
Denitrogenation of tosylhydrazones: synthesis of aryl alkyl sulfones catalyzed by a phenalenyl-based molecule†
Shiv
Kumar‡
,
Paramita
Datta‡
,
Anup
Bhunia
* and
Swadhin K.
Mandal
 *
*
Department of Chemical Sciences, Indian Institute of Science Education and Research – Kolkata, Mohanpur-741246, India. E-mail: bhunia1998@gmail.com; swadhin.mandal@iiserkol.ac.in
First published on 8th November 2023
Abstract
Sulfur-containing organic molecules such as sulfones are prevalent in pharmaceuticals and agrochemicals. Herein, we report the first transition-metal-free process for in situ denitrogenation of tosylhydrazones to produce a diverse array of aryl alkyl sulfones. We have used phenalenyl-based odd alternant hydrocarbon as a photoredox catalyst for denitrogenation of tosylhydrazones, where the phenalenyl acts as a potent oxidant to form a sulfinate radical intermediate from a sulfinate anion. We performed radical trapping, in situ electron paramagnetic resonance (EPR), and fluorescence quenching studies to elucidate the plausible mechanism. This method shows wide functional-group tolerance, with applications in late-stage modification of various natural products with good to high yields. The protocol provides an easy method for the synthesis of sulfones.
Introduction
Sulfones are core to various synthetic intermediates1–3 and pharmacophores,4,5 and are functional keystones in materials science applications6,7 and agrochemicals.8 Their metabolic and physicochemical stability, as well as distinctive chemical reactivity transmitted by the sulfonyl group, have resulted in a growing number of applications that require facile and expeditious synthetic access to structurally diverse sulfone-containing molecules.9–11 For example, molecules used to treat a range of diseases, such as lung cancer,12 arthritis,13 migraines, and prostate cancer,14 all feature sulfone units (Scheme 1a). Therefore, the synthesis of sulfones has attracted appreciable attention in recent years.15–17 A variety of methods are available for the synthesis of sulfones, including several transition-metal catalyzed approaches.18–20 Sulfones are commonly synthesized either via oxidation of the corresponding sulfide or sulfoxide with various metal catalysts (W, Ti, or Ru) in the presence of excess amounts of oxidizing reagents,21–25 or via alkylation of sulfinate salts.26 However, both methods suffer serious drawbacks due to the use of large excesses (3–4 equiv.) of oxidizing reagents. In addition, the starting material is often a thiol, many of which are foul-smelling. Moreover, many of these classical syntheses are either multi-step or engage harsh and non-selective reagents, which remarkably decreases their utility. Along with this, the synthesis of sulfones commonly involves a reaction with excess sulfur dioxide, a harmful gas.27 Importantly, synthetic manipulations with sulfur dioxide require high-pressure equipment or preparation of a stock solution in CH3CN, DMSO, or DMF at a low temperature (+4 °C).17,28,29 These drawbacks have inspired a search for reagents that can serve as reasonable and safe sulfur-dioxide equivalents.30,31 Although DABSO is a bench-stable reagent, it is expensive and generates a stoichiometric amount of amine, which limits its practical applicability. Therefore, the development of an efficient and green method that can generate a variety of sulfone sources under mild reaction conditions is an optimal goal.To date, only a few methods have been reported for accessing aryl alkyl sulfones by utilizing tosylhydrazones as the source of the sulfur dioxide unit. In addition, tosylhydrazones are enormously important intermediates in organic synthesis, considering their role as precursors for the in situ synthesis of diazo compounds32–34 and metal carbenoids35,36 in various transition-metal catalyzed XH (X = C, N, O) insertion reactions,37,38 cyclopropanations,39,40etc. In this regard, substantial progress in metal carbenoid intermediates has been demonstrated by the groups of Yu,41 Valdés,42 and Zhao43 using catalysts based on transition metals, such as copper and iron, to access aryl alkyl sulfones via denitrogenation of tosylhydrazones (Scheme 1b, top). More recently, a photocatalytic strategy using an Ir catalyst44 has been developed to achieve denitrogenative sulfonylation of tosylhydrazones to furnish aryl alkyl sulfones (Scheme 1b, bottom). The proposed intermediate of the reaction, a carbene radical cation generated from carbene by a photooxidant, is an elusive intermediate due to the higher electrophilicity of the carbene intermediate. Our investigation began with a search for a transition metal-free alternative for efficient denitrogenation of tosylhydrazones to rapidly access value-added aryl alkyl sulfones. In this regard, we introduce a redox-active phenalenyl-based molecule as a catalyst. For over six decades, the odd alternant hydrocarbon phenalenyl (PLY) has been well-known for its applicability in constructing highly conducting materials based on its neutral radical state,45,46 its ability to act as a spin simulator,47,48 and its applicability in spin electronic devices,49 taking advantage of its easily accessible empty nonbonding molecular orbital (NBMO).50 Over the last decade, our group has successfully developed a number of catalytic redox processes of the PLY system through a chemo-redox pathway under homogeneous51–53 and heterogeneous conditions.54 Furthermore, very recently, our group reported the photochemical55 properties of PLY. In continuation of our program, we further sought to explore the application of PLY systems in photoredox catalysis, in particular for the development of the first transition-metal-free catalytic denitrogenation of tosylhydrazones to synthesize aryl alkyl sulfones.
In this work, we report a transition-metal-free denitrogenative transformation of tosylhydrazones to aryl alkyl sulfones (Scheme 1c). The tosylhydrazone undergoes base-mediated fragmentation to form a sulfinate radical and a (diazomethyl)arene, which eventually undergoes photoinduced denitrogenation to form a transient carbene intermediate. The in situ generated carbene intermediate and sulfinate radical then recombine to deliver the alkyl sulfone molecule. This redox-neutral protocol exhibits broad substrate scope and wide functional-group compatibility, enabling the late-stage modification of complex natural products and biologically active molecules. To probe the reactive intermediates involved in the catalytic steps, a series of investigations, including radical inhibition, trapping of intermediates, in situ electron paramagnetic resonance (EPR) studies and steady-state spectroscopic studies, have been performed.
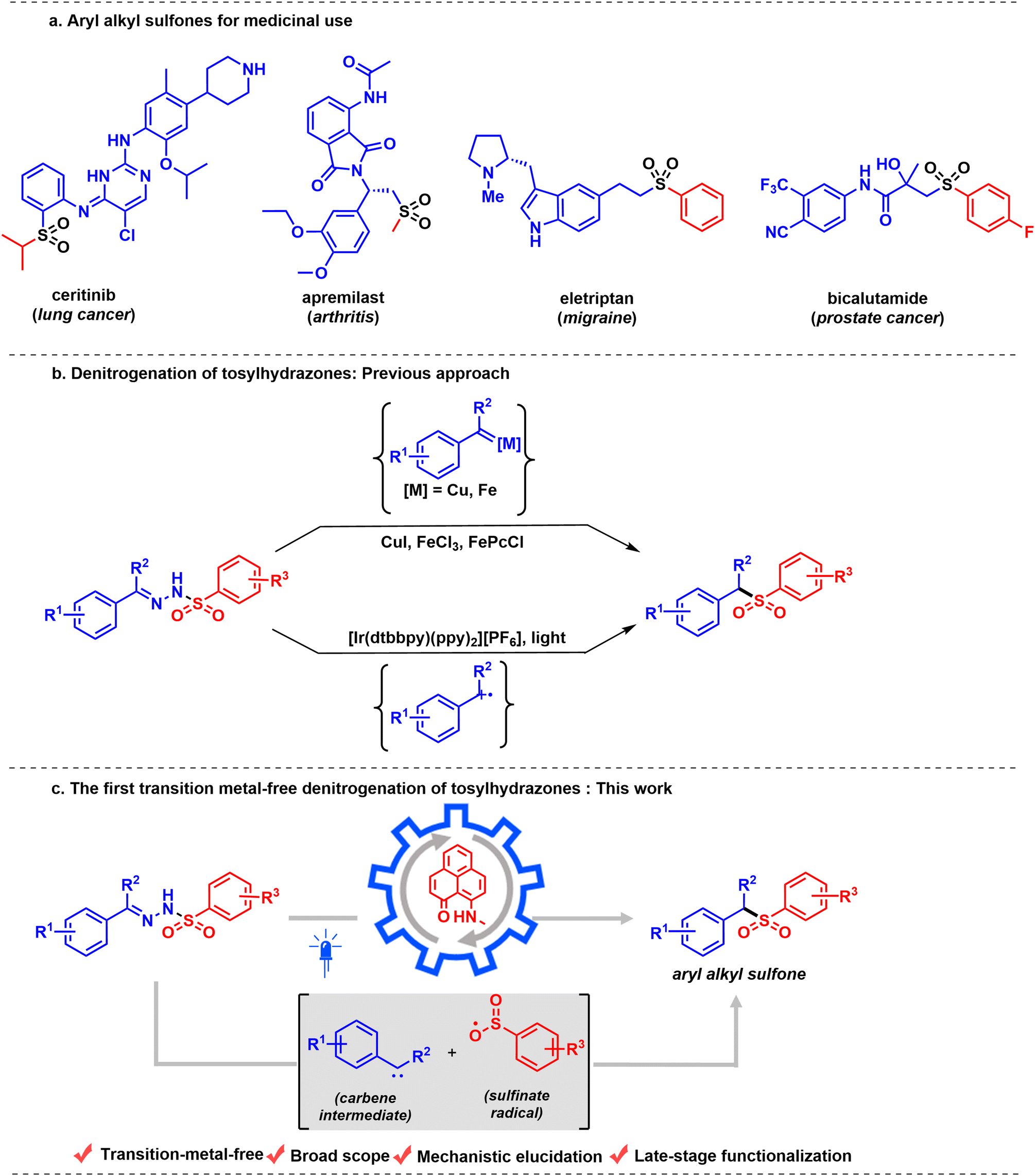 | ||
| Scheme 1 Strategies for synthesis of aryl alkyl sulfones via denitrogenative sulfonylation of tosylhydrazones. (a) Aryl alkyl sulfones for medicinal use. (b) Denitrogenation of tosylhydrazones: previous approaches. (c) This work: the first transition-metal-free denitrogenation of tosylhydrazones using a PLY-based photocatalyst. | ||
Results and discussion
At first, the reaction optimization was conducted with tosylhydrazone 1a as the substrate (Table 1). After several trials, we were pleased to note that the photoirradiation of a mixture of 1a, PLY(N,O) (5 mol%) and K2HPO4 (3.0 equiv.) in DMF (1.5 mL, 0.2 M) under argon atmosphere with a blue light-emitting diode (λ = 456 nm, Kessil lamp, 40 W) for 12 h provided aryl alkyl sulfone 2a in 87% yield (entry 1, Table 1), along with 2a′ (6%). The N-alkylated tosylhydrazone (2a′) was the side product obtained due to the insertion of the in situ generated carbene into the N–H bond of the substrate (for details, see the mechanistic part, Scheme 3b). PLY(N,O) turned out to be the ideal catalyst (Table 1, entry 1) among the other photocatalysts examined (see Table 1), under irradiation with light of 456 nm wavelength. The reaction was slightly less efficient (77%) at a shorter wavelength (440 nm, entry 2). Other PLY derivatives such as PLY(O,O) gave inferior results (70%, entry 3, Table 1). In addition, a range of well-established organic photocatalytic systems, namely 4CzIPN (entry 4), an acridinium salt (entry 5), 10-phenylphenothiazine (PTH, entry 6), eosin Y (entry 7) and rose bengal (entry 8), also led to lesser yields as compared to PLY(N,O) under the optimized reaction conditions (entry 1, Table 1).56,57 Changing the solvent to DMSO resulted in a lesser yield (64%, entry 9). The reaction without PLY(N,O), while keeping other parameters unchanged from entry 1, resulted in no product formation (entry 10). A control experiment ruled out the ground-state reaction of the PLY photocatalyst in the absence of light (entry 11, Table 1). Moreover, the omission of the base resulted in a drastically reduced yield (12%, entry 12). Finally, a diminished yield was observed when the reaction was carried out in open air (entry 13), pointing to the detrimental effect of oxygen (for detailed optimization studies, see the ESI†).| a Reaction conditions: tosylhydrazone (1a) (0.3 mmol, 1 equiv.), PLY(N,O) (5 mol%), K2HPO4 (3 equiv.), DMF (1.5 mL, 0.2 M), for 12 h under blue LED irradiation (456 nm). The yields are determined by 1H-NMR analysis (CDCl3) of crude products using 1,4-dimethoxybenzene as the internal standard. b Isolated yield. |
|---|
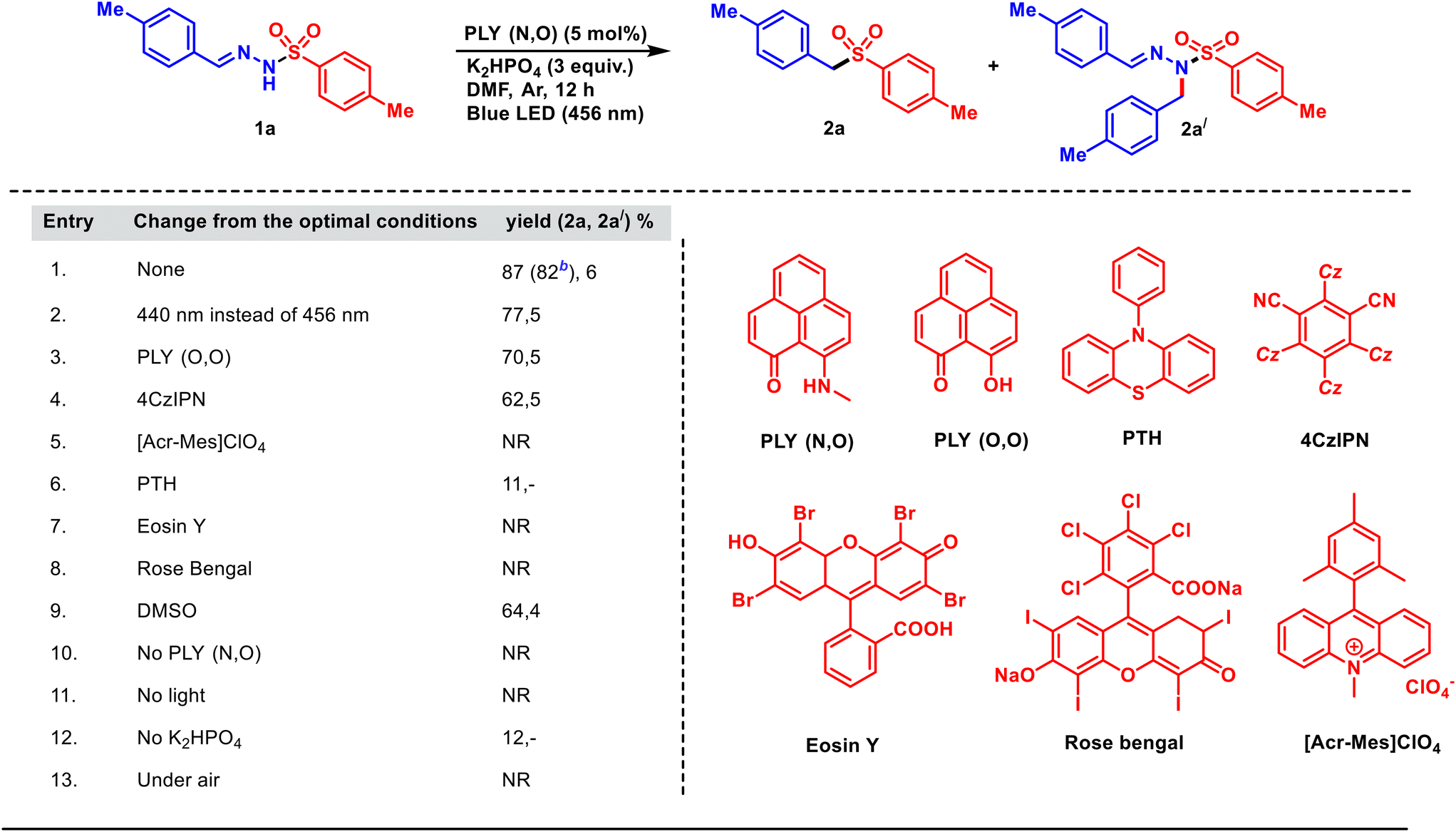
|
The simplicity of the direct denitrogenative sulfonylation of tosylhydrazones under transition-metal-free conditions provided us an opportunity to carry out further evaluation of the substrate scope, which revealed broad compatibility and efficiency of the sulfonylation with a wide array of functionalized tosylhydrazones (e.g., 1a–1aa, Scheme 2). Tosylhydrazones bearing electron-donating alkyl and alkoxy groups at the para-position were converted to sulfones (2a–2e) in good to excellent yields (67–82%). Under similar reaction conditions, 3,4-substituted tosylhydrazone (1f) furnished a good isolated yield (66%) of the corresponding substituted sulfone (2f). The standard catalytic reaction protocol was also applicable for the more sterically encumbered 2-methyl tosylhydrazone derivative (1g), which provided a good yield (2g, 58%). Further, we investigated whether the position of the substituents in the tosylhydrazones had any influence on the outcome of the transition-metal-free denitrogenative sulfonylation process. Accordingly, the 3-methyl tosylhydrazone derivative (1h) was treated under identical reaction conditions, and the corresponding sulfone (2h) was isolated in 59% yield. Notably, the 4-phenyl tosylhydrazone derivative (1i), under the optimized reaction conditions, furnished a good yield of the corresponding product (2i, 66%). Polycyclic derivatives were also compatible with this catalytic method, with the alpha-naphthaldehyde- and 9-anthracenecarboxaldehyde-substituted tosylhydrazone derivatives (1j and 1k) producing the corresponding sulfones (2j and 2k) in moderate yields (38–61%). Furthermore, electron-withdrawing substituents at the para-position were also well tolerated, including fluoro (1l), chloro (1m), bromo (1n) and trifluoromethyl (1o) groups, producing moderate to good yields of the corresponding aryl alkyl sulfones (2l–2o, 54–66%). Furthermore, a tosyl hydrazone derivative bearing an electron-withdrawing substituent at the ortho-position (1p) was also well-tolerated (2p, 47%), indicating that the protocol is not particularly sensitive to steric hindrance.
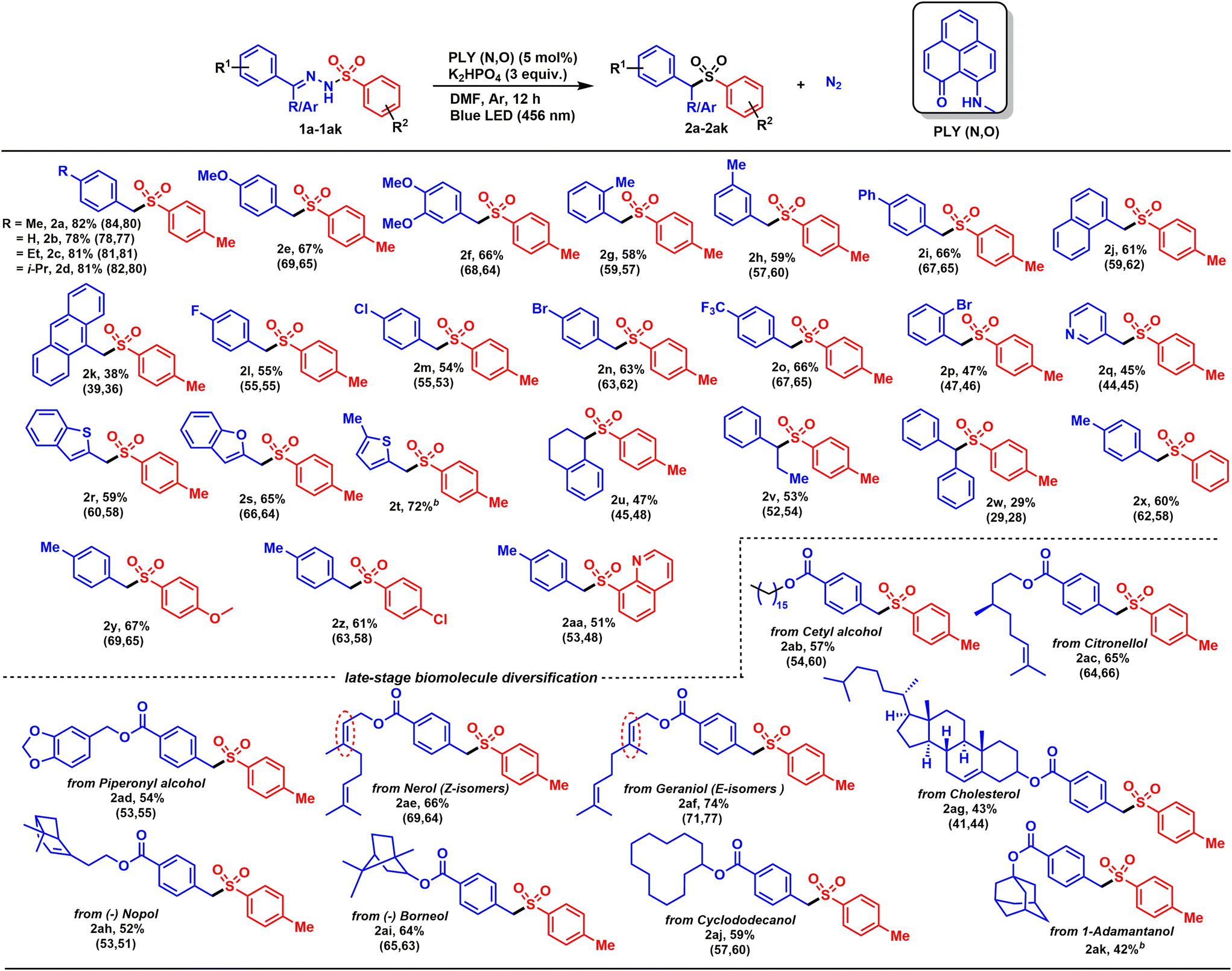 | ||
| Scheme 2 Scope of the visible light-induced PLY(N,O)-catalyzed denitrogenation of tosylhydrazones:a synthesis of aryl alkyl sulfones. aReaction conditions: tosylhydrazones (1a–ak) (0.3 mmol, 1 equiv.), PLY(N,O) (5 mol%), K2HPO4 (3 equiv.), DMF (1.5 mL, 0.2 M), for 12 h under blue-LED irradiation (456 nm). Average isolated yields from two catalytic runs (yields of independent catalytic runs are shown in the parentheses) are presented. bYield obtained from one catalytic run. | ||
To our delight, the scope of sulfone synthesis can be extended to a wide array of heterocyclic tosylhydrazones. A nitrogenous heterocycle with a pyridine moiety provided the corresponding sulfone (2q) in a moderate yield (45%). Similarly, other heterocyclic motifs can also be readily applied, including benzothiophene (1r), benzofuran (1s), and 5-methyl thiophene (1t) substituted tosylhydrazones, giving moderate to good yields (59–72%). To document the generality of the denitrogentive sulfonylation reaction, we treated tosylhydrazones derived from both cyclic and acyclic ketones, such as tetralone (1u), propiophenone (1v), and benzophenone (1w), under the optimized reaction conditions. All the substrates worked efficiently to derive the desired products (2u–2w) in moderate to good yields (29–53%). In addition, substituent variation at the aryl ring of the sulfonyl partner from neutral (Ph, 1x), to electron rich (OMe, 1y), electron deficient (Cl, 1z), and even heterocyclic compound (quinoline, 1aa) was tolerated well to give the corresponding products in good yields (2x–2aa, 51–67%).
The scope and functional group tolerance of the reaction was further examined with more structurally complex substrates including natural products and active pharmaceutical ingredients (Scheme 2). Notably, a primary aliphatic alcohol, namely a cetyl-alcohol-derived tosylhydrazone (1ab) was also a viable candidate, giving a 57% yield of the corresponding product (2ab). Meanwhile, a (±)-citronellol-derived tosylhydrazone (1ac), with an isolated tri-substituted double bond, remains intact in the final product (2ac), giving a satisfactory yield (65%). Given the importance of diverse natural products, tosylhydrazone (1ad) (derived from piperonyl alcohol, commonly used as an anti-oxidant) delivered a 54% yield of 2ad. In addition, nerol and geraniol-based tosylhydrazones were effectively used in this transformation, and moderate to good yields (2ae and 2af, 66–74%) were obtained. Likewise, a cholesterol-derived tosylhydrazone (1ag) was also a well-tolerated substrate and a moderate yield (43%) of the corresponding sulfone (2ag) was obtained. Further to this, derivatives of biologically relevant nopol and borneol-substituted tosylhydrazones (1ah and 1ai) were also efficiently converted to the corresponding sulfones (2ah and 2ai), highlighting the broad scope of the reaction. Furthermore, the sulfone derivative of a cyclododecanol-derived tosylhydrazone (2aj, 59%) was produced, revealing a straightforward route to biologically important aryl alkyl sulfone derivative. Next, to realize the general applicability of the catalytic protocol, a more inert aliphatic-alcohol-derived tosylhydrazone was explored. In addition to this, tosylhydrazone obtained from 1-adamantanol, treated under the standard reaction conditions, provided a 42% isolated yield of the corresponding product (2ak).
Next, to shed light on the reaction mechanism, a series of control experiments (Scheme 3) and spectroscopic investigations (Fig. 1) were conducted. The in situ deprotonation of 1a in the presence of K2HPO4 resulted in the formation of the diazo intermediate 3 (Scheme 3a), which was characterized by 1H NMR and HRMS.58 This experiment unarguably validates the formation of the diazo fragment in the presence of only base.59,60 Furthermore, during optimization, a carbene insertion product (2a′), was obtained as a minor product (Scheme 3b), which was characterized by NMR (1H, 13C) and HRMS. The formation of 2a′ can be explained by denitrogenation of 3 in the presence of light to generate a carbene,61–63 which undergoes an insertion reaction into the N–H bond of the substrate. Furthermore, for the generation of 2a′ from 1a, the presence of a blue LED and base are sufficient, and it does not require the PLY(N,O) catalyst (see the ESI†). These control experiments suggest that base-mediated formation of the diazo intermediate 3 triggers light-induced in situ carbene generation, which does not require the presence of the catalyst.
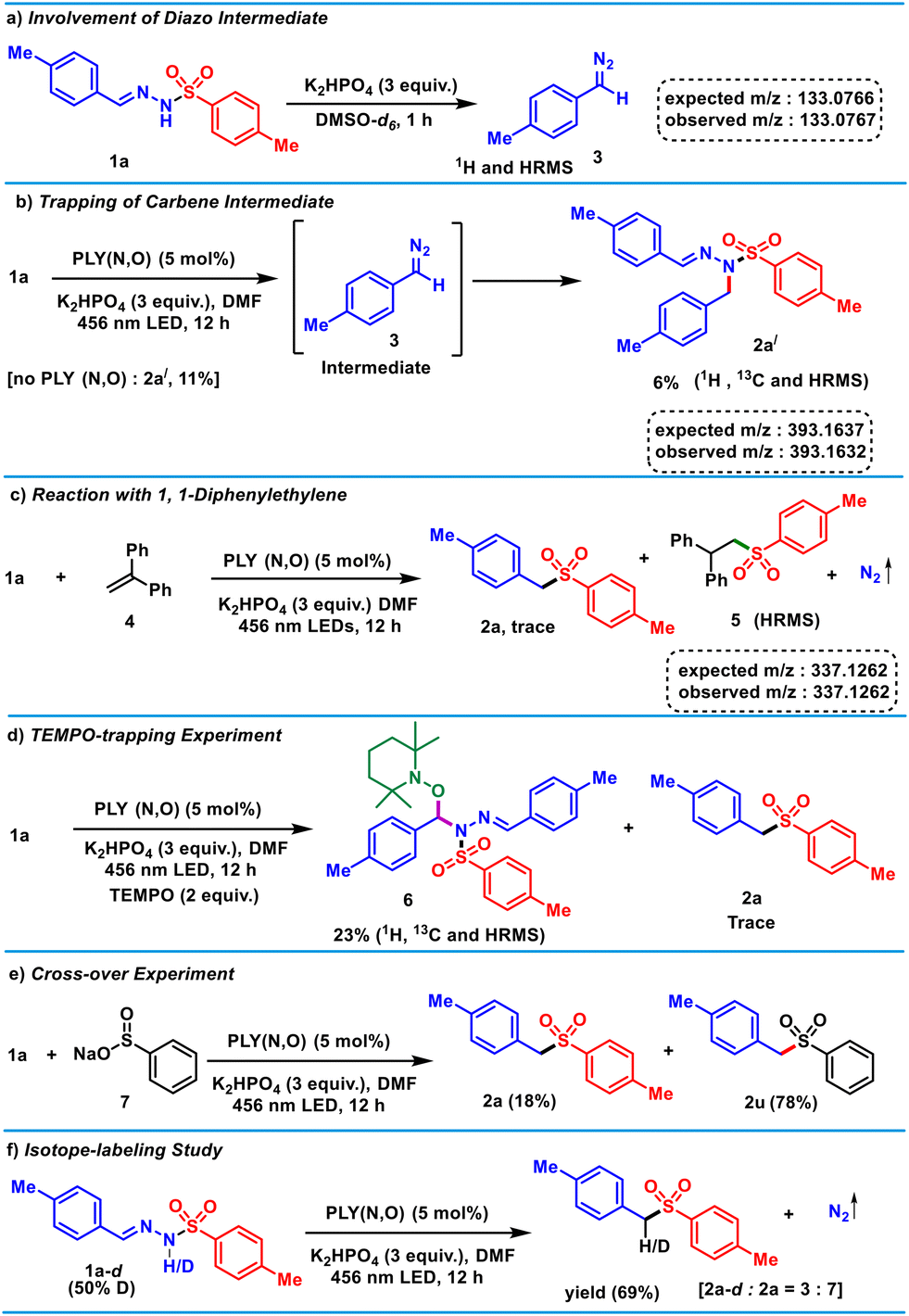 | ||
| Scheme 3 Control experiments and mechanistic investigations. | ||
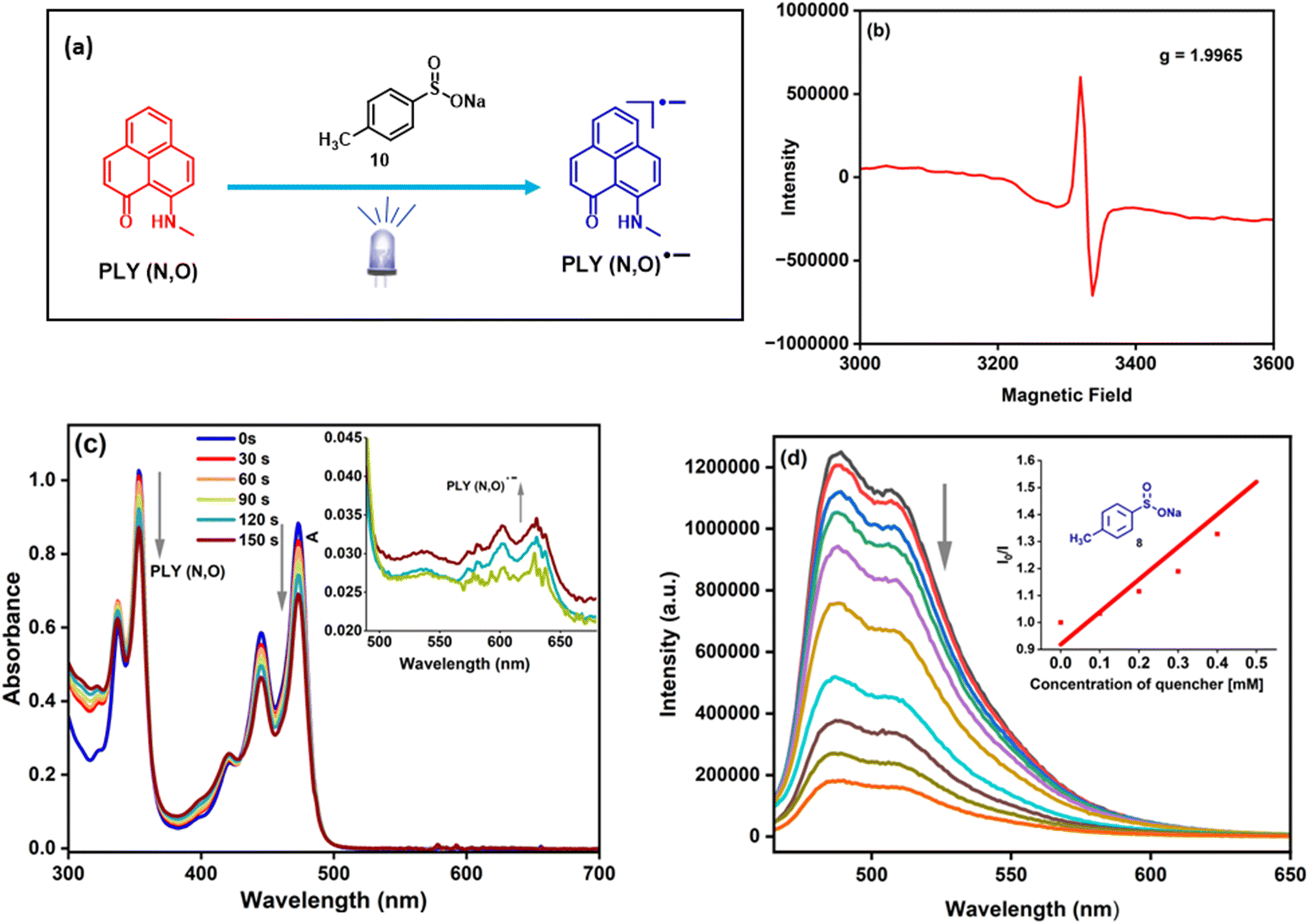 | ||
| Fig. 1 (a) Photo-induced generation of the PLY radical anion. (b) The EPR spectrum of the PLY(N,O) radical anion. (c) Absorption spectra of the in situ generated PLY(N,O) radical anion. (d) Fluorescence quenching study between PLY(N,O) and 8 (inset: Stern–Volmer luminescence quenching experiment). | ||
In addition, the use of the radical acceptor diphenylethylene (4) with 1a under standard reaction conditions provided the desired product 2a in <5% yield along with the sulfinate-radical-trapped product 5 (Scheme 3c), which was detected via HRMS [m/z 337.1262 (observed), 337.1262 (expected)]. This observation suggests the generation of a sulfinate radical during the PLY(N,O)-catalyzed denitrogenative sulfonylation reaction. Moreover, the formation of 2a was completely suppressed in the presence of 2.0 equivalents of TEMPO, which hinted at a radical-mediated pathway. The TEMPO-trapped hydrazone-tethered O-benzylhydroxylamine (6) was formed via a benzyl carbenoid (Scheme 3d),64 which was detected by 1H and 13C NMR spectroscopy, as well as HRMS [m/z 570.2762 (observed), 570.2766 (expected)]. Furthermore, a cross-over experiment with substrate 1a and benzenesulfinic acid sodium salt 7 under the standard conditions led to the formation of alkyl sulfones 2a and 2u in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.3 ratio, suggesting the sulfinate anion was oxidized in the presence of the PLY to form the sulfinate radical and the reaction proceeded via a stepwise mechanism (Scheme 3e).
:4.3 ratio, suggesting the sulfinate anion was oxidized in the presence of the PLY to form the sulfinate radical and the reaction proceeded via a stepwise mechanism (Scheme 3e).
Next, isotope-labelling studies were performed to investigate the hydrogen source of the product. First, denitrogenative sulfonylation of tosylhydrazone 1a was examined under standard reaction conditions in the presence of DMSO-d6 as the solvent, yet no deuterium was incorporated (see the ESI†). However, the monodeuterated product 2a-d along with 2a were formed in a ratio of 1:3 in the presence of CD3OD (see the ESI† for details). This result indicates that CD3OD provides a proton at the benzylic position of sulfone 2a in the photocatalytic reaction (see the ESI†). Along this line, when D-containing 1a-d was used under the optimized conditions, 2a-d and 2a were formed in a ratio of 3:7 (Scheme 3f).
Next, we focused on the spectroscopic investigations of PLY by performing further control experiments. A mixture of PLY(N,O) and sulfinate (in DMF at 77 K) in the absence of light irradiation remains EPR-silent. However, after blue-LED illumination, a sharp signal was observed in the EPR spectrum with g = 1.996, which supports the generation of a PLY radical anion55,65 (Fig. 1a and b). These observations demonstrate a photoinduced electron transfer (PET) between sodium p-toluenesulfinate and the photoexcited state of PLY(N,O). This is further corroborated by an in situ absorption experiment of sodium sulfinate and PLY(N,O) under light irradiation (456 nm) for different time intervals (Fig. 1c). The absorption peaks corresponding to free PLY(N,O) undergo a sharp decline in intensity over time, followed by the appearance of a new band in the region of 550–650 nm, consistent with the generation of PLY-based radical species.55 This finding indicates the interaction between sodium sulfinate and PLY(N,O) upon irradiation, resulting in a PET from sodium sulfinate to the excited state of PLY(N,O), forming the PLY-based radical anion. Furthermore, Stern–Volmer quenching experiments (Fig. 1d) revealed that sodium p-toluenesulfinate (8) could quench the excited state of PLY(N,O) with a much higher rate constant in comparison to other reagents, such as a diazo compound, 1a and K2HPO4 (see the ESI† for details). These results suggest that the reaction proceeds through a reductive quenching process, in which the sulfinate anion undergoes oxidation by the photoexcited state of PLY(N,O). Finally, the quantum yield determination (Φ = 0.14) for this model denitrogenative sulfonylation reaction of tosylhydrazones disproved a chain mechanism (see the ESI† for details).
Based on the above experiments, a plausible mechanism is depicted in Scheme 4. The deprotonation of 1a in the presence of K2HPO4 takes place to produce nitrogen anion intermediate 9 followed by desulfonylation to provide sulfinate anion 10 and diazo intermediate 3.58 Next, in the presence of light, denitrogenation of intermediate 3 affords the corresponding carbene intermediate 12.62 In addition, light irradiation promotes PLY(N,O) to the excited state, which then oxidizes sulfinate anion 10, through a PET process, generating PLY(N,O)˙− and sulfinate radical intermediate 11. The resulting sulfinate radical intermediate 11 combines with 12, generating the benzylic radical species 13. The benzyl radical of intermediate 13 has been trapped with TEMPO and the corresponding mass of the compound was detected by HRMS (see the ESI† for details). In the next step, single electron transfer from the reduced PLY(N,O)˙− to intermediate 13 produces benzyl anion 14. Finally, the protonation of anion 14 furnishes the desired aryl alkyl sulfone product 2a.
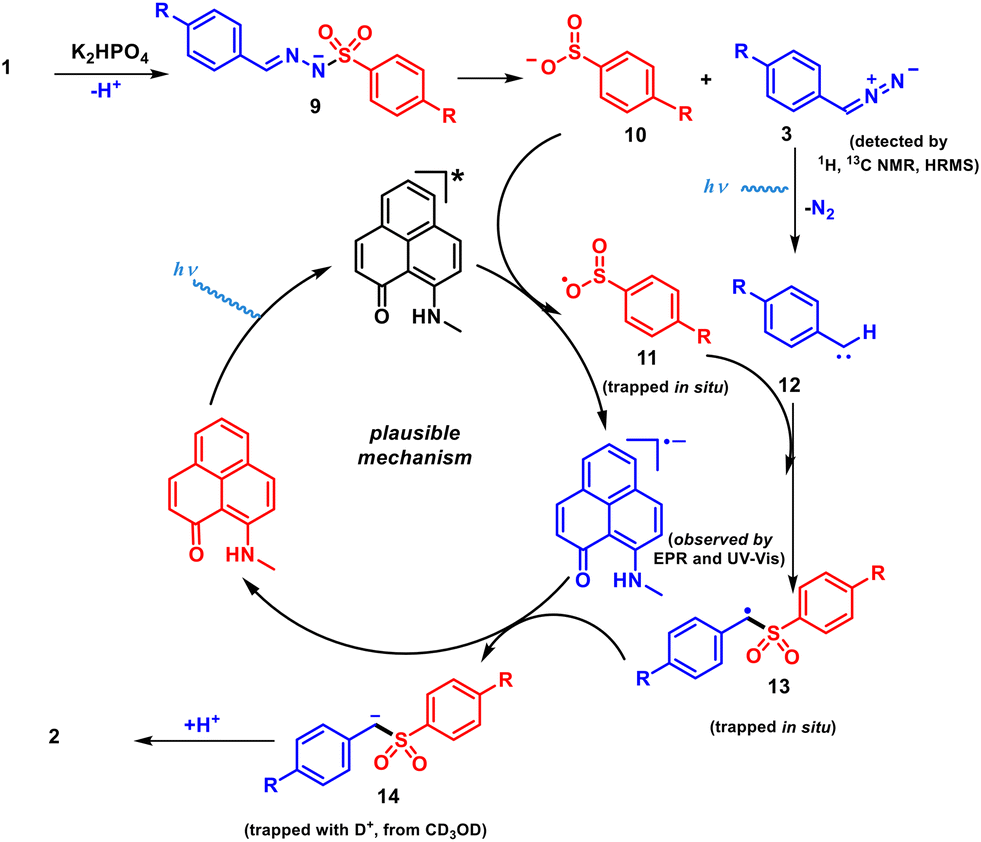 | ||
| Scheme 4 Proposed reaction mechanism. | ||
The synthetic utility of the aryl alkyl sulfones has been demonstrated by deuterium labelling of the active methylene group (2a-d2). In addition, the dichloro-substituted sulfone 15 has been derived from 2a using hexachloroethane. Both reactions proceed via the base-induced deprotonation and substitution of two protons of the active methylene group. Moreover, the active methylene position has been functionalized with an aldehyde to form the β-hydroxy sulfone derivative 16 with a moderate yield and diastereoselectivity. Activated aryl alkyl sulfones are well-known reagents for vinylation reactions. Treatment of sulfone 2a with benzyl alcohol in the presence of a strong base, namely NaH, resulted in the formation of stilbene 17 (see the ESI†) (Scheme 5).
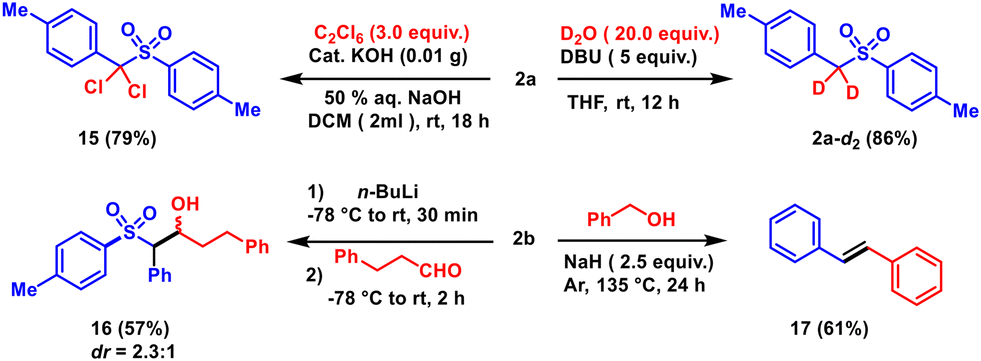 | ||
| Scheme 5 Synthetic applications. | ||
Conclusion
In summary, we report a highly efficient, straightforward, transition-metal-free, photocatalytic process for the denitrogenative sulfonylation of tosylhydrazones, producing a diverse range of aryl alkyl sulfones, catalyzed by a redox-active phenalenyl molecule. This protocol allows the transformation of various tosylhydrazones with a broad functional group tolerance. Furthermore, a variety of biologically active compounds could be functionalized using this protocol. The mechanistic investigation indicated the involvement of sulfinate radicals and diazo intermediates through a radical-mediated pathway and this is supported by various control experiments, an in situ EPR study, and a fluorescence quenching study. Overall, this protocol is a significant addition to the existing protocols for the synthesis of aryl alkyl sulfones.Data availability
All experimental procedures, characterization details, copies of NMR spectra for all compounds, and other data related to this article have been uploaded as part of the ESI.†Author contributions
Original approaches for this result was conceived by SKM. The experiments were designed by PD, AB, and SKM. All the experiments were carried out by SK and PD. The manuscript was written by PD with contributions from all authors. All authors have given approval to the final version of the manuscript. SKM and AB supervised the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the SERB, India (Grant No. CRG/2022/000471) for financial support. Anup Bhunia thanks the DST SERB for the Ramanujan Fellowship Grant (RJF/2020/000099). S. K. thanks IISER – Kolkata for a research fellowship. P. D. thanks the CSIR (09/921(0232)/2019)-EMR-I, for providing a fellowship during research tenure.Notes and references
- S. Patai, The Chemistry of Sulfinic Acids, Esters and Their Derivatives, John Wiley & Sons, Ltd, New Jersey, 1990 Search PubMed.
- C. Aïssa, Eur. J. Org. Chem., 2009, 1831–1844 CrossRef.
- X.-Q. Chu, D. Ge, Y.-Y. Cui, Z.-L. Shen and C.-J. Li, Chem. Rev., 2021, 121, 12548–12680 CrossRef CAS PubMed.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed.
- K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef.
- A. Kausar, S. Zulfiqar and M. I. Sarwar, Polym. Rev., 2014, 54, 185–267 CrossRef CAS.
- W. K. Bell, B. M. Rawlings, B. K. Long, R. C. Webb, B. K. Keitz, L. Häußling and C. G. Willson, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2769–2775 CrossRef CAS.
- A. Plant, J. E. Boehmer, J. Black and T. D. Sparks, WO 2006024820, 2006, Chem. Abst., 2006, 144, 274262v.
- M. W. Johnson, S. W. Bagley, N. P. Mankad, R. G. Bergman, V. Mascitti and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 4404–4407 CrossRef CAS PubMed.
- E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2014, 53, 10204–10208 CrossRef CAS PubMed.
- S. Jin, G. C. Haug, R. Trevino, V. D. Nguyen, H. D. Arman and O. V. Larionov, Chem. Sci., 2021, 12, 13914–13921 RSC.
- T. H. Marsilje, W. Pei, B. Chen, W. Lu, T. Uno, Y. Jin, T. Jiang, S. Kim, N. Li, M. Warmuth, Y. Sarkisova, F. Sun, A. Steffy, A. C. Pferdekamper, A. G. Li, S. B. Joseph, Y. Kim, B. Liu, T. Tuntland, X. Cui, N. S. Gray, R. Steensma, Y. Wan, J. Jiang, G. Chopiuk, J. Li, W. P. Gordon, W. Richmond, K. Johnson, J. Chang, T. Groessl, Y.-Q. He, A. Phimister, A. Aycinena, C. C. Lee, B. Bursulaya, D. S. Karanewsky, H. M. Seidel, J. L. Harris and P.-Y. Michellys, J. Med. Chem., 2013, 56, 5675–5690 CrossRef CAS.
- H. W. Man, P. Schafer, L. M. Wong, R. T. Patterson, L. G. Corral, H. Raymon, K. Blease, J. Leisten, M. A. Shirley, Y. Tang and D. M. Babusis, J. Med. Chem., 2009, 52, 1522–1524 CrossRef CAS PubMed.
- H. Tucker and G. J. Chesterson, J. Med. Chem., 1988, 31, 885–887 CrossRef CAS PubMed.
- G. Manolikakes, N.-W. Liu and S. Liang, Synthesis, 2016, 48, 1939–1973 CrossRef.
- G. Qiu, K. Zhou, L. Gao and J. Wu, Org. Chem. Front., 2018, 5, 691–705 RSC.
- S. P. Blum, K. Hofman, G. Manolikakes and S. R. Waldvogel, Chem. Commun., 2021, 57, 8236–8249 RSC.
- O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS PubMed.
- X. Zhao, E. Dimitrijevic and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 3466–3467 CrossRef CAS.
- M. Ueda and J. F. Hartwig, Org. Lett., 2010, 12, 92–94 CrossRef CAS.
- B. M. Trost and R. Braslau, J. Org. Chem., 1988, 53, 532–537 CrossRef CAS.
- K. Sato, M. Hyodo, M. Aoki, X.-Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469–2476 CrossRef CAS.
- P. H. J. Carlsen, T. Katsuki, V. S. Martin and K. B. Sharpless, J. Org. Chem., 1981, 46, 3936–3938 CrossRef CAS.
- C. M. Rodriguez, J. M. Ode, J. M. Palazon and V. S. Martin, Tetrahedron, 1992, 48, 3571–3576 CrossRef CAS.
- K. P. Bryliakov and E. P. Talsi, Eur. J. Org. Chem., 2008, 3369–3376 CrossRef CAS.
- Y. Ju, D. Kumar and R. S. Varma, J. Org. Chem., 2006, 71, 6697–6700 CrossRef CAS PubMed.
- X.-B. Wang, J.-B. Du and H. Cui, Life Sci., 2014, 98, 63–67 CrossRef CAS.
- W. F. Giauque and C. C. Stephenson, J. Am. Chem. Soc., 1938, 60, 1389–1394 CrossRef CAS.
- J. Luginina, J. Uzulena, D. Posevins and M. Turks, Eur. J. Org. Chem., 2016, 1760–1771 CrossRef CAS.
- A. S. Deeming, E. J. Emmett, C. S. Richards-Taylor and M. C. Wills, Synthesis, 2014, 46, 2701–2710 CrossRef.
- E. J. Emmett and M. C. Wills, Asian J. Org. Chem., 2015, 4, 602–611 CrossRef CAS.
- J. R. Fulton, V. K. Aggarwal and J. De Vicente, Eur. J. Org. Chem., 2005, 2005, 1479–1492 CrossRef.
- E. Levesque, S. T. Laporte and A. B. Charette, Angew. Chem., Int. Ed., 2017, 56, 837–841 CrossRef CAS.
- D. Arunprasath, B. Devi Bala and G. Sekar, Adv. Synth. Catal., 2019, 361, 1172–1207 CrossRef CAS.
- W. R. Bamford and T. S. Steven, J. Chem. Soc., 1952, 4735–4740 RSC.
- G. M. Kaufman, J. A. Smith and G. G. Vender Stouw, J. Am. Chem. Soc., 1965, 87, 935–937 CrossRef CAS.
- E. Aller, D. S. Brown, G. G. Cox and D. J. Miller, J. Org. Chem., 1995, 60, 4449–4460 CrossRef CAS.
- E. Cuevas-Yanez, J. M. Serrano, G. Huerta, J. M. Muchowski and R. Cruz-Almanza, Tetrahedron, 2004, 60, 9391–9396 CrossRef CAS.
- V. K. Aggarwal, J. de Vicente and R. V. Bonnert, Org. Lett., 2001, 3, 2785–2788 CrossRef CAS PubMed.
- L. A. Adams, V. K. Aggarwal, R. V. Bonnert, B. Bressel, R. J. Cox, J. Shepherd, J. de Vicente and M. Walter, J. Org. Chem., 2003, 68, 9433–9440 CrossRef CAS PubMed.
- X.-W. Feng, J. Wang, J. Zhang, J. Yang, N. Wang and X.-Q. Yu, Org. Lett., 2010, 12, 4408–4411 CrossRef CAS PubMed.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Eur. J. Org. Chem., 2011, 1520–1526 CrossRef CAS.
- J.-L. Zhao, S.-H. Guo, J. Qiu, X.-F. Gou, C.-W. Hua and B. Chen, Tetrahedron Lett., 2016, 57, 2375–2378 CrossRef CAS.
- X. Huang, X. Chen, H. Xie, Z. Tan, H. Jiang and W. Zeng, Org. Lett., 2021, 23, 6784–6788 CrossRef CAS PubMed.
- S. K. Pal, M. E. Itkis, F. S. Tham, R. W. Reed, R. T. Oakley and R. C. Haddon, Science, 2005, 309, 281–284 CrossRef CAS PubMed.
- S. K. Mandal, S. Samanta, M. E. Itkis, D. W. Jensen, R. W. Reed, R. T. Oakley, F. S. Tham, B. Donnadieu and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 1982–1994 CrossRef CAS PubMed.
- V. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197–204 CrossRef.
- A. Ueda, S. Suzuki, K. Yoshida, K. Fukui, K. Sato, T. Takui, K. Nakasuji and Y. Morita, Angew. Chem., Int. Ed., 2013, 52, 4795–4799 CrossRef CAS PubMed.
- K. V. Raman, A. M. Kamerbeek, A. Mukherjee, N. Atodiresei, T. K. Sen, R. Lazic, V. Caciuc, R. Michel, D. Stalke, S. K. Mandal, S. Blugel, M. Munzenberg and J. S. Moodera, Nature, 2013, 493, 509–513 CrossRef CAS PubMed.
- R. C. Haddon, Nature, 1975, 256, 394–396 CrossRef CAS.
- A. Mukherjee, S. C. Sau and S. K. Mandal, Acc. Chem. Res., 2017, 50, 1679–1691 CrossRef CAS PubMed.
- J. Ahmed and S. K. Mandal, Chem. Rev., 2022, 122, 11369–11431 CrossRef CAS PubMed.
- S. Sil, A. S. Bhaskaran, S. Chakraborty, B. Singh, R. Kuniyil and S. K. Mandal, J. Am. Chem. Soc., 2022, 144, 22611–22621 CrossRef CAS PubMed.
- A. Biswas, A. Bhunia and S. K. Mandal, Chem. Sci., 2023, 14, 2606–2615 RSC.
- P. Datta, T. Goswami, N. Kandoth, A. Banik, J. Ahmed, A. S. Bhaskaran, R. Saha, R. Kuniyil, H. N. Ghosh and S. K. Mandal, ChemPhotoChem, 2023, 7, e202300033 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- A. VegaPenaloza, J. Mateos and X. Companyo, Angew. Chem., Int. Ed., 2021, 60, 1082–1097 CrossRef CAS PubMed.
- Y. Wang, X. Wen, X. Cui, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2017, 139, 1049–1052 CrossRef CAS PubMed.
- Z. Liu, P. Sivaguru, G. Zanoni and X.-N. Bi, Acc. Chem. Res., 2022, 55, 1763–1781 CrossRef CAS PubMed.
- P. Xu, W. Li and C. Zhu, Acc. Chem. Res., 2018, 51, 484–495 CrossRef CAS.
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS.
- J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9, 8895–8918 CrossRef CAS.
- Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833–6847 RSC.
- R. Chen, Y. Zhao, S. Fang, W. Long, H. Sun and X. Wan, Org. Lett., 2017, 19, 5896–5899 CrossRef CAS PubMed.
- J. Ahmed, P. Sreejyothi, G. Vijaykumar, A. Jose, M. Raj and S. K. Mandal, Chem. Sci., 2017, 8, 7798–7806 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01194a |
| ‡ PD and SK contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |