Energy Mater., 2018, 8, 1–7 Search PubMed.
- W. Dong, H. Liu, X. Liu, H. Wang, X. Li and L. Tian, Int. J. Hydrogen Energy, 2021, 46, 9360–9370 CrossRef CAS.
- N. H. Attanayake, A. C. Thenuwara, A. Patra, Y. V. Aulin, T. M. Tran, H. Chakraborty, E. Borguet, M. L. Klein, J. P. Perdew and D. R. Strongin, ACS Energy Lett., 2018, 3, 7–13 CrossRef CAS.
- S. Das, G. Swain and K. Parida, Mater. Chem. Front., 2021, 5, 2143–2172 RSC.
- H. Cao, Z. Bai, Y. Li, Z. Xiao, X. Zhang and G. Li, ACS Sustainable Chem. Eng., 2020, 8, 7343–7352 CrossRef CAS.
- K. C. Lalithambika, K. Shanmugapriya and S. Sriram, Appl. Phys. A: Mater. Sci. Process., 2019, 125, 1–8 CrossRef.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef.
- J. Lee, J. Yang, S. G. Kwon and T. Hyeon, Nat. Rev. Mater., 2016, 1, 16034 CrossRef CAS.
- D. Wang, Z. Pan, Z. Wu, Z. Wang and Z. Liu, J. Power Sources, 2014, 264, 229–234 CrossRef CAS.
- C. H. Sharma, A. P. Surendran, A. Varghese and M. Thalakulam, Sci. Rep., 2018, 8, 12463 CrossRef.
- M. Wu, J. Zhan, K. Wu, Z. Li, L. Wang, B. Geng, L. Wang and D. Pan, J. Mater. Chem. A, 2017, 5, 14061–14069 RSC.
- A. Hasani, Q. Van Le, M. Tekalgne, M. J. Choi, T. H. Lee, S. Y. Kim and H. W. Jang, NPG Asia Mater., 2019, 11(1), 47 CrossRef CAS.
- M. Suleman, S. Lee, M. Kim, V. H. Nguyen, M. Riaz, N. Nasir, S. Kumar, H. M. Park, J. Jung and Y. Seo, ACS Omega, 2022, 7, 30074–30086 CrossRef CAS.
- K. Zhang, B. M. Bersch, F. Zhang, N. C. Briggs, S. Subramanian, K. Xu, M. Chubarov, K. Wang, J. O. Lerach, J. M. Redwing, S. K. Fullerton-Shirey, M. Terrones and J. A. Robinson, ACS Appl. Mater. Interfaces, 2018, 10, 40831–40837 CrossRef CAS.
- W. Zhan, X. Yong, W. Haolin, W. Ruixue, N. Tang, Z. Yongjie, S. Jing, J. Teng, Z. Ying, L. Yimin, Y. Mei, W. Weidong, Z. Qing, M. Xiaohua and H. Yue, Nanotechnology, 2017, 28, 325602 CrossRef.
- M. Yi and C. Zhang, RSC Adv., 2018, 8, 9564–9573 RSC.
- L. Fei, S. Lei, W. B. Zhang, W. Lu, Z. Lin, C. H. Lam, Y. Chai and Y. Wang, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- X. Tong, Y. Qi, J. Chen, N. Wang and Q. Xu, ChemNanoMat, 2017, 3, 466–471 CrossRef CAS.
- D. Y. Hwang, K. H. Choi, J. E. Park and D. H. Suh, Nanoscale, 2017, 9, 503–508 RSC.
- Z. Zhang, Y. Dong, G. Liu, J. Li, H. Sun, H. Luo and S. Liu, Colloids Surf., A, 2020, 589, 124431 CrossRef CAS.
- S. Kumar, M. Goswami, N. Singh, H. Siddiqui, H. C. Prasad, M. Ashiq, R. Khan, S. Natarajan and S. Kumar, J. Energy Storage, 2023, 66, 107407 CrossRef.
- A. P. Murthy, J. Theerthagiri, J. Madhavan and K. Murugan, Phys. Chem. Chem. Phys., 2017, 19, 1988–1998 RSC.
- J. He, L. Song, J. Yan, N. Kang, Y. Zhang and W. Wang, Metals, 2017, 7, 211 CrossRef.
- T. ul Haq, S. A. Mansour, A. Munir and Y. Haik, Adv. Funct. Mater., 2020, 30, 1–11 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |