
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
rNets: a standalone package to visualize reaction networks†
Sergio
Pablo-García
 *abc,
Raúl
Pérez-Soto
d,
Albert
Sabadell-Rendón
e,
Diego
Garay-Ruiz
e,
Vladyslav
Nosylevskyi
f and
Núria
López
e
*abc,
Raúl
Pérez-Soto
d,
Albert
Sabadell-Rendón
e,
Diego
Garay-Ruiz
e,
Vladyslav
Nosylevskyi
f and
Núria
López
e
aDepartment of Chemistry, University of Toronto, Lash Miller Chemical Laboratories, 80 St. George Street, Toronto, ON M5S 3H6, Canada. E-mail: spgarcica@gmail.com
bDepartment of Computer Science, University of Toronto, Sandford Fleming Building, 40 St. George Street, Toronto, ON M5S 2E4, Canada
cVector Institute for Artificial Intelligence, 661 University Ave. Suite 710, Toronto, ON M5G 1M1, Canada
dDepartment of Chemistry, Colorado State University, 1301 Center Avenue, Ft. Collins, CO 80523-1872, USA
eInstitute of Chemical Research of Catalonia (ICIQ-CERCA), The Barcelona Institute of Science and Technology (BIST), Av. Països Catalans 16, 43007, Tarragona, Spain
fIndependent Researcher, Ukraine
First published on 19th June 2024
Abstract
In the study of chemical processes, visualizing reaction networks is pivotal for identifying crucial compounds and transformations. Traditional methods, such as network schematics and reaction path linear plots, often struggle to effectively represent complex reaction networks due to their size and intricate connectivity. Alternatives capable of leading with complexity include graph methods, but they are not user-friendly, lacking simplicity and modularity, which hinders their integration with widely-used research software. This work introduces rNets an innovative tool designed for the efficient visualization of reaction networks with a user-friendly interface, modularity, and seamless integration with existing software packages. The effectiveness of rNets is demonstrated through its application in analyzing three catalytic reactions, showcasing its potential to significantly enhance research both in homogeneous and heterogeneous catalysis fields. This tool not only simplifies the visualization process but also opens new avenues for exploring complex reaction networks in diverse research contexts.
1 Introduction
Catalyst development plays a pivotal role in enhancing industrial chemical production by optimizing processes to reduce material, financial, and environmental costs.1,2 The conventional approach to develop these catalysts relies on a labor-intensive cycle of design, making, and testing of numerous candidates to find the most effective ones for specific reactions. As the number of candidates grows, this method becomes increasingly time-consuming and resource-intensive.2–9 With pressing environmental challenges, the need for more efficient strategies is evident, prompting the integration of density functional theory (DFT) simulations into the discovery process. DFT simulations offer valuable theoretical insights and can act as a decision-making tool to predict reactions' outcomes more accurately.10–18 However, effectively utilizing these insights requires not only prediction but also the visualization of the complex networks formed by reactions, a significant challenge in both homogeneous and heterogeneous catalysis due to their branched and complex nature.19,20Traditional visualization techniques, such as reaction path plots, leverage ab initio data to provide a two-dimensional view of the network's thermodynamics and, when including activation energies, its kinetic behavior. While effective for small, straightforward reactive systems, these plots fall short in representing the intricate, branched networks characteristic of homogeneous and heterogeneous catalysis. This comes as a direct consequence of employing a linear reaction coordinate (sequential process) as the x-coordinate of the plot. A more robust alternative is the representation of reaction mechanisms as reaction network graphs,6,19,20 encoding the reaction intermediates as nodes and the reactions which interconnect them, as edges. More specifically, reaction networks are formed by elementary reactions, which are chemical transformations in which one or more reactants evolve into products in a single reaction step.1
This conceptualization of chemical processes is shared not only by both homogeneous and heterogeneous catalysis,6 but also by any non-catalytic cascade of chemical reactions. Typically, these more complex networks have been illustrated through manually drawn graphs, tracing the reaction mechanism, as depicted in the left part of Fig. 1. The most used tools to trace these reaction mechanisms and linear energy diagrams manually are Adobe Illustrator,17,21 OriginLab,18,22,23 Inkscape,15,24,25 Matplotlib,26–28 and ChemDraw,10–12,29 among others. From there, it is possible to switch to a representation that is more akin to graphs in other domains of science, explicitly showing nodes as circles and edges as full connections between them (right side of Fig. 1), using tools such as Graphviz.30
 | ||
| Fig. 1 Schematics of a reaction network graph. Left, reaction-centered representation, using reaction arrows to depict processes and labels for intermediates. Right, complete graph-based representation with intermediates as nodes and reactions as edges. | ||
However, this manual approach to graphing reaction mechanisms poses significant limitations. First, the representation cost escalates with the number of compounds, challenging the depiction of networks that suffer from combinatorial explosion,4,19,31 which is an increasingly common situation due to the generalization of the use of automated mechanism discovery algorithms.4,32–43 Additionally, unlike reaction paths, these graphs do not visually convey thermodynamic and kinetic information. Alternative and promising methods for automatically encoding reactions with these considerations have been proposed,44–46 but they often face constraints that hinder their widespread adoption by the computational chemistry community, such as being tied to specific software, requiring multiple dependencies, or using complex input formats. From these examples, ReNView44 is primarily focused on the visualization of microkinetic simulations in heterogeneous catalysis, coupled with the OpenMKM package.47 Then, CaRMeN45 is mainly a platform for the automation of parameter exploration tasks in microkinetic simulations, offering specific visualization for the simulated reaction networks. Finally, SCAN46 provides a database with web visualization, including network analysis, mainly focused on organic chemistry discovered through AFIR.32,33
To overcome these limitations, this work introduces a standalone, package-agnostic software designed to efficiently visualize the thermodynamic and kinetic properties of reaction networks, rNets. rNets is built on three core design principles: (i) ease of use for both programmers and non-programmers, (ii) readiness for integration with various DFT and microkinetic (MK) packages, and (iii) minimal dependency requirements to simplify deployment and enhance robustness. To demonstrate the capabilities of our code, we have integrated rNets with two distinct microkinetic modeling packages, showcasing its effectiveness in visualizing the complex reactions associated with both homogeneous and heterogeneous catalysts. Moreover, we have also coupled rNets with a knowledge-graph based representation of reaction networks, integrated within the ioChem-BD48,49 database for computational chemistry data. This approach not only streamlines the analysis of intricate catalytic networks but also bridges the gap between theoretical simulations and practical applications, offering a more comprehensive and accessible tool for researchers in the field of computational chemistry. Through rNets, we aim to facilitate a more rapid, automated and informed catalyst discovery process.
2 Methods
rNets, is a software package written in Python 3.12, that aims to provide an extensible and modular tool for visualizing the thermodynamic and kinetic properties of reaction networks. The choice of Python for its development is a nod to the language's widespread use and acceptance within the scientific community. rNets stands out for its ease of use, minimalistic design, and its capability to seamlessly integrate with external codes. The core functionality of rNets involves surrogating the visualization of reaction graphs to Graphviz, a software able to parsing dot files and rendering them in various formats. Thus, rNets can be characterized as a metaprogramming package, able to translate reaction networks into the dot language for visualization, with Graphviz as its sole external dependency. For the present work Graphviz 9.0.0 has been used. Fig. 2 summarizes the logic behind rNets.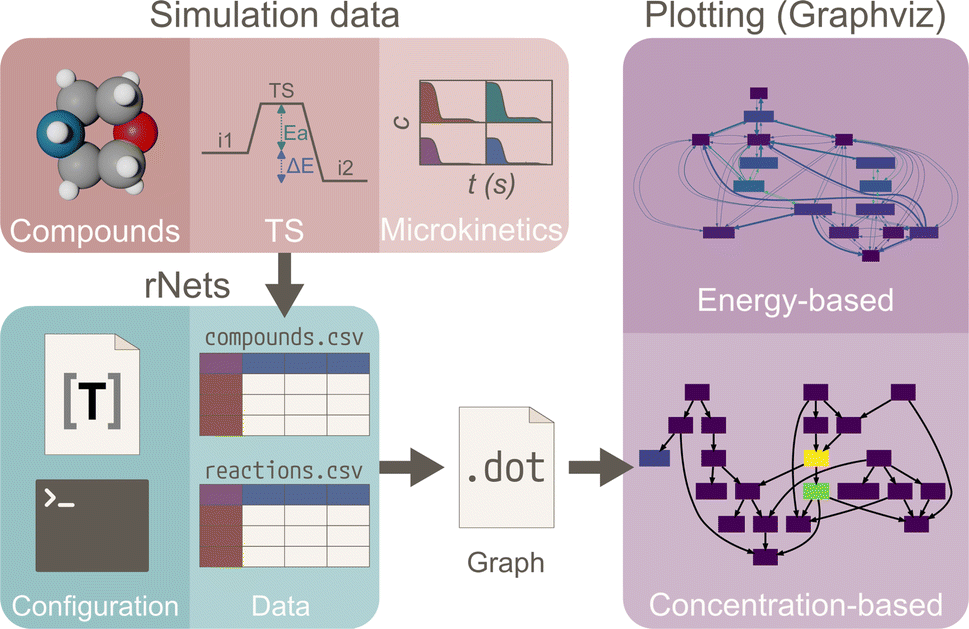 | ||
| Fig. 2 Representation of the workflow followed by rNets. First, the energies of the compounds, transition states and/or concentrations are collected into. CSV data files from simulations. Together with the configuration files, rNets is used to generate a dot file containing the plotting instructions for the graph. Finally, Graphviz is used to compute the energy or concentration-based plot. | ||
However, the rapid development of Python packages for chemistry has been accompanied by increasing complexity in managing software dependencies, a crucial aspect for ensuring consistent and accurate execution of chemical software. Traditional solutions like package managers and virtual environments have only partially addressed these issues, often struggling with the integration of multiple packages that have conflicting dependencies within a single Python distribution. This has resulted in a tendency towards isolated Python packages, each with its own set of dependencies, reducing their modular capabilities. In contrast, rNets has been meticulously crafted as a standalone package, devoid of these dependency entanglements. By requiring only Graphviz for plotting, rNets effectively maximizes its portability and modularity. This strategic design choice, along with its practical implications, are further explored in the following subsections.
2.1 Input parsing
Theoretical studies of reaction networks frequently rely on ab initio methods to compute thermodynamic and kinetic properties, such as ground-state energies of compounds and activation energies of reactions. These properties are typically collated into simple, spreadsheet-like tables for easy parsing and analysis. Reaction path plots, a popular visualization method, excel in depicting these tables, especially for simple mechanisms. Recognizing the simplicity and widespread use of table formats in the research community, rNets has been tailored to interface seamlessly with input files resembling these tables. This design strategy is aimed at facilitating the adoption of rNets, especially among chemists with limited programming expertise. rNets utilizes two primary file types to define a reaction network: the “compounds” file, detailing the compounds, and the “reactions” file, outlining the reactions. The chosen file format for both is comma-separated values (CSV), balancing simplicity, versatility, and FAIRness.The structure of these files is straightforward: the first line contains column names (in no specific order) that define the data represented in each column, followed by lines that provide the data for individual compounds or reactions. To enhance user flexibility, column names are categorized as either “required” or “optional”. The “required” columns constitute the essential data needed to define a compound or a reaction. For instance, the “compounds” file mandates the  and
and  columns, whereas the “reactions” file requires at least one
columns, whereas the “reactions” file requires at least one  and one
and one  column. During the parsing process, rNets checks for the presence of these required columns in the input files. Conversely, the “optional” columns allow for additional, user-specific customizations to each compound or reaction. This flexibility enables users to tailor the analysis to their specific needs. Tables S1–S2† summarize this information, offering users a quick reference guide for efficient data organization and input into rNets, also included in the code repository.
column. During the parsing process, rNets checks for the presence of these required columns in the input files. Conversely, the “optional” columns allow for additional, user-specific customizations to each compound or reaction. This flexibility enables users to tailor the analysis to their specific needs. Tables S1–S2† summarize this information, offering users a quick reference guide for efficient data organization and input into rNets, also included in the code repository.
2.2 Graphical representation of reaction networks
In rNets, the reaction network extracted from the input files is encoded graphically. This representation involves depicting compounds as nodes and reactions as edges within a graph. The nodes, representing compounds, are illustrated as square boxes, each labeled at the center with the compound's name, derived by default from the “ ” column in the “compounds” file. To augment the visual appeal and clarity, users have the option to modify the label format using the “optional”
” column in the “compounds” file. To augment the visual appeal and clarity, users have the option to modify the label format using the “optional”  column in the input file. Here, format modifiers can be applied, separated by a colon (“:”). These modifiers include b for bold, i for italic, and u for underlined text, allowing for a customized and informative visual presentation. Further refining the representation, each edge in the graph symbolizes a reaction connecting two nodes. The default directionality of these reactions is forward, denoted as “−>”. However, users have the option to alter this directionality to backward (“<−”) or bidirectional (“<−>”) by specifying their preference in the “optional”
column in the input file. Here, format modifiers can be applied, separated by a colon (“:”). These modifiers include b for bold, i for italic, and u for underlined text, allowing for a customized and informative visual presentation. Further refining the representation, each edge in the graph symbolizes a reaction connecting two nodes. The default directionality of these reactions is forward, denoted as “−>”. However, users have the option to alter this directionality to backward (“<−”) or bidirectional (“<−>”) by specifying their preference in the “optional”  column name in the “reactions” file. rNets comes with two different graphical encodings: “thermo” representing the thermodynamic values of the compounds and the computed kinetic constants and “kinetic” focusing on the concentrations of the compounds and the net rates of the reactions. These encodings are described in the following sections.
column name in the “reactions” file. rNets comes with two different graphical encodings: “thermo” representing the thermodynamic values of the compounds and the computed kinetic constants and “kinetic” focusing on the concentrations of the compounds and the net rates of the reactions. These encodings are described in the following sections.
rNets is capable of visually encoding reaction kinetics when transition state energies are provided in the “reactions” file. These energies, listed under the “optional”  column, enable rNets to dynamically adjust the width of the arrows. This adjustment is proportional to the pseudo-kinetic constants derived from the specified energies, following the methodology outlined in Note S1.† This feature allows for a more nuanced and informative representation of the reaction network, highlighting the kinetic relationships between different components.
column, enable rNets to dynamically adjust the width of the arrows. This adjustment is proportional to the pseudo-kinetic constants derived from the specified energies, following the methodology outlined in Note S1.† This feature allows for a more nuanced and informative representation of the reaction network, highlighting the kinetic relationships between different components.
 and energy
and energy  metrics are specified for all network entities at a chosen time step, rNets illustrate the reaction dynamics. Node backgrounds depict compound concentrations, using a color gradient normalized for immediate clarity, and arrow widths signify reaction rates—wider arrows indicate faster reactions. Directionality is inherently shown by the net reaction rate, without the need for explicit
metrics are specified for all network entities at a chosen time step, rNets illustrate the reaction dynamics. Node backgrounds depict compound concentrations, using a color gradient normalized for immediate clarity, and arrow widths signify reaction rates—wider arrows indicate faster reactions. Directionality is inherently shown by the net reaction rate, without the need for explicit  markers. This design, with concentration and rate normalization, offers a direct, unified view of the reaction network's kinetics and dynamics at any simulated time step. Normalization of concentration and rate values ensures consistent visualization scales. For detailed computational methods behind these values, see Note S2.†
markers. This design, with concentration and rate normalization, offers a direct, unified view of the reaction network's kinetics and dynamics at any simulated time step. Normalization of concentration and rate values ensures consistent visualization scales. For detailed computational methods behind these values, see Note S2.†
2.3 Enhanced component visibility
rNets incorporates a feature that addresses the common need in reaction network visualization to selectively focus on certain parts of the network. Researchers often find themselves in situations where some compounds or reactions may not be relevant to their current analysis or where they might require to highlight specific segments of the network. However, even when certain elements are not the focus, their energies play a crucial role, especially in representing kinetic properties, and thus cannot be entirely omitted from the dataset. As a solution, rNets introduces the column functionality. This feature grants users the flexibility to either hide or dim specific compounds or reactions within the graphical depiction of the network. When the
column functionality. This feature grants users the flexibility to either hide or dim specific compounds or reactions within the graphical depiction of the network. When the  column for a given element is set to
column for a given element is set to  , that particular compound or reaction will not appear in the visual representation of the reaction graph. Importantly, this does not remove its influence from the overall network; it continues to contribute to the normalization of network energies. For compounds, this means they are still considered in the computation of kinetic constants for related reactions.
, that particular compound or reaction will not appear in the visual representation of the reaction graph. Importantly, this does not remove its influence from the overall network; it continues to contribute to the normalization of network energies. For compounds, this means they are still considered in the computation of kinetic constants for related reactions.
In instances where complete removal of an element from the visual field is not desired, setting the  column to
column to  offers an alternative. This option achromatizes the node or edge associated with the element, effectively making it less prominent without entirely eliminating it from the visual network. This greying out of elements allows for a nuanced approach to visualization, enabling users to de-emphasize certain aspects of the network while maintaining a holistic view.
offers an alternative. This option achromatizes the node or edge associated with the element, effectively making it less prominent without entirely eliminating it from the visual network. This greying out of elements allows for a nuanced approach to visualization, enabling users to de-emphasize certain aspects of the network while maintaining a holistic view.
This  column feature in rNets provides a powerful and flexible tool for users, allowing them to tailor the network visualization to their specific research needs. It ensures that the graphical representation of the network remains comprehensive yet clear, focusing on elements of interest without losing sight of the overall context of the network.
column feature in rNets provides a powerful and flexible tool for users, allowing them to tailor the network visualization to their specific research needs. It ensures that the graphical representation of the network remains comprehensive yet clear, focusing on elements of interest without losing sight of the overall context of the network.
2.4 Customization and user interface
rNets equips the users with extensive customization capabilities, allowing for the personalization of network visualizations. Users can adjust different plot options, such as the maximum edge width and color scheme, tailoring the visual output to their preferences. These options can be configured through two primary methods: (i) a Tom's obvious minimal language (TOML) file that specifies the desired values, and (ii) direct command-line interface (CLI) arguments for on-the-fly adjustments.The hierarchical structure of option precedence ensures flexibility and control; settings defined in the configuration file override the default values coded within rNets, while CLI arguments take precedence over both configuration settings and defaults. This design allows users to establish a baseline configuration file reflecting their standard visualization preferences, with the flexibility to use CLI arguments for temporary or experiment-specific modifications. rNets rigorously checks the validity of user-defined values to maintain stability and reliability. The available configuration options are described in Tables S3–S5† and an example of a configuration file can be found in Note S5† and in the code repository. For advanced customization, the  option enables the specification of Graphviz parameters for various elements of the network visualization. Additional details on utilizing this feature are available in Note S6.†
option enables the specification of Graphviz parameters for various elements of the network visualization. Additional details on utilizing this feature are available in Note S6.†
In addition to its standalone functionality, rNets is designed for seamless integration as a Python library within broader Python projects. This capability is comprehensively documented, ensuring users can effectively incorporate rNets into their workflows.
3 Results and discussion
In this section, we present three case studies, tackling both homogeneous and heterogeneous catalysis, to demonstrate the capability and versatility of rNets for solving the aforementioned challenges in this field of study. Further details about the relevant simulation details are available in the ESI (Note S7–S9).†The inspection of the energies of reaction networks provides valuable insights about chemical systems. Indeed, although experimental observations can be correlated to energies, it is challenging to provide a one-to-one mapping with properties such as selectivity, yield, or conversion,10,51 which are essential to control a given chemical process. In this context, microkinetic simulations are a valuable tool to bridge this gap52 and allow an actual quantitative comparison with experimental results.
3.1 Energy-based examples
This complexity and the critical role of corrections were exemplified in our previous investigation of imine condensation between benzaldehyde and n-butylamine in aprotic media.58 In that study we observed how a bias of 13.4 kJ mol−1 per computed structure coupled with a reference state correction (from 1 atm to 1 M) was able to bridge the gap between the DFT-derived microkinetic simulation and the experimental concentration vs. time values. The rNets' representation of both, the uncorrected system and the corrected system are included in the ESI (Fig. S1),† while a simplified representation is included in Fig. 3.
 | ||
| Fig. 3 Simplified reaction network of the imine condensation in DCM between benzaldehyde (A) and n-butylamine (N). The adduct of “X” and “Y” is represented as “[X + Y]”. Water, the hemiaminal intermediate and the product Imine are represented as “W”, “H” and “I” respectively. The magnitude of the energy barriers for each elementary step is represented by the color and the thickness of the arrows, while the color of the nodes represents the stability of the intermediates. The color scale goes from dark, corresponding to low energies (thus high stability) and barriers to light colors, indicating unstable intermediates and high barriers. (a) Corresponds reaction network without any correction and (b) to the same network but with standard state correction and a bias of 0.14 eV per DFT simulation. | ||
The full reaction network is composed of a total of 28 species and 52 reversible reactions, which in its majority corresponds to bimolecular association processes, leading to an intricate network. A preliminary examination of the plot in Fig. 3b readily reveals two prominent alterations within the network: a notably darker overall tone and an increased presence of thicker edges, indicating significant changes. Further scrutiny of the network highlights specific variations in the appearances of species “A” and “I”, which correspond to benzaldehyde and N-butyl-1-phenylmethanimine, respectively. Notably, these species exhibit a lighter shade in Fig. 3b compared to the rest of the network, suggesting differential effects of the applied corrections on these particular reaction components. The different adducts (i.e. the adduct of “A” and “W” is represented as “[A + W]”) have become darker, indicating that they are now more stable. As a consequence, the overall reaction becomes more dynamic, as the generalized increase in the thickness of the edges indicates. Some reactions are still slow, such as the uncatalyzed hemiaminal dehydration (corresponding to the edges between “H” and “[I + W]”, in green the full reaction network shown at Fig. S3 and S4†). This interpretation aligns well with the results of the microkinetic simulations carried out in the original work where the simulations with the uncorrected system (Fig. 3a), led to no imine formation, while the simulations of the corrected system were consistent with previous experimental results.
CO2(g) + H2(g) ⇌ CO(g) + H2O(g)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) RWGS reaction RWGS reaction | (1) |
| CO2(g) + 3H2(g) ⇌ CH3OH(g) + H2O(g)MeOH reaction | (2) |
| CO2(g) + 4H2(g) ⇌ CH4(g) + 2H2O(g)Methanation reaction | (3) |
In Fig. 4 we report as an example the simplified reaction network of CO2 hydrogenation on Pd(111) neglecting the formation of methane, involving 21 elementary steps, 16 intermediates, and 5 gas-phase species (labelling at Table S7†). The network starts with the adsorption and dissociation of H2 (i1) on the surface (i0). Then, CO2 is adsorbed (i2), and the reaction evolves through the oxygen hydrogenation of CO2, resulting in COOH* (i3), leading to the dissociation of i3 into CO* and OH* fragments, and the posterior elimination of OH* via water formation and desorption (intermediates i5 and i6).60,63 At this point, the reaction can evolve towards two different pathways: CO* desorption (i6 to CO(g) and i0), culminating the RWGS reaction, or CO* sequential hydrogenation to generate methanol (from i7 to i8 or i13). We use the last elementary steps as examples to understand the information contained in Fig. 4. The color of the i8 intermediate (COH* + H*) is brighter than i7 (CO* + 2H*) meaning that this step is thermodynamically unfavored, as for i13 formation (CHO* + H*). The kinetic equilibrium between the initial and the final steps is expressed with the thickness and the color of the arrows (edges) in forward or backward directions. Thus, according to Fig. 4, CO* hydrogenation pathway is less favored than the RWGS route both thermodynamically and kinetically.
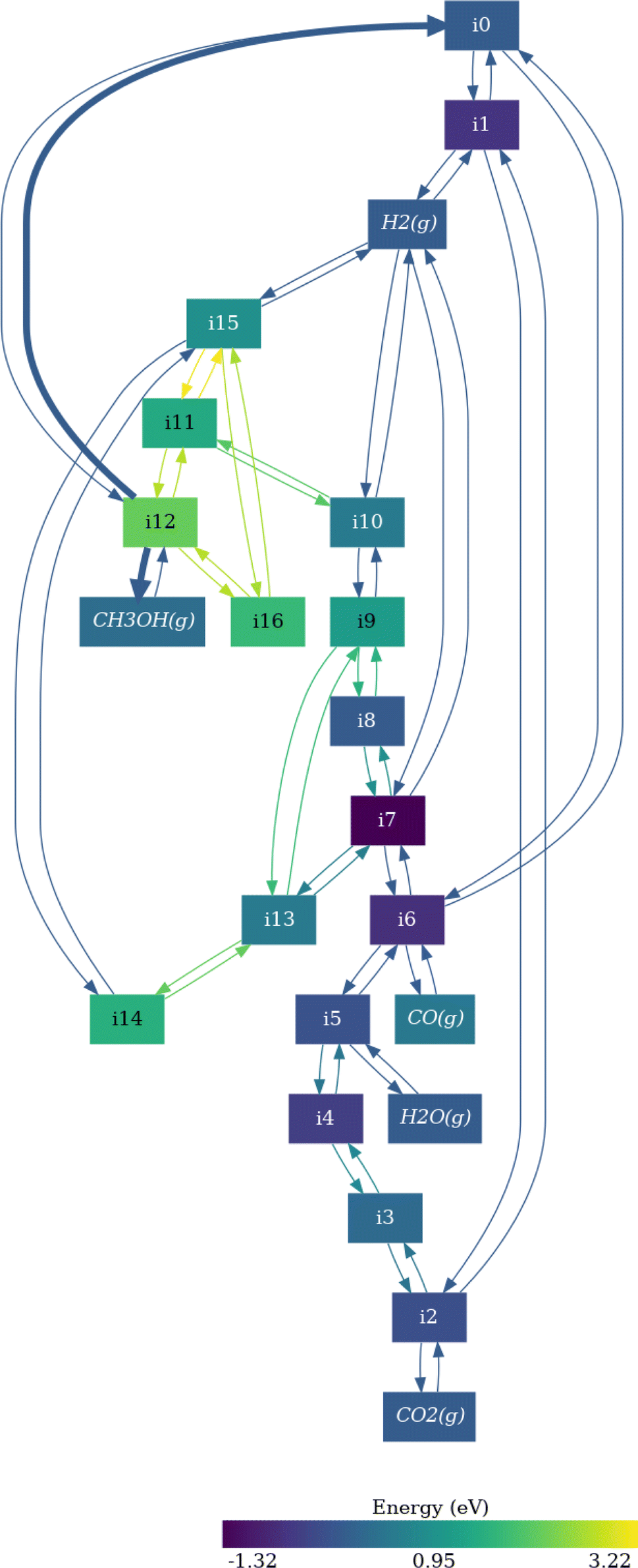 | ||
| Fig. 4 Visualization of the reaction network for CO2 hydrogenation on Pd(111). The intermediates are labeled in Table S3.† The magnitude of the energy barriers for each elementary step is represented by the color and the thickness of the arrows, while the color of the nodes represent the stability of the intermediates. Bright colors imply unstable intermediates or high activation energies. | ||
Following the CO* hydrogenation pathway through COH* + H* intermediate (i8), the next step is the generation of CHOH* (i9). Then, i9 is sequentially hydrogenated to obtain methanol (i12) via CHOH* + 2H* and CH2OH + H* intermediates (i10 and i11 respectively). Alternatively, CO* can be also hydrogenated through CHO* + H* (i13), followed by the generation of CH2O*, CH2O* + 2H*, and CH3O* (i14, i15, and i16, respectively). However, the formation of CH2O intermediate (i14) is kinetically less probable than CHOH*, and thus, the reaction would evolve through the i9, i10, i11, and i12 pathway according to Fig. 4. In any case, the formation of COH* is thermodynamically more favored than CHO*, since i13 is brighter than i8, and thus, methanol generation is more prone to evolve through the i8 route. All these observations are in line with previous computational and experimental works.18,60
The ioChem-BD platform48,49 provides a robust solution for the first part of the problem, translating these diverse output files into an unique format (chemical markup language, CML) and providing a framework in which completely curated datasets can be shared with the community, enhancing reproducibility and interoperability. Regarding the relationships between data elements, the OntoRXN ontology65 was introduced as an organization scheme following the principles of semantic data, which established the core entities that are required to properly define a CRN and map it to the properties gathered from quantum chemical methods. Through the combination of ioChem-BD and OntoRXN, it becomes possible to define knowledge graphs (CRN-KGs): non-relational databases which contain all the information about a given reaction network. A CRN-KG, then, can be used as a standard format to process and analyze data, extracting the properties and relationships of interest through queries in the SPARQL format.
One of the foundational applications of CRN-KGs was indeed the automated setup of microkinetic models, processing the individual elementary steps encoded in the knowledge graphs and determining the corresponding reaction barriers and rate constants. In this context, rNets provides a solid foundation to streamline the visualization of the simplified CRN gathered from the CRN-KG and to follow up with the introduction of standards for computational chemical reaction networks. Additionally, we can consider this pipeline as a relatively straightforward connection between rNets and the increasing number of computational results available in ioChem-BD. As a test system, we have consider the halide-catalyzed regiospecific fixation of carbon dioxide in a cyclooctene-based epoxy alcohol,66 for which the results of microkinetic simulations were in very good agreement with experimental yields and selectivities.
Despite its simplicity in species and reactions, this system offers a practical example for kinetic simulations in homogeneous catalysis and reveals unique mechanistic insights due to the cyclooctene moiety's versatility.
A naive direct application of rNets to this mechanism resulted in a reaction network that was challenging to interpret, as shown in Fig. S2.† This complexity arose from modeling most chemical reaction network (CRN) processes as transitions from bimolecular reactants to bimolecular products, which often resulted in the mapping of two nodes to another pair of nodes, cluttering the visual representation with numerous arrows. This scenario, however, showcases the ideal context for demonstrating the visualization enhancement capabilities of rNets. While some of the species in the network (namely, the counterion TMA, the bromide salt TMABr and carbon dioxide) are required to balance energies and appropriately compute barriers and rate constants, they might be omitted from the reaction network in order to focus on the actual rearrangement of the epoxy alcohol to the cyclic carbonate and the different pathways through it which might occur. Therefore, setting the  property of these entities to
property of these entities to  sheerly improves the readability of the produced graph (Fig. 5), while keeping compound and reaction energies consistent.
sheerly improves the readability of the produced graph (Fig. 5), while keeping compound and reaction energies consistent.
 | ||
| Fig. 5 Visualization of the reaction network for CO2 fixation on a epoxy alcohol cyclooctene derivative. The magnitude of the energy barriers for each elementary step is represented by the color and the thickness of the arrows, while the color of the nodes represents the stability of the intermediates. | ||
These three examples highlight the versatility of rNets to handle the complexity of reaction networks in both homogeneous and heterogeneous catalysis, as well as different schemes for data organization. There is a clear synergy between tools for automated data generation (AMUSE), and tools for the standardization of the produced information (ioChem-BD and CRN-KGs). rNets would conform an essential part of this coupled workflow, simplifying and unifying the visualization of the likely very complex systems studied through this kind of pipelines. Furthermore, the possibility of performing microkinetic simulations as part of this workflow opens the door to in situ visualizations of the evolution of the concentrations of the different species in the reaction network, as we will detail in the following section.
3.2 Concentration-based examples
We have followed the same methodology as in our original paper, and recomputed the potential energies through SP simulations using the adjusted radii computed using DRACO. We recomputed the free energies and added a reference state correction from 1 atm to 1 M.
With the resulting set of energies, we conducted a microkinetic simulation, which, revealing minimal imine formation, suggests the 13.4 kJ mol−1 correction transcends mere inaccuracies in electronic energy level cavity treatments. In our previous work we observed that upon the addition of other corrections, quasi-harmonic corrections, the concentration profile became completely incompatible with the experimental one, even when we followed the same approach and added a constant bias to the energy of each calculation. We decided to also apply it for the DRACO-corrected energies, the total concentration of imine is shown in Fig. 6. To our surprise, compared with our previous results, upon a correction of 17.6 kJ mol−1 the concentration profile showed an acceptable correlation with the experimental data. This may indicate that at least for this specific example, the correction introduced with DRACO is proportional to the molecularity of the species. Thus, it introduces a shift to the energies of each computed species that for our example is a negative value which leads to a higher correction. Next, we proceeded to visualize the concentration vs. time evolution of the 17.6 kJ mol−1 corrected system using rNets, with the aim of getting more insights into significant changes in the reaction pathway to confirm the previous statement. Fig. 7a and b show respectively the simplified network (comparable to Fig. 3) at 10 min and 40 min respectively. The equivalent images with the full network can be found in the ESI† and a GIF with the evolution over time is also attached.
 | ||
| Fig. 6 Total concentration of imine vs. time of different microkinetic simulations upon constantly biased energies. The bias applied to each simulation is indicated in kJ mol−1 over the curve as well as in the colorbar legend. The experimental points are represented as black circles and were extracted from69 following the procedure explained in our previous work.58 | ||
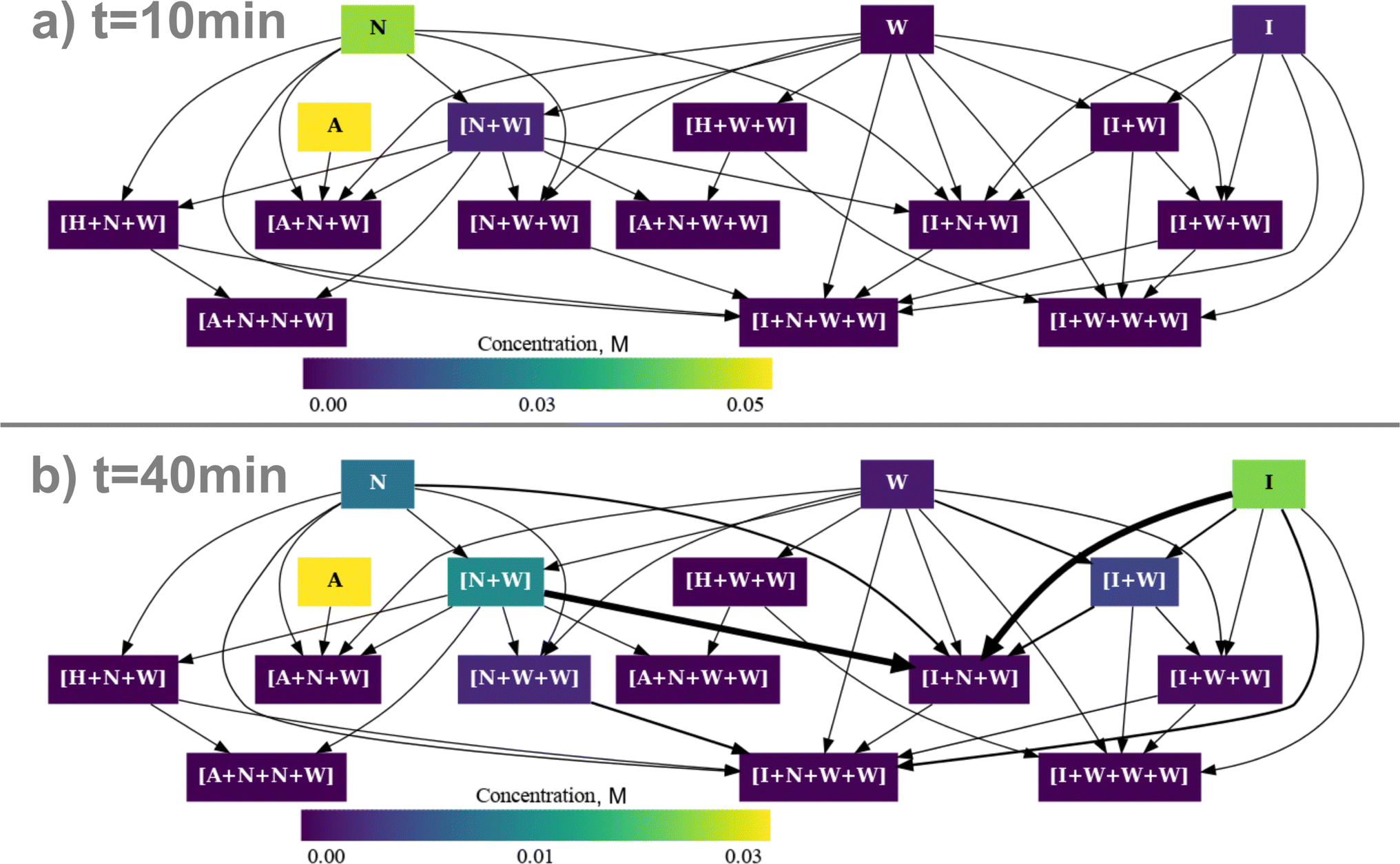 | ||
| Fig. 7 Simplified reaction network of the imine condensation in DCM between benzaldehyde and n-butylamine. The intermediates are labeled following the same nomenclature as in our previous work.58 The thickness of the arrows represents the net rate for each elementary step, and the orientation of the arrow indicates the direction of the reaction, while the color of the nodes represents the concentration of the intermediates. A dark color represents a low or null concentration while a bright color corresponds to a high concentration. | ||
At 10 min, Fig. 7a, the initial water (W) impurity is displaced towards forming an adduct ([N + W]) with the n-butylamine (N) which can act as a proton shuttle. As the concentration of n-butylamine and the benzaldehyde (A) the formation of the [A + N] adduct is very dynamic (showing a thicker reaction arrow) but as it is not thermodynamically favored it either reacts to form the hemiaminal or dissociates. As the reaction progresses, Fig. 7b, the concentration of water increases as it is a product of the reaction, but instead of staying as an adduct with the imine (I) it is displaced to coordinate with the amine. As the amine is consumed more water is available increasing the formation of the [N + W + W] adduct, which is an intermediate in the water dimer ([W + W]) proton shuttle catalysis.
:CO2 ratio equals to 4, in line with the typical range of experimental operation conditions for CO2 hydrogenation.59,60 The thickness of the edges (arrows) corresponds to the magnitude of the rate calculated with rNets from the coverages obtained with AMUSE of each elementary step, and the direction indicates whether the reaction is displaced to reactants or products. In Fig. 8, it is clearly shown that all the kinetic equilibria are displaced to reactants and that only CO* and 2H* (i7 intermediate, θi7 = 0.999) is formed, poisoning the surface. This data supports quantitatively the qualitative analysis obtained with Fig. 4. Thus, rNets has proven to be a useful tool not only for visualization but, more importantly, for the analysis of the results. In addition, rNets can be also used to directly follow the time evolution of a microkinetic simulation, as shown in the ESI† Gif, simplifying the analysis of the results.
 | ||
| Fig. 8 Visualization of the reaction network for CO2 hydrogenation on Pd(111) considering the rates. The intermediates are labeled in Table S3.† The thickness of the arrows represents the net rate for each elementary step, and the orientation of the arrow indicates the direction of the reaction, while the color of the nodes represents the concentration of the intermediates. | ||
4 Conclusions
Through various application scenarios, rNets has proven to be a highly effective tool for the visualization of thermodynamic and kinetic properties within catalytic reaction networks. Its design is both minimalist and modular, ensuring straightforward integration with kinetic analysis software. This design choice offers users previews of reactions as well as comprehensive microkinetic analyses. Furthermore, rNets' user-friendly, table-based input system significantly reduces the learning curve, making it readily accessible even to chemists with limited programming expertise. As a pivotal advancement in open-source visualization tools for the chemistry sector, rNets exemplifies the power of simplifying the representation of complex data, thereby broadening its accessibility and utility in the community.Data availability
The data used in this work is publicly available on the ioChem-BD platform. Imine condensation: geometry optimizations [https://doi.org/10.19061/iochem-bd-1-154] and single-point simulations [https://doi.org/10.19061/iochem-bd-1-308] with DRACO-based parameters. Regiospecific CO2 fixation [https://doi.org/10.19061/iochem-bd-1-141]. CO2 hydrogenation on Pd(111) [https://doi.org/10.19061/iochem-bd-1-43]. The input files and the intermediate dot files generated by rNets to build the figures of this work can be found as an examples in our code repository. Additionally, these examples have been integrated within the project documentation. The source code of rNets is publicly available at Github [https://github.com/spgarcica/rNets], including comprehensive documentation built with Sphinx 7.2.6 [https://spgarcica.github.io/rNets]. To assure reproducibility, the version of the code used (24.06) to compute the examples of this manuscript has been uploaded to Zenodo [https://doi.org/10.5281/zenodo.11463949]. rNets has been tested on Windows 11 (Build 1000.22700.1003.0), Ubuntu 20.04, Gentoo x64, and Arch x64. The package rNets can be also found on PyPi [https://pypi.org/project/rNets/]. The source code of pykinetic and AMUSE can be found on their respective Github repositories [https://github.com/rperezsoto/pykinetic, https://github.com/LopezGroup-ICIQ/amuse]. Additionally, a package of pykinetic can be found on PyPi [https://pypi.org/project/pykinetic/] and Zenodo.68 The OntoRXN ontology [https://gitlab.com/dgarayr/ontorxn] and the Python library ontorxn-tools [https://gitlab.com/dgarayr/ontorxn_tools] to generate knowledge graphs are available on GitLab.Author contributions
S. P.-G., conceptualization, methodology, software, writing – original draft, supervision, project administration. R. P.-S., validation, investigation, data curation, writing – original draft, visualization. A. S.-R., validation, investigation, data curation, writing – original draft, visualization. D. G.-R., validation, investigation, data curation, writing – original draft, visualization. V. N., conceptualization, methodology, software, writing review & editing. N. L., writing review & editing, funding acquisition.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Subaward by University of Minnesota, Project title: Development of Machine Learning and Molecular Simulation Approaches to Accelerate the Discovery of Porous Materials for Energy-Relevant Applications under Award Number DE-SC0023454 (UMN Subaward A010026303). A. S.-R. and N. L. thank TotalEnergies (contract reference CT00001052) and the Spanish Ministry of Science and Innovation (PID2021-122516OB-I00) for funding, and Dr K. Kaźmierczak, F. Euzenat, D. Curulla-Ferré, N. Montroussier, and F. Cannizaro for the discussion, and the Barcelona Supercomputing Center for support and resources. D. G.-R. thanks the Spanish Ministry of Science and Innovation (reference PID2020-112806RB-I00) and European Union NextGenerationEU/PRTR (reference TED2021-132850B-I00) for funding.References
- I. Chorkendorff and J. W. Niemantsverdriet, Concepts of Modern Catalysis and Kinetics; Wiley, Weinheim, 2003, DOI:10.1002/3527602658.
- M. Salciccioli, M. Stamatakis, S. Caratzoulas and D. G. Vlachos, A review of multiscale modeling of metal-catalyzed reactions: Mechanism development for complexity and emergent behavior, Chem. Eng. Sci., 2011, 66, 4319–4355, DOI:10.1016/j.ces.2011.05.050.
- S. Mitchell, E. Vorobyeva and J. Pérez-Ramírez, The multifaceted reactivity of single-atom heterogeneous catalysts, Angew. Chem., Int. Ed., 2018, 57, 15316–15329, DOI:10.1002/anie.201806936.
- Z. W. Ulissi, A. J. Medford, T. Bligaard and J. K. Nørskov, To address surface reaction network complexity using scaling relations machine learning and DFT calculations, Nat. Commun., 2017, 8, 14621, DOI:10.1038/ncomms14621.
- J. T. Margraf, H. Jung, C. Scheurer and K. Reuter, Exploring catalytic reaction networks with machine learning, Nat. Catal., 2023, 1–10, DOI:10.1038/s41929-022-00896-y.
- S. Pablo-García, R. García-Muelas, A. Sabadell-Rendón and N. López, Dimensionality reduction of complex reaction networks in heterogeneous catalysis: From l inear-scaling relationships to statistical learning techniques, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1540, DOI:10.1002/wcms.1540.
- A. Bruix, J. T. Margraf, M. Andersen and K. Reuter, First-principles-based multiscale modelling of heterogeneous catalysis, Nat. Catal., 2019, 2, 659–670, DOI:10.1038/s41929-019-0298-3.
- F. Della Monica and A. W. Kleij, Mechanistic Guidelines in Nonreductive Conversion of CO2: The Case of Cyclic Carbonates, Catal. Sci. Technol., 2020, 10, 3483–3501, 10.1039/D0CY00544D.
- A. Kumar, P. Daw and D. Milstein, Homogeneous Catalysis for Sustainable Energy: Hydrogen and Methanol Economies, Fuels from Biomass, and Related Topics, Chem. Rev., 2022, 122, 385–441, DOI:10.1021/acs.chemrev.1c00412.
- M. Besora and F. Maseras, Microkinetic Modeling in Homogeneous Catalysis, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1–13, DOI:10.1002/wcms.1372.
- J. N. Harvey, F. Himo, F. Maseras and L. Perrin, Scope and Challenge of Computational Methods for Studying Mechanism and Reactivity in Homogeneous Catalysis, ACS Catal., 2019, 9, 6803–6813, DOI:10.1021/acscatal.9b01537.
- R. Huang, J. Rintjema, J. González-Fabra, E. Martín, E. C. Escudero-Adán, C. Bo, A. Urakawa and A. W. Kleij, Deciphering Key Intermediates in the Transformation of Carbon Dioxide into Heterocyclic Products, Nat. Catal., 2019, 2, 62–70, DOI:10.1038/s41929-018-0189-z.
- J. A. Hueffel, T. Sperger, I. Funes-Ardoiz, J. S. Ward, K. Rissanen and F. Schoenebeck, Accelerated Dinuclear Palladium Catalyst Identification through Unsupervised Machine Learning, Science, 2021, 374, 1134–1140, DOI:10.1126/science.abj0999.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, Origin of the overpotential for oxygen reduction at a fuel-cell cathode, J. Phys. Chem. B, 2004, 108, 17886–17892, DOI:10.1021/jp047349j.
- M. S. Frei, C. Mondelli, R. García-Muelas, J. Morales-Vidal, M. Philipp, O. V. Safonova, N. López, J. A. Stewart, D. C. Ferré and J. Pérez-Ramírez, Nanostructure of nickel-promoted indium oxide catalysts drives selectivity in CO2 hydrogenation, Nat. Commun., 2021, 12, 1960, DOI:10.1038/s41467-021-22224-x.
- K. Kaźmierczak, R. K. Ramamoorthy, A. Moisset, G. Viau, A. Viola, M. Giraud, J. Peron, L. Sicard, J.-Y. Piquemal and M. Besson, et al.). Importance of the decoration in shaped cobalt nanoparticles in the acceptor-less secondary alcohol dehydrogenation, Catal. Sci. Technol., 2020, 10, 4923–4937, 10.1039/D0CY00390E.
- F. Cannizzaro, E. J. Hensen and I. A. Filot, The Promoting Role of Ni on In2O3 for CO2 Hydrogenation to Methanol, ACS Catal., 2023, 13, 1875–1892, DOI:10.1021/acscatal.2c04872.
- R. García-Muelas and N. López, Statistical learning goes beyond the d-band model providing the thermochemistry of adsorbates on transition metals, Nat. Commun., 2019, 10, 4687, DOI:10.1038/s41467-019-12709-1.
- S. Pablo-García, F. L. P. Veenstra, L. R. L. Ting, R. García-Muelas, F. Dattila, A. J. Martín, B. S. Yeo, J. Pérez-Ramírez and N. López, Mechanistic routes toward C3 products in copper-catalysed CO2 electroreduction, Catal. Sci. Technol., 2022, 12, 409–417, 10.1039/d1cy01423d.
- D. Garay-Ruiz, M. Álvarez-Moreno, C. Bo and E. Martínez-Núñez, New tools for taming complex reaction networks: the unimolecular decomposition of indole revisited, ACS Phys. Chem. Au, 2022, 2, 225–236, DOI:10.1021/acsphyschemau.1c00051.
- Adobe Illustrator, 2024, https://www.adobe.com, accessed: -02-07.
- OriginLab, 2024, https://www.originlab.com, accessed: -02-07.
- M. S. Frei, M. Capdevila-Cortada, R. García-Muelas, C. Mondelli, N. López, J. A. Stewart, D. C. Ferré and J. Perez-Ramirez, Mechanism and microkinetics of methanol synthesis via CO2 hydrogenation on indium oxide, J. Catal., 2018, 361, 313–321, DOI:10.1016/j.jcat.2018.03.014.
- Inkscape, 2024, https://inkscape.org, accessed: -02-07.
- S. Pablo-García, A. Sabadell-Rendón, A. J. Saadun, S. Morandi, J. Pérez-Ramírez and N. López, Generalizing Performance Equations in Heterogeneous Catalysis from Hybrid Data and Statistical Learning, ACS Catal., 2022, 12, 1581–1594, DOI:10.1021/acscatal.1c04345.
- J. D. Hunter, Matplotlib: A 2D graphics environment, Comput. Sci. Eng., 2007, 9, 90–95, DOI:10.1109/MCSE.2007.55.
- D. Garay-Ruiz and C. Bo, Rationalizing the Mechanism of Peroxyformate Decomposition: Computational Insights To Understand Solvent Influence, Chem.–Eur. J., 2021, 27, 11618–11626, DOI:10.1002/chem.202100755.
- E. Petrus, D. Garay-Ruiz, M. Reiher and C. Bo, Multi-Time-Scale Simulation of Complex Reactive Mixtures: How Do Polyoxometalates Form?, J. Am. Chem. Soc., 2023, 145, 18920–18930, DOI:10.1021/jacs.3c05514.
- ChemDraw, 2024, https://revvitysignals.com/products/research/chemdraw, accessed: 02-07.
- E. R. Gansner and S. C. North, An Open Graph Visualization System and Its Applications to Software Engineering, Softw. Pract. Exper., 2000, 30, 1203–1233, DOI:10.1002/1097-024X(200009)30:11<1203::AID-SPE338>3.0.CO;2-N.
- Q. Li, R. García-Muelas and N. López, Microkinetics of Alcohol Reforming for H2 Production From a Fair Density Functional Theory Database, Nat. Commun., 2018, 9, 526, DOI:10.1038/s41467-018-02884-y.
- S. Maeda, K. Ohno and K. Morokuma, Systematic Exploration of the Mechanism of Chemical Reactions: The Global Reaction Route Mapping (GRRM) Strategy Using the ADDF and AFIR Methods, Phys. Chem. Chem. Phys., 2013, 15, 3683, 10.1039/c3cp44063j.
- S. Maeda, Y. Harabuchi, H. Hayashi and T. Mita, Toward Ab Initio Reaction Discovery Using the Artificial Force Induced Reaction Method, Annu. Rev. Phys. Chem., 2023, 74, 287–311, DOI:10.1146/annurev-physchem-102822-101025.
- E. Martínez-Núñez, An Automated Method to Find Transition States Using Chemical Dynamics Simulations, J. Comput. Chem., 2015, 36, 222–234, DOI:10.1002/jcc.23790.
- E. Martínez-Núñez, G. L. Barnes, D. R. Glowacki, S. Kopec, D. Peláez, A. Rodríguez, R. Rodríguez-Fernández, R. J. Shannon, J. J. P. Stewart, P. G. Tahoces and S. A. Vazquez, AutoMeKin2021 : An Open-source Program for Automated Reaction Discovery, J. Comput. Chem., 2021, 42, 2036–2048, DOI:10.1002/jcc.26734.
- Y. Kim, J. W. Kim, Z. Kim and W. Y. Kim, Efficient Prediction of Reaction Paths through Molecular Graph and Reaction Network Analysis, Chem. Sci., 2018, 9, 825–835, 10.1039/C7SC03628K.
- Y. Guan, V. M. Ingman, B. J. Rooks and S. E. Wheeler, AARON: An Automated Reaction Optimizer for New Catalysts, J. Chem. Theory Comput., 2018, 14, 5249–5261, DOI:10.1021/acs.jctc.8b00578.
- L.-P. Wang, A. Titov, R. McGibbon, F. Liu, V. S. Pande and T. J. Martínez, Discovering Chemistry with an Ab Initio Nanoreactor, Nat. Chem., 2014, 6, 1044–1048, DOI:10.1038/nchem.2099.
- J. P. Unsleber, S. A. Grimmel and M. Reiher, Chemoton 2.0: Autonomous Exploration of Chemical Reaction Networks, J. Chem. Theory Comput., 2022, 18, 5393–5409, DOI:10.1021/acs.jctc.2c00193.
- S. Vernuccio and L. J. Broadbelt, Discerning complex reaction networks using automated generators, AICHE J., 2019, 65, e16663, DOI:10.1002/aic.16663.
- C. F. Goldsmith and R. H. West, Automatic Generation of Microkinetic Mechanisms for Heterogeneous Catalysis, J. Phys. Chem. C, 2017, 121, 9970–9981, DOI:10.1021/acs.jpcc.7b02133.
- C. W. Gao, J. W. Allen, W. H. Green and R. H. West, Reaction Mechanism Generator: Automatic Construction of Chemical Kinetic Mechanisms, Comput. Phys. Commun., 2016, 203, 212–225, DOI:10.1016/j.cpc.2016.02.013.
- A. Hashemi, S. Bougueroua, M.-P. Gaigeot and E. A. Pidko, ReNeGate: A Reaction Network Graph-Theoretical Tool for Automated Mechanistic Studies in Computational Homogeneous Catalysis, J. Chem. Theory Comput., 2022, 18, 7470–7482, DOI:10.1021/acs.jctc.2c00404.
- U. Gupta and D. G. Vlachos, Reaction Network Viewer (ReNView): An open-source framework for reaction path visualization of chemical reaction systems, SoftwareX, 2020, 11, 100442, DOI:10.1016/j.softx.2020.100442.
- R. Chacko, H. Gossler, S. Angeli and O. Deutschmann, Interconnected Digital Solutions to Accelerate Modeling of the Reaction Kinetics in Catalysis, ChemCatChem, 2023, e202301355, DOI:10.1002/cctc.202301355.
- M. Kuwahara, Y. Harabuchi, S. Maeda, J. Fujima and K. Takahashi, Searching chemical action and network (SCAN): an interactive chemical reaction path network platform, Digital Discovery, 2023, 2, 1104–1111, 10.1039/D3DD00026E.
- B. Medasani, S. Kasiraju and D. G. Vlachos, OpenMKM: An Open-Source C++ Multiscale Modeling Simulator for Homogeneous and Heterogeneous Catalytic Reactions, J. Chem. Inf. Model., 2023, 63, 3377–3391, DOI:10.1021/acs.jcim.3c00088.
- M. Álvarez-Moreno, C. de Graaf, N. López, F. Maseras, J. M. Poblet and C. Bo, Managing the Computational Chemistry Big Data Problem: The ioChem-BD Platform, J. Complementary Integr. Med., 2015, 55, 95–103, DOI:10.1021/ci500593j.
- C. Bo, F. Maseras and N. López, The role of computational results databases in accelerating the discovery of catalysts, Nat. Catal., 2018, 1, 809–810, DOI:10.1038/s41929-018-0176-4.
- S. Garnier, N. Ross, R. Rudis, A. P. Camargo, M. Sciaini, and C. Schererviridis(Lite) - Colorblind-Friendly Color Maps for R, viridis package version 0.6.4, 2023, DOI:10.5281/zenodo.4679423.
- A. Sabadell-Rendon, K. Kazmierczak, S. Morandi, F. Euzenat, D. Curulla-Ferre and N. Lopez, Automated MUltiscale simulation environment, Digital Discovery, 2023, 2, 1721–1732, 10.1039/D3DD00163F.
- A. H. Motagamwala and J. A. Dumesic, Microkinetic modeling: a tool for rational catalyst design, Chem. Rev., 2020, 121, 1049–1076, DOI:10.1021/acs.chemrev.0c00394.
- H. Ryu, J. Park, H. K. Kim, J. Y. Park, S.-T. Kim and M.-H. Baik, Pitfalls in Computational Modeling of Chemical Reactions and How To Avoid Them, Organomet, 2018, 37, 3228–3239, DOI:10.1021/acs.organomet.8b00456.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation, J. Phys. Chem. B, 2011, 115, 14556–14562, DOI:10.1021/jp205508z.
- G. Luchini, J. V. Alegre-Requena, I. Funes-Ardoiz and R. S. Paton, GoodVibes: automated thermochemistry for heterogeneous computational chemistry data, F1000Research, 2020, 9, 291, DOI:10.12688/f1000research.22758.1.
- F. Jensen, Basis Set Superposition Errors Are Partly Basis Set Imbalances, J. Chem. Theory Comput., 2024, 20, 767–774, DOI:10.1021/acs.jctc.3c01156.
- J. González-Fabra, F. Castro-Gómez, W. M. C. Sameera, G. Nyman, A. W. Kleij and C. Bo, Entropic corrections for the evaluation of the catalytic activity in the Al(III) catalysed formation of cyclic carbonates from CO2 and epoxides, Catal. Sci. Technol., 2019, 9, 5433–5440, 10.1039/C9CY01285K.
- R. Pérez-Soto, M. Besora and F. Maseras, The Challenge of Reproducing with Calculations Raw Experimental Kinetic Data for an Organic Reaction, Org. Lett., 2020, 22, 2873–2877, DOI:10.1021/acs.orglett.0c00367.
- M. I. Alam, R. Cheula, G. Moroni, L. Nardi and M. Maestri, Mechanistic and multiscale aspects of thermo-catalytic CO2 conversion to C1 products, Catal. Sci. Technol., 2021, 11, 6601–6629, 10.1039/D1CY00922B.
- M. S. Frei, C. Mondelli, R. García-Muelas, K. S. Kley, B. Puértolas, N. López, O. V. Safonova, J. A. Stewart, D. Curulla Ferré and J. Pérez-Ramírez, Atomic-scale engineering of indium oxide promotion by palladium for methanol production via CO2 hydrogenation, Nat. Commun., 2019, 10, 3377, DOI:10.1038/s41467-019-11349-9.
- F. Villagra-Soza, S. Godoy, A. Karelovic and R. Jiménez, Scrutinizing the mechanism of CO2 hydrogenation over Ni, CO and bimetallic NiCo surfaces: Isotopic measurements, operando-FTIR experiments and kinetics modelling, J. Catal., 2022, 414, 1–15, DOI:10.1016/j.jcat.2022.08.018.
- R. C. Catapan, A. A. Oliveira, Y. Chen and D. G. Vlachos, DFT study of the water–gas shift reaction and coke formation on Ni (111) and Ni (211) surfaces, J. Phys. Chem. C, 2012, 116, 20281–20291, DOI:10.1021/jp302488f.
- L. Dietz, S. Piccinin and M. Maestri, Mechanistic Insights into CO2 activation via reverse water–gas shift on metal surfaces, J. Phys. Chem. C, 2015, 119, 4959–4966, DOI:10.1021/jp512962c.
- M. D. Wilkinson, et al.). The FAIR Guiding Principles for Scientific Data Management and Stewardship, Sci. Data, 2016, 3, 160018, DOI:10.1038/sdata.2016.18.
- D. Garay-Ruiz and C. Bo, Chemical Reaction Network Knowledge Graphs : The OntoRXN Ontology, J. Cheminf., 2022, 14, 29, DOI:10.1186/s13321-022-00610-x.
- C. Maquilón, B. Limburg, V. Laserna, D. Garay-Ruiz, J. González-Fabra, C. Bo, M. Martínez Belmonte, E. C. Escudero-Adán and A. W. Kleij, Effect of an Al(III) Complex on the Regio-and Stereoisomeric Formation of Bicyclic Organic Carbonates, Organometallics, 2020, 39, 1642–1651, DOI:10.1021/acs.organomet.9b00773.
- C. Plett, M. Stahn, M. Bursch, J.-M. Mewes and S. Grimme, Improving Quantum Chemical Solvation Models by Dynamic Radii Adjustment for Continuum Solvation (DRACO), J. Phys. Chem. Lett., 2024, 15, 2462–2469, DOI:10.1021/acs.jpclett.3c03551.
- R. Pérez-Soto, S. Pablo-García, M. Besora and F. MaserasPykinetic: v0.1.0, 2023, DOI:10.5281/zenodo.8053050.
- M. Ciaccia, R. Cacciapaglia, P. Mencarelli, L. Mandolini and S. D. Stefano, Fast transimination in organic solvents in the absence of proton and metal catalysts. A key to imine metathesis catalyzed by primary amines under mild conditions, Chem. Sci., 2013, 4, 2253–2261, 10.1039/C3SC50277E.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dd00087k |
| This journal is © The Royal Society of Chemistry 2024 |