
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automated approaches, reaction parameterisation, and data science in organometallic chemistry and catalysis: towards improving synthetic chemistry and accelerating mechanistic understanding
Stuart C.
Smith
 ,
Christopher S.
Horbaczewskyj
,
Theo F. N.
Tanner
,
Jacob J.
Walder
and
Ian J. S.
Fairlamb
*
,
Christopher S.
Horbaczewskyj
,
Theo F. N.
Tanner
,
Jacob J.
Walder
and
Ian J. S.
Fairlamb
*
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: ian.fairlamb@york.ac.uk
First published on 24th May 2024
Abstract
Automation technologies and data science techniques have been successfully applied to optimisation and discovery activities in the chemical sciences for decades. As the sophistication of these techniques and technologies have evolved, so too has the ambition to expand their scope of application to problems of significant synthetic difficulty. Of these applications, some of the most challenging involve investigation of chemical mechanism in organometallic processes (with particular emphasis on air- and moisture-sensitive processes), particularly with the reagent and/or catalyst used. We discuss herein the development of enabling methodologies to allow the study of these challenging systems and highlight some important applications of these technologies in problems of considerable interest to applied synthetic chemists.
 Stuart C. Smith | Stuart C. Smith is a doctoral candidate reading for a PhD in Chemistry at the University of York. He holds an MSci. degree in Chemistry with Medicinal Chemistry from the University of Glasgow. His current research interests include synthetic organic methodology development (with an emphasis on catalysis and mechanism), computational chemistry, automation, data science, and bench synthesis. In particular, his research focusses on automating exploration of the chemical space of Csp2-Csp3 Suzuki–Miyaura cross-couplings, in conjunction with statistical and machine learning approaches, to facilitate reaction development and aid mechanistic understanding of this challenging reaction for industrial pharmaceutical applications. |
 Christopher S. Horbaczewskyj | Christopher S. Horbaczewskyj is the Technical Specialist at the University of York's Chemistry Automation Laboratory. Chris obtained his MChem degree from the Manchester Metropolitan University (2014). He obtained his PhD in chemistry from the University of Leeds. Chris is applying current technologies, using various optimisation techniques, to gather mechanistic insight into palladium-catalysed cross-couplings. He is developing new automation systems for the rapid screening and analysis of manganese-carbonyl photo-catalysed reactions using pump-probe laser spectroscopy, in collaboration with Prof. Jason Lynam (and wider team in York), and the Central Laser Facility (Rutherford Laboratories, UK). |
 Theo F. N. Tanner | Theo F. N. Tanner is an Associate Lecturer in Chemistry at the University of York. After obtaining an MChem degree at the University of York, he went on to study for a PhD in inorganic mechanistic chemistry in the same department. His PhD investigations focused on the mechanism of the Pauson–Khand reaction, applying a range of statistical, spectroscopic and theoretical investigations to further mechanistic understanding. Theo's research interests include descriptor-based statistical modelling of complex chemical processes, the identification of mechanistically relevant species using in situ and ultrafast spectroscopic measurements, and the quantum chemical modelling of reaction energy surfaces. |
 Jacob J. Walder | Jacob J. Walder is a PhD candidate at the University of York. Jake completed his MChem degree at the University of York in 2022, before beginning his postgraduate studies in the group of Prof. Ian Fairlamb later that year. His research focuses on understanding the mechanisms of competing side reactions in Suzuki–Miyaura cross-couplings of heteroaromatic compounds using high-throughput experimentation and data analysis. Jake's research interests include synthetic organic and organometallic chemistry, mechanistic studies, and chemical automation. |
 Ian J. S. Fairlamb | Ian J.S. Fairlamb did his BSc/PhD in Manchester (MMU, UK), with Dr Julia Dickinson (1993–99). He was a post-doctoral researcher with Prof. Guy Lloyd-Jones in Bristol (UK) (2000–1). His independent work began in York (UK) in 2001. He is a past recipient of a Royal Society URF (2004–12); promoted to Personal Chair in Organic Chemistry (2010). Research awards include the 2003 RSC Meldola Medal, 2007 Astra Zeneca younger researcher award, 2016 RSC Corday-Morgan Prize and Medal, 2019 Process Chemistry GSK-Pfizer-Syngenta Award and 2021 RSC Horizons Prize (with Lynam, York). He leads a team of ∼20 researchers working in mechanistic chemistry, automation and rich data analysis. |
1 Introduction
In the last 30 years, the use of robotic platforms in both industry and academia has increased greatly.1,2 Their use, in combination with traditional synthetic techniques,3,4 offers a range of benefits, including the potential to improve laboratory efficiency,5 output,6 and experimental reproducibility; a contemporary goal is to allow complete reproducibility between different laboratories internationally.7,8 Automated systems are prevalent in the pharmaceutical industry, and commonly employ both flow9,10 and batch conditions11,12 for synthesis. Additionally, different models have been developed via mining of the chemical literature, allowing for the prediction of reaction yields and further optimisation of benchtop chemistry.13Integration of automated approaches to synthetic chemistry and investigations of chemical mechanisms is not a modern idea. Magnetic stirrer hotplates that are used in nearly every laboratory around the world have been in circulation since the start of the 20th century and the first fully-automated synthetic platform for solid phase peptide synthesis was reported in 1966 by Merrifield and Stewart.14 This work greatly accelerated and simplified the process of peptide synthesis. Further innovations followed this work throughout the 1970s, including the development of a computer-controlled system for kinetic investigation of the reaction between a semi-carbazide and 2,6-dichlorophenolindophenol15 and the introduction of a closed-loop chemical synthesis system by Berkoff and coworkers in 1978.16 Closed-loop systems often consist of an experimental set-up or “loop” in which the results from a previous experiment directly inform inputs into subsequent experiments.17 In this case, the system consisted of a stirred reactor connected to reservoirs containing solutions of reagents that were added via pumps. The vessel was attached to a reverse-phase liquid chromatographic column for analysis and was controlled by a computer-system employing a simplex algorithm that changed conditions based on previous results in order to optimise product formation.
In the early 1980s, a new method of reaction optimisation was introduced by Furka – combinatorial chemistry.18 This technique was initially developed as an efficient route to peptide formation on a solid-support, achieved by splitting the resin into equal portions (k) to which k building blocks are added. The individual portions are then mixed, allowing for the elongation of the peptide chain. Since then, combinatorial chemistry has been employed in different forms in the drug discovery process.19 In recent years, employment of synthesis-based combinatorial chemistry has decreased, with computational in silico techniques being more widely utilised.20,21 The “Pool and Split” method is a variation of traditional combinatorial approaches which is capable of preparing millions of compounds. Compounds are first pooled and screened for catalytic activity before hits are split into their active components gradually making the combinations less complex. This method, and traditional combinatorial chemistry in general, was gradually phased out by the introduction of rational library design, facilitated by High-Throughput Experimentation (HTE). The combination of the “Pool and Split” method and HTE allows multiple reactions to be performed and multiple hypotheses evaluated, in parallel.22,23
High-Throughput Screening (HTS) was originally developed during investigation of antibiotic production in different Streptomycete libraries under a variety of fermentation conditions and involves automated screening of targets in a binding assay, and allows for many compounds to be rapidly screened in a cost-effective manner.24 HTS technology was later applied in chemical contexts to perform reactions in parallel to optimise a particular process, for example, optimising conditions for a specific reaction.25 HTE involves performing a large number of reactions simultaneously. One of the first reported uses of HTE was by Pfizer in 1986, revolutionising their screening process – increasing the output from 20–50 reactions that were performed on a 1 mL scale in separate reaction vessels to between 1000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 by utilising apparatus such as 96-well plates, on 100–150 μL scale. Since its first reported use, HTE has become increasingly prevalent and was not limited to drug development. For example, Burgess et al. employed HTE for catalyst screening for a specific C–H insertion reaction.26 The researchers used a microtiter plate (MTP) to screen 96 different reaction conditions, on a 100 μL scale. Each reaction was filtered through Celite and analysed via HPLC, allowing for the data to be collected quickly and efficiently (in under a week), and hits were investigated using benchtop techniques on a larger scale. Cooper and colleagues demonstrated the significance of HTE approaches in reaction discovery involving photochemicals27 as well as in the development of photocatalysts for hydrogen peroxide production.28
000 by utilising apparatus such as 96-well plates, on 100–150 μL scale. Since its first reported use, HTE has become increasingly prevalent and was not limited to drug development. For example, Burgess et al. employed HTE for catalyst screening for a specific C–H insertion reaction.26 The researchers used a microtiter plate (MTP) to screen 96 different reaction conditions, on a 100 μL scale. Each reaction was filtered through Celite and analysed via HPLC, allowing for the data to be collected quickly and efficiently (in under a week), and hits were investigated using benchtop techniques on a larger scale. Cooper and colleagues demonstrated the significance of HTE approaches in reaction discovery involving photochemicals27 as well as in the development of photocatalysts for hydrogen peroxide production.28
By the turn of the millennium, HTE became more common in all aspects of the chemical industry, including pharmaceuticals and agrochemicals. Multiple commercial robotic systems have since been developed that are able to perform synthesis, purification, and characterisation.29 These developments included the creation of bespoke systems for synthetic chemistry, with examples reported by Burke and co-workers.30–34 Cronin and co-workers developed robotic platforms for use in advanced synthetic chemistry.35–43 Other researchers have focused on the development of flow systems in the analysis of organometallic catalysis including Bode,44–47 Lapkin,48 and Burés, and Larossa.49
Apart from accelerating the execution of synthesis tasks, automation and data science have the potential to fundamentally change how we execute and understand chemistry. Some of the best examples of this include a closed-loop optimisation system applied to improvement of Suzuki–Miyaura cross-coupling (SMCC) reaction conditions developed by Burke,34 the ‘chemputer’ by Cronin,36 flow chemical apparatus by Bourne for kinetic analysis,50 and the application of evolutionary algorithms by Jensen to de-novo drug design.51
These are but a few examples of what can be achieved, and with the rate at which such systems are being developed, the rest of the decade could show a huge shift in the way research in advanced synthesis and catalysis is carried out (Fig. 1).
 | ||
| Fig. 1 Timeline of some key advancements in laboratory automation. | ||
Catalysis is a cornerstone of the chemical industry, contributing to 35% of the world's GDP,52 with the bulk of these processes employing organometallic compounds consisting of precious platinum group metals. Since catalysis is crucial to the synthesis of a range of feedstocks and products required by the pharmaceutical, agrochemical, and petrochemical industrial sectors, mechanistic understanding of different catalytic systems is of paramount importance. In particular, research in this area focuses on development of catalytic transformations with reduced catalyst loading, high product selectivity, a greener and more sustainable reaction outcome, and a lower cost of final purified product.
Recent advances in the fields of inorganic and organometallic chemistry have included the incorporation of robotic synthesis platforms, machine learning (ML) and sophisticated data analysis for mechanistic studies. We note the examples of this reported by Jensen and co-workers who incorporated de novo-based drug design methods to facilitate the synthesis of organometallic building blocks.51,53,54
Typical mechanistic studies in organometallic chemistry involve stoichiometric experiments, kinetic analysis, and intermediate trapping and characterisation. Recent research in this area has focused on improving the reproducibility of these studies via analysis of a large number of reaction outcomes and decreasing the time required for these types of studies – traditional mechanistic studies are notoriously time-consuming,55 as demonstrated in traditional mechanistic studies by Blackmond et al. which utilised in situ IR spectroscopy and calorimetric methods for kinetic investigations.56–59 Complementary automated approaches have been developed by Varela60 and Bourne, who incorporated flow systems to measure the kinetics of various reactions.61–64
ML methods have more recently been employed for the optimisation of reaction conditions and prediction of products.65,66 One of the most important tools related to ML is parameterisation which can give valuable insight into chemical mechanisms. For example, Fey and co-workers have demonstrated the application of computational descriptors of different phosphorus-based ligands67–74 for catalysis and mechanistic studies.75–82 Other notable work in ligand parameterisation comes from the group of Sigman, in which the authors parameterised ligands in order to explain their reactivity.83–90 One of the limitations of these methods is that they do not deal with metal catalyst speciation, an important complication of Pd-catalysed chemistry which is rarely accounted for in reaction models.
The use of HTE in mechanistic studies can result in a large amount of data being generated. Rigorous inferences can be made from these datasets by incorporating methods of data science and statistics into analytical workflows. This can allow for a greater understanding of a given catalytic system by uncovering mechanistic effects that would not be noticeable via traditional analyses. A technique that has been successful is Principal Component Analysis (PCA), a dimensionality reduction method that aims to reduce the number of variables in a dataset while still maintaining as much information as possible. Further in-depth information on the implementation of PCA has been reported by Hotelling91 and Sigman.92,93
Some key statistically-led chemical investigations have been conducted by Hein et al., in which they employed techniques such as non-linear regression to analyse reaction data.94–96 One example of this was to take the UV absorptions of various compounds and calculate their concentrations using non-linear regression analysis in the absence of response factors (molar absorption coefficient and path length of the Beer–Lambert Law).97 Alongside this, Sigman and co-workers have employed data-driven optimisation for various reactions and conditions.98–103.
Despite the wealth of examples and innovations that have been reported in the field,104,105 it appears that many laboratories are still hesitant to incorporate modern automation platforms to complement their synthetic work. This is not unsurprising – some of this technology, such as bespoke robotic systems – require expensive maintenance and are priced beyond what many academic laboratories can afford. However, we hope that this review will demonstrate how these, and other technologies, can be readily applied to traditional and applied organometallic chemistry, aligned with synthetic chemistry applications (Fig. 2).
 | ||
| Fig. 2 Outline of the work discussed in this review. | ||
2 Organometallic reaction automation – from simple to more complex methodologies
The earliest modern standard robots emerged in the 1950s as programmable manipulation arms. As computational power and the sophistication of related technologies increased during the 1970s, robotic systems became more widely adopted, before their widespread implementation in commercial settings, e.g. in the automotive industry and petrochemical industries.106–108 As these technologies have become cheaper and more accessible, they have become firmly integrated into academic and research institutions.109 The modern synthetic laboratory is a complex web of computer-controlled devices including heating and stirring apparatus, liquid handling units, various analytical techniques and, computer-controlled robotics. This network of instrumentation offers a range of benefits, including improvements in experimental efficiency, reducing human error, and improving overall experimental reproducibility.110Gaining these benefits is not without issue – robotic platforms typically require large capital investments and dedicated technical expertise to implement. Additionally, certain processes can be problematic to automate e.g., accurate transfer of electrostatic solids and viscous solutions. The development of lower cost technologies has been a key concern in the field in recent years, and several platforms have been developed to this end, including OpenTrons, Tecan Robots, Hamilton, and Mosquito robotics. These technologies are rapidly improving and typically offer significant potential for integration of difficult processes into standard workflows. Even though there is a substantial range of platforms available, it should be emphasised that it is not necessary to automate every step in an experimental campaign.
There are many ways to automate chemical reactions: simple reaction condition control (heating and cooling, stirring or substance transfer); the use of robotic platforms for solid and liquid handling and process control; the application of computer algorithms or the utilisation of optimisation software to the implementation of in situ analytical technologies.111 Each of these approaches offers the potential to improve understanding of chemical reactions, or to increase the efficiency of traditional synthetic methods (Fig. 3).
 | ||
| Fig. 3 General workflow diagram of modern approaches to mechanistic investigation of palladium catalysed cross-coupling reactions. The process begins by generating a grid of experiments. Data points of each of these are then gathered before the data is analysed. From this, new mechanistic insights can be gathered relating to the reaction under scrutiny. | ||
2.1 Automating benchtop or fume hood organometallic reactions
Simple technologies, such as magnetic stirrer hotplates, offer a simple method to increase the throughput of a synthetic process. Rudimentary, specialised syringe pumps have allowed the addition of solutions at regular time intervals over long periods. This represents only the first portion of a synthetic procedure, with other steps typically being completed manually, e.g., purging reaction vessels with inert gases, workup, and purification.The Chemputer (Fig. 4), developed by Cronin et. al.36 adapts traditional laboratory glassware by adding pumps, sensors, and switches so that multiple reaction steps can occur consecutively in an automated fashion, facilitated by an in-house developed Chemical Description Language, XDL. This language permits translation of discrete chemical tasks (reagent addition, reaction, extractions etc.) into machine-executable operations (turn on stirrer hot plate, increase flow rate of pump driver, heat reaction vessel etc). Although this platform has largely been used to synthesise and purify organic compounds, it has the potential to be adapted (employing inert atmospheric conditions) to create inorganic and organometallic compounds.
 | ||
| Fig. 4 Image of the chemputer developed by Cronin and coworkers. It consists of a backbone of switches, valves and pumps, a reactor unit, filtration and separator units, and a rotary evaporator.36 | ||
The chemputer has been used independently to enable a fully automated synthesis of Ag nanoparticles (NPs),113 where a change in Ag concentration could reliably and reproducibly generate the correct size of NP; 2.2 nm ± 1.0 nm (2.23 mg mL−1) and 5.6 nm ± 3.8 nm (12.59 mg mL−1), and in reasonable yields (30–35% and 41–53%, respectively).
This system shows the capability of new technologies to adapt existing frameworks and modernise laboratories. Indeed, this system could be applied to many routine laboratory transformations in order to make them more efficient and reproducible. However, not all reactions or workflows may be applicable to this form of reactor e.g., air sensitive reactions. It is possible to use roaming robots to achieve what a chemist normally does around the lab, particularly if the robot can access fume hoods, samples, and instrumentation. This has mainly been utilised for materials chemistry research and the development of organic photocatalysts.114
An adaption of the chemputer has been developed which allows a fully automated approach to air-sensitive chemistry while using standard lab glassware, which has been called the Schlenkputer.115 This system makes use of a standard gas/vacuum handling unit and a liquid handling unit alongside other standard glassware. A number of motors, switches, and valves were used to control the tap positions in the Schlenk line and controlled by XDL commands to open the vacuum. A liquid handling setup was also established and used in conjunction with the Schlenk system to remotely execute challenging inorganic batch transformations. Initial stages of this work led Bell and coworkers to assess the ability of the platform to inert the liquid handling system and the glassware employed. This was facilitated by the titanocene species, [Cp2TiIII(MeCN)2]+, a widely used colour-metric indicator which is highly sensitive to O2 (which turns from blue to yellow upon O2 exposure). These tests showed that their Schlenkputer was an inert system, but also highlighted the requirement to pre-wash parts of the liquid handling system which could not be connected to the Schlenk system. Further successful tests allowed the authors to synthesise titanocene from its constituent components and facilitated the synthesis of highly air-sensitive Ce(N(SiMe3)2)3, highly moisture sensitive Lewis acid tris(pentafluorophenyl)borane, and a MgI dimer (via Na0 reduction). This example illustrates the significant opportunities that emerging automation technologies offer to facilitate challenging organometallic chemistry.
 | ||
| Fig. 5 A simple flow system setup using two syringe pumps, a T-connector, a reactor coil, a pressure regulator, and a collection vial. The reaction can easily flow into a sample device for on-line sampling, or manual samples can be taken for at-line or off-line sampling. Adding computer control to the system and temperature regulation will make this setup an automated flow system. | ||
Most reaction paradigms can be adapted for a flow process and there are many examples making use of photochemical or electrochemical methods. Continuous flow systems allow quick and easy system cleaning between reactions with a solvent flush. Additionally, reactors can be pressurised to allow solvents to be used far above their boiling points and solids – which are not easy to dissolve – can be supported on a column.
Highly exothermic reactions can be performed more safely in a flow system. This is mainly due to the small channel dimensions which allow quick and efficient heat dissipation, reducing the risk of a ‘runaway’ reaction. This method has been rigorously evaluated with lithiation reactions,116–118 where typical conditions in a batch necessitate temperatures of −78 °C and slow addition of reagents. Lithiations have also been shown to be possible in flow, at higher temperatures (∼25 °C), compared to more traditional methods. This demonstrates that mixing and heat transfer in a flow reactor allows much greater reaction control than in batch systems.118 The reactor setup required three peristaltic pumps, two T-pieces and lengths of PTFE tubing. The system employed was simplistic yet highly effective and easy to adopt in any laboratory. Integration of reaction analytical technologies and computer control (via a user interface or algorithms) greatly improves flow processes, allowing the creation of a closed-loop optimisation platform and significantly expanding the scope of application of flow systems.
Continuous flow systems equipped with in-line UV-Vis spectroscopic analysis have been employed in the synthesis and self-optimisation (maximising conversion) of AuNPs. This system demonstrates how readily optimisation algorithms can be applied to in-line gathered data to achieve impressive conversions (of up to 95% in this case). Generated data was then used to create kinetic models and was subsequently validated using batch reactions.119
The effectiveness of an automated, droplet-flow device has been demonstrated via exploration and optimisation of a Pd-catalysed SMCC reaction.120 Droplet microfluidic technologies allow discrete variables to be added into optimisation procedures and behave similarly to nano-scale batch reactors. The flow system uses a droplet sample injector, in which the droplet is transported by a flow of argon before injection of an aliquot of base initiator into the reaction droplet. Droplet systems allow increased throughput within many, small, individual reactors with rapid mixing. This is achieved via chaotic advection and provides increased reproducibility when compared with single reactions executed in continuous flow.121 The newly activated droplet travels through a temperature-controlled tubular reactor and is later quenched to allow sampling – individually using refractive index sensors for accurate timing – before HPLC analysis. Reaction conditions {substrate (4), boronic acid (2), pre-catalysts (2), ligands (7), temperature (30–110 °C), reaction time (1–10 min), and catalyst loading (0.5–2.5 mol%)} were screened iteratively and the HPLC data interpreted by the algorithm to generate the next iteration of reaction conditions.
The range of conditions tested led to the identification of optimal conditions (maximum reaction yield and turnover number) for each substrate evaluated (Fig. 6). The breadth and depth of data gathered from screening campaigns of this type can help to find relationships that are more difficult to find from manual one-variable-at-a-time (OVAT) methods. Key mechanistic insights developed from this study included the performance of ligands in the oxidative addition step with aryl halides, and the global effect of the ligand on the overall reaction yield. Different ligands enabled decreased reaction times, increased product yields, and reduced protodeborylation side-reactions. The outcome can be explained by promotion of faster oxidative addition and transmetallation steps, and concomitant downregulation of side-product pathways. The employment of pinacol boronates were shown to efficiently allow coupling with aryl halides. However, some combinations of catalyst and ligand led to the free organoboronic acid outcompeting rapid protodeborylation.
 | ||
| Fig. 6 Representation of the optimal conditions found for various combinations of aryl halides and boronic acid.120 | ||
This combination of continuous flow methodology and HTE permitted reactions to be performed under more intense reaction conditions (elevated temperature and pressure), while gathering many data points. Perera et al.10 used a flow platform, integrated with UPLC-MS, to perform 5760 reactions at nanomole-scale (Fig. 7) over several days (ca. 1500 reactions per day), using two UPLC-MS instruments working synergistically. A full profile of each reaction was gathered, allowing a profile of substrate, internal standard, and reaction products to be developed. The reactor itself was a simple tubular coil heated to 100 °C (pressurised at 100 bar) with methanol as a carrier solvent. Subsequently, gathered data was analysed off-line (1500 datapoints h−1). Given the size of the dataset, it may have been beneficial to program analysis and visualisation of the dataset. Of the substrates explored, 6-chloroquinoline was the worst performing and least robust transformation (tolerating only XPhos and SPhos as ligands). The indazole substrate gave the highest density of high conversion conditions over the other more electrophilic substrates. Trifluoroborate salts were shown to be the least efficient boronate partner, suggesting that the 1 minute residence time is insufficient. With such a wealth of data, it is possible to determine other trends by clustering the data in terms of yield (>85%) and outlining conditions which work regardless of the substrate chosen (XPhos or SPhos, and MeCN). Any conditions, or models gathered from large bodies of work like this do need to be rigorously assessed (particularly when applied to scaled-up processes). Technical experimental replicates – which are facilitated by HTE approaches – are therefore critical.
 | ||
| Fig. 7 Flow reactor setup for the work performed by Perera et al.10 A total of 5760 reactions were performed to evaluate a host of reaction conditions: 7 substrates, 4 coupling partners, 12 ligands, 8 bases, and 4 solvent systems. Other reaction conditions were held constant. The heat map diagram (re-created from the published raw data) shows a quarter of the results (1536) for boronic acid 2a. | ||
This system not only allowed screening of many combinations of conditions at small scale (to find optimum reaction conditions) but provided key mechanistic insights into the reaction for a variety of substrates. Performing flow reactions in this way is highly valuable, despite the inability of flow platforms to easily handle all reaction conditions or reagent states, e.g., solid reagents, reactions which precipitate solids or dual solvent biphasic systems. The broad range of approaches to automated synthesis theoretically permits exploration of any reaction space.
2.2 Automation of organometallic reactions using robotic platforms and multiplexed plates
High throughput platforms permit a range of conditions to be trialled simultaneously. A large range of reaction plates sizes have been developed (with some plates capable of housing several thousand wells).122–126 Larger input spaces lead to greater volumes of data, which can easily introduce a bottleneck to determining reaction outcomes. This can be solved by integrating programs to manage data and assist with mass-data interpretation.Many reaction conditions can be screened using a high throughput platform, including a variety of discrete and continuous variables – including base, catalyst, ligand, solvent, temperature, reaction time, and stoichiometries. Manual HTE is possible using traditional lab approaches, in which solids and solutions are prepared and subsequently transferred by hand to each reaction well. This methodology has been used to complete 384 SMCC reactions to create a useful map of many combinations of conditions. This work highlighted optimal operating conditions, as well as how each parameter affected reaction yield.127 There are many other examples of HTE using manual, streamlined approaches, e.g., the use of solid transfer scoops or multi-necked micropipettes.8
Robotic platforms also make traditionally time-consuming reactions (e.g. gathering kinetic data) significantly easier and less demanding.128 Collecting kinetic data for reactions provides key insights into mechanistic features. For example, it is possible to use a liquid handling unit (e.g., Chemspeed Swing) to transfer reaction aliquots to a sampling device to gather ex situ data at regular time intervals to generate kinetic profiles for different pre-catalyst species (four) during a SMCC reaction.
An initiator was added to each stirred reaction solution and samples were taken at set time points over a specific period (5/20 min intervals, 400 per min reaction time). Each sample was analysed immediately after preparation. The comparison to off-line sampling used the same samples left for 20 h before re-sampling. The work highlights the dramatic difference in performing sampling immediately after analysis vs. delayed analysis after sample aging/degradation. It also shows the ease with which the robotic platform can gather kinetic data (over an 8 hours period) to provide accurate profiles.
HTE approaches permit the development of simple workflows, but this methodology can also be applied to much larger experimental design spaces to accelerate the discovery of general reaction conditions. For example, the overall workflow methodology employed by Angello and coworkers34 involved a data mining procedure to first screen for appropriate substrates from a catalogue of ca. 5400 (hetero)aryl halides. The successful candidates were combined with 54 MIDA† boronates to generate an initial design space of 2688 potential products. Examining a space of this size was deemed unfeasible in this case, however, algorithmic testing of each of these sets was performed using solvent, base, pre-catalyst, ligand, and temperature each with a range of associated values. Prior reaction knowledge and human-led decisions led to a series of eleven substrate pairs being chosen for further examination.34
The robotic system performed reactions using the eleven substrate pairs, each under seven different reaction conditions, using seven pre-catalysts. These were compared to a literature procedure129 using common conditions: 5:1 dioxane/H2O, 60 °C, K3PO4, and the Pd SPhos G4 pre-catalyst. These reactions were performed as “seeding” experiments to train the algorithm and to help identify redundant conditions. Two ligands (PCy3 and dppf) and Pd2(dba)3 were removed as poor performing conditions, leading to a reaction space of 528 reactions (Fig. 8).
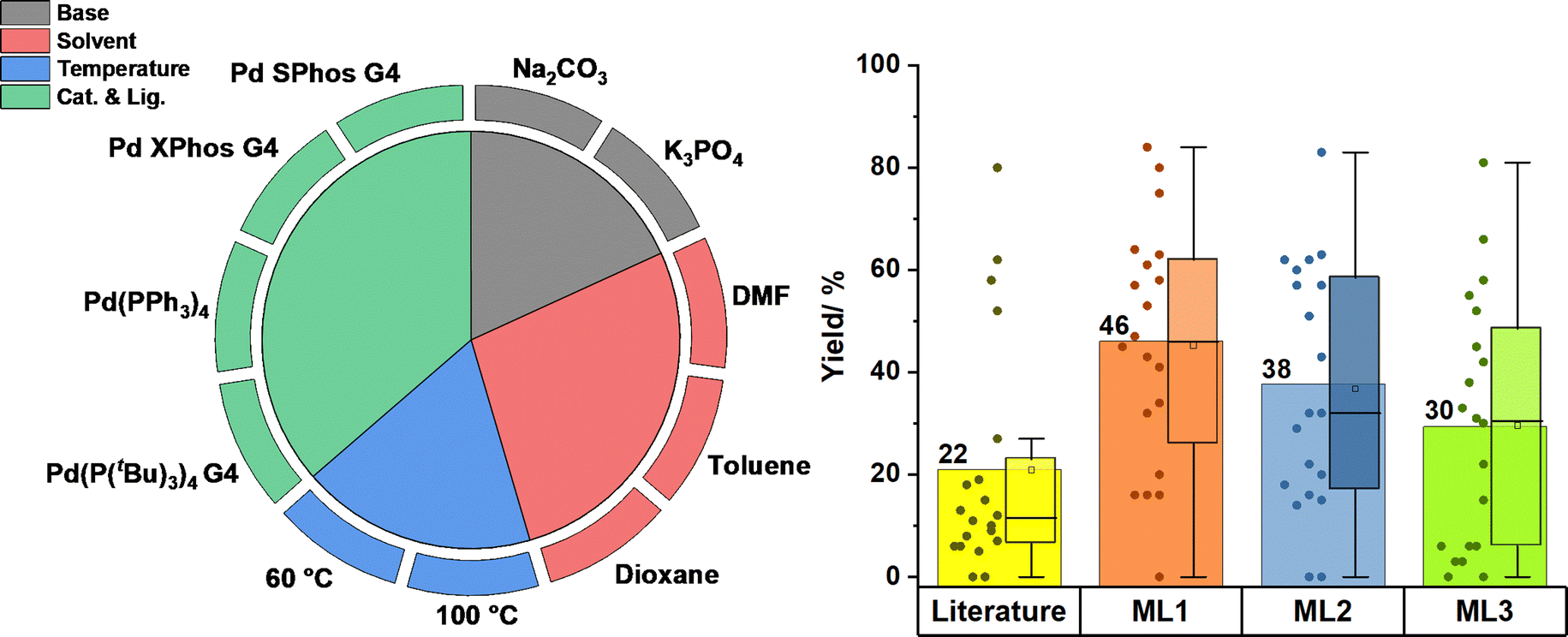 | ||
| Fig. 8 Left: summary of reaction conditions performed using HTE during an SMCC reaction of arylboronic acids and heteroarylmethyl chlorides. Right: performance of the top ML conditions compared to literature benchmark conditions.34 | ||
Subsequently, a Bayesian algorithm used the “seed” data and reaction conditions list to provide batches of experiments, organised into an intelligent priority queue. These experiments were executed on the robotic system and data was gathered via LCMS. Analysis of this data led to algorithmic generation of the next iteration of experimental conditions, and this process was repeated for a total of five rounds. The top three algorithmically found general conditions, which were applied to twenty other substrate pairs, chosen by another algorithm, and compared to previously reported general conditions. All condition sets performed well for nineteen substrate pairs (5–85%), with only one pair having no detectable yield. The algorithm identified conditions which performed better than the previously reported conditions, with yields improving by two-fold. The procedural selection of variables in this study could have been more reliant on the algorithm rather than having human intervention. This removes a reinforcement bias in which researchers rely on inputting conditions which they consider – consciously or subconsciously – more likely to work. This can potentially lead to missed opportunities to uncover novel reactivity.
These technologies have allowed the number of reactions performed to be vastly increased. The volume of data gathered would be intractable without the introduction of supporting analytical technologies. The use of analytical instrumentation has been noted in many of the examples above and is often fully integrated into synthetic workflows. This allows reactions and analysis to be completed with minimal human intervention.
2.3 Automated analytical instrumentation: integration and closed-loop generation
Analytical instrumentation can be easily integrated or adapted into many workflows to allow reaction data to be gathered immediately or with little time delay and minimal sample preparation. Automation has already been widely employed in analytical applications where autosamplers are extensively used. Additionally, powerful algorithms are routinely employed during sample analysis, e.g., solvent suppression algorithms in NMR spectrometry,130 and the wealth of sophisticated technology used in analytical instruments.There are a multitude of analytical instruments to choose from, each providing unique information about the chemical system. When used in tandem, these technologies can assist in compiling a picture of the mechanistic detail of a given transformation. In combination with sampling devices and control software, it is possible to create a system which can run a reaction, gather analytical data, and process or organise the data into a useable format. Many mechanistic investigations have made use of analytical instrumentation to analyse reaction aliquots without significant sample preparation or intervention which can be termed either in-line, on-line, at-line, or off-line.131
In/on-line analysis ensures reaction mass balance is more consistent and leads to increased accuracy and reproducibility between replicates. At/off-line analysis (a change of sample conditions i.e. dilution or quenching) can potentially influence the reaction aliquot. Dilution will only affect the reaction concentration, while quenching may change the sample conditions – such as catalyst speciation. In turn, this may misrepresent details of the mechanism under investigation. In-line, on-line, and often at-line measurements give a more realistic look at the reaction being performed.
Reaction analysis measurements can also be performed using specially designed miniaturised analytical instruments or using traditional full-sized instruments which can be adapted to monitor reactions. There are facilities in the United Kingdom which can aid the monitoring of chemical reactions using common analytical techniques, notably the DReaM facility132 at the University of Bath and the ROAR facility133 at Imperial College London. Many large facilities internationally also aide the probing of reaction details by supplying more powerful electromagnetic radiation techniques: RAL, in Oxfordshire, UK;134,135 the ESRF in Grenoble, France;136 the SSRF in Shanghai, China,137 and the KCSRN in Moscow, Russia.138
A recent example of spectroscopic reaction monitoring was provided by Lynam and coworkers,139 who studied the speciation and kinetics of a light activated manganese-catalysed C–H bond functionalisation using Time Resolved Infrared spectroscopy (TRIR). Picosecond species observation was demonstrated in this work, executed at RAL, providing key insights into the mechanistic pathway of the metal-catalyst. The study shows that immediately after sample irradiation and ligand dissociation, a solvated complex is formed. This suggests that diffusion control was critical to speciation and the formation of alkyne manganese complexes. This is a powerful in situ technique to understand photo-initiated chemical reactions, although very specialist and expensive. Miniature instruments are cheaper, space efficient and often faster than their larger counterparts, although resolution and sensitivity are more limited.
In/on-line analysis facilitates mechanistic understanding by providing evidence of troublesome reaction species often missed when analysing reaction samples off-line. An organozinc-catalysed Mannich reaction used in-line infrared spectroscopy (ReactIR) to probe the specific pathway of the amine.140 Primary amines were found to proceed through an imine intermediate and could be significantly enhanced by the addition of organozinc reagent. The formation of secondary amines was shown to proceed via a hemiaminal intermediate and organozinc reagents were found to accelerate formation of the intermediate species. In/on-line analysis provides rich data, but in-line analysis is often the quickest method for gathering data.
New analytical technologies are routinely being introduced to allow rapid sample screening and reduce the likelihood of sample contamination. Acoustic levitation techniques are becoming increasingly popular and are capable of performing high-throughput sample analysis to gather large datasets more rapidly. Ultrasound at ca. 40 kHz suspends small and easily manipulable liquid droplets which function as reaction vessels and can be directly analysed. This aligns perfectly with HTE which are often run on small scales. Acoustic Droplet Ejection – Open Port Interface (ADE-OPI),141–144 allows nanolitres (1–20 nL) of sample aliquots to be analysed rapidly using a variety of techniques, e.g., mass spectrometry (MS). A 348-well plate can be sampled in ca. 10 minutes, directly after a HTE screen rapidly providing a wealth of data (Fig. 9). Other similar methods which include MS (MALDI,145,146 AMI147,148 or DESI149,150) are also applicable in HTE workflows, but require extra steps in sample handling (lengthening the time between reaction and analysis). Adding rapid in/on-line analysis vastly improves workflows and allows rapid generation of large datasets. However, this will add to the bottleneck of having a backlog of data, unless subsequent programs are integrated to manage data and assist with mass-data interpretation.
 | ||
| Fig. 9 Diagram of an ADE-OPI-MS rapid data gathering system. Acoustic energy ejects a small droplet upwards which is transported to the mass spectrometer, where reaction outputs can be measured. This technique is capable of analysing ca. three samples per second.142 | ||
Integration of in/on-line analysis is key in the development of closed-loop synthetic workflows. A closed loop is created by integrating software or an algorithm with a synthetic platform, allowing reaction conditions to be regulated and monitored easily. However, many still require the user to manually feed in conditions which the robotic system can then cycle through, needing to be updated by the operator every time or using a manual termination procedure.6,151–154 Employing computer algorithms to intelligently control, monitor, and change conditions adds additional complexity and cannot only be achieved algorithmically. Closed loop systems can be created from batch, flow, and high-throughput systems. Alongside these, in silico techniques are now becoming far more advanced and can aid researchers in a variety of ways, including improving sampling control and facilitating data analysis.
2.4 Summary
Choosing suitable technologies to facilitate execution of a given synthetic route can seem challenging. Each approach offers specific advantages and disadvantages which may align their application more favourably with certain reaction spaces. The inherent modularity of many of these approaches vastly increases the scope of reactions which can be automated. New and simple engineering skills may need to be learned to keep platforms in good operating condition. As many of these platforms are still being developed, a typical workflow may include significant manual handling of substances or manual data handling. Generally, the field aims to automate routine, mundane or time-consuming tasks and to lower the risks associated with highly hazardous materials.Automating traditional bench methods can allow researchers who do not have access to high capital funding to integrate and modernise laboratories. This is also an effective way of performing more specialist chemistries without human intervention. If reaction intensification or algorithmic optimisation methods are required, then continuous flow systems can be employed. Flow chemistry lends itself to solution chemistry or easily flowable slurries and has the advantage over other methods at being able to intensify reaction conditions due to the ease of putting the system under pressure. Air-, moisture-, and light-sensitive chemistries can be performed if the correct reactors are chosen. However, many of these reactors cannot handle solids anywhere in the reaction process due to the small dimensions of reactor pathways. Specialist reactors can be purchased, or current ones adapted, but either of these options greatly increase the capital costs or require further expertise. Furthermore, large liquid handling platforms or commercial systems can be expensive but offer a wide range of reactors capable of handling a variety of chemistries and which can be adapted to function for specific reactions. Reactions which may not be suitable for flow systems may be performed using liquid handling platforms. These platforms are largely used for performing high-throughput studies where many categorical factors may have an effect on the reaction system.
Process analytical technology is fundamental in the setup of an efficient workflow. Although largely integrated already, in/on-line analysis are invaluable in the analysis of organometallic reactions. Rapid synthetic execution demands equally fast analysis to prevent a bottleneck. In-line, on-line, or at-line methods are all capable of facilitating organometallic synthetic or mechanistic workflows. Commercial software packages are available for most in/on-line analysis to prevent the formation of a data analysis bottleneck.
Although these technologies have been used for many years in application to organic chemistry, they are still in their infancy for more specialist chemical reactions, particularly catalysis and other reactions which are oxygen/moisture sensitive. New methods specifically developed for these means are becoming increasingly prevalent and, we predict, will form the basis of investigations in the field of organometallic synthesis and mechanistic investigation.
3 Rich data analysis and statistical in mechanistic investigations
Traditional mechanistic investigations of organometallic catalytic cycles typically involve use of a limited number of mechanistic tools (including in situ/ex situ analysis, intermediate trapping, and analysis of reaction kinetics) to make chemical inferences. With the increasing availability of low-cost laboratory automation equipment, it is becoming feasible for more laboratories to consider medium-to high-throughput synthesis and mechanistic activities. This is attractive since it is now possible to mechanistically probe systems rapidly and leverage the statistical power associated with large datasets. In this manner, it has been demonstrated that it is possible to extract subtle mechanistic effects impacting a system in a more rigorous and time-effective way than could be offered by a traditional bench investigation.To fully understand the mechanisms of catalytic processes for organometallic compounds, it is important to analyse substantial amounts of data. This necessitates employment of tools from a range of fields including data science and statistics. Rich data analysis (RDA) refers to the analysis of multiple data sources to gain a comprehensive understanding of a particular system. In the context of catalytic processes for organometallic compounds, RDA can be applied to large datasets from multiple sources, including spectroscopic data, reaction kinetics and thermodynamics data. These data can be statistically analysed in ways which maximise the insight that can be obtained. This section highlights several key examples which exemplify applications of these tools to the analysis of problems of significant mechanistic interest.
3.1 Rich data analysis
Hein and coworkers have recently described three data categories which are key in elucidating mechanism and making chemical inference – fundamental properties, reaction outcomes and reaction mechanics.155 They emphasise the importance of molecular descriptors of reactivity – including substrate electronics for organic reactivity and physicochemical properties such as lipophilicity for medicinal chemistry – and how this can be linked to chemical outcomes. Solubility was highlighted as a parameter which is critical for mechanistic chemistry, but for which there is a dearth of reliable literature data available.156 They then proceeded to describe the development of in-house platforms to generate computational and experimental solubility data via a closed-loop approach. The interplay of fundamental molecular properties, reaction outcomes and mechanics, and the statistical and automation tools highlighted in this review are summarised in Fig. 10. | ||
| Fig. 10 Graphical representation of a workflow to gather large datasets in the push towards mechanistic understanding of organometallic reactions driven by automation, optimisation, and data science. Each element works in combination with all others to funnel towards the end goal of detailed insights into reactions involving organometallic species. | ||
Fundamental property databases have been successfully employed in a number of applications including computationally-guided drug design157 and rational solvent selection,158 as well as enabling ML studies. Structure- and electronics-based QM databases, such as ANI-1 (ref. 159) and GDB-17 (ref. 160) are frequently adopted to great advantage. It is often computationally expensive to calculate complex physical parameters such as solubility using ab initio methods like Density Functional Theory (DFT). This led the authors to develop predictive models, including a quantitative structure–activity relationship (QSAR) method. The utility of HTE approaches was also highlighted in this work through the development of a solubility screening workflow to generate large libraries of solubility data rapidly.
While HTE approaches have enabled generation of large datasets for reaction optimisation,161–163 the data input to these is scope-limited and requires user-selection of input variables based on expert chemical knowledge.164 This represents a clear potential for experimentalist bias and the potential for overlooking unexpected outcomes. Resources such as the Open Reaction Database165 aim to further expand the quality and breadth of data available through open access routes for appropriate training models.
Reaction outcomes are variable and often hinge on the interplay of complex and subtle interactions in high-dimensional input space. It is often difficult, therefore, to generate multivariate ML models due to the lack of availability of sufficiently complex and massive datasets. This problem often leads to the combination of several datasets from multiple literature sources and HTE to allow the development of predictive models for optimisation. Regression analysis has been employed successfully in a number of optimisation applications (including prediction of optimal catalyst, ligand, and substrates in specific systems).166–168
A key drawback of regression analysis, however, is that it often requires prior mechanistic knowledge about the reaction to select relevant descriptors. Doyle and coworkers have demonstrated the power of ML algorithms in the absence of such prior knowledge with a sufficiently large dataset. They examined a Pd-catalysed Buchwald–Hartwig cross-coupling of 4-methylaniline with aryl halides using several potential inhibitors (Scheme 1).
 | ||
| Scheme 1 A summary of the input space examined by Doyle and coworkers.169 | ||
Of the 4608 experiments conducted in 1536-well plates, 30% failed to produce any product, yet, a spread of yields were observed across the remaining plates. This allowed Doyle to begin parameterising this dataset via extraction of mechanistically relevant descriptors – atomic, molecular, and vibrational – before beginning training a machine-learning model. They showed that a random forest model – a number of independent decision trees whose aggregate classification on regression and prediction problems are more accurate than any single tree in isolation – trained on an ultra-HTE dataset could successfully predict the influence of additives in synthesis without the need to pre-select descriptors. This work required development of a dataset of over 4000 entries, and highlights some of the key challenges of research in this area. The challenges associated with management of a dataset of this size was highlighted by Frey and Willoughby in 2022.170 The model resulting from this database cannot be extrapolated to other types of chemistry, which would require bespoke investigations of their own. Each new system under investigation requires bespoke experimental generation of a suitable dataset, appropriate analysis, and data management processes (which is an often overlooked and non-trivial aspect of this class of investigation).
Basic data on the formation and consumption of reaction components over time provides key mechanistic insight into reaction kinetics and thermodynamics. For example, such data has allowed the development of ML kinetic models built to study simple mechanisms such as cycloadditions,171 SN2 substitutions,172 and E2 eliminations.173 More complex systems require increasingly complex mechanistic investigations. In situ analytical technologies allow quantitative resolution of many reaction components over a wide dynamic range, providing mechanistic insight. Analysis of this type represents the ideal in mechanistic studies: however, most systems are not amenable to current in situ technology. Many challenges prevent the common usage of these technologies as primary tools of investigation, including air/moisture sensitivity, extreme conditions, and heterogeneity issues. In response, researchers often develop and employ model reactions as facsimiles of their system which are amenable to in situ analysis. It is often difficult to quantify the degree to which the use of such model systems impacts the conclusions of a mechanistic investigation. It is imperative, therefore, for the field to focus on the development of new analytical technologies for the investigation of complex chemical systems in real-time (vide supra).
3.2 Design of experiments
Chemical space, referring to the total property space spanned by all theoretically possible compounds and molecules, is vast. In order to explore this space efficiently to find new compounds and novel reactivity, as well as to avoid convergence on local minima during optimisation campaigns, it is necessary to employ statistical tools. Design of Experiments, DOE, is a statistical approach to the design of chemical investigations which aims to explore a broad chemical input space parsimoniously, by formulating statistically rigorous models of an output variable observed at the extremities of an input space and interpolating to allow predictions to be made about the space in general (Fig. 11). In his seminal work in this area, “The Design of Experiments”,174 Fisher emphasised the importance of controls, randomisation, replication, and the use of multifactorial experiments. The opportunity to interrogate a chemical system by simultaneously varying a number of input factors and to use statistically rigorous models to derive chemical inference is attractive. It results in reduction of the number of experiments necessary to assay a complex chemical space, as well as reducing the likelihood of converging on false optima (e.g., a local rather than a global energetic minimum), and provides the opportunity to resolve factor interactions. Even in low-resolution factor screening designs, with the potential for multiple-factor confounding, DOE aids in decision-making and in the planning of further optimisation studies. As DOE data is analysed statistically across a whole study, using multiple linear regressions, the error throughout the regression model can be estimated without the need for the many replicate experiments required of traditional OVAT approaches. These benefits are summarised in Fig. 11. | ||
| Fig. 11 A comparison of OVAT and DOE approaches. (a) OVAT design involving sequential optimisations in different input dimensions. An optimum is found in the first dimension, which is then taken as fixed in other input dimensions. (b) DOE approaches involve definition of an experimental input space with extreme values between which statistically rigorous interpolation is possible. This allows identification of a maximum at any point within the design space. A factor screen is used to determine which input variables are worth investigating in more detailed designs. (c) Response surface optimisations allow rigorous optimisation of an output variable and are suitable for producing models with quadratic terms. Red indicates a high level of the response variable, and blue represents a low level. | ||
In 2019, Maurer and coworkers described the application of DOE approaches to the optimisation of copper-mediated 18F-fluorination reactions of arylstannanes.176 This chemistry is of significant interest in the development of medical devices, where copper-mediated radiofluorination reactivity has provided access to Positron Emission Tomography (PET) probes. 18F is highly suitable for these applications due to its decay mode (97% positron emission with low tissue penetration), high specific activity and short half-life. Physical properties of fluoride, however, render radiofluorination challenging.177,178 By sequential application of DOE approaches, Maurer was able to successfully develop reactions to synthesise novel PET probes via radiofluorination, and gain significant mechanistic insight into these processes. Fig. 12 summarises the chemical problems and the key experimental designs employed by Maurer and coworkers in this study.
 | ||
| Fig. 12 Summary of the radiofluorination input space explored in this design.176 | ||
A main factor screening with a Resolution V + fractional factor design (FFD) was employed to determine the most important factors in the input space of 18F-fluorination of 4-tributyl stannylbiphenyl. This design took loading of Cu(OTf)2 (1–4 eq.), pyridine loading (4–30 eq.), DMA loading (400–1000 μL), temperature (100–140 °C) and atmosphere (argon vs. air) as its input space. This design can resolve main effects, two-factor interactions and indicates whether there is model curvature present. The factors in the input space are summarised in Fig. 13, with radiochemical conversion of the reaction (%RCC) as determined by radioTLC taken as the response variable.
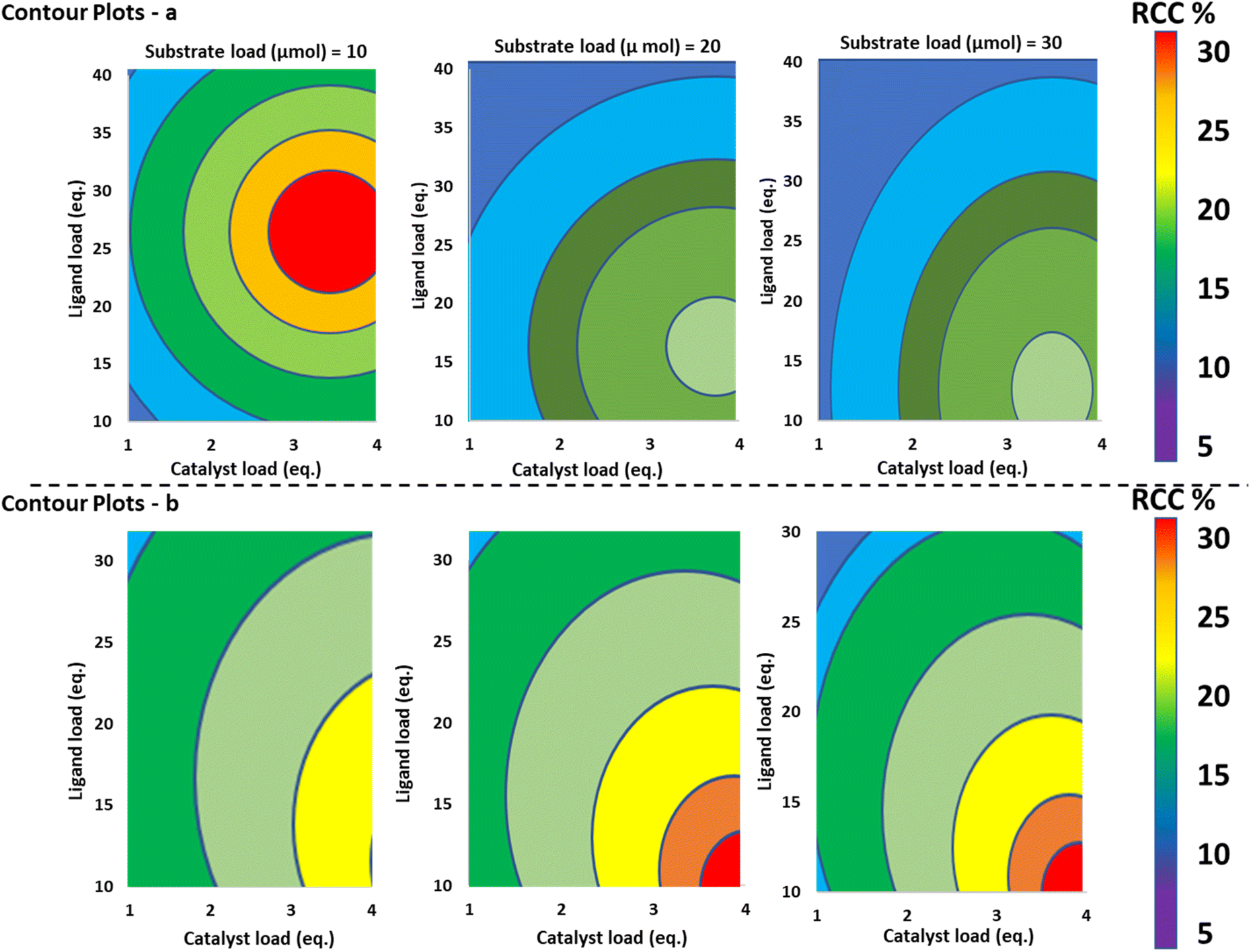 | ||
| Fig. 13 Contour plot summaries of Maurer and coworkers' main findings. Catalyst load, ligand load and substrate load were investigated for both the FFD and the CCD investigations. Red indicates a high level of the response variable, and blue represents a low level. This figure has been recreated and modified from Sci. Rep., 2019, 9, 11370 – copyright Nature [ref. 175]. Licensed under a Creative Commons CC BY licence. | ||
Following reaction data collection, the response dataset was fitted using multiple linear regressions (MLR) and checked for outliers and model quality to obtain a normal distribution of data. Factor significance was estimated by the normal coefficient of the corresponding model term. Only Cu(OTf)2 and pyridine loading were found to be significant, and model curvature was observed. It was determined that a Response Surface Optimisation (RSO) would be required to determine which quadratic terms would be required to improve model fit.
Analysis of the model obtained from this factor screening DOE suggested that, when using stoichiometric quantities of copper(II) triflate, the choice of atmosphere was insignificant. It was experimentally confirmed that when catalytic quantities of copper(II) triflate were used, oxidative atmospheres are required to promote complex oxidation to Cu(III) and to regenerate the catalyst. When stoichiometric quantities of copper(II) triflate are used, the reaction can be performed under argon as the oxidation of the inactive Cu(II) complex to the active Cu(III) complex is mediated by non-ligated Cu(II) through a single electron transfer.
Having identified important factors in the DOE screening (Fig. 13), the authors proceeded to construct a more detailed orthogonal central composite design RSO study to optimise this radiosynthesis. Cu(OTf)2 loading (1–4 eq.), pyridine loading (10–40 eq.), and substrate loading (10–30 μmol) were chosen as the input space to this model based on factor screen (with solvent volume, reaction temperature, atmosphere, and random variables having been discarded as unimportant).
All three main factors were found to be significant, and the experiment also resolved quadratic behaviours for both catalyst and pyridine loading factors. A factor interaction between pyridine and substrate loading was also resolved and included in the model. Strong quadratic behaviours were found for both copper(II) triflate and pyridine loading, and a strong negative factor interaction was detected between the equivalents of pyridine and the amount of substrate used. Fig. 13 shows the response surface across the investigated ranges, providing an optimal set of conditions: 3.5 equivalents of catalyst, 25 equivalents of pyridine, and 10 μmol substrate. Three validation runs were executed under the optimised conditions, affording high yields and reproducible results.
The findings of this investigation were applied to the synthesis of [18F]pFBnOH [18F]6, an important radiochemical building block. A RSO was constructed to optimise its synthesis using a Box Behnken Design (BBD), with substrate loading, catalyst loading, and pyridine loading taken as the input space. Catalyst loading and pyridine loading were found to be significant factors and pyridine behaved quadratically in the model, while precursor loading was found to be insignificant. The RSO showed the greatest response at high catalyst and low pyridine loadings and these findings were verified in additional validation experiments which afforded [18F]6 with a %RCC of 58 ± 5.3% in a single step – representing a significant improvement on previously published attempts at this synthesis.
This investigation represents an excellent application of the tools of DOE methodology to a problem of significant chemical interest. The combination of an initial factor screening in combination with several iterations of various DOE designs allowed rapid convergence on significant mechanistic understanding and synthetic improvements in an efficient manner.176
3.3 Data dimensionality reduction and its application in catalytic processes
Data dimensionality reduction is a technique used to reduce the number of variables in a dataset to those most responsible for observed variation in a chosen output variable. This process makes large datasets easier to analyse and interpret. In the context of catalytic processes, data dimensionality reduction techniques such as Principal Component Analysis (PCA) are commonly used to identify important variables and simplify the data. Projection techniques like PCA involve creating Principal Component axes which are linear combinations of several mechanistically relevant descriptors in a way which maximises the total variation which can be described using the minimum number of mutually orthogonal PC axes. In this way, the physicochemical factors which are responsible for variation in an output variable (e.g., selectivity, purity, and yield) can be readily identified. Fig. 14 summarises the process of PCA for the iris native-R dataset. | ||
| Fig. 14 Visual representation of the process of data dimensionality reduction on the native R dataset, iris. The plot shown is generated via the process of Principal Components Analysis. (i) Data standardisation. (ii) Calculation of covariance matrix to obtain eigenvectors descriptive of the data. (iii) Eigenvector sorting to allow choice of k eigenvectors corresponding to largest k eigenvalues (where k is the number of dimensions of the new subspace). (iv) Construction of projection matrix W from selected k eigenvectors. (v) Transformation of original dataset via W to obtain k-dimensional feature subspace. The arrow represents loading extraction from principal components to generate mathematical models of subspace with mechanistically relevant descriptors. | ||
Durand and Fey have recently described how databases of DFT-calculated steric and electronic descriptors can be built for organometallic catalysts and have demonstrated how these can be used in the mapping, interpretation, and predication of catalyst properties and reactivities.76 Structural and computational chemistry is often employed to elucidate the coordination and reactivity of organometallic catalysts. The Ligand Knowledge Base (LKB) examines novel ligands and provides relevant mechanistic insights in this space. Fey and coworkers have published extensively on the application of such databases to the analysis of ligand effects and development of predictive statistical models.69–76,179–181 A key challenge highlighted in this work lies in the visualisation of chemical space defined by a multivariate database. PCA was the main statistical projection approach employed in this study to help determine key descriptors impacting the prediction of ligand and catalyst effects in organometallic catalysis. Statistical robustness is highly variable in projection techniques such as PCA. Small changes in the input dataset typically change the generated model. Fey and coworkers assumed a random and representative sample in their LKB, which may be difficult to justify. Where outliers are observed, it is difficult to know whether they reflect meaningful chemical information or not. Despite the challenges associated with interpretation of such outliers the LKB has been shown to have significant predictive power.
The ligand knowledge base for phosphines (LKB-P)182 explored descriptors from representative complex/ligand adducts and used free ligand optimisation calculations to produce a variety of mechanistically relevant descriptors. Following geometry optimisations via computational approaches (vide infra), twenty-eight descriptors were defined which were ligand responsive and computationally inexpensive. These descriptors were also shown to be highly correlated, making PCA useful for visualisation of the datasets. Although the orientation and compositions of the PCs are variable with input, chemically-intuitive clustering of ligands is observed and sustained when the map is expanded to include new ligands. The maps show relationships between ligand structure and catalyst properties, allowing researchers to choose ligands as appropriate to their catalytic application. The upper panel of Fig. 15 shows the principal component score plot for ligands in LKB-P. The lower panel shows the PCA analysis of the expanded LKB for chelating bisphosphines.
 | ||
| Fig. 15 Results of PCA analysis of the initial LKB (upper panel), and the expanded LKB containing chelating bisphosphines.182 The legends of each plot indicate the general substituent of each phosphine. PC axes were oriented to capture the most variation possible in each dataset. This figure has been recreated and modified from Fey and coworkers. | ||
The LKB has been successfully applied to problems of catalytic interest, including to hydroformylation of 1-heptene (for which the ubiquitous PPh3 was identified as the best ligand) and hydrocyanation of 3-pentenenitrile (for which P(O-o-Tol)3 was identified as optimal). It has therefore been demonstrated that this map can suggest regions of ligand space which are active for a given catalytic application.
3.4 Summary
The use of RDA, statistics, and data dimensionality reduction is critical in investigating the mechanism of catalytic processes for organometallic compounds. By analysing data from multiple sources and reducing the dimensionality of the data, researchers can gain a deeper understanding of reaction mechanism and improve the efficiency and efficacy of catalytic processes. We suffer significantly from current limitations of analytical technologies, which are often incompatible with systems of significant chemical interest (e.g. systems which are heterogeneous, air-sensitive, and moisture-sensitive). It is imperative, therefore, that the field continues to concentrate intellectual resource in the development and improvement of accessible in situ analytical techniques to facilitate time-resolved observation of fundamental mechanistic processes.DOE approaches have been demonstrated in a variety of academic and industrial settings to significantly accelerate reaction optimisation. This technique of experimental design is particularly powerful when used in conjunction with high-throughput experimentation approaches. The possibility of parsing larger volumes of chemical space in reduced timeframes not only significantly accelerates convergence on local minima in a thermodynamic landscape, but also offers the possibility, when used in an unbiased manner, of converging on true global minima outside of previously defined input spaces. It is advisable, therefore, to cast the net wide during optimisation activities involving DOE approaches.
Data dimensionality reduction is absolutely crucial in the majority of complex chemical applications, which have large and multidimensional input space and often produce a high volume of rich-data. Tools such as PCA offer the opportunity to make sense of complex datasets. This is usually an essential pre-processing component prior to the implementation of ML methods and allows suitable attenuation of noise from variables of limited predictive and interpolative value. The statistical tools discussed in this section are at their most powerful when they are used in conjunction with one another, leading to the commonly implemented “closed-loop” model of automated iterative self-optimisation routines.
4 Parameterisation in mechanistic studies
4.1 Parameter definition and selection
Parameterisation of chemical species provides quantitative numeric descriptors for use in data analysis. This parameterisation is required due to RDA techniques requiring quantitative input data. The parameters discussed in this section serve as methods to represent chemical structure and characteristics as discrete quantities, for use in these analytical methods.There are, of course, a range of techniques to describe chemical species, and they are too numerous to be discussed here. This section focuses on the generation of parameters to describe phosphine ligands used in common metal-mediated reactions. Phosphine ligands are ubiquitous across many areas of chemistry, so a set of parameters describing them is broadly applicable. Furthermore, ligand variation scopes can give valuable mechanistic insight, and parameterisation of the ligands allows for more sophisticated RDA-type approaches to mechanistic investigations.
Selection of suitable parameters for the system in question is key when conducting a parameter-based study. Poorly chosen parameters may have no meaningful correlation with observed experimental results. At best, such parameters provide no benefit to the analysis and at worst, they actively obfuscate important relationships.
Historically, the first methods of parameterisation were gathered from direct relationships of chemical substructure to reaction outcome. Perhaps the most famous of these are the Hammett parameters.183–185 In these landmark works, Hammett explored the effects of the variation of functional groups on the rate of reactions, and equilibrium constants, of various benzoic acid derivatives. These comparisons led to a parameter being assigned to each substituent, expressing a parameter (θ) as the logarithm of the ratio between the equilibrium constants of substituted and non-substituted benzoic acids. The parameter quantifies the effect on rate for each substituent, and therefore the activating or deactivating character of the substituent for the reaction in question. This Hammett parameter (σ) acts as an electronic descriptor for each substituent.
While this style of descriptor is more often considered in relation to physical organic chemistry, as opposed to the mechanistic organometallic chemistry covered in this review, the Hammett parameter has been used to great effect within the latter field. Perhaps the highest profile of these examples is Jacobsen's work in identifying the causes of enantioselectivity in the epoxidation of alkenes using MnIII(Salen) catalysts. Jacobsen and co-workers were able to correlate the logarithm of the enantiomeric ratio with the Hammett σpara parameter of substituents on the Salen ligand backbone, thereby confirming that substituent electronic effects were highly influential in determining product stereochemistry.186 This approach is a good example of how ligand functionalisation can be used to build sub-parameters.
Importantly, Hammett's parameters were derived purely on a group-by-group basis from measured reaction outcomes. Thus, any new functional group of interest would have to be experimentally verified relative to the unsubstituted benzoic acid to generate the σ parameter for the group. This approach, while providing experimentally verified parameter results, is also not as efficient as the more modern methods of deriving parameters from crystallographic or calculated structures.
The Tolman cone angle is a method of describing the steric bulk of a phosphine ligand using the angle (θ) at the tip of a cone drawn from the metal centre which encompasses the entire phosphine group (Fig. 16A).187,188 This has the advantage of describing the steric effect from the perspective of the metal centre, where steric effects are most likely to affect mechanistic processes. Less advantageously, this one-dimensional parameter does not account well for ligands which deviate from the standard PR3 model. For instance, bidentate phosphines cannot be easily described using a standard Tolman cone angle. Furthermore, there has been discussion into how certain assumptions built into Tolman's model have led to inaccuracies in its handling of even relatively structurally simple phosphines.189
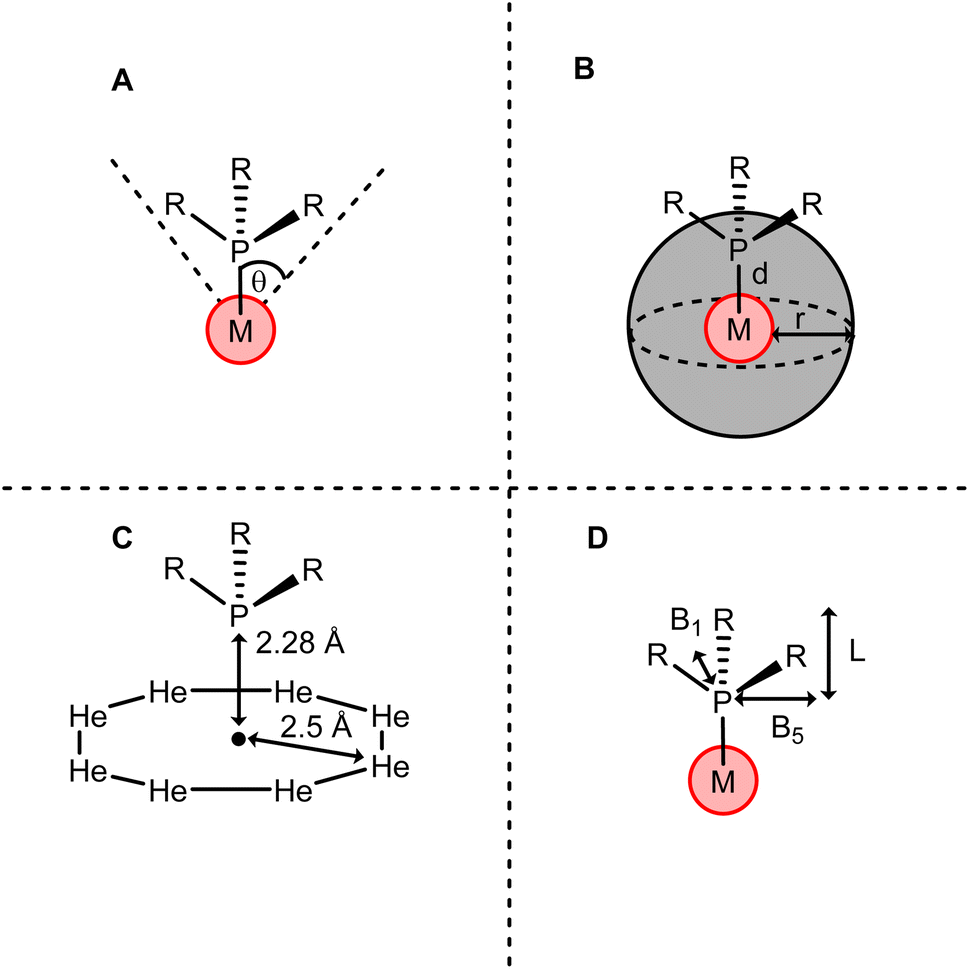 | ||
| Fig. 16 Representation of steric calculation methods for a phosphine ligand PR3 by (A) Tolman cone angle, (B) percentage buried volume, (C) He8 steric and (D) sterimol parameters. | ||
More recently, ligands have been described sterically using percentage buried volume (%Vbur).190 This parameter is defined as the proportion of the volume of a sphere of a set radius occupied by the ligand, with the centre of the sphere located on the metal centre (Fig. 16B). While initially developed for N-heterocyclic carbenes (NHCs), this method can also be applied to phosphines.191,192 The key advantage of this approach is that more structurally complex phosphines are able to be described. Clavier and Nolan use this technique to provide the first steric parameterisation of the Buchwald-type biaryldialkyl phosphines.193 Wu and Doyle191 showed that the best model to describe reactivity for a range of phosphine ligands of varying complexity is a mixed-parameter approach combining θ and %Vbur in a multicomponent linear regression model. θ is competent to describe ligand size far from the metal centre. Conversely, %Vbur is more heavily weighted towards steric bulk close to the metal centre, and so does not always take ligand size far away from the metal into account. The authors note that this leads to %Vbur not representing ligands which display remote steric hindrance, an effect which can have important impacts on mechanism. By using a mixed model, Wu and Doyle were able to combine the strengths of both parameters, providing a rounded picture of the ligands' steric properties.
Fey and co-workers proposed a new steric parameter, He8, which calculates the energy of the phosphine a set distance away from a ring of eight helium atoms constrained on a plane (Fig. 16C). They described that this approach minimises the contribution from the van der Waals effects of the phosphorus atom and focuses on contributions from the substituents. This approach can be adapted to create conformations of helium atoms tailored to the bulk shapes of certain substituents. For instance, Fey later described the steric characteristics of bidentate ligands using a wedge of helium atoms, rather than a ring.194
When considering ligands which deviate strongly from the standard symmetric PR3 model, accounting for structural anisotropy is desirable. To this end, the Sterimol parameters were developed. These parameters describe the steric character of a ligand in the form of multiple dimensional parameters, describing the ligand separately by length L, minimum width B1 and maximum width B5 (Fig. 16D). These multidimensional parameter sets have been used to analyse asymmetric mechanisms, providing insight that other mono-dimensional parameters could not.195–197
These metrics are sensitive to the conformation of the input structure used to calculate them. It is not always guaranteed that the conformation of the ligand obtained from crystallographic or computational geometries is the same as during the reaction pathway. More recently, work has been undertaken by Paton and co-workers to obtain a range of Sterimol values across conformer space for a given ligand.198
In addition to steric information, the electronic parameters of ligands in electronic systems play a pivotal role in determining activity and mechanism. The earliest methods of quantifying electronic effects of ligands in inorganic systems were derived from spectroscopic data collected from model complexes. The Tolman Electronic Parameter (TEP)187 uses the energy of the A1 stretching mode of Ni(CO)3L in dichloromethane as a method of determining the electronic contribution of the ligand to the complex (Fig. 17). Similarly to Hammett parameters, this approach requires the collection of physical spectroscopic data for each ligand, making the screening of large ligand libraries inefficient. More recently, there has been discussion on the utility of the TEP, and its ability to adequately describe metal–ligand bonding strength. Work has been undertaken to compare the donation strengths and properties of a wide range of 2-electron donor ligands by Gusev, where the standard Ni–CO frequencies of the original TEP are compared with other ligand properties, including iridium cyclopentadienyl carbonyl analogues.199 Gusev demonstrated that DFT-derived Ni–CO stretching frequencies correlate extremely well with experimentally derived values, allowing for accurate and precise parameter determination of new phosphine examples using purely theoretical methods. More importantly, however, Gusev compared the observed CO stretching frequencies of a range of ligand types (including NHCs, trialkyl phosphines and water) bound to iridium and osmium metal complex environments. It was shown that while the TEP is a descriptive parameter for phosphine ligands, it is not as adept at comparing ligands across classes, i.e., comparing many phosphines and NHC ligands.
 | ||
| Fig. 17 The A1 symmetric CO stretching mode of Ni(CO)3L complexes, used to determine the Tolman Electronic Parameter (TEP). | ||
This highlights an important consideration when selecting appropriate parameters. While a parameter may be reliable within a certain region of chemical space, it may not provide good comparisons between diverse examples. In this case, due to the complexities of metal–ligand bonding, the TEP can only provide a full description of the metal electronic environment in a small subset of cases. This point is explored in depth by Cremer and co-workers, who suggest a computationally-derived parameter, the metal–ligand electronic parameter (MLEP), to overcome these issues and provide a metric for metal–ligand interaction strength across a broad region of chemical space.200
With modern access to a high level of theoretical quantum chemistry techniques, the calculation of electronic parameters from crystallographically-derived or computationally optimised structures is more achievable than ever. This enables the calculation of parameters for large databases of ligands, which can be subsequently employed in optimisation routines and downstream data analysis.
Fey and co-workers demonstrated the power of this approach with their LKB (vide supra).69–76,179–181,201,202 The aim of the work was to map ligand space, providing a set of descriptors for chemically relevant phosphine ligands which can provide parametric data for use in data analysis. Importantly, the use of solely theoretically-derived parameters allows for a far wider range of ligands than can be realistically obtained in traditional physical studies.
This study emphasised the scalability of parameterisation, with chosen parameters able to be synthesised at relatively low computational costs. Results of calculations which require greater computational resources, such as frequency analysis, were not used as parameters for this reason.
The frontier molecular orbitals (HOMO and LUMO) of the unbound phosphine species can be calculated, giving an intrinsic electronic parameter. Frontier molecular orbitals provide insight into the reactivity of species. A high-energy HOMO increases the electron-donating power of a species. Similarly, a low-lying LUMO is better able to accept electrons. By quantifying these abilities, the electronic contributions of the coordinated ligand can be modelled. Frontier molecular orbital analysis has been used as a tool to investigate the reactive behaviour of several catalytically-relevant ligands.203,204
By calculating and comparing the relative energies of the protonated and deprotonated ligand, ([LH]+ and [L] respectively), the proton affinity of the ligand can be established. These proton affinities have been used to describe the binding behaviour of ligands to metal centres.205,206 In a related parameter, the lone pair energy of the phosphine has also been used as a metric of electronic donation strength from the ligand to the metal centre.207
The electronic character of a molecule can be expressed by considering the charge localised on each atom. These charges are calculated by distributing the electron density of a molecule into discrete atomic contributions.208 Calculation of point charges of atoms can be achieved through several methods. Mulliken charges are the earliest example of such a calculation.209 This method of population analysis is often computed as standard in computational packages such as Gaussian, but Mulliken's method can often return nonsensical orbital occupations that are greater than two, or less than zero.210,211 This, coupled with the sensitivity of the resulting calculated charges to basis set and functional selection212,213 has led to other metrics of charge assignment being developed. The natural population analysis (NPA) method, developed as an alternative to Mulliken charges, seeks to address some of the issues found with the method.214,215 By introducing weighting that reduces the contribution of low-occupancy orbitals (which are more sensitive to basis set variation), the resulting analysis is more reliable and standardised across methods. Similarly, and at around the same time, the natural hybrid orbitals approach was developed.216 By making use of orbital hybridisation theory, this approach can return more chemically relevant results. Over time, this methodology was refined into the natural bond orbital (NBO) analysis program.217,218 The NBO analysis suite is able to provide several useful electronic metrics, including atom charges, bond indexes and orbital energies.219 These have been used as descriptors for statistical modelling in different fields.220–224
Finally, empirical parameterisation techniques have enjoyed considerable success when applied to solvent optimisation activities in organometallic catalytic contexts.225,226 These approaches typically involve empirical measurements of fundamental properties of the solvent related to polarity, e.g. the solvatochromic properties associated with Kamlet–Abboud–Taft parameterisation: π*, a measure of solvent polarisability; α, a measure of solvent Hydrogen Bond Donors (HBD) acidities; and β, a measure of Hydrogen Bond Acceptors (HBA) basicities.227 These parameters can then be used to either map out chemical space of solvents to identify greener alternatives,228 or applied as descriptors in ML studies.229
4.2 Machine learning in mechanistic understanding
More recently, advances in ML techniques and their increased ease of use and accessibility have led to their increased employment in many new areas. The most recognised challenge to the implementation of ML in chemistry is that to develop a system capable of producing meaningful and insightful output, a large amount of high-quality and broad-scope training data is required. As chemical experimental data is, relatively speaking, slow to collect and often discontinuous in procedure across different research groups and environments, it can be challenging to construct such a training set for synthetic chemistry. Similarly, ML algorithms in chemistry can be highly chemical-space dependent; an algorithm may experience a noted loss in accuracy when attempting to predict outcomes based on inputs dissimilar to its training dataset.230Furthermore, while a well-trained ML model can possess powerful predictive capabilities, it will not necessarily provide insight as to why these predictions are being made. When attempting to elucidate the mechanisms of chemical processes, this can be a problematic tendency, as without this information it can be difficult to draw mechanistic conclusions from the model output.
A full treatment of the current state of the art of ML in chemistry at large is beyond the scope of this review and has been covered recently in other publications.231,232 Instead this section will focus on recent implementation of ML techniques to applications in organometallic chemistry.
There have been several notable advances in the application of ML techniques to organometallic catalysis. For example, Cronin and coworkers developed a ML approach to predict reactivity, and then applied it to the Suzuki–Miyaura cross-coupling reaction.233 By considering the findings of Perera and co-workers’ high-throughput screening,10 it was shown that a model trained on just 10% of the available reaction data was able to accurately predict reactivity for the remaining 90%.
Hartwig and co-workers reported a method for the prediction of site selectivity of borylation reactions.234 The method employed here is described as a “hybrid ML approach”, where a combination of linear and nonlinear regression models are used alongside ML to build a robust predictive system. This approach uses new tools to provide in-depth parameters and statistics for unseen examples. By calculating semi-empirical quantum mechanical transition state energy approximations, then refining the approximated energies using linear regression, a site-selectivity prediction can be made. Importantly, the Rogers–Tanimoto similarity ratings were used to compare unseen inputs to the training set, and dynamically mix the regression function corrections in an attempt to provide the best combined model possible for each supplied structure. When an input structure was similar to the training set, a primarily ML based model could be applied, with little linear regression correction. When a dissimilar input structure is supplied, which lies outside of the chemical space envelope of the training dataset, linear regression is emphasised in the predictive method instead. It was noted that this approach moves to address the chemical space extrapolation issues commonly found when applying ML approaches to chemical problems.
From a mechanistic perspective, there have been examples of ML capabilities applied to existing mechanistic understanding to enhance the gathering of insights. Bures and Larossa described a new system for handling measured kinetic data for reactions.49 They remark that although the technological capabilities for measuring kinetics have advanced quickly, the analysis techniques for handling these measured data have not been able to keep pace at quite the same rate. This work details a method of abstracting the handling of kinetic information away from the system of rate law derivations and orders in reagents, and instead embracing a machine-learning based classification approach. By classifying a range of organic mechanisms and generating sample data for training, the authors built a deep learning model which was able to autonomously classify new kinetic data into one of the predetermined mechanisms. A series of case studies for various reactions showed that these mechanistic assignments were reliable when compared to previous mechanistic investigations. Importantly, the methods by which the ML process determines the mechanism is decoupled from traditional kinetic modelling approaches, instead using a holistic and integrated approach which connects directly from input data to output mechanism.
It has been convincingly demonstrated that ML representations are applicable to property predictions of transition metal complexes. Corminboeuf and von Lilienfeld have reported significant acceleration of descriptor screening of 18602 homogeneous catalysts – a large library based on Pt, Pd, Ni, Cu, Ag, and Au, combined with 91 ligands – for SMCC reactions.235 This screening was conducted on the basis of identification of thermodynamically plausible systems via analysis of molecular volcano plots – graphs of significance vs. fold-change – with respect to computationally inexpensive energetic descriptors. The representations examined in this study were a variant of the sorted Coulomb Matrix,236 the Bag of Bonds,237 and the Spectrum of London and Axilrod–Teller–Muto potential.238 These representations were employed to generate a description of the oxidative step directly from the SMILES structure (without the need to provide accurate molecular geometry as model input). Following conversion from SMILES to coordinates, the authors mapped their input representation to a corresponding continuous label value using kernel ridge regression (which uses the kernel trick to compress multidimensional data into a linear feature space). The quality of generated models was evaluated by separating the data into training and test sets and calculating the mean absolute error of prediction on the out-of-sample test set (with appropriately randomised cross-validation) The input space was narrowed to a set of 37 interesting and low-cost (<10 USD mol−1) complexes featuring palladium and copper with a variety of ligands. This work represents an exciting application of ML representations and molecular volcano plots to the discovery of novel chemical reactivity in a computationally-inexpensive manner.
Molecular volcano plots have enjoyed additional application in the assessment and prediction of catalytic efficiency in organometallic systems. Turnover frequency (TOF) and turnover number (TON) are commonly reported as indicators of catalytic efficiency. Most computational studies of catalytic free-energy landscapes only indicate the relative stabilisation of intermediates, and associated barriers to transition states. Corminboeuf and coworkers239 have demonstrated the dual application of linear scaling relationships with the energy span model – a theoretical model connecting steady-state kinetics240 with Eyring transition state theory241 – to create volcano plots. These plots have been shown to rapidly correlate the free energy associated with a given step in a given catalytic cycle with a computationally-inexpensive descriptor variable. This information, when fed into the AUTOF software package developed by Uhe, Kozuch, and Shaik242–244 could be used to estimate the TOF of a given catalyst. Plotting TOF values over a range of descriptors led to the generation of a TOF volcano plot, which could be used to rapidly screen catalysts on the basis of predicted activity. The authors exemplified their developed workflow by rapid identification of novel metal/pincer-ligand catalytic systems for CO2 hydrogenation to formate. Several systems – Os(PONOP/–CO), Co(PONOP/–Cl), Ir(NNN–Cl), and Rh(PONOP/–Cl) – were identified as theoretically competent of altering the energetics of the catalytic cycle via the equatorial Cl– or CO– ligands and through the connecting atoms of the pincer ligand. In turn, these systems were predicted to be highly active in CO2 reductions. Jung and coworkers have additionally demonstrated the application of neural network and kernel ridge regression methods to the prediction of CO adsorption energies on the surface of CO2 reduction alloy catalysts.245 By application of an active learning approach, the authors were able to produce a model with a mean error of 0.05 eV and identified Cu3Y@Cu* and Cu3Sc@Cu* as highly active and low-cost electrochemical CO2 reduction catalysts (which were competent to produce CO with an overpotential approximately 1 V lower than a typical Au catalyst).
4.3 Summary
Selection of suitable parameters is essential to building models which can provide realistic descriptions of chemical systems. The parameters must be chemically relevant to the system. There are additional concerns which should also be considered when selecting parameters. Sigman and co-workers discuss these concerns in more detail in building multivariate linear regression models.246Some parameters, for a given set of ligands, may display small differences across the set. When this range of values is very small, the parameter will not be greatly affected by variation of the ligand. Furthermore, if this parameter is used in a scaled model, it will be much more sensitive to random noise arising from calculation methods. The amplification of this noise can lead to the model quality being negatively impacted. Generally, if a parameter has a proportionally small range of values across the sample population, it should be excluded from the model.
It may be attractive to fit as many parameters as possible to the data, to provide the model with as much information as possible. This can lead to overfitting, which can cause the model to become specialised to the random noise in the data, leading to good predictions for the training set, but poor transferability to unseen examples.247 While this can be overcome with more sophisticated validation techniques,248 the problem is best avoided by selecting only parameters which are necessary to build a complete and descriptive model.
Another argument against the selection of too many parameters is the effect of inter-correlated parameters on model outputs. The regression coefficients of models supplied correlated parameters can be skewed by the correlation between the factors.249 To avoid this issue, it is advisable to perform cross-correlation analysis on the proposed descriptor set prior to model building. Any parameters which are found by this effect to be strongly correlated should be re-evaluated. The parameters may be able to be condensed into a combined parameter, or one might be discarded in favour of the other.
5 Conclusions
The use of rich data analysis, statistical analysis, and data dimensionality reduction is critical in investigating the mechanisms of catalytic processes for organometallic compounds. By analysing data from multiple sources and reducing the dimensionality of the data, researchers can gain a deeper understanding of the reaction mechanisms and improve the efficiency and efficacy of catalytic processes. We suffer significantly from the current limitations of common analytical techniques, which are often incompatible with systems of significant chemical interest (e.g. systems which are heterogeneous, air-sensitive and/or moisture-sensitive). It is imperative, therefore, that the field continues to concentrate intellectual resources on the development and improvement of accessible in situ analytical techniques to facilitate time-resolved observation of fundamental mechanistic processes.DOE approaches have been demonstrated in both academic and industrial settings to significantly accelerate reaction optimisation. This technique of experimental design is particularly powerful when utilised in conjunction with HTE approaches. The possibility of parsing larger volumes of chemical space in reduced timeframes not only significantly accelerates convergence on local minima in a thermodynamic landscape, but also offers the possibility, when used in an unbiased manner, of converging on true global minima outside of previously defined input spaces. It is advisable, therefore, to cast the net wide during reaction optimisation activities involving DOE approaches.
Data dimensionality reduction is broadly helpful in the majority of complex chemical applications with large and multidimensional input space, often producing a high volume of rich data. Tools such as PCA offer the opportunity to make sense of seemingly incomprehensible datasets.250 Parameterisation offers quantitative ways to describe chemical variation to form input for these statistical tools. This is a critical part of data workflows prior to the implementation of ML methods and allows distinction of noise from variables of predictive and interpolative value.
The evolving interdisciplinarity of the chemistry community means that it is getting easier for mechanistic organometallic researchers to take full advantage of statistical methods. Perhaps of greater challenge, however, is the need to convince the chemistry community at large of the applicability, versatility, and significance of automated systems, routines, and associated data science tools to organometallic chemistry. We believe that, by providing additional demonstration of their application in the solution of problems of significant chemical interest, researchers employing these techniques are both laying strong foundations for future studies and helping to inspire confidence in the field at large.
Abbreviations
| ADE-OPI | Acoustic droplet ejection-open port interface |
| AMI | Acoustic mist ionization mass spectrometry |
| BBD | Box–Behnken design |
| CCD | Central composite design |
| ROAR | Centre for rapid analysis of online reactions |
| DFT | Density functional theory |
| DOE | Design of experiments |
| DESI | Desorption electrospray ionization mass spectrometry |
| DLS | Dynamic light scattering |
| DReaM | Dynamic reaction monitoring facility |
| FFD | Fractional factorial design |
| GDP | Gross domestic product |
| HOMO | Highest occupied molecular orbital |
| HPLC | High-performance liquid chromatography |
| HTE | High-throughput experimentation |
| HTS | High-throughput screening |
| IR | Infrared spectroscopy |
| LKB | Ligand knowledge base |
| LC-MS | Liquid chromatography mass spectrometry |
| LUMO | Lowest unoccupied molecular orbital |
| ML | Machine learning |
| MALDI | Matrix-assisted laser desorption ionization |
| MLR | Multiple linear regressions |
| NPs | Nanoparticles |
| NBO | Natural bond orbital analysis |
| NPA | Natural population analysis |
| NMR | Nuclear magnetic resonance spectroscopy |
| OVAT | One-variable-at-a-time |
| %Vbur | Percentage buried volume |
| PTFE | Poly(tetrafluoroethylene) |
| PET | Positron emission tomography |
| PCA | Principal component analysis |
| QSAR | Quantitative structure–activity relationship |
| QM | Quantum mechanical |
| %RCC | Radiochemical conversion |
| radioTLC | Radiolabeled thin layer chromatography |
| RSO | Response surface optimisation |
| RDA | Rich data analysis |
| SVD | Singular value decomposition |
| SAXS | Small-angle X-ray scattering |
| SMCC | Suzuki–Miyaura cross-coupling |
| TEM | Transmission electron microscopy |
| UPLC-MS | Ultra high-performance liquid chromatography mass spectrometry |
| UV-Vis | Ultra-violet visible spectroscopy |
Data availability
All the material included within this review on the above topic is cited in the primary literature. Any data used was supplied in Supporting Information documents (for primary publications) and any associated raw data shared through data repositories. In some cases, the published data has been re-processed so that an equivalent or related figure can be used in the review article.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Figures and Schemes requiring the reprocessing of published data were reproduced by C. S. H.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to the EPSRC for funding (C. S. H. and I. J. S. F.; EP/S009965/1 and EP/W031914/1) and to the Royal Society for an Industry Fellowship (I. J. S. F.). AstraZeneca and GSK have provided PhD studentship co-funding for S. C. S. and J. J. W. respectively. We thank Prof. Jason Lynam from the Department of Chemistry in York and Dr Neil Scott for their comments and valuable input into this review article. We also thank Dr George Clarke for his input into the potential material that could be included in this review paper. We are grateful to the EPSRC, Royal Society and industry funders (AstraZeneca and GSK) for supporting our research efforts in this area.References
- W. Gao, P. Raghavan and C. Coley, Nat. Commun., 2022, 13, 1075 CrossRef CAS PubMed.
- B. Burger, P. Maffettone, V. Gusev, C. Aitchson, Y. Bai, X. Wang, X. Li, B. M. Alston, R. Clowes, N. Rankin, B. Harris, R. S. Spick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
- T. Doi, S. Fuse, S. Miyamoto, K. Nakai, D. Sasuga and T. Takahashi, Chem.–Asian J., 2006, 1(3), 370–383 CrossRef CAS PubMed.
- T. Cernak, N. J. Gesmundo, K. Dykstra, Y. Yu, Z. Wu, Z. Shi, P. Vachal, D. Sperbeck, S. He, B. A. Murphy, L. Sonatore, S. Williams, M. Madeira, A. Verras, M. Reiter, C. H. Lee, J. Cuff, E. C. Sherer, J. Kuethe, S. Goble, N. Perrotto, S. Pinto, D.-M. Shen, R. Nargund, J. Balkovec, R. J. DeVita and S. D. Dreher, J. Med. Chem., 2017, 60(9), 3594–3605 CrossRef CAS PubMed.
- T. Ha, D. Lee, Y. Kwon, M. S. Park, S. Lee, J. Jang, B. Choi, H. Jeon, J. Kim, H. Choi, H.-T. Seo, W. Choi, W. Hong, Y. J. Park, J. Jang, J. Cho, B. Kim, H. Kwon, G. Kim, W. S. Oh, J. W. Kim, J. Choi, M. Min, A. Jeon, Y. Jung, E. Kim, H. Lee and Y.-S. Choi, Sci. Adv., 2023, 9, 44 Search PubMed.
- M. Christensen, L. P. E. Yunker, P. Shiri, T. Zepel, P. L. Prieto, S. Grunert, F. Bork and J. E. Hein, Chem. Sci., 2021, 12, 15473–15490 RSC.
- K. Jablonka, L. Patina and B. Smit, Nat. Chem., 2022, 14, 365–376 CrossRef CAS PubMed.
- A. Cook, R. Clément and S. Newman, Nat. Protoc., 2021, 16, 1152–1169 CrossRef CAS PubMed.
- L. Malet-Sanz and F. Susanne, J. Med. Chem., 2012, 55, 4062–4098 CrossRef CAS PubMed.
- D. Perera, J. W. Tucker, S. Brahmbhatt, C. J. Helal, A. Chong, W. Farrell, P. Richardson and N. W. Sach, Science, 2018, 359, 429–434 CrossRef CAS PubMed.
- M. Alonso, S. Cañ, F. Delgado, M. Serrano, A. Diéguez-Vázquez and J. E. Gómez, Org. Lett., 2023, 25, 771–776 CrossRef CAS PubMed.
- E. S. Isbrandt, R. J. Sullivan and S. G. Newman, Angew. Chem., Int. Ed., 2019, 58, 7180–7191 CrossRef CAS PubMed.
- F. S. Emami, A. Vahid, E. K. Wylie, S. Szymkuć, P. Dittwald, K. Molga and B. A. Grzybowski, Angew. Chem., Int. Ed., 2015, 54, 10797–10801 CrossRef CAS PubMed.
- R. B. Merrifield, J. M. Stewart and N. Jernberg, J. Am. Chem. Soc., 1965, 61, 692 Search PubMed.
- S. N. Denting and H. L. Pardue, Anal. Chem., 1970, 42, 1466–1467 CrossRef.
- H. Winicov, J. Schainbaum, J. Buckley, G. Longino, J. Hill and C. E. Berkoff, Anal. Chim. Acta, 1978, 103, 469–476 CrossRef CAS.
- P. M. Attia, A. Grover, N. Jin, K. A. Severson, T. M. Markov, Y.-H. Liao, M. H. Chen, B. Cheong, N. Perkins, Z. Yang, P. K. Herring, M. Aykol, S. J. Harris, R. D. Braatz, S. Ermon and W. C. Chueh, Nature, 2020, 578, 397–402 CrossRef CAS PubMed.
- A. Furka, Drug Discovery Today, 2002, 7, 1–4 CrossRef PubMed.
- R. Liu, X. Li and K. S. Lam, Curr. Opin. Chem. Biol., 2017, 38, 117–126 CrossRef CAS PubMed.
- I. Casciuc, A. Osypenko, B. Kozibroda, D. Horvath, G. Marcou, F. Bonachera, A. Varnek and J.-M. Lehn, ACS Cent. Sci., 2022, 8(6), 804–813 CrossRef CAS PubMed.
- R. Potyrailo, K. Rajan, K. Stoewe, I. Takeuchi, B. Chisholm and H. Lam, ACS Comb. Sci., 2011, 13(6), 579–633 CrossRef CAS PubMed.
- J. Fordham, P. Kollmus, M. Cavegn, R. Schneider and M. Santagostino, J. Org. Chem., 2022, 87, 4400–4414 CrossRef CAS PubMed.
- R. Steimbach, P. Kollmus and M. Santagostino, J. Org. Chem., 2021, 86, 1528–1539 CrossRef CAS PubMed.
- D. A. Pereira and J. A. Williams, Br. J. Pharmacol., 2007, 152, 53–61 CrossRef CAS PubMed.
- S. M. Mennen, C. Alhambra, C. L. Allen, M. Barberis, S. Berritt, T. A. Brandt, A. D. Campbell, A. H. Cherney, M. Christensen, D. B. Damon, J. E. de Diego, S. García-Cerrada, P. García-Losada, J. Janey, D. C. Leitch, L. Li, F. Liu, P. C. Lobben, D. W. C. MacMillan, J. Magano, E. McInturff, S. Monfette, R. J. Post, D. Schultz, B. J. Sitter, J. M. Stevens, I. I. Strambeanu, J. Twilton, K. Wang and M. A. Zajac, Org. Process Res. Dev., 2019, 23, 1213–1242 CrossRef CAS.
- K. Burgess, H.-J. Lim, A. M. Porte and G. A. Sulikowski, Angew. Chem., Int. Ed., 1996, 35, 119–223 CrossRef.
- S. Vijayakrishnan, J. W. Ward and A. I. Cooper, ACS Catal., 2022, 12, 10057–10064 CrossRef CAS.
- W. Zhao, P. Yan, B. Li, M. Bahri, L. Liu, X. Zhou, R. Clowes, N. D. Browning, Y. Wu, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9902–9909 CrossRef CAS PubMed.
- T. Dimitrov, C. Kreisbeck, J. S. Becker, A. Aspuru-Guzik and S. K. Saikin, ACS Appl. Mater. Interfaces, 2019, 11, 24825–24836 CrossRef CAS PubMed.
- D. J. Blair, S. Chitti, M. Trobe, D. M. Kostyra, H. M. S. Haley, R. L. Hansen, S. G. Ballmer, T. J. Woods, W. Wang, V. Mubayi, M. J. Schmidt, R. W. Pipal, G. F. Morehouse, A. M. E. P. Ray, D. L. Gray, A. L. Gill and M. D. Burke, Nature, 2022, 604, 92–97 CrossRef CAS PubMed.
- E. M. Woerly, J. Roy and M. D. Burke, Nat. Chem., 2014, 6, 484–491 CrossRef CAS PubMed.
- J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. E. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Science, 2015, 347, 1221–1226 CrossRef CAS PubMed.
- J. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297–2307 CrossRef CAS PubMed.
- N. H. Angello, V. Rathore, W. Beker, A. Wołos, E. R. Jira, R. Roszak, T. C. Wu, C. M. Schroeder, A. Aspuru-Guzik, B. A. Grzybowski and M. D. Burke, Science, 2022, 378, 399–405 CrossRef CAS PubMed.
- A. Bubliauskas, D. J. Blair, H. Powell-Davies, P. J. Kitson, M. D. Burke and L. Cronin, Angew. Chem., Int. Ed., 2022, 134, e2021161 CrossRef.
- S. Steiner, J. Wolf, S. Glatzel, A. Andreou, J. M. Granda, G. Keenan, T. Hinkley, G. Aragon-Camarasa, P. J. Kitson, D. Angelone and L. Cronin, Science, 2019, 363, 1–8 CrossRef PubMed.
- V. Sans, L. Porwol, V. Dragone and L. Cronin, Chem. Sci., 2015, 6, 1258–1264 RSC.
- L. Porwol, D. J. Kowalski, A. Henson, D.-L. Long, N. L. Bell and L. Cronin, Angew. Chem., 2020, 132, 11352–11357 CrossRef.
- J. S. Manzano, W. Hou, S. S. Zalesskiy, P. Frei, H. Wang, P. J. Kitson and L. Cronin, Nat. Chem., 2022, 14, 1311–1318 CrossRef CAS PubMed.
- P. S. Gromski, J. M. Granda and L. Cronin, Trends Chem., 2020, 2, 4–12 CrossRef CAS.
- C. J. Richmond, H. N. Miras, A. R. de la Oliva, H. Zang, V. Sans, L. Paramonov, C. Makatsoris, R. Inglis, E. K. Brechin, D.-L. Long and L. Cronin, Nat. Chem., 2012, 4, 1037–1043 CrossRef CAS PubMed.
- P. J. Kitson, G. Marie, J. P. Francoia, S. S. Zalesskiy, R. C. Sigerson, J. S. Mathieson and L. Cronin, Science, 2018, 359, 314–319 CrossRef CAS PubMed.
- D. J. Kowalski, C. M. MacGregor, D.-L. Long, N. L. Bell and L. Cronin, J. Am. Chem. Soc., 2023, 145, 2332–2341 CrossRef CAS PubMed.
- M. K. Jackl, L. Legnani, B. Morandi and J. W. Bode, Org. Lett., 2017, 19, 4696–4699 CrossRef CAS PubMed.
- T. Jiang, S. Bordi, A. E. McMillan, K.-Y. Chen, F. Saito, P. L. Nichols, B. M. Wanner and J. W. Bode, Chem. Sci., 2021, 12, 6977–6982 RSC.
- I. Kreituss and J. W. Bode, Nat. Chem., 2017, 9, 446–452 CrossRef CAS.
- A. E. McMillan, W. W. X. Wu, P. L. Nichols, B. M. Wanner and J. W. Bode, Chem. Sci., 2022, 13, 14292–14299 RSC.
- J. Zakrzewski, P. Yaseneva, C. J. Taylor, M. J. Gaunt and A. A. Lapkin, Org. Process Res. Dev., 2023, 27, 649–658 CrossRef CAS.
- J. Burés and I. Larrosa, Nature, 2023, 613, 689–695 CrossRef PubMed.
- C. A. Hone, N. Holmes, G. R. Akien, R. A. Bourne and F. L. Muller, React. Chem. Eng., 2017, 2, 103–108 RSC.
- Y. Chu, W. Heyndrickx, G. Occhipinti, V. R. Jensen and B. K. Alsberg, J. Am. Chem. Soc., 2012, 134, 8885–8895 CrossRef CAS PubMed.
- Z. Ma and F. Zaera, Encyclopaedia of Inorganic Chemistry, John Wiley & Sons Ltd, 2006 Search PubMed.
- M. Foscato, V. Venkatraman, G. Occhipinti, B. K. Alsberg and V. R. Jensen, J. Chem. Inf. Model., 2014, 54, 1919–1931 CrossRef CAS PubMed.
- M. Foscato, G. Occhipinti, V. Venkatraman, B. K. Alsberg and V. R. Jensen, J. Chem. Inf. Model., 2014, 54, 767–780 CrossRef CAS PubMed.
- T. A. Young, J. J. Silcock, A. J. Sterling and F. Duarte, Angew. Chem., Int. Ed., 2021, 60, 4266–4274 CrossRef CAS PubMed.
- R. D. Baxter and D. G. Blackmond, Tetrahedron, 2013, 69, 5604–5608 CrossRef CAS.
- A. G. O'Brien, O. R. Luca, P. S. Baran and D. G. Blackmond, React. Chem. Eng., 2016, 1, 90–95 RSC.
- B. Wei, J. C. Sharland, P. Lin, S. M. Wilkerson-Hill, F. A. Fullilove, S. McKinnon, D. G. Blackmond and H. M. L. Davies, ACS Catal., 2020, 10, 1161–1170 CrossRef CAS.
- B. Wei, J. C. Sharland, D. G. Blackmond, D. G. Musaev and H. M. L. Davies, ACS Catal., 2022, 12, 13400–13410 CrossRef CAS PubMed.
- J. A. Varela, S. A. Vázquez and E. Martínez-Núñez, Chem. Sci., 2017, 8, 3843–3851 RSC.
- C. A. Hone, A. Boyd, A. O'Kearney-McMullan, R. A. Bourne and F. L. Muller, React. Chem. Eng., 2019, 4, 1565–1570 RSC.
- C. J. Taylor, H. Seki, F. M. Dannheim, M. J. Willis, G. Clemens, B. A. Taylor, T. W. Chamberlain and R. A. Bourne, React. Chem. Eng., 2021, 6, 1404–1411 RSC.
- C. J. Taylor, J. A. Manson, G. Clemens, B. A. Taylor, T. W. Chamberlain and R. A. Bourne, React. Chem. Eng., 2022, 7, 1037–1046 RSC.
- C. J. Taylor, M. Booth, J. A. Manson, M. J. Willis, G. Clemens, B. A. Taylor, T. W. Chamberlain and R. A. Bourne, Chem. Eng. J., 2021, 413, 1385–8947 CrossRef.
- M. Saebi, B. Nan, J. E. Herr, J. Wahlers, Z. Guo, A. M. Zurański, T. Kogej, P.-O. Norrby, A. G. Doyle, N. V. Chawla and O. Wiest, Chem. Sci., 2023, 14, 4997–5005 RSC.
- P. M. Pflüger and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 18860–18865 CrossRef PubMed.
- N. Fey, A. Koumi, A. V. Malkov, J. D. Moseley, B. N. Nguyen, S. N. G. Tyler and C. E. Willans, Dalton Trans., 2020, 49, 8169–8178 RSC.
- N. Fey, J. N. Harvey, G. C. Lloyd-Jones, P. Murray, A. G. Orpen, R. Osborne and M. Purdie, Organometallics, 2008, 27, 1372–1383 CrossRef CAS.
- N. Fey, Dalton Trans., 2010, 39, 296–310 RSC.
- N. Fey, M. F. Haddow, J. N. Harvey, C. L. McMullin and A. G. Orpen, Dalton Trans., 2009, 39, 8183–8196 RSC.
- D. J. Durand and N. Fey, Chem. Rev., 2019, 119, 6561–6594 CrossRef CAS PubMed.
- N. Fey, A. G. Orpen and J. N. Harvey, Coord. Chem. Rev., 2009, 253, 704–722 CrossRef CAS.
- N. Fey, A. C. Tsipis, S. E. Harris, J. N. Harvey, A. G. Orpen and R. A. Mansson, Chem.–Eur. J., 2005, 12, 291–302 CrossRef PubMed.
- C. L. McMullin, B. Rühle, M. Besora, A. G. Orpen, J. N. Harvey and N. Fey, J. Mol. Catal. A: Chem., 2010, 324, 48–55 CrossRef CAS.
- N. Fey, Chem. Cent. J., 2015, 9, 1–10 CrossRef CAS PubMed.
- D. J. Durand and N. Fey, Acc. Chem. Res., 2021, 54, 837–848 CrossRef CAS PubMed.
- C. L. McMullin, N. Fey and J. N. Harvey, Dalton Trans., 2014, 43, 13545–13556 RSC.
- A. Lai, J. Clifton, P. L. Diaconescu and N. Fey, Chem. Commun., 2019, 55, 7021–7024 RSC.
- A. W. McFord, C. P. Butts, N. Fey and R. W. Alder, J. Am. Chem. Soc., 2021, 143, 13573–13578 CrossRef CAS PubMed.
- J. Jover and N. Fey, Chem.–Asian J., 2014, 9, 1714–1723 CrossRef CAS PubMed.
- A. I. Green, C. P. Tinworth, S. Warriner, A. Nelson and N. Fey, Chem.–Eur. J., 2021, 27, 2402–2409 CrossRef CAS PubMed.
- N. Fey and J. M. Lynam, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1590 CAS.
- K. C. Harper, E. N. Bess and M. S. Sigman, Nat. Chem., 2012, 4, 366–374 CrossRef CAS PubMed.
- Z. L. Niemeyer, A. Milo, D. P. Hickey and M. S. Sigman, Nat. Chem., 2016, 8, 610–617 CrossRef CAS PubMed.
- Z. L. Niemeyer, S. Pindi, D. A. Khrakovsky, C. N. Kuzniewski, C. M. Hong, L. A. Joyce, M. S. Sigman and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 12943–12946 CrossRef CAS PubMed.
- J. Y. Guo, Y. Minko, C. B. Santiago and M. S. Sigman, ACS Catal., 2017, 7, 4144–4151 CrossRef CAS.
- M. Orlandi, J. A. S. Coelho, M. J. Hilton, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 6803–6806 CrossRef CAS PubMed.
- T. Gensch, S. R. Smith, T. J. Colacot, Y. N. Timsina, G. Xu, B. W. Glasspoole and M. S. Sigman, ACS Catal., 2022, 12, 7773–7780 CrossRef CAS.
- C. Sandford, L. R. Fries, T. E. Ball, S. D. Minteer and M. S. Sigman, J. Am. Chem. Soc., 2019, 141, 18877–18889 CrossRef CAS PubMed.
- T. Gensch, G. Dos, P. Gomes, P. Friederich, E. Peters, T. Gaudin, R. Pollice, K. Jorner, A. Nigam, M. Lindner-D’addario, M. S. Sigman and A. Aspuru-Guzik, J. Am. Chem. Soc., 2022, 144, 1205–1217 CrossRef CAS PubMed.
- H. Hotelling, Biometrika, 1936, 28, 321–377 CrossRef.
- J. J. Dotson, L. van Dijk, J. C. Timmerman, S. Grosslight, R. C. Walroth, F. Gosselin, K. Püntener, K. A. Mack and M. S. Sigman, J. Am. Chem. Soc., 2023, 145, 110–121 CrossRef CAS PubMed.
- J. Xu, S. Grosslight, K. A. Mack, S. C. Nguyen, K. Clagg, N.-K. Lim, J. C. Timmerman, J. Shen, N. A. White, L. E. Sirois, C. Han, H. Zhang, M. S. Sigman and F. Gosselin, J. Am. Chem. Soc., 2022, 144, 20955–20963 CrossRef CAS PubMed.
- T. Maschmeyer, L. P. E. Yunker and J. E. Hein, React. Chem. Eng., 2022, 7, 1061–1072 RSC.
- J. A. Daponte, Y. Guo, R. T. Ruck and J. E. Hein, ACS Catal., 2019, 9, 11484–11491 CrossRef CAS.
- J. Liu, Y. Sato, F. Yang, A. J. Kukor and J. E. Hein, Chemistry–Methods, 2022, 2, e202200009 CrossRef CAS.
- M. C. Deem and J. E. Hein, J. Org. Chem., 2023, 88, 1292–1297 CrossRef CAS PubMed.
- S. Akita, J.-Y. Guo, F. W. Seidel, M. S. Sigman and K. Nozaki, Organometallics, 2022, 41, 3185–3196 CrossRef CAS.
- M. Christensen, L. P. E. Yunker, F. Adedeji, F. Häse, L. M. Roch, T. Gensch, G. dos, P. Gomes, T. Zepel, M. S. Sigman, A. Aspuru-Guzik and J. E. Hein, Commun. Chem., 2021, 4, 1–12 CrossRef PubMed.
- J. P. Reid, R. S. J. Proctor, M. S. Sigman and R. J. Phipps, J. Am. Chem. Soc., 2019, 141, 19178–19185 CrossRef CAS PubMed.
- J. Werth and M. S. Sigman, ACS Catal., 2021, 11, 3916–3922 CrossRef CAS PubMed.
- S. H. Newman-Stonebraker, S. R. Smith, E. Borowski, E. Peters, T. Gensch, H. C. Johnson, M. S. Sigman and A. G. Doyle, Science, 2021, 374, 301–308 CrossRef CAS PubMed.
- T. Tang, E. Jones, T. Wild, A. Hazra, S. D. Minteer and M. S. Sigman, J. Am. Chem. Soc., 2022, 144, 20056–20066 CrossRef CAS PubMed.
- A. Trunschke, Catal. Sci. Technol., 2022, 12, 3650–3669 RSC.
- C. L. Allen, D. C. Leitch, M. S. Anson and M. A. Zajac, Nat. Catal., 2019, 2, 2–4 CrossRef.
- C. Brändli, P. Maiwald and J. Schröer, Chimia, 2003, 57, 284 CrossRef.
- K. Hitomi, Technovation, 1994, 14, 121–128 CrossRef.
- K. Olsen, SLAS Technol., 2012, 17, 469–480 CrossRef PubMed.
- P. Christopher, ACS Energy Lett., 2020, 5, 2737–2738 CrossRef CAS.
- Y. Al Naam, S. Elsafi, M. Al Jahdali, R. Al Shaman, B. Al-Qurouni, E. Al Zahrani and J. Healthc, Leadersh., 2022, 14, 55–62 Search PubMed.
- I. Holland and J. A. Davies, Front. Bioeng. Biotechnol., 2020, 8, 571777 CrossRef PubMed.
- A. Bowler, S. Bakalis and N. Watson, Chem. Eng. Res. Des., 2020, 153, 463–495 CrossRef CAS.
- J. B. Wolf, T. M. Stawski, G. J. Smales, A. F. Thünemann and F. Emmerling, Sci. Rep., 2022, 12, 5769 CrossRef CAS PubMed.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
- N. L. Bell, F. Boser, A. Bubliauskas, D. R. Wilcox, V. S. Luna and L. Cronin, Nat. Chem. Eng., 2024, 1, 180–189 CrossRef.
- F. F. Mulks, B. Pinho, A. W. J. Platten, M. R. Andalibi, A. J. Expósito, K. J. Edler, E. Hevia and L. Torrente-Murciano, Chem, 2022, 8, 3382–3394 CAS.
- B. Pieber, T. Glasnov and C. O. Kappe, RSC Adv., 2014, 4, 13430 RSC.
- J. Y. F. Wong, J. M. Tobin, F. Vilela and G. Barker, Chem.–Eur. J., 2019, 25, 12439–12445 CrossRef CAS PubMed.
- B. L. Hall, C. J. Taylor, R. Labes, A. F. Massey, R. Menzel, R. A. Bourne and T. W. Chamberlain, Chem. Commun., 2021, 57, 4926–4929 RSC.
- B. J. Reizman, Y.-M. Wang, S. L. Buchwald and K. F. Jensen, React. Chem. Eng., 2016, 1, 658–666 RSC.
- N. Nam-Trung, in Encyclopaedia of Microfluidics and Nanofluidics, Springer US, Boston, MA, 2008, pp. 423–427 Search PubMed.
- A. B. Santanilla, E. L. Regalado, T. Pereira, M. Shevlin, K. Bateman, L.-C. Campeau, J. Schneeweis, S. Berritt, Z.-C. Shi, P. Nantermet, Y. Liu, R. Helmy, C. J. Welch, P. Vachal, I. W. Davies, T. Cernak and S. D. Dreher, Science, 2014, 347, 49–53 CrossRef PubMed.
- A. Gioello, A. Piccinno, A. M. Lozza and B. Cerra, J. Med. Chem., 2020, 63, 6624–6647 CrossRef PubMed.
- B. Mahjour, R. Zhang, Y. Shen, A. McGrath, R. Zhao, O. G. Mohamed, Y. Lin, Z. Zhang, J. L. Douthwaite, A. Tripathi and T. Cernak, Nat. Commun., 2023, 14, 3924 CrossRef CAS PubMed.
- B. Mahjour, J. Hoffstadt and T. Cernak, Org. Process Res. Dev., 2023, 27, 1510–1516 CrossRef CAS.
- B. Mahjour, Y. Shen and T. Cernak, Acc. Chem. Res., 2021, 54, 2337–2346 CrossRef CAS PubMed.
- J. R. Schmink and M. T. Tudge, Tetrahedron Lett., 2013, 54, 15–20 CrossRef CAS.
- M. Christensen, F. Adedeji, S. Grosser, K. Zawatzky, Y. Ji, J. Liu, J. A. Jurica, J. R. Naber and J. E. Hein, React. Chem. Eng., 2019, 4, 1555–1558 RSC.
- D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961–6963 CrossRef CAS PubMed.
- W. Kew, N. G. A. Bell, I. Goodall and D. Uhrín, Magn. Reson. Chem., 2017, 55, 785–796 CrossRef CAS PubMed.
- K. A. Bakeev, Process Analytical Technology: Spectroscopic Tools and Implementation Strategies for the Chemical and Pharmaceutical Industries, Wiley, 2010 Search PubMed.
- A. Saib, A. Bara-Esataun, O. J. Harper, D. B. G. Berry, I. A. Thomlinson, R. Broomfield-Tagg, J. P. Lowe, C. L. Lyall and U. Hintermair, React. Chem. Eng., 2021, 9, 1548–1573 RSC.
- L. Schrecker, J. Dickhaut, C. Holtze, P. Staehle, M. Vranceanu, K. Hellgardt and K. K. (Mimi) Hii, React. Chem. Eng., 2023, 8, 41–46 RSC.
- L. A. Hammarback, A. L. Bishop, C. Jordan, G. Athavan, J. B. Eastwood, T. J. Burden, J. T. W. Bray, F. Clarke, A. Robinson, J.-P. Krieger, A. Whitwood, I. P. Clark, M. Towrie, J. M. Lynam and I. J. S. Fairlamb, ACS Catal., 2022, 12, 1532–1544 CrossRef CAS.
- T. J. Burden, K. P. R. Fernandez, M. Kagoro, J. B. Eastwood, T. F. N. Tanner, A. C. Whitwood, I. P. Clark, M. Towrie, J. Krieger, J. M. Lynam and I. J. S. Fairlamb, Chem.–Eur. J., 2023, 29, e202203038 CrossRef CAS PubMed.
- M. Carosso, E. Vottero, A. Lazzarini, S. Morandi, M. Manzoli, K. A. Lomachenko, M. J. Ruiz, R. Pellegrini, C. Lamberti, A. Piovano and E. Groppo, ACS Catal., 2019, 9, 7124–7136 CrossRef CAS.
- R. Chang, Y. P. Hong, S. Axnanda, B. Mao, N. Jabeen, S. Wang, R. Tai and Z. Liu, Curr. Appl. Phys., 2012, 12, 1292–1296 CrossRef.
- U. V. Ancharova, V. P. Pakharukova, A. A. Matvienko and S. V. Tsybulya, J. Struct. Chem., 2015, 56, 1076–1083 CrossRef CAS.
- J. B. Eastwood, L. A. Hammarback, T. J. Burden, I. P. Clark, M. Towrie, A. Robinson, I. J. S. Fairlamb and J. M. Lynam, Organometallics, 2023, 42, 1766–1773 CrossRef CAS PubMed.
- E. Le Gall, S. Sengmany, C. Hauréna, E. Léonel and T. Martens, J. Organomet. Chem., 2013, 736, 27–35 CrossRef CAS.
- K. J. DiRico, W. Hua, C. Liu, J. W. Tucker, A. S. Ratnayake, M. E. Flanagan, M. D. Troutman, M. C. Noe and H. Zhang, ACS Med. Chem. Lett., 2020, 11, 1101–1110 CrossRef CAS PubMed.
- H. Zhang, C. Liu, W. Hua, L. P. Ghislain, J. Liu, L. Aschenbrenner, S. Noell, K. J. Dirico, L. F. Lanyon, C. M. Steppan, M. West, D. W. Arnold, T. R. Covey, S. S. Datwani and M. D. Troutman, Anal. Chem., 2021, 93, 10850–10861 CrossRef CAS PubMed.
- Y. Wang, S. Shaabani, M. Ahmadianmoghaddam, L. Gao, R. Xu, K. Kurpiewska, J. Kalinowska-Tluscik, J. Olechno, R. Ellson, M. Kossenjans, V. Helan, M. Groves and A. Dömling, ACS Cent. Sci., 2019, 5, 451–457 CrossRef CAS PubMed.
- J. Leiterer, U. Panne, A. F. Thünemann and S. M. Weidner, Anal. Methods, 2011, 3, 70–73 RSC.
- A. Bednařík, M. Machálková, E. Moskovets, K. Coufalíková, P. Krásenský, P. Houška, J. Kroupa, J. Navrátilová, J. Šmarda and J. Preisler, J. Am. Soc. Mass Spectrom., 2019, 30, 289–298 CrossRef PubMed.
- B. M. Prentice, C. W. Chumbley and R. M. Caprioli, J. Mass Spectrom., 2015, 50, 703–710 CrossRef CAS PubMed.
- I. Sinclair, M. Bachman, D. Addison, M. Rohman, D. C. Murray, G. Davies, E. Mouchet, M. E. Tonge, R. G. Stearns, L. Ghislain, S. S. Datwani, L. Majlof, E. Hall, G. R. Jones, E. Hoyes, J. Olechno, R. N. Ellson, P. E. Barran, S. D. Pringle, M. R. Morris and J. Wingfield, Anal. Chem., 2019, 91, 3790–3794 CrossRef CAS PubMed.
- I. Sinclair, G. Davies and H. Semple, Expert Opin. Drug Discovery, 2019, 14, 609–617 CrossRef CAS PubMed.
- J. Tillner, V. Wu, E. A. Jones, S. D. Pringle, T. Karancsi, A. Dannhorn, K. Veselkov, J. S. McKenzie and Z. Takats, J. Am. Soc. Mass Spectrom., 2017, 28, 2090–2098 CrossRef CAS PubMed.
- B. P. Loren, H. S. Ewan, L. Avramova, C. R. Ferreira, T. J. P. Sobreira, K. Yammine, H. Liao, R. G. Cooks and D. H. Thompson, Sci. Rep., 2019, 9, 14745 CrossRef PubMed.
- N. Collins, D. Stout, J.-P. Lim, J. P. Malerich, J. D. White, P. B. Madrid, M. Latendresse, D. Krieger, J. Szeto, V.-A. Vu, K. Rucker, M. Deleo, Y. Gorfu, M. Krummenacker, L. A. Hokama, P. Karp and S. Mallya, Org. Process Res. Dev., 2020, 24, 2064–2077 CrossRef CAS.
- A. M. Schweidtmann, A. D. Clayton, N. Holmes, E. Bradford, R. A. Bourne and A. A. Lapkin, Chem. Eng. J., 2018, 352, 277–282 CrossRef CAS.
- D. M. Parry, ACS Med. Chem. Lett., 2019, 10, 848–856 CrossRef CAS PubMed.
- C. P. Breen, A. M. K. Nambiar, T. F. Jamison and K. F. Jensen, Trends Chem., 2021, 3, 373–386 CrossRef.
- Y. Shi, P. L. Prieto, T. Zepel, S. Grunert and J. E. Hein, Acc. Chem. Res., 2021, 54, 546–555 CrossRef CAS PubMed.
- J. Qiu and J. Albrecht, Org. Process Res. Dev., 2018, 22, 829–835 CrossRef CAS.
- K. Mansouri, C. M. Grulke, R. S. Judson and A. J. Williams, J. Cheminf., 2018, 10, 10 Search PubMed.
- Y. Amar, A. M. Schweidtmann, P. Deutsch, L. Cao and A. Lapkin, Chem. Sci., 2019, 10, 6697–6706 RSC.
- J. S. Smith, O. Isayev and A. E. Roitberg, Sci. Data, 2017, 4, 170193 CrossRef CAS PubMed.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Sci. Data, 2014, 1, 140022 CrossRef CAS PubMed.
- L. Ruddigkeit, R. van Deursen, L. C. Blum and J.-L. Reymond, J. Chem. Inf. Model., 2012, 52, 2864–2875 CrossRef CAS PubMed.
- A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed.
- M. Shevlin, M. R. Friedfeld, H. Sheng, N. A. Pierson, J. M. Hoyt, L.-C. Campeau and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 3562–3569 CrossRef CAS PubMed.
- M. S. Sigman, K. C. Harper, E. N. Bess and A. Milo, Acc. Chem. Res., 2016, 49, 1292–1301 CrossRef CAS PubMed.
- S. M. Kearnes, M. R. Maser, M. Wleklinski, A. Kast, A. G. Doyle, S. D. Dreher, J. M. Hawkins, K. F. Jensen and C. W. Coley, J. Am. Chem. Soc., 2021, 143, 18820–18826 CrossRef CAS PubMed.
- C.-C. Tsai, C. Sandford, T. Wu, B. Chen, M. S. Sigman and F. D. Toste, Angew. Chem., Int. Ed., 2020, 59, 14647–14655 CrossRef CAS PubMed.
- A. Milo, E. N. Bess and M. S. Sigman, Nature, 2014, 507, 210–214 CrossRef CAS PubMed.
- A. Milo, A. J. Neel, F. D. Toste and M. S. Sigman, Science, 2015, 347, 737–743 CrossRef CAS PubMed.
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Science, 2018, 360, 186–190 CrossRef CAS PubMed.
- C. Willoughby and J. G. Frey, Digit. Discov., 2022, 1, 183–194 RSC.
- J. M. Ravasco and J. A. S. Coelho, J. Am. Chem. Soc., 2020, 142, 4235–4241 CrossRef CAS PubMed.
- T. Gimadiev, T. Madzhidov, I. Tetko, R. Nugmanov, I. Casciuc, O. Klimchuk, A. Bodrov, P. Polishchuk, I. Antipin and A. Varnek, Mol. Inf., 2019, 38, 1800104 CrossRef PubMed.
- T. I. Madzhidov, A. V. Bodrov, T. R. Gimadiev, R. I. Nugmanov, I. S. Antipin and A. A. Varnek, J. Struct. Chem., 2015, 56, 1227–1234 CrossRef CAS.
- R. A. Fisher, The Design of Experiments, Oliver & Boyd, 1935 Search PubMed.
- J. D. Moseley and P. M. Murray, J. Chem. Technol. Biotechnol., 2014, 89, 623–632 CrossRef CAS.
- G. D. Bowden, B. J. Pichler and A. Maurer, Sci. Rep., 2019, 9, 1–10 CrossRef CAS PubMed.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- A. F. Brooks, J. J. Topczewski, N. Ichiishi, M. S. Sanford and P. J. H. Scott, Chem. Sci., 2014, 5, 4545–4553 RSC.
- R. A. Mansson, A. H. Welsh, N. Fey and A. G. Orpen, J. Chem. Inf. Model., 2006, 46, 2591–2600 CrossRef CAS PubMed.
- N. Fey, M. Garland, J. P. Hopewell, C. L. McMullin, S. Mastroianni, A. G. Orpen and P. G. Pringle, Angew. Chem., Int. Ed., 2012, 51, 118–122 CrossRef CAS PubMed.
- J. Jover, N. Fey, M. Purdie, G. C. Lloyd-Jones and J. N. Harvey, J. Mol. Catal. A: Chem., 2010, 324, 39–47 CrossRef CAS.
- J. Jover, N. Fey, J. N. Harvey, G. C. Lloyd-Jones, A. G. Orpen, G. J. J. Owen-Smith, P. Murray, D. R. J. Hose, R. Osborne and M. Purdie, Organometallics, 2012, 31, 5302–5306 CrossRef CAS PubMed.
- L. P. Hammett, Chem. Rev., 1935, 17, 125–136 CrossRef CAS.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- L. P. Hammett, Trans. Faraday Soc., 1938, 34, 156 RSC.
- E. N. Jacobsen, W. Zhang and M. L. Guler, J. Am. Chem. Soc., 1991, 113, 6703–6704 CrossRef CAS.
- C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2956–2965 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- K. Bunten, Coord. Chem. Rev., 2002, 233–234, 41–51 CrossRef CAS.
- A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, Organometallics, 2003, 22, 4322–4326 CrossRef CAS.
- K. Wu and A. G. Doyle, Nat. Chem., 2017, 9, 779–784 CrossRef CAS PubMed.
- M. Marín, J. J. Moreno, M. M. Alcaide, E. Álvarez, J. López-Serrano, J. Campos, M. C. Nicasio and E. Carmona, J. Organomet. Chem., 2019, 896, 120–128 CrossRef.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- J. Jover and N. Fey, Dalton Trans., 2013, 42, 172–181 RSC.
- K. C. Harper, E. N. Bess and M. S. Sigman, Nat. Chem., 2012, 4, 366–374 CrossRef CAS PubMed.
- R. Ardkhean, P. M. C. Roth, R. M. Maksymowicz, A. Curran, Q. Peng, R. S. Paton and S. P. Fletcher, ACS Catal., 2017, 7, 6729–6737 CrossRef CAS.
- R. Ardkhean, M. Mortimore, R. S. Paton and S. P. Fletcher, Chem. Sci., 2018, 9, 2628–2632 RSC.
- A. V. Brethomé, S. P. Fletcher and R. S. Paton, ACS Catal., 2019, 9, 2313–2323 CrossRef.
- D. G. Gusev, Organometallics, 2009, 28, 763–770 CrossRef CAS.
- D. Setiawan, R. Kalescky, E. Kraka and D. Cremer, Inorg. Chem., 2016, 55, 2332–2344 CrossRef CAS PubMed.
- J. Jover, N. Fey, J. N. Harvey, G. C. Lloyd-Jones, A. G. Orpen, G. J. J. Owen-Smith, P. Murray, D. R. J. Hose, R. Osborne and M. Purdie, Organometallics, 2010, 29, 6245–6258 CrossRef CAS.
- N. Fey, S. Papadouli, P. G. Pringle, A. Ficks, J. T. Fleming, L. J. Higham, J. F. Wallis, D. Carmichael, N. Mézailles and C. Müller, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 706–714 CrossRef CAS.
- C. Tabares-Mendoza and P. Guadarrama, J. Organomet. Chem., 2006, 691, 2978–2986 CrossRef CAS.
- H. Kargar, M. Fallah-Mehrjardi, R. Behjatmanesh-Ardakani, M. Bahadori, M. Moghadam, M. Ashfaq, K. S. Munawar and M. N. Tahir, Inorg. Chem. Commun., 2022, 142, 109697 CrossRef CAS.
- M. Duan, L. Zhu, X. Qi, Z. Yu, Y. Li, R. Bai and Y. Lan, Sci. Rep., 2017, 7, 7619 CrossRef PubMed.
- P. Jerabek, L. Vondung and P. Schwerdtfeger, Chem.–Eur. J., 2018, 24, 6047–6051 CrossRef CAS PubMed.
- R. Kumar, G. Li, V. A. Gallardo, A. Li, J. Milton, J. J. Nash and H. I. Kenttämaa, J. Am. Chem. Soc., 2020, 142, 8679–8687 CrossRef PubMed.
- H. M. Senn, D. V. Deubel, P. E. Blöchl, A. Togni and G. Frenking, J. Mol. Struct.: THEOCHEM, 2000, 506, 233–242 CrossRef CAS.
- E. R. Davidson and A. E. Clark, Int. J. Quantum Chem., 2022, 122, e26860 CrossRef CAS.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- J. Baker, Theor. Chim. Acta, 1985, 68, 221–229 CrossRef CAS.
- J. S. Gómez-Jeria, J. Chil. Chem. Soc., 2009, 54, 482–485 Search PubMed.
- H. P. Lüthi, J. H. Ammeter, J. Almlöf and K. Faegri, J. Chem. Phys., 1982, 77, 2002–2009 CrossRef.
- F. Martin and H. Zipse, J. Comput. Chem., 2005, 26, 97–105 CrossRef CAS PubMed.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- F. Weinhold, C. R. Landis and E. D. Glendening, Int. Rev. Phys. Chem., 2016, 35, 399–440 Search PubMed.
- V. R. Saunders and I. H. Hillier, Int. J. Quantum Chem., 1973, 7, 699–705 CrossRef.
- T. Leyssens, D. Peeters, A. G. Orpen and J. N. Harvey, Organometallics, 2007, 26, 2637–2645 CrossRef CAS.
- E. A. Eno, H. Louis, T. O. Unimuke, T. E. Gber, J. A. Akpanke, I. O. Amodu, A.-L. E. Manicum and O. E. Offiong, Chem. Phys. Impact, 2022, 5, 100105 CrossRef.
- J. Wei, M. Li, J. Ding, W. Dai, Q. Yang, Y. Feng, C. Yang, W. Yang, Y. Zheng, M.-Y. Wang and X. Ma, ChemCatChem, 2022, 14, e202200423 CrossRef CAS.
- T. Ikariya and Y. Kayaki, Catal. Surv. Jpn., 2000, 4, 39–50 CrossRef CAS.
- M. A. Klingshirn, R. D. Rogers and K. H. Shaughnessy, J. Organomet. Chem., 2005, 690, 3620–3626 CrossRef CAS.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS.
- T. Islam, Md. Z. Islam Sarker, A. H. Uddin, K. B. Yunus, R. Prasad, Md. A. R. Mia and S. Ferdosh, Anal. Chem. Lett., 2020, 10, 550–561 CrossRef CAS.
- V. Venkatraman and K. C. Lethesh, Front. Chem., 2019, 7, 605 CrossRef CAS PubMed.
- Z.-W. Zhao, M. del Cueto and A. Troisi, Digit. Discov., 2022, 1, 266–276 RSC.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
- M. Meuwly, Chem. Rev., 2021, 121, 10218–10239 CrossRef CAS PubMed.
- J. M. Granda, L. Donina, V. Dragone, D.-L. Long and L. Cronin, Nature, 2018, 559, 377–381 CrossRef CAS PubMed.
- E. Caldeweyher, M. Elkin, G. Gheibi, M. Johansson, C. Sköld, P.-O. Norrby and J. Hartwig, J. Am. Chem. Soc., 2023, 145, 17367–17376 CrossRef CAS PubMed.
- B. Meyer, B. Sawatlon, S. Heinen, O. A. von Lillenfeld and C. Corminboeuf, Chem. Sci., 2018, 9, 7069–7077 RSC.
- M. Rupp, A. Tkatchenko, K.-R. Müller and O. A. von Lilienfeld, Phys. Rev. Lett., 2012, 108, 058301 CrossRef PubMed.
- K. Hansen, F. Biegler, R. Ramakrishnan, W. Pronobis, O. A. Von Lilienfeld, K.-R. Müller and A. Tkatchenko, J. Phys. Chem. Lett., 2015, 6, 2326–2331 CrossRef CAS PubMed.
- B. Huang and O. A. Von Lilienfeld, arXiv, 2020, preprint, arXiv:1707.04146, DOI:10.48550/arXiv.1707.04146.
- M. D. Wodrich, B. Sawatlon, E. Solel, S. Kozuch and C. Corminboeuf, ACS Catal., 2019, 6, 5716–5725 CrossRef.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS.
- S. Kozuch and S. Shaik, J. Am. Chem. Soc., 2006, 128, 3355–3365 CrossRef CAS PubMed.
- S. Kozuch and S. Shaik, J. Phys. Chem. A, 2008, 112, 6032–6041 CrossRef CAS PubMed.
- A. Uhe, S. Kozuch and S. Shaik, J. Comput. Chem., 2011, 32, 978–985 CrossRef CAS PubMed.
- J. Noh, S. Back, J. Kim and Y. Jung, Chem. Sci., 2018, 9, 5152–5159 RSC.
- C. B. Santiago, J.-Y. Guo and M. S. Sigman, Chem. Sci., 2018, 9, 2398–2412 RSC.
- D. M. Hawkins, J. Chem. Inf. Comput. Sci., 2004, 44, 1–12 CrossRef CAS PubMed.
- C.-Y. Chang, M.-T. Hsu, E. X. Esposito and Y. J. Tseng, J. Chem. Inf. Model., 2013, 53, 958–971 CrossRef CAS PubMed.
- P. Geladi, Chemom. Intell. Lab. Syst., 2002, 60, 211–224 CrossRef CAS.
- G. E. Clarke, J. D. Firth, L. A. Ledingham, C. S. Horbaczewskyj, R. A. Bourne, J. T. W. Bray, P. L. Martin, J. B. Eastwood, R. Campbell, A. Pagett, D. J. MacQuarrie, J. M. Slattery, J. M. Lynam, A. C. Whitwood, J. Milani, S. Hart, J. Wilson and I. J. S. Fairlamb, Nat. Commun., 2024, 15, 3968 CrossRef PubMed.
Footnote |
| † (Hetero)aryl N-methyliminodiacetic acid. |
| This journal is © The Royal Society of Chemistry 2024 |