
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anchoring highly dispersed metal nanoparticles by strong electrostatic adsorption (SEA) on a dealuminated beta zeolite for catalysis†
Run
Zou
ab,
Gabriel A.
Bramley
 c,
Shanshan
Xu
a,
Sarayute
Chansai
a,
Monik
Panchal
d,
Huanhao
Chen
e,
Yangtao
Zhou
b,
Pan
Gao
f,
Guangjin
Hou
f,
Stuart M.
Holmes
a,
Christopher
Hardacre
a,
Yilai
Jiao
*b,
Andrew J.
Logsdail
*c and
Xiaolei
Fan
*ag
c,
Shanshan
Xu
a,
Sarayute
Chansai
a,
Monik
Panchal
d,
Huanhao
Chen
e,
Yangtao
Zhou
b,
Pan
Gao
f,
Guangjin
Hou
f,
Stuart M.
Holmes
a,
Christopher
Hardacre
a,
Yilai
Jiao
*b,
Andrew J.
Logsdail
*c and
Xiaolei
Fan
*ag
aDepartment of Chemical Engineering, School of Engineering, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: Xiaolei.fan@manchester.ac.uk
bShenyang National Laboratory for Materials Science, Institute of Metal Research Chinese Academy of Sciences, 72 Wenhua Road, Shenyang, 110016, China. E-mail: Yljiao@imr.ac.cn
cCardiff Catalysis Institute, School of Chemistry, Cardiff University, Park Place, Cardiff, CF103AT, Wales, UK. E-mail: LogsdailA@cardiff.ac.uk
dDepartment of Chemistry, Durham University, Stockton Road, DH1 3LE, UK
eState Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemical Engineering, Nanjing Tech University, Nanjing 211816, China
fState Key Laboratory of Catalysis, National Laboratory for Clean Energy, 2011-Collaborative Innovation Center of Chemistry for Energy Materials, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Zhongshan Road 457, Dalian 116023, China
gNottingham Ningbo China Beacons of Excellence Research and Innovation Institute, University of Nottingham Ningbo China, 211 Xingguang Road, Ningbo 315100, China
First published on 27th November 2023
Abstract
Zeolites with defects can be combined with appropriate synthetic protocols to beneficially stabilise metallic clusters and nanoparticles (NPs). In this work, highly dispersed Ni NPs were prepared on a defect-rich dealuminated beta (deAl-beta) zeolite through strong electrostatic adsorption (SEA) synthesis, which enabled strong interactions between the electronegative deAl-beta and cationic metal ammine complexes (e.g., Ni(NH3)62+) via the framework silanol nests. Ni NPs with diameters of 1.9 ± 0.2 nm were formed after SEA and reduction in H2 at 500 °C and showed good activity in CO2 methanation (i.e., specific reaction rate of 3.92 × 10−4 mol s−1 gNi−1 and methane selectivity of 99.8% at 400 °C under GHSV of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1). The mechanism of the SEA synthetic process was elucidated by ex situ XAFS, in situ DRIFTS, and DFT. XAFS of the as-prepared Ni catalysts (i.e., unreduced) indicates that SEA leads to the exchange of anions in Ni precursors (e.g., Cl− and NO3−) to form Ni(OH)2, while in situ DRIFTS of catalyst reduction shows a significant decrease in the signal of IR bands assigned to the silanol nests (at ∼960 cm−1), which could be ascribed to the strong interaction between Ni(OH)2 and silanol nests via SEA. DFT calculations show that metallic complexes bind more strongly to charged defect sites compared to neutral silanol nest defects (up to 150 kJ mol−1), confirming the enhanced interaction between metallic complexes and zeolitic supports under SEA synthesis conditions. The results provide new opportunities for preparing highly dispersed metal catalysts using defect-rich zeolitic carriers for catalysis.
000 mL g−1 h−1). The mechanism of the SEA synthetic process was elucidated by ex situ XAFS, in situ DRIFTS, and DFT. XAFS of the as-prepared Ni catalysts (i.e., unreduced) indicates that SEA leads to the exchange of anions in Ni precursors (e.g., Cl− and NO3−) to form Ni(OH)2, while in situ DRIFTS of catalyst reduction shows a significant decrease in the signal of IR bands assigned to the silanol nests (at ∼960 cm−1), which could be ascribed to the strong interaction between Ni(OH)2 and silanol nests via SEA. DFT calculations show that metallic complexes bind more strongly to charged defect sites compared to neutral silanol nest defects (up to 150 kJ mol−1), confirming the enhanced interaction between metallic complexes and zeolitic supports under SEA synthesis conditions. The results provide new opportunities for preparing highly dispersed metal catalysts using defect-rich zeolitic carriers for catalysis.
1. Introduction
Supported ultrasmall metal and/or metal oxide nanoparticles (NPs) are an important class of catalysts applied in various fields such as emission control and C1 chemistry.1,2 Ultrasmall NPs (typically smaller than 5 nm)3 can be highly active in catalysis due to the high density of active sites exposed to reactants, and therefore significant research efforts have been made to engineer new catalysts of this nature.4,5 Metal NPs are commonly supported on inorganic oxides (such as alumina, silica, titania and ceria) or on highly porous materials (such as zeolites and metal–organic frameworks, MOFs) and the interaction between the catalyst and the support is vital to control the size and dispersion of the resulting supported metal NPs, with the size key to the subsequent catalytic activity and selectivity. However, for ultrasmall NPs supported on highly porous materials, it is challenging to control the metal size and dispersion when the conventional impregnation methods are employed.6To address the challenge of size-controlled nanoparticle synthesis, the strong electrostatic adsorption (SEA) process was proposed to fabricate highly dispersed metal species on various supports (such as SiO2,7 Al2O3,8,9 carbon10 and zeolites11,12), where the protonation/deprotonation of surface hydroxyl groups leads to increased interaction strength between support and precursor. Zeolites are ideal supports for ultrasmall NPs due to their high porosity, frameworks with different pore sizes and topologies, good stability, and the number of available synthetic routes to include metallic phases.13–15 In combination, these properties offer opportunities to engineer specific host–guest interactions, which allows the preparation of highly dispersed supported metal NPs. Attempts to combine SEA with zeolitic carriers have resulted in highly dispersed noble metal species like Pt NPs on ZSM-22 and ZSM-5 zeolites.11,12 For example, Niu et al. prepared Pt clusters (of ∼1 nm, 1 wt%) on hollow silicalite-1 zeolite by SEA using Pt(NH3)4(NO3)2 as the precursor in an alkaline aqueous solution (pH of 11.5, adjusted by NH4OH) for deep hydrogenation of polycyclic aromatic hydrocarbons.11 The resulting Pt catalyst possesses electron-deficient Ptδ+ species due to the strong metal-support interactions, leading to a good deep hydrogenation performance. Ning et al. employed the SEA synthesis to prepare highly dispersed Pt clusters (∼1.2 nm, 0.3 wt%, Pt(NH3)4Cl2 as the precursor in an alkaline aqueous solution at pH 9 adjusted by NH4OH), which showed better conversion and selectivity in n-dodecane isomerization than the control catalyst prepared by impregnation.12 Previous studies have demonstrated the potential of combining of SEA and zeolitic supports, yet the mechanisms relating to the interactions between metal precursors and zeolites during SEA are not fully understood, requiring further investigation to progress the synthetic approach.
Post-synthetic dealumination of zeolite frameworks can improve the hydrothermal stability and catalytic activity.16 After dealumination of a zeolite (e.g., by acid treatment), silanol defects, such as silanol nests, are produced, which could be employed for functionalisation of dealuminated zeolites with metallic species for catalytic applications. Taking dealuminated beta zeolite (deAl-beta) as the example, incorporation of single atoms (e.g., Sn (ref. 17) and Zn (ref. 18)) and stabilisation of metal clusters and NPs (e.g., CuOx,19,20 NiOx (ref. 21–23) and CoOx (ref. 24 and 25)) has been demonstrated by the silanols nests in the framework and/or at extraframework sites. The anchoring of the highly dispersed metal species on deAl-beta is strongly affected by the silanol groups, as well as the synthesis methods. For instance, Ni/deAl-beta (sizes of the Ni NPs <1 nm, 1 wt%, Ni(acac)2 as the precursor in n-pentane) was developed for C2H4 hydrogenation, and the EXAFS characterisation of the Ni coordination environment suggested that the Ni species occupied the silanol nests in deAl-beta with a distorted tetrahedral geometry.22 In the study by Gac et al.,23 the supported Ni NPs catalysts were prepared on both pristine H-beta (silicon to alumina ratio, SAR = 12) and deAl-beta (SAR = 1000) by impregnation (using Ni(NO3)2 as the precursor with metal loading of ∼10 wt%), and the latter promoted the formation of smaller Ni NPs (particle diameters of ∼7.3 nm) than the pristine H-beta (with the resulting Ni NPs of ∼22.5 nm), leading to the improved performance in catalytic CO2 methanation. Comparatively, conventional impregnation in aqueous systems is prone to encourage the formation of large metal NPs, possibly due to the poor guest–host interactions.
Here, we report a detailed study of the effect of SEA (between a silanol nest-rich deAl-beta zeolite and cationic metal precursors) on the preparation of highly dispersed ultrasmall metal NPs on deAl-beta. To demonstrate the effectiveness of SEA for promoting Ni dispersion, a comparative study using the SEA and conventional impregnation (IM) was conducted (using NiCl2 as the precursor) with the resulting catalysts comprehensively characterised to obtain the relevant physicochemical properties for comparison. The mechanism of the SEA synthesis was elucidated by the results from various characterisation using X-ray absorption spectroscopy (XAS), in situ hydrogen temperature-programmed reduction coupled mass spectroscopy (H2-TPR-MS) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), as well as density functional theory (DFT) simulations. Finally, the advantage of the Ni catalysts prepared by the SEA synthesis was demonstrated by the model catalytic system of CO2 methanation.
2. Results and discussion
2.1 Characterisations of highly dispersed metal nanoparticles on deAl-beta
Dealumination of the pristine H-beta (SAR = 12) using concentrated HNO3 (13 mol L−1, 1 g zeolite per 20 mL solution) led to significant Al removal (SAR > 1000, by ICP) and formation of abundant silanol nests in deAl-beta (as evidenced by 1H NMR, Fig. S1†), agreeing with previous findings.26–28 We first explored the SEA synthesis for preparing the Ni catalyst on deAl-beta using NiCl2 as the precursor (herein denoted as Ni–Cl@deAl-beta-SEA). The NiCl2 precursor is known as non-ideal for catalyst preparation due to the detrimental effect of residual chlorine on catalytic performance,29,30 as exemplified by Ni catalysts based on various supports such as SiO2, Al2O3, CeO2 and TiO2.31–34 In the SEA synthesis, NiCl2 was dissolved in deionised water with the pH adjusted by NH4OH to ∼12 to encourage formation of hexaamminenickel chloride (Ni(NH3)6Cl2), as evidenced by a colour change of the solution from the light green of aqueous NiCl2 to light blue of Ni(NH3)6Cl2 (Fig. S2†) and the electronegative deAl-beta (i.e., the macroanion, due to the low isoelectric point of zeolite35,36). Consequently, the strong electrostatic adsorption between the precursor and zeolite prevents metal sintering during the direct thermal reduction in H2 at 500 °C (without calcination) and enables the formation of small Ni NPs in Ni–Cl@deAl-beta-SEA. Detailed experimental and characterisation approaches are presented in the ESI.†After direct H2 reduction, HAADF-STEM and HRTEM (Fig. 1a–d) reveals that the diameters of the Ni NPs on Ni–Cl@deAl-beta-SEA are 3.5 ± 0.4 nm, with a dispersion of ∼29% according to the calculations in ESI.† For the same material subjected to the impregnation synthetic method, forming Ni–Cl/deAl-beta-IM, the result is significantly larger with NP diameters of 23.3 ± 4.2 nm, with a dispersion of ∼4%. SEM-EDS mapping images (Fig. S3†) also shows noticeable Ni particle aggregation for Ni–Cl/deAl-beta-IM whilst Ni–Cl@deAl-beta-SEA shows homogenous Ni distribution. The results suggest that, when metal chlorides are used as precursors for catalyst preparation, SEA may intrinsically mitigate the adverse effect caused by residual Cl on metal chloride-derived catalysts, i.e., metal aggregation.
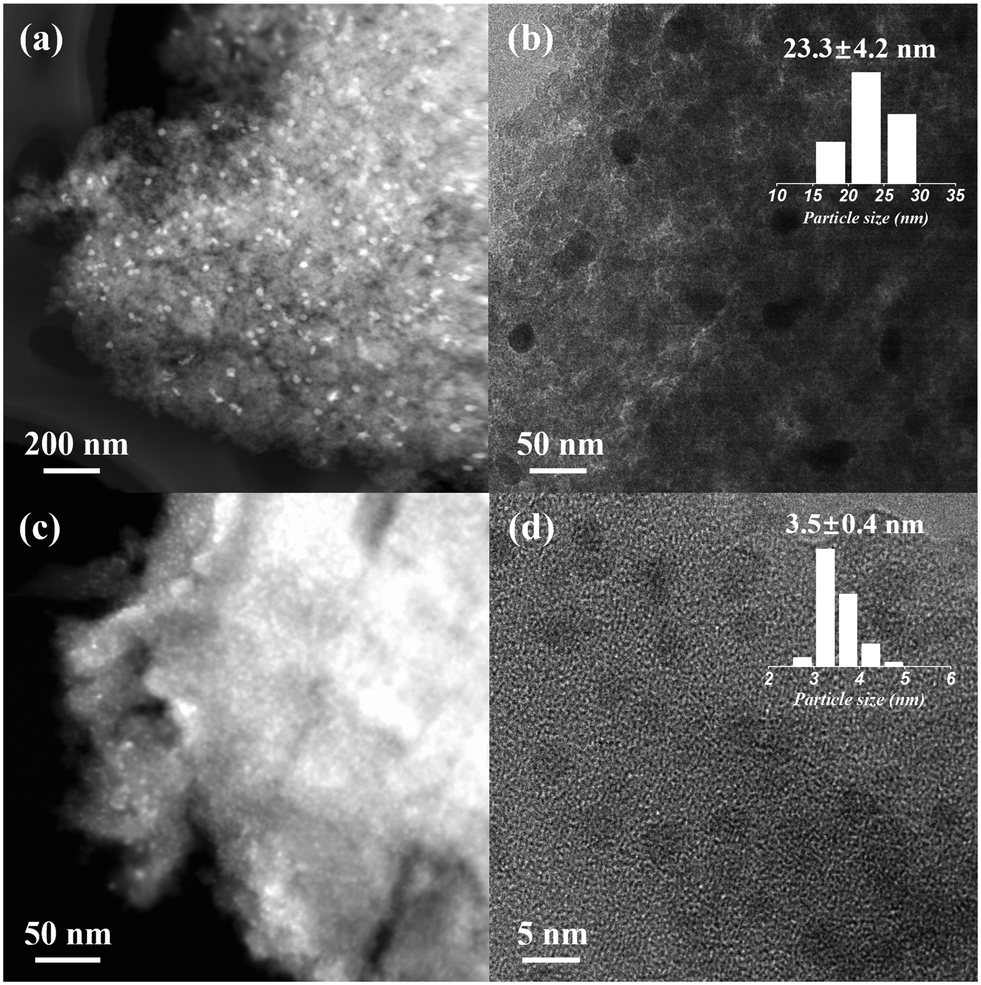 | ||
| Fig. 1 HAADF-STEM and HRTEM micrographs of (a and b) Ni–Cl/deAl-beta-IM and (c and d) Ni–Cl@deAl-beta-SEA. The particle size distribution of the studied catalysts was based on statistics of counting ca. 100 particles from relevant HRTEM images (using ImageJ). | ||
XRD analysis (Fig. 2a) verifies the presence of highly dispersed Ni NPs in Ni–Cl@deAl-beta-SEA. The characteristic diffraction peaks of metallic Ni phases (at 2θ = 44.7, 51.9 and 76.6°,37 JCPDS No. 01-070-0989) were insignificant in the XRD pattern of Ni–Cl@deAl-beta-SEA despite the relatively high ∼8 wt% Ni loading detected by ICP (Table S1†). Conversely, intense diffraction peaks of metallic Ni crystalline were measured for Ni–Cl/deAl-beta-IM, suggesting large Ni particles were formed. The diameter of Ni crystallites was estimated to be ∼18 nm, by application of the Scherrer equation on the peaks at 2θ = 44.7, 51.9 and 76.6°, which agrees with the HRTEM.38 Nitrogen (N2) physisorption analysis (Fig. 2b, Table S1†) shows a significant reduction in microporosity for Ni–Cl@deAl-beta-SEA, with a specific micropore area, Smicro, of 184 m2 g−1, compared to 361 m2 g−1 for the bare deAl-beta support (note that the mesoporosity of the support was preserved after the metal deposition). The mesopore diameter distribution for Ni–Cl@deAl-beta-SEA centres at ∼7 nm, whilst that of deAl-beta is ∼10 nm (Fig. 2b, inset); the difference in mesopore sizes corresponds to the diameter of the small Ni NPs in Ni–Cl@deAl-beta-SEA (i.e., ∼3.5 nm). Considering that the deAl-beta is in the form of nanocrystals with intercrystalline mesopores (Fig. S4†), small Ni NPs should reside in the mesoporous domains of the deAl-beta, causing the partial blockage of the intrinsic micropores of the deAl-beta. In contrast, Ni–Cl/deAl-beta-IM show properties comparable to deAl-beta (e.g., SBET, 545 vs. 582 m2 g−1 of deAl-beta, Table S1†), indicating that the large Ni particles were mainly located on the external surface, which agrees also with the HRTEM results.
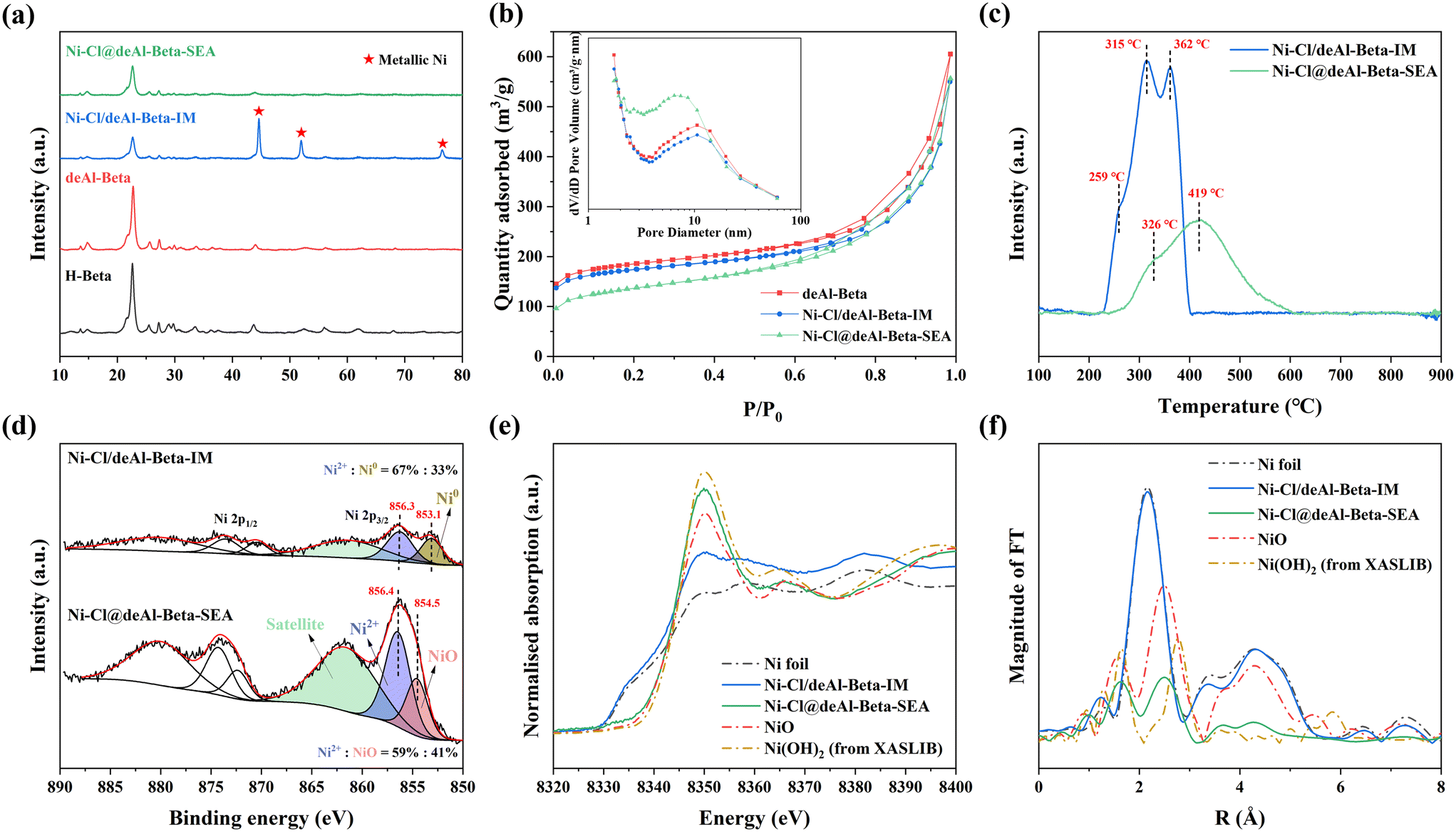 | ||
| Fig. 2 (a) XRD patterns and (b) N2 physisorption isotherms of the beta zeolite supports and the associated Ni catalysts; (c) H2-TPR (of the as-prepared Ni catalysts after drying at 120 °C for 4 h), (d) XPS profiles, (e) XANES and (f) Fourier transform of k3-weighted EXAFS spectra of the reduced Ni–Cl@deAl-beta-SEA and Ni–Cl/deAl-beta-IM. Note that the data of Ni(OH)2 is referred to the XASLIB database. | ||
The reducibility of Ni–Cl@deAl-beta-SEA and Ni–Cl/deAl-beta-IM was studied by H2-TPR (Fig. 2c), revealing that Ni–Cl/deAl-beta-IM could be reduced easily at lower temperatures, with sharp reduction peaks, especially at 315 and 362 °C. The reduction peaks are tentatively attributed to the reduction of Ni2+ species at the outer surface,39 where relatively weak guest-host interactions exist. Ni–Cl@deAl-beta-SEA were comparatively difficult to reduce, showing a broad reduction peak at 200–600 °C that suggests the relatively strong guest–host interaction in Ni–Cl@deAl-beta-SEA enabled by the SEA synthesis. Similar reduction behaviours were reported previously by Wang et al. for relevant Ni/SiO2 catalysts.40 The peak area of Ni–Cl/deAl-beta-IM (normalised by the mass of the sample) is 1.5 times that of Ni–Cl@deAl-beta-SEA, corresponding to the ratio of the actual Ni loadings on the two catalysts (i.e., ratio of ∼1.48, the Ni loading in Ni–Cl/deAl-beta-IM and Ni–Cl@deAl-beta-SEA is 11.71 and 7.92 wt%, respectively, Table S1†). In addition, H2-TPR characterisation suggests that thermal reduction at 500 °C could reduce the two catalysts satisfactorily.
The surface chemical state of Ni was probed by ex situ XPS and XAS. XPS elemental analysis shows that the surface Ni/Si atomic ratio in Ni–Cl@deAl-beta-SEA is higher than Ni–Cl/deAl-beta-IM, viz. ∼0.074 vs. ∼0.01 (calculation method is shown in ESI†). Moreover, the peak intensity of the Ni 2p spectra of Ni–Cl@deAl-beta-SEA is higher than that of Ni–Cl/deAl-beta-IM (Fig. 2d). According to previous studies,41,42 the differences observed in the results suggest that the Ni NPs are better dispersed on Ni–Cl@deAl-beta-SEA. The deconvolution of the Ni 2p3/2 spectra shows that Ni–Cl@deAl-beta-SEA is dominated by three peaks with binding energy (B.E.) of 854.6 and 856.6 eV, along with a broad shake-up satellite peak at 862 eV, corresponding to Ni2+ species.41–43 Conversely, two B.E. peaks of 853.1 and 856.3 eV, corresponding to Ni0 (proportion of ∼33%) and Ni2+ cations (proportion of ∼67%), were found in Ni–Cl/deAl-beta-IM, consistent with the XRD results above, i.e., metallic Ni exists. To obtain detailed structural information of Ni species, Ni K-edge XANES and EXAFS characterisation was conducted. The Ni K-edge XANES spectra (Fig. 2e) shows that the white line position of Ni–Cl@deAl-beta-SEA is higher than the Ni–Cl/deAl-beta-IM and Ni foil, indicating the oxidation state is higher in the SEA synthesised Ni catalyst. The chemical form of Ni species in the SEA and IM synthesised Ni catalysts was further analysed by EXAFS (Fig. 2f). Fourier transform of k3-weighted EXAFS spectra indicate that the Ni–Cl@deAl-beta-SEA and Ni–Cl/deAl-beta-IM materials show features like NiO and Ni foil standards, respectively, agreeing with the results of the XPS. In summary, our results suggest that the highly dispersed Ni NPs in Ni–Cl@deAl-beta-SEA are in a higher oxidation state than in Ni–Cl/deAl-beta-IM. In view of the fact that the Ni–Cl@deAl-beta-SEA should be reduced after reduction at 500 °C, the mere presence of NiO species could only be due to complete re-oxidisation of the smaller NPs when exposed to air during the ex situ characterisations.
2.2 Mechanism of SEA synthesis on deAl-beta
To understand the chemical state of Ni species in the as-prepared Ni catalysts (i.e., unreduced) from the IM and SEA synthesis, XAFS was applied (Fig. 3). From the Ni K-edge XANES spectra (Fig. 3a), the as-prepared Ni–Cl/deAl-beta-IM shows similar features to the NiCl2 standard, whilst the Ni–Cl@deAl-beta-SEA displays different characteristics (e.g., at ∼8360 and ∼8365 eV), indicating the presence of another Ni form. With support from the k3-weighted EXAFS, the as-prepared Ni–Cl/deAl-beta-IM is confirmed as a NiCl2 state, whilst the Ni–Cl@deAl-beta-SEA is identical to the Ni(OH)2 standard. The result indicates that the Cl− could be exchanged by OH− during the SEA synthesis, which enables its segregation from the catalyst (note that the catalyst is centrifuged after SEA process), and alleviates the undesirable effect of Cl− on reduced metal dispersion. In addition, significant Cl− was detected by adding silver nitrate solution (0.1 mol L−1) to the solution after SEA (which was separated from the solid product by centrifugation), showing the formation of the white precipitate (AgCl), as shown in Fig. S5.† Also, SEM-EDS analysis of the as-prepared catalysts (Fig. S3†) shows that the residual chlorine on Ni–Cl@deAl-beta-SEA was notably lower than on Ni–Cl/deAl-beta-IM, viz. ∼0.1 vs. ∼1.2 wt%, confirming that residual chlorine could be efficiently segregated from the catalyst surface during SEA before reduction.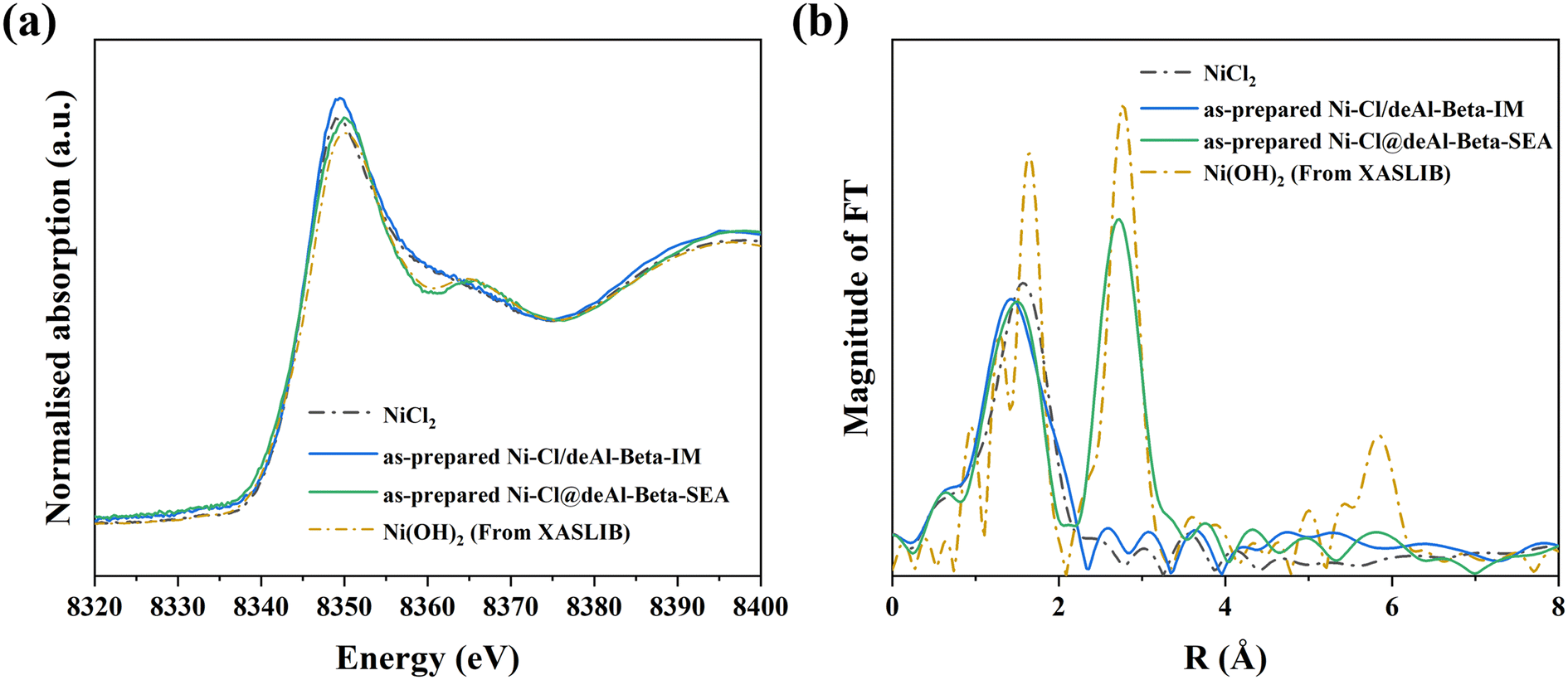 | ||
| Fig. 3 Ni K-edge XANES analysis (a) of Ni form in as-prepared Ni–Cl/deAl-beta-IM and Ni–Cl@deAl-beta-SEA (after drying at 120 °C for 4 h) and (b) the Fourier transform of k3-weighted EXAFS spectra. | ||
The species evolution of the Ni catalysts prepared by the IM and SEA were further studied by in situ H2-TPR-MS (Fig. S6†). The MS profiles of the Ni–Cl/deAl-beta-IM (Fig. S6a†) reduction show considerable H2 uptake, leading to water and HCl formation (m/z = 18 and 38) at 356 °C and 379 °C, respectively. Comparatively, noticeable variation in the H2 consumption of Ni–Cl@deAl-beta-SEA (Fig. S6b†) was shown at 460 °C but being less significant than that of Ni–Cl/deAl-beta-IM. No HCl was detected for the reduction of Ni–Cl@deAl-beta-SEA, confirming that the Cl− was segregated from the catalyst after the SEA synthesis and not present in the final material. Notably, water formation was remarkable at >200 °C during the reduction of Ni–Cl@deAl-beta-SEA, which could be primarily assigned to the reaction of Ni(OH)2, since the water production from condensation of silanol nest only starts at >400 °C and was less significant (Fig. S6c†).
In situ DRIFTS was employed to study the evolution of hydroxyl groups in the catalysts during reduction from 100 to 400 °C at 10 °C min−1 (Fig. 4). The isolated Si–OH in deAl-beta (at ∼3727 cm−1) is red-shifted and broadened after Ni loading by IM and SEA, with the latter showing more significant changes. The red-shift and broadening of isolated Si–OH in zeolitic materials has been reported as perhaps related to the hydrogen-bonding perturbation with guest substances;44,45 here, we speculate that the phenomenon observed in Ni–Cl@deAl-beta-SEA could be derived from the interaction with Ni(OH)2. The broad adsorption band of the silanol nest and/or H2O, with a maximum at ∼3500 cm−1, shows a gradual decrease in intensity with the increase of temperature in all samples, which is probably due to the metal–silanol nest interaction and/or removal of the adsorbed water. Notably, the adsorption band at ∼960 cm−1, which is considered to be from the silanol defects due to the removal of framework Al,46–48 reduces in intensity more in Ni–Cl@deAl-beta-SEA than Ni–Cl/deAl-beta-IM. Thus, the fundings above suggest that in the model system the Ni(OH)2 species were formed during SEA and interacted with the silanol nests in deAl-beta for anchoring Ni species on the zeolitic support. In addition, the signal at ∼3601 cm−1 was found for pristine H-beta (instead of the dealuminated ones), being ascribed to the Brønsted acid sites in the H-beta.49
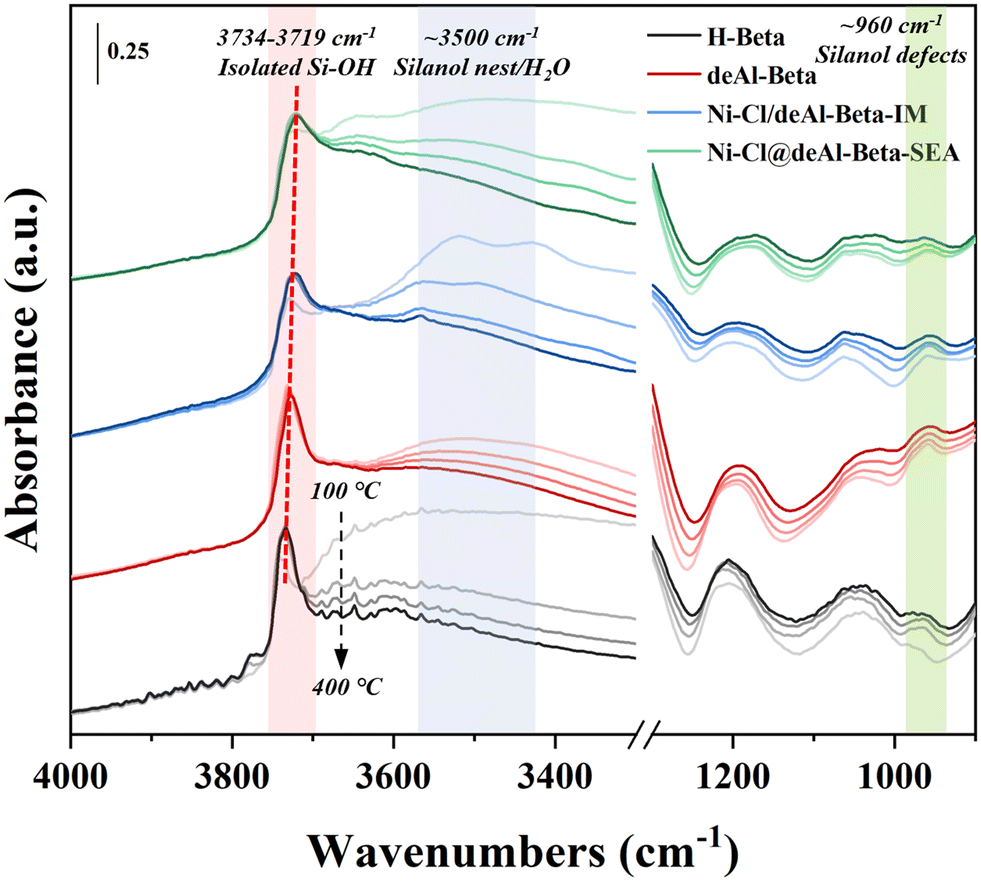 | ||
| Fig. 4 In situ DRIFTS studies on reduction of Ni catalysts prepared by IM and IM with NiCl2 as precursor. | ||
2.3 Validation of SEA with different Ni precursors and deAl-beta supports
Thus far, the SEA synthesis showed the ability to exchange Cl− to form Ni(OH)2 in the as-prepared Ni catalyst, which then interacts with the silanol nest and stabilises during reduction. However, it remains unclear whether the increase in metal dispersion was due to the Cl− removal or the metal stabilisation by the silanol nest, or the combination of both. Therefore, Ni catalysts synthesised using Ni(NO3)2 as the precursor were prepared (i.e., Ni–NO3@deAl-beta-SEA), and relevant properties were characterised and discussed in ESI† (Fig. S7 and Table S1, including XRD, N2 physisorption, XPS and in situ DRIFTS of catalyst reduction). Ni–NO3@deAl-beta-SEA (Ni loading of ca. 7 wt%) showed smaller Ni NPs and narrower diameter distribution compared to the counterpart prepared by impregnation, i.e., Ni–NO3/deAl-beta-IM (1.9 ± 0.2 vs. 3.3 ± 1.2 nm, Fig. 5a and b), proving that the SEA synthesis is generic regardless of the types of Ni precursors. Also, it is worth noting that the Ni–NO3@deAl-beta-SEA catalyst presents one of the smallest supported Ni NPs in comparison with the relevant state-of-the-arts (with the Ni NPs sizes ranging from 2.6 to 18 nm, prepared by different syntheses such as ligand-protected encapsulation and ion exchange-deposition–precipitation, Table S2†). | ||
| Fig. 5 TEM images of Ni catalysts prepared using Ni(NO3)2 as the precursor. (a) Ni–NO3@deAl-beta-SEA, (b) Ni–NO3/deAl-beta-IM and (c) Ni–NO3@calcinated deAl-beta-SEA. The particle size distribution of the three catalysts was based on statistics of counting ca. 100 particles from relevant HRTEM images (using ImageJ). | ||
To study the role of the silanol defects in the SEA synthesis, a Ni catalyst was prepared using the calcinated deAl-beta, which has less hydroxyl groups (as evidenced by TGA, DRIFTS, XRD and N2 physisorption characterisation, Fig. S8a–f†). The results show that the resulting Ni–NO3@calcinated deAl-beta-SEA catalyst has slightly larger Ni NPs (Fig. 5c) and less reduction in porosity (Table S1†), indicating the importance of abundant silanol groups in the stabilisation of highly dispersed Ni NPs during SEA.
The Ni catalysts prepared with Ni(NO3)2 were further characterised by in situ CO-DRIFTS to study the types of Ni species (metallic and/or metal oxide, Fig. S9†). Ni–NO3@deAl-beta-SEA presents a symmetrical band at 2036 cm−1 assigning to the CO linearly adsorbed on metallic Ni atom, whilst the Ni–NO3/deAl-beta-IM and Ni–NO3@calcinated deAl-beta-SEA catalysts display more heterogeneous bands correlating to both CO adsorption on metallic (at ∼2053 cm−1) and oxidised Ni species (at ∼2116 cm−1). In summary, SEA enables effective utilisation of the silanol groups in deAl-beta to encourage the formation of highly dispersed metallic Ni NPs, which is expected to benefit the catalysis where small metallic NPs are preferred.
2.4 DFT simulation
To demonstrate the presence of strong electrostatic interactions between the zeolite framework and the metallic complexes, DFT calculations were performed to obtain the adsorption energy for metal complexes (including NiCl2, Ni(NO3)2 and Ni(OH)2) on a fully protonated (IM) and a singly and/or doubly deprotonated silanol nests (SEA) within deAl-beta (Table 1). Results show that the stability of the adsorbate complex generally increases with the degree of deprotonation of the silanol nest. For example, the adsorption energy of Ni(NO3)2 strengthened from −66 to −71 and −198 kJ mol−1 for the fully protonated, singly deprotonated and doubly deprotonated silanol nests, respectively. The enhanced interaction between metal complexes and zeolite framework is also indicated by the distance between Ni and closest silanol oxygen (Fig. 6), which are 3.78, 2.02, and 1.86 Å for fully protonated (Fig. 6a), singly deprotonated (Fig. 6b) and doubly deprotonated silanol nest (Fig. 6c). In the fully protonated silanol nest, the Ni(NO3)2 adsorbs weakly to the silanol site and lies in the centre of the zeolite cage, while the Ni(NO3)2 interacts strongly with the dangling oxygen group in the singly deprotonated silanol nest, but the metal cation generally lies outside of the silanol nest. Furthermore, for the doubly deprotonated silanol nest, the metal centre coordinates between the two negatively charged oxygen fragments, which overall come into far closer contact with the dealuminated sites (Fig. 6c). The combination of adsorption energetics and geometry imply that the metal centres form strong electrostatic interactions with the deprotonated silanol framework oxygens, which is expected to promote higher dispersion of the Ni species throughout the zeolitic framework.| Ligand | Fully protonated (IM) | Singly deprotonated (SEA) | Doubly deprotonated (SEA) |
|---|---|---|---|
| NiCl2 | −52 | −110 | −207 |
| Ni(NO3)2 | −66 | −71 | −198 |
| Ni(OH)2 | −113 | −71 | −190 |
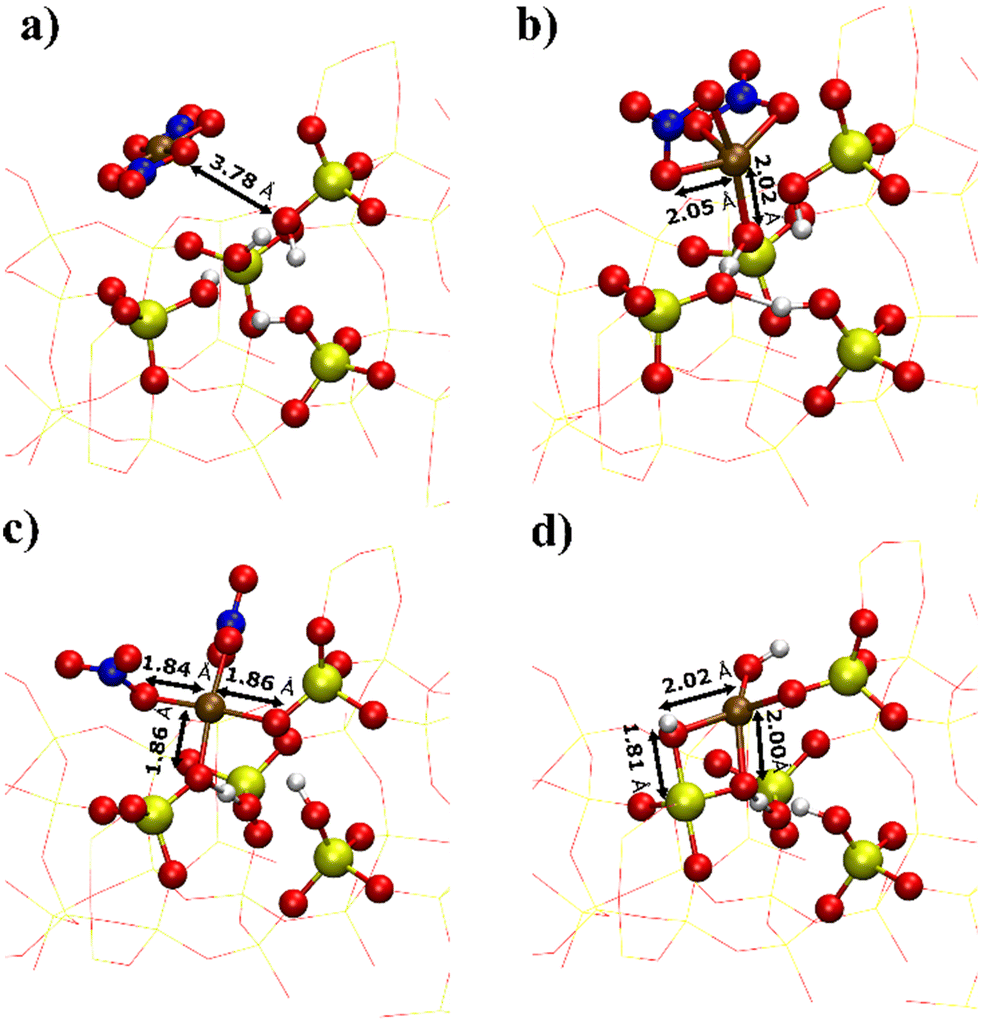 | ||
| Fig. 6 Optimised geometries of metallic complexes adsorbed on the deAl-beta framework: Ni(NO3)2 adsorption to the silanol nest at (a) full protonation (IM), (b) first degree of deprotonation, (c) second degree of deprotonation, and (d) Ni(OH)2 adsorption at the doubly deprotonated silanol nest. | ||
XANES analysis (Fig. 3) has shown that Ni(OH)2 is the dominant complex in the as-prepared Ni–Cl@deAl-beta-SEA, as opposed to Ni–Cl/deAl-beta-IM, where NiCl2 remains the main mode of the metallic complexation. It is therefore proposed that OH− from the basic aqueous solution exchanges with the Cl− ligands of the adsorbed Ni centre in the SEA process. To confirm this, a set of simulations were conducted to measure the energetic changes involved with ion exchange, where the exchange of two/one OH− ions for a given ligand coordinated to the adsorption complex, NiX2 (X = NO3, Cl), is expressed in eqn (1) and (2).
| 2OH− + deAl-beta-NiX2 → deAl-beta-Ni(OH)2 + 2X− | (1) |
| OH− + deAl-beta-NiX → deAl-beta-Ni(OH) + X− | (2) |
| Reaction | Singly deprotonated (charge = −1) | Doubly deprotonated (charge = −2) |
|---|---|---|
| 2OH− + deAl-beta-NiCl2 → deAl-beta-Ni(OH)2 + 2Cl− | −294 | −303 |
| 2OH− + deAl-beta-Ni(NO3)2 → deAl-beta-Ni(OH)2 + 2NO3− | −352 | −331 |
| OH− + deAl-beta-NiCl → deAl-beta-NiOH + Cl− | −204 | −145 |
| OH− + deAl-beta-Ni(NO3) → deAl-beta-NiOH + NO3− | +316 | −178 |
| 2OH− + NiCl2 → Ni(OH)2 + 2Cl− | −49 | |
| 2OH− + Ni(NO3)2 → Ni(OH)2 + 2NO3− | −338 | |
The above results qualitatively illustrate that the formation of the adsorbed deAl-beta-Ni(OH)2 complex is generally favourable. To further verify the results, calculations were performed with an implicit solvation model (3D-RISM) to incorporate an aqueous reference state for the ligands, allowing demonstration as to why this exchange process is observed for SEA but not for IM. Ion exchange is expressed as the eqn (3) in a solvent, (where X = NO3).
| 2OH− + NiX2 → Ni(OH)2 + 2X− | (3) |
Finally, we consider bonding interactions that occur for deAl-beta-Ni(OH)2, where an OH− ligand is shared between a silicon in the silanol nest and the Ni centre (e.g.,Fig. 6d). This structure occurs for multiple starting geometries, despite the repositioning of the OH− ligand around the Ni2+ centre. However, [Ni(NH3)6(OH)2](aq) was not observed in as-prepared Ni–Cl/deAl-beta-IM, meaning the adsorption of Ni(OH)2 was not considered a significant route within the IM process. Furthermore, XANES analysis showed that the formation of Ni(OH)2 likely follows a ligand exchange route from either adsorbed NiCl2 or Ni(NO3)2, and in situ DRIFTS studies show more significant red-shift and broadening of the isolated silanol band during SEA synthesis. In combination with the spectroscopic evidence, for adsorbed Ni(OH)2 during SEA synthesis, the cross-centre bonding of the OH species calculated for adsorbed Ni(OH)2 during SEA synthesis may be responsible for the observed broadening of the isolated silanol band.
2.5 Catalytic performances of highly dispersed nickel catalysts for CO2 methanation
To demonstrate the advantage of the ultrasmall Ni catalysts prepared by SEA compared to preparations with IM, catalytic CO2 methanation over the relevant catalysts was conducted (at 350–400 °C and GHSV of 30000 mL g−1 h−1, total flowrate of 50 mL min−1, CO2/H2/Ar = 1/4/5, over ∼0.1 g catalyst), and results were summarised in Table 3. Ni–Cl@deAl-beta-SEA showed considerably better performance than Ni–Cl/deAl-beta-IM, with much higher specific reaction rate (rCO2) and CH4 selectivity (SCH4) at the two studied reaction temperatures (e.g., rCO2: 3.38 vs. 0.81 × 10−4 mol s−1 gNi−1 and SCH4: 99.7 vs. 23.8% at 400 °C), suggesting that the Cl poisoning was avoided by the SEA synthesis.32 Notably, Ni–NO3@deAl-beta-SEA with the ultrasmall Ni NPs demonstrates the comparatively best performance in CO2 methanation among catalysts under investigation (Table 3), as well as the state-of-the-art catalysts (such as Ni supported on SiO2, Al2O3, ZrO2 and other zeolitic supports, Table S3†), with rCO2 of 3.11 and 3.92 × 10−4 mol s−1 gNi−1 and SCH4 of >97% at 350 and 400 °C, respectively. The Ni–NO3@calcinated deAl-beta-SEA catalyst showed slightly lower activity (e.g., rCO2 of 3.63 × 10−4 mol s−1 gNi−1 at 400 °C) than Ni–NO3@deAl-beta-SEA, whilst the Ni-NO3/deAl-beta-IM displayed a lower specific reaction rate (e.g., rCO2 of 2.86 × 10−4 mol s−1 gNi−1 at 400 °C) than the catalysts prepared by SEA. The results from the catalytic studies demonstrate that SEA can promote the utilisation of metal active species compared with IM, mainly due to the improved metal dispersion. Longevity test (Fig. S10†) of Ni–NO3@deAl-beta-SEA at 400 °C for 40 h showed that the catalyst maintained high conversion (∼75%) and Ni dispersion (i.e., diameters of 2.2 ± 0.3 nm, by post-reaction TEM, Fig. S11†), suggesting the good stability of the highly dispersed Ni NPs prepared by SEA.
000 mL g−1 h−1, total flowrate of 50 mL min−1, CO2/H2/Ar = 1/4/5, over ∼0.1 g catalyst)
| Catalyst | Ni loading (%) | X CO2 (%) | S CH4 (%) | r CO2 (×10−4 mol s−1 gNi−1) | |||
|---|---|---|---|---|---|---|---|
| a Note that the results of XCO2, SCH4 and rCO2 at 350 and 400 °C were shown, respectively. b The standard deviation of the average was calculated using at least three independent measurements. | |||||||
| Ni–Cl/deAl-beta-IM | 11.71 | 6.8 (±0.2)b | 25.3 (±0) | 0 | 23.8 (±0.3) | 0.22 (±0.01) | 0.81 (±0) |
| Ni–Cl@deAl-beta-SEA | 7.92 | 45.3 (±1.4) | 72.0 (±1.1) | 96.2 (±0.4) | 99.7 (±0.1) | 2.13 (±0.06) | 3.38 (±0.05) |
| Ni–NO3/deAl-beta-IM | 10.16 | 61.8 (±2.1) | 78.0 (±0.1) | 99.4 (±0.2) | 99.9 (±0) | 2.26 (±0.08) | 2.86 (±0) |
| Ni–NO3@calcinated deAl-beta-SEA | 7.67 | 58.1 (±0.7) | 74.9 (±0) | 97.6 (±0.2) | 99.6 (±0) | 2.82 (±0.03) | 3.63 (±0) |
| Ni–NO3@deAl-beta-SEA | 7.12 | 59.6 (±1.9) | 75.0 (±0.18) | 97.8 (±0.7) | 99.8 (±0) | 3.11 (±0.1) | 3.92 (±0.01) |
3. Conclusions
A strategy to prepare highly dispersed metal NPs on dealuminated beta zeolite (deAl-beta) has been realised, in which the SEA synthetic protocol enhances the interaction between the electronegative zeolitic framework and cationic metal precursors. The SEA method beneficially stabilises the supported metal species during thermal reduction, which leads to the formation of ultrasmall supported metallic NPs. As a result, the supported Ni catalysts synthesised with NiCl2 as the precursor have a high Ni loading of ∼8 wt% and dispersion of ∼29%, as well as ultrasmall NP diameters of 3.5 ± 0.4 nm. In contrast, the conventional impregnation synthesis showed significant metal aggregation with Ni NP diameters of 23.3 ± 4.2 nm and low metal dispersion of ∼4%. The Ni catalysts prepared by SEA show high activity in CO2 methanation at low temperatures, which we attribute to the high dispersion of the active species. Importantly, mechanistic studies show that the SEA synthesis enhances the interaction between silanol defects on the deAl-beta zeolite and the metal precursor, which contributed to the stabilisation of metal NPs on the carrier during thermal reduction in H2. The results are supported by DFT simulations, where the bonding at the deprotonated silanol sites (SEA) is calculated to be up to ∼150 kJ mol−1 stronger than the fully protonated (IM) silanol nest. The proposed SEA synthesis protocol can be a general method for developing high-quality zeolite-supported metal nanoparticles for various catalytic applications.Data availability
Data supporting the DFT simulations of this study are openly available from the NOMAD repository at DOI: https://doi.org/10.17172/NOMAD/2023.10.03-1.Author contributions
Run Zou: conceptualisation, investigation, formal analysis, visualization, writing – original draft; Gabriell Bramley: software, investigation, formal analysis, visualization, writing – original draft; Shanshan Xu: methodology, validation, writing – review & editing; Sarayute Chansai: methodology, investigation, resources, writing – review & editing; Monik Panchal: methodology, investigation, writing – review & editing; Huanhao Chen: investigation, resources; Yangtao Zhou: methodology, resources; Pan Gao: methodology, investigation; Guangjin Hou: methodology, resources; Stuart M. Holmes: resources, writing – review & editing; Christopher Hardacre: resources, supervision, writing – review & editing; Yilai Jiao: resources, supervision, writing – review & editing; Andrew Logsdail: supervision, funding acquisition, resources, writing – review & editing; Xiaolei Fan: conceptualization, supervision, resources, funding acquisition, project administration, writing – review & editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 872102. R.Z. thanks the financial support from the China Scholarship Council (file no. 201906740020) and The University of Manchester for supporting his PhD research at the University of Manchester in the United Kingdom. A. J. L. and G. B. acknowledge funding by the UKRI Future Leaders Fellowship program (MR/T018372/1). HPC facilities were provided by Advanced Research Computing at Cardiff (ARCCA) under the Supercomputing Wales (SCW) project, which is part-funded by the European Regional Development Fund (ERDF) via Welsh Government. M. P. acknowledges Durham X-ray Absorption Facility supported by the Engineering and Physical Sciences Research Council (EP/V029053/1).References
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- A. K. Datye and M. Votsmeier, Nat. Mater., 2021, 20, 1049–1059 CrossRef CAS PubMed.
- N. Wang, Q. Sun and J. Yu, Adv. Mater., 2019, 31, e1803966 CrossRef PubMed.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS PubMed.
- C. Gao, F. Lyu and Y. Yin, Chem. Rev., 2021, 121, 834–881 CrossRef CAS PubMed.
- L. Liu and A. Corma, Nat. Rev. Mater., 2021, 6, 244–263 CrossRef CAS.
- Q. L. A. Wong, S. Griffin, A. Nicholls and J. R. Regalbuto, Science, 2017, 358, 1427–1430 CrossRef PubMed.
- X. Zhang, D. Wang, X. Chen, L. Meng and C. Liang, Energy Fuels, 2022, 36, 2775–2786 CrossRef CAS.
- J. Zecevic, G. Vanbutsele, K. P. de Jong and J. A. Martens, Nature, 2015, 528, 245–248 CrossRef CAS PubMed.
- S. Lambert, N. Job, L. Dsouza, M. Pereira, R. Pirard, B. Heinrichs, J. Figueiredo, J. Pirard and J. Regalbuto, J. Catal., 2009, 261, 23–33 CrossRef CAS.
- X. Niu, R. Zhao, Y. Han, X. Zhang and Q. Wang, Fuel, 2022, 326, 125021 CrossRef CAS.
- Q. Ning, H. Zhang, Y. He, Z. Chen, S. Liu and J. Ren, New J. Chem., 2019, 43, 13967–13978 RSC.
- Q. Sun, N. Wang and J. Yu, Adv. Mater., 2021, e2104442 CrossRef PubMed.
- Y. Wang, C. Wang, L. Wang, L. Wang and F. S. Xiao, Acc. Chem. Res., 2021, 54, 2579–2590 CrossRef CAS PubMed.
- S. Xu, T. J. A. Slater, H. Huang, Y. Zhou, Y. Jiao, C. M. A. Parlett, S. Guan, S. Chansai, S. Xu, X. Wang, C. Hardacre and X. Fan, Chem. Eng. J., 2022, 446, 137439 CrossRef CAS.
- R. Simancas, A. Chokkalingam, S. P. Elangovan, Z. Liu, T. Sano, K. Iyoki, T. Wakihara and T. Okubo, Chem. Sci., 2021, 12, 7677–7695 RSC.
- C. Hammond, S. Conrad and I. Hermans, Angew. Chem., Int. Ed., 2012, 51, 11736–11739 CrossRef CAS PubMed.
- L. Qi, Y. Zhang, M. A. Conrad, C. K. Russell, J. Miller and A. T. Bell, J. Am. Chem. Soc., 2020, 142, 14674–14687 CrossRef CAS PubMed.
- D. Yu, W. Dai, G. Wu, N. Guan and L. Li, Chin. J. Catal., 2019, 40, 1375–1384 CrossRef CAS.
- A. Rokicińska, P. Majerska, M. Drozdek, S. Jarczewski, L. Valentin, J. Chen, A. Slabon, S. Dzwigaj and P. Kuśtrowski, Appl. Surf. Sci., 2021, 546, 149148 CrossRef.
- X. Zhou, S. Wu, Y. Luo, L. Zhu and D. He, Energy Fuels, 2023, 37, 450–458 CrossRef CAS.
- M. Meloni, J. Hong, A. S. Hoffman, S. Holton, A. Kulkarni, S. R. Bare and R. C. Runnebaum, J. Phys. Chem. C, 2022, 126, 21213–21222 CrossRef CAS.
- W. Gac, W. Zawadzki, G. Słowik, M. Kuśmierz and S. Dzwigaj, Appl. Surf. Sci., 2021, 564, 150421 CrossRef CAS.
- A. Rokicińska, M. Drozdek, B. Dudek, B. Gil, P. Michorczyk, D. Brouri, S. Dzwigaj and P. Kuśtrowski, Appl. Catal., B, 2017, 212, 59–67 CrossRef.
- R. Sadek, K. Chalupka-Spiewak, J.-M. Krafft, Y. Millot, L. Valentin, S. Casale, J. Gurgul and S. Dzwigaj, Catalysts, 2022, 12, 1644 CrossRef CAS.
- W. N. P. van der Graaff, G. Li, B. Mezari, E. A. Pidko and E. J. M. Hensen, ChemCatChem, 2015, 7, 1152–1160 CrossRef CAS.
- J. Dijkmans, D. Gabriëls, M. Dusselier, F. de Clippel, P. Vanelderen, K. Houthoofd, A. Malfliet, Y. Pontikes and B. F. Sels, Green Chem., 2013, 15, 2777–2785 RSC.
- F. Yi, Y. Chen, Z. Tao, C. Hu, X. Yi, A. Zheng, X. Wen, Y. Yun, Y. Yang and Y. Li, J. Catal., 2019, 380, 204–214 CrossRef CAS.
- M. Li, F. Bi, Y. Xu, P. Hao, K. Xiang, Y. Zhang, S. Chen, J. Guo, X. Guo and W. Ding, ACS Catal., 2019, 9, 11676–11684 CrossRef CAS.
- X. Zhu, B. Cheng, J. Yu and W. Ho, Appl. Surf. Sci., 2016, 364, 808–814 CrossRef CAS.
- K. Urasaki, Y. Tanpo, Y. Nagashima, R. Kikuchi and S. Satokawa, Appl. Catal., A, 2013, 452, 174–178 CrossRef CAS.
- X. Wen, L. Xu, M. Chen, Y. Shi, C. Lv, Y. Cui, X. Wu, G. Cheng, C.-E. Wu, Z. Miao, F. Wang and X. Hu, Appl. Catal., B, 2021, 297, 120486 CrossRef CAS.
- Z. Yu, X. Hu, P. Jia, Z. Zhang, D. Dong, G. Hu, S. Hu, Y. Wang and J. Xiang, Appl. Catal., B, 2018, 237, 538–553 CrossRef CAS.
- B. Wang, Y. Xiong, Y. Han, J. Hong, Y. Zhang, J. Li, F. Jing and W. Chu, Appl. Catal., B, 2019, 249, 257–265 CrossRef CAS.
- Y. Tian, H. Duan, B. Zhang, S. Gong, Z. Lu, L. Dai, C. Qiao, G. Liu and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 21713–21717 CrossRef CAS PubMed.
- Y.-H. Chen, C.-Y. Mou and B.-Z. Wan, Appl. Catal., B, 2017, 218, 506–514 CrossRef CAS.
- Y. Shi, L. Wang, M. Wu and F. Wang, Appl. Catal., B, 2023, 337, 122927 CrossRef CAS.
- J. S. J. Hargreaves, Catal., Struct. React., 2016, 2, 33–37 CAS.
- A. Quindimil, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos and J. R. González-Velasco, Appl. Catal., B, 2018, 238, 393–403 CrossRef CAS.
- F. Wang, K. Han, L. Xu, H. Yu and W. Shi, Ind. Eng. Chem. Res., 2021, 60, 3324–3333 CrossRef CAS.
- Y. Chen, B. Qiu, Y. Liu and Y. Zhang, Appl. Catal., B, 2020, 269, 118801 CrossRef CAS.
- X. Jia, X. Zhang, N. Rui, X. Hu and C.-J. Liu, Appl. Catal., B, 2019, 244, 159–169 CrossRef CAS.
- J. Huang, Y. Yan, S. Saqline, W. Liu and B. Liu, Appl. Catal., B, 2020, 275, 119109 CrossRef CAS.
- J. Chang, X. Song, C. Yu, J. Yu, Y. Ding, C. Yao, Z. Zhao and J. Qiu, Adv. Funct. Mater., 2020, 30, 2006270 CrossRef CAS.
- Z. Luan and J. A. Fournier, Microporous Mesoporous Mater., 2005, 79, 235–240 CrossRef CAS.
- L. Xie, Y. Chai, L. Sun, W. Dai, G. Wu, N. Guan and L. Li, J. Energy Chem., 2021, 57, 92–98 CrossRef CAS.
- S. Dzwigaj, P. Massiani, A. Davidson and M. Che, J. Mol. Catal. A: Chem., 2000, 155, 169–182 CrossRef CAS.
- D. Scarano, A. Zecchina, S. Bordiga, F. Geobaldo and G. Spoto, J. Chem. Soc., Faraday Trans., 1993, 89, 4123–4130 RSC.
- J. McCaig and H. H. Lamb, Catalysts, 2022, 12, 824 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ESI is available and includes full details of post-synthetic modification of zeolites, catalyst synthesis, computation details, catalytic experiments, characterization techniques, presentation and brief discussion of the supplementary characterization data, and comparison of the physiochemical properties and catalytic performance of the developed catalysts with the relevant state-of-the-arts. See DOI: https://doi.org/10.1039/d3cy01334k |
| This journal is © The Royal Society of Chemistry 2024 |