
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilizing Pd–cerium oxide–aluminum oxide catalysts for methane oxidation by reduction pretreatments†
Anil C.
Banerjee
 *a,
Laura
Proaño
b,
Alexis
Alvarez
a,
Imani
Rogers
a,
Jihyeon
Park
a,
Maddison
Montgomery
a,
Mehmet Z.
Billor
c,
Bert M.
Weckhuysen
d and
Matteo
Monai
*d
*a,
Laura
Proaño
b,
Alexis
Alvarez
a,
Imani
Rogers
a,
Jihyeon
Park
a,
Maddison
Montgomery
a,
Mehmet Z.
Billor
c,
Bert M.
Weckhuysen
d and
Matteo
Monai
*d
aDepartment of Chemistry, Columbus State University, Columbus, GA 31907, USA. E-mail: banerjee_anil@columbusstate.edu; Tel: +17065693030
bChemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
cDepartment of Geosciences, Auburn University, Auburn, AL 36849, USA
dInorganic Chemistry and Catalysis Group, Debye Institute for Nanomaterials Science and Institute for Sustainable and Circular Chemistry, Utrecht University, Universiteitsweg 99, Utrecht, CG 3584, The Netherlands. E-mail: m.monai@uu.nl
First published on 29th November 2023
Abstract
Pd–CeO2 based catalysts are state-of-the-art for methane oxidation, but deactivate due to Pd nanoparticle growth at high temperature. While encapsulation by CeO2 shells was reported to increase the stability of Pd nanoparticles (NPs), the established synthesis methods are not easily scalable. Here, we report the synthesis of PdO/CeOx/Al2O3 and CeOx/PdO/Al2O3 catalysts with improved stability for methane oxidation by sequential impregnation of Pd and Ce precursors on an Al2O3 support followed by reduction. The reduced catalysts displayed higher methane oxidation activity, and a lowering of the light-off temperature compared to the fresh calcined catalysts. High resolution transmission electron microscopy (HRTEM) and energy dispersive spectroscopy (EDS) chemical imaging of the reduced catalysts indicated a redispersion of cerium oxide on the support surface and anchoring of Pd nanoparticles by cerium oxide. After aging at high temperature (850 °C), the activity of the reduced CeOx/PdO/Al2O3 catalysts remained high, while the activity of the calcined catalysts dropped significantly. The results showed that simple reduction pretreatments could improve the catalytic activity and stability of the catalysts at both low and high reaction temperatures through changing Pd–Ce interactions, restructuring of the surface and changes in the nature and types of adsorbed species, as shown by HRTEM, EDS and operando diffuse reflectance infrared Fourier transform spectroscopy. We believe that the proposed strategy can be used as a scalable alternative to embedding of Pd NPs by CeO2 in core-shell systems for enhanced thermal stability in exhaust after treatment applications.
1. Introduction
Oxidation of methane under lean conditions in the presence of excess air is used to generate heat and electricity for industry and households, while natural gas fueled vehicles (NGV) operate near stoichiometric conditions. The exhaust from these facilities and diesel engines gives out unburned methane and carbon dioxide and contributes to air pollution. Reviews1–6 on catalytic oxidation of methane have highlighted the progress and challenges on the development of catalysts during the last forty years. Farrauto7 highlighted the challenges of finding the most suitable catalyst material for the oxidation of methane that works at low temperatures and helps to address methane emissions in automobile exhaust gas abatement systems, namely high-temperature stability against sintering and resistance to poisons, such as S-containing molecules.Among the automobile pollution control catalysts, platinum and palladium metals on aluminum oxide8–14 and cerium oxide15–17 supports have been researched extensively for methane oxidation due to their high activity. CeO2 is a known promoter of methane oxidation activity, favoring oxidation via redox cycling, Pd oxidation state modification, and oxygen activation. Since combining Al2O3 and CeO2 has the advantage of increasing the surface area, while maintaining the promotion effect of CeO2, several research studies have also reported on the catalytic oxidation of methane by palladium catalysts on dual aluminum oxide–cerium oxide supports prepared by different methods.18–24
Even for state-of-the-art Pd catalysts, the deactivation at high temperatures due to sintering and or aggregation of metal active sites remains an issue.7,23,25 To improve the thermal stability of Pd by a physical barrier, encapsulation21,23,25 and atom trapping26–28 with CeO2 were targeted, using different approaches. Even though encapsulation is a good strategy, it needs advanced catalyst synthesis methods, which are not easily scalable. It is thus worthwhile to explore whether encapsulation or anchoring of Pd by CeO2 can be achieved using a more potentially scalable method. Here, we describe a sequential impregnation method to synthesize two Pd catalysts on an aluminum oxide-cerium oxide dual support, and provide evidence on how reduction induced Pd anchoring on CeOx and lead to improved catalytic activity and stability for methane oxidation under lean, dry and high temperature operating conditions.
2. Experimental
2.1. Catalyst preparation
The catalyst materials were prepared by a modified sequential incipient wetness impregnation method14,24 followed by calcination at 850 °C. The impregnation sequence in the as-prepared catalysts is shown in Fig. 1. γ-Al2O3 (Sigma-Aldrich, 99.9%; particle size <50 nm; surface area >40 m2 g−1), Ce(NO3)3·6H2O (Sigma-Aldrich) and Pd(NO3)2·xH2O (Sigma-Aldrich) were used as precursors in the preparation of the catalyst materials. | ||
| Fig. 1 Schematic showing the impregnation sequence in the as-prepared Pd/CeOx/Al2O3 (Pd/Ce/Al-F) and CeOx/Pd/Al2O3 (Ce/Pd/Al-F) catalysts (F = fresh). | ||
For the preparation of the CeOx/γ-Al2O3 (Ce/Al) dual support, 3.036 g of cerium nitrate hexahydrate was dissolved in 2.0 mL of distilled water and transferred to an injection syringe. A slurry of solid 6.406 g γ-Al2O3 (pre-calcined at 850 °C) was made with 20 mL of distilled water in a vortex tube. The precursor solution was added to the slurry of the support in the vortex tube by a syringe pump at 20 μL min−1, and simultaneously vortexed at a speed of 900 rpm for 3 h. The slurry was dried at 110 °C for 12 h, then calcined at 850 °C for 5 h, and finally crushed to get particle sizes 53–75 μm.
For the preparation of a Pd/CeOx/γ-Al2O3 catalyst, 3.004 g solid cerium oxide–alumina dual support (Ce/Al) calcined at 850 °C were added to a vortex tube followed by addition of 5.0 mL of distilled water and vortexed to make a slurry. An aqueous solution of 0.3891 g palladium nitrate hydrate (36.4% Pd) in 1.5 mL of distilled water was transferred to an injection syringe. The palladium nitrate hydrate solution was added to the slurry of the dual support and the rest of the procedure was the same as described earlier. This Pd/CeOx/γ-Al2O3 catalyst calcined at 850 °C was designated as Pd/Ce/Al-F.
A CeOx/Pd/γ-Al2O3 catalyst was prepared by impregnating an aqueous solution of cerium nitrate hexahydrate to a slurry of Pd/γ-Al2O3. An aqueous solution of 0.989 g palladium nitrate hydrate (36.4% Pd) was added to 7.64 g γ-Al2O3 (pre-calcined at 850 °C) following the method described above to make Pd/γ-Al2O3 calcined at 850 °C. A sample of 2.14 g Pd/γ-Al2O3 850 °C was placed in a vortex tube along with 4.0 mL of distilled water and vortexed until a slurry was formed. An aqueous solution of 0.951 g cerium nitrate hexahydrate was made in 1.0 mL of distilled water and transferred to an injection syringe. The rest of the procedure was the same. This Ce/Pd/γ-Al2O3 catalyst calcined at 850 °C was designated as Ce/Pd/Al-F.
The spent catalysts (Pd/Ce/Al-S and Ce/Pd/Al-S) were prepared by reacting the fresh calcined catalysts with a gas mixture of 1% CH4, 4% O2 and balance of nitrogen at 500 °C for 1 h. The reduced catalysts (Pd/Ce/Al-R and Ce/Pd/Al-R) were made by in situ reduction of the calcined catalysts (Pd/Ce/Al-F and Ce/Pd/Al-F) in a flow of hydrogen (10% hydrogen, 90% nitrogen) at 500 °C for 1 h. Then the in situ reduced catalysts were exposed to a gas mixture of 1% CH4, 4% O2 and balance of nitrogen at 850 °C for 5 h to get the reduced/aged catalysts (Pd/Ce/Al-R-a and Ce/Pd/Al-R-a). The reduced catalysts (Pd/Ce/Al-R and Ce/Pd/Al-R) were reacted with the same gas mixture at 500 °C for 1 h to get the reduced/spent (R-S) catalyst. The fresh catalysts (Pd/Ce/Al-F and Ce/Pd/Al-F) were reacted similarly with the gas mixture at 850 °C for 5 h to make the aged catalysts (Pd/Ce/Al-a and Ce/Pd/Al-a).
The targeted content of Pd was 4.5 wt% in all the catalysts, and the cerium content was 12.2 wt% in Pd/Ce/Al-F and Ce/Pd/Al-F catalysts and 12.9 wt% in the Ce/Al dual support. The Pd wt% was kept high for characterization of Pd species. A high calcination temperature of 850 °C was chosen to test the activity and stability of the catalysts under elevated temperature conditions in a catalytic converter.
2.2. Physicochemical characterization
Elemental analysis of Pd and Ce was conducted using inductively coupled plasma optical emission spectrometry (ICP-OES) in a Perkin Elmer 5300 V spectrometer. The samples were prepared with a sodium peroxide fusion due to high % of Al2O3, followed by dissolution in water and acidification.Pulse CO chemisorption was performed in Micromeritics AutoChem II 2920 (Micromeritics Corporation). The catalyst (80 mg) was first reduced with hydrogen at 450 °C and then pulses of 10% CO/He were injected. The dispersion and particle size of the catalyst were calculated from the CO uptake, wt% of Pd loading, and assuming a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio between the absorbed and Pd-active site. For temperature-programmed reduction (TPR) with hydrogen, the catalyst (50 mg) was heated to 200 °C at a ramp rate of 5 °C min−1 and flow of 20 mL min−1 of He, held for 2 h, and then cooled to 50 °C in a 20 mL min−1 flow of 10% H2/He. The catalyst was then reduced by increasing the temperature to 800 °C at a ramp rate of 5 °C min−1 under a flow of 10% H2/He (20 mL min−1). The effluent gas flow was fed through a cold trap of liquid nitrogen and acetone and then into a thermal conductivity detector (TCD) to determine hydrogen uptake. Calculated H2 consumption was estimated using the Pd wt% load and a stoichiometry factor of 2.
:1 ratio between the absorbed and Pd-active site. For temperature-programmed reduction (TPR) with hydrogen, the catalyst (50 mg) was heated to 200 °C at a ramp rate of 5 °C min−1 and flow of 20 mL min−1 of He, held for 2 h, and then cooled to 50 °C in a 20 mL min−1 flow of 10% H2/He. The catalyst was then reduced by increasing the temperature to 800 °C at a ramp rate of 5 °C min−1 under a flow of 10% H2/He (20 mL min−1). The effluent gas flow was fed through a cold trap of liquid nitrogen and acetone and then into a thermal conductivity detector (TCD) to determine hydrogen uptake. Calculated H2 consumption was estimated using the Pd wt% load and a stoichiometry factor of 2.
Ex situ X-ray photoelectron spectroscopy (XPS) analysis was performed with a Thermo K-Alpha spectrometer (Thermo Fisher Scientific), using a monochromatic Al Kα radiation source. Spectra were measured in the regions of Pd 3d, O 1s, and Ce 3d. The X-ray beam spot size was 400 μm and the spectrometer was operated at 50 eV pass energy and 50 ms dwell time for each step size of 0.1 eV. Charge referencing was performed from the C 1s region, referenced to the adventitious carbon (at 284.8 eV) and peak deconvolution was performed using the Thermo Avantage v5.9921 software. The samples were dropped on a holy carbon-coated Cu grid.
A Hitachi HD2700 aberration-corrected scanning transmission electron microscope was used to record high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images. The X-ray energy dispersive spectroscopy (EDS) elemental mapping was acquired using a FEI Tecnai F30 (scanning) transmission electron microscope equipped with an Oxford EDS detector.
X-ray diffraction (XRD) data was obtained from a Bruker D2 Phaser Tabletop diffractometer using Cu-Kα radiation at 30 kV and 10 mA, step size 0.02 (2θ)/second and 10–90° (2θ).
Operando diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was conducted in a Harrick high temperature DRIFTS cell, using a Bruker Tensor 37 FT-IR spectrometer equipped with a MCT detector. The catalyst powders were sieved to the 150–75μm fraction and 20mg of powder was loaded in the DRIFTS cell for testing in CH4 oxidation. The total gas flow rate was kept at 50 mL min−1, using a N2 carrier, 0.5 mL min−1 CH4 and 2 mL min−1 O2, the temperature was ramped from 25 to 450 °C, with a 5 °C min−1 ramp, and a 1 h hold at 450 °C, after which the catalysts were cooled down under reaction conditions to 25 °C. After the first run, the catalysts were reduced in situ at 450 °C for 1h under 50% H2/N2, then the catalyst was tested again as in the first run. All gases were introduced through Bronkhorst EL-FLOW Mass Flow Controllers. During the experiments, DRIFTS spectra were collected with a time resolution of 1 spectrum per min.
2.3. Catalytic experiments
The catalytic performance measurements were performed following the procedure reported in our previous work.14 The methane oxidation reaction was conducted in a temperature-controlled fixed-bed catalytic reactor packed with 0.10 g of the catalyst. When the desired temperature was reached, the flow of nitrogen was stopped and a reacting gas mixture (1.0 vol% CH4, 4.0 vol% O2, N2 balance) was passed through the catalyst bed at a flow rate of 150 cm3 min−1 for 1 h. After the reaction time, a sample of 1.0 cm3 of the gas mixture from the reactor outlet was collected in a Luer-Lock gas-tight syringe and injected into a gas chromatograph (Hewlett-Packard 6850) fitted with a ‘Carbon Plot’ capillary column (Agilent) and a flame ionization detector. All activity experiments were conducted under steady state conditions with a gas hourly space velocity (GHSV) of 90000 h−1. The methane conversion (% v/v) was calculated from the vol% CH4 at 25 °C and 1.0 atm in the gas mixture before and after the reaction. The experimental error in activity measurements was within 3–5%.
The experiments with the reduced and aged catalysts were conducted using the same procedure after in situ reduction with 10% hydrogen at 500 °C for 1 h and in situ aging with the reacting gas mixture (CH4 1 vol%, O2 4 vol/%, N2 balance) at 850 °C for 5 h. For the determination of activation energy, activity measurements at temperatures corresponding to methane conversion below 15% were conducted under steady state conditions and in the kinetic region and absence of mass transfer limitations. The activation energies were calculated from the Arrhenius plot. The catalytic cycle experiments with the reduced and aged catalysts (Pd/Ce/Al-R-a and Ce/Pd/Al-R-a) were conducted by measuring the activity at three 30 min time intervals for each isotherm (500 °C, 700 °C, and 850 °C). The ignition (heating)–extinction (cooling) experiments were conducted with the reduced (Pd/Ce/Al-R and Ce/Pd/Al-R) catalysts to get three consecutive ignition–extinction curves for methane oxidation reaction for each reduced catalyst at 250–600 °C under the steady state conditions.
The rate and turn-over frequency (TOF) were calculated using the following equations.29
| rCH4 = (Cf·X·v)/(WPd)(mol gPd−1 s−1) | (1) |
| TOF = (Cf·X·v)/(WPd·MPd−1·D)(s) | (2) |
3. Results and Discussion
Two catalyst materials, Pd/CeOx/γ-Al2O3 (Pd/Ce/Al-F) and CeOx/Pd/γ-Al2O3 (Ce/Pd/Al-F), were prepared by a sequential incipient wetness impregnation method followed by calcination at 850 °C. Inductively coupled plasma optical emission spectrometry (ICP-OES) analysis revealed a content of 4.7 (±0.47) wt% Pd and 10.6 (±1.06) wt% Ce in the catalysts, which was close to the targeted content of Pd and Ce (4.5 wt% and 12.2 wt% respectively). The catalysts were tested for the methane oxidation reaction in a fixed-bed catalytic reactor (Fig. 2). The methane light-off temperature and kinetic parameters of the as-synthesized catalysts under study are shown in Table 1. The Pd/Ce/Al-F catalyst exhibited higher methane conversion, lower light-off temperature and lower apparent activation energy compared to the Ce/Pd/Al-F catalyst (Fig. 2, Table 1). The lower methane activity of Ce/Pd/Al-F could be due to larger particle size (Table 2) and less accessible PdO particles probably due to partial embedment on the cerium oxide support (see Fig. 3). The results showcase the effect of sequential impregnation on catalytic activity and properties. | ||
| Fig. 2 (A) CH4 conversion as a function of temperature (light-off curves); (B) apparent activation energy values (Ea, kJ mol−1); (C) Time-on stream activity of over Pd/CeOx/γAl2O3 (Pd/Ce/Al) and CeOx/Pd/γ-Al2O3 (Ce/Pd/Al) catalysts for methane oxidation (F = fresh, R = reduced). Conditions: (A, B and C) with CH4 1 vol%, O2 4 vol/%, N2 balance; GHSV 90000 h−1, (B) at conversions below 15% in the kinetic regions; internal mass transfer limitations were estimated to be negligible according to the Weisz-Prater criterion (Table S7†); (C) at 390 and 350 °C for fresh and reduced and catalysts, respectively. | ||
| Catalyst | T 50 (°C) | r CH4 (10−5 mol gPd−1 s−1) | TOFa (10−2 s–1) | E a (kJ mol−1) |
|---|---|---|---|---|
|
a
r
CH4 and TOF were estimated at 350 °C corresponding to low methane conversion and surface Pd atoms measured by CO chemisorption experiments based on 1:1 Pd:CO stoichiometry.
b Calculated from the Arrhenius plots in Fig. 2.
|
||||
| Pd/Ce/Al-F | 420 | 2.4 | 3.9 | 48 |
| Pd/Ce/Al-R | 380 | 7.1 | 9.8 | 132 |
| Ce/Pd/Al-F | 520 | 1.2 | 2.8 | 96 |
| Ce/Pd/Al-R | 390 | 4.5 | 10.1 | 108 |
| Catalysts | Dispersiona (%) | Particle diametera (nm) | Dispersionb (%) | Particle diameterb (nm) |
|---|---|---|---|---|
|
a Estimated from CO chemisorption based on 1:1 Pd:CO stoichiometry.
b Estimated from CO chemisorption based on 2:1 Pd:CO stoichiometry.
|
||||
| Pd/Ce/Al-F | 6.5 | 17 | 13.1 | 9 |
| Pd/Ce/Al-F-S | 11.1 | 10 | 22.3 | 5 |
| Pd/Ce/Al-R | 7.7 | 15 | 15.0 | 7 |
| Pd/Ce/Al-R-S | 9.3 | 12 | 18.6 | 6 |
| Ce/Pd/Al-F | 4.4 | 25 | 8.8 | 12 |
| Ce/Pd/Al-F-S | 11.8 | 10 | 23.6 | 5 |
| Ce/Pd/Al-R | 4.7 | 24 | 9.4 | 12 |
| Ce/Pd/Al-R-S | 11.8 | 9 | 23.6 | 5 |
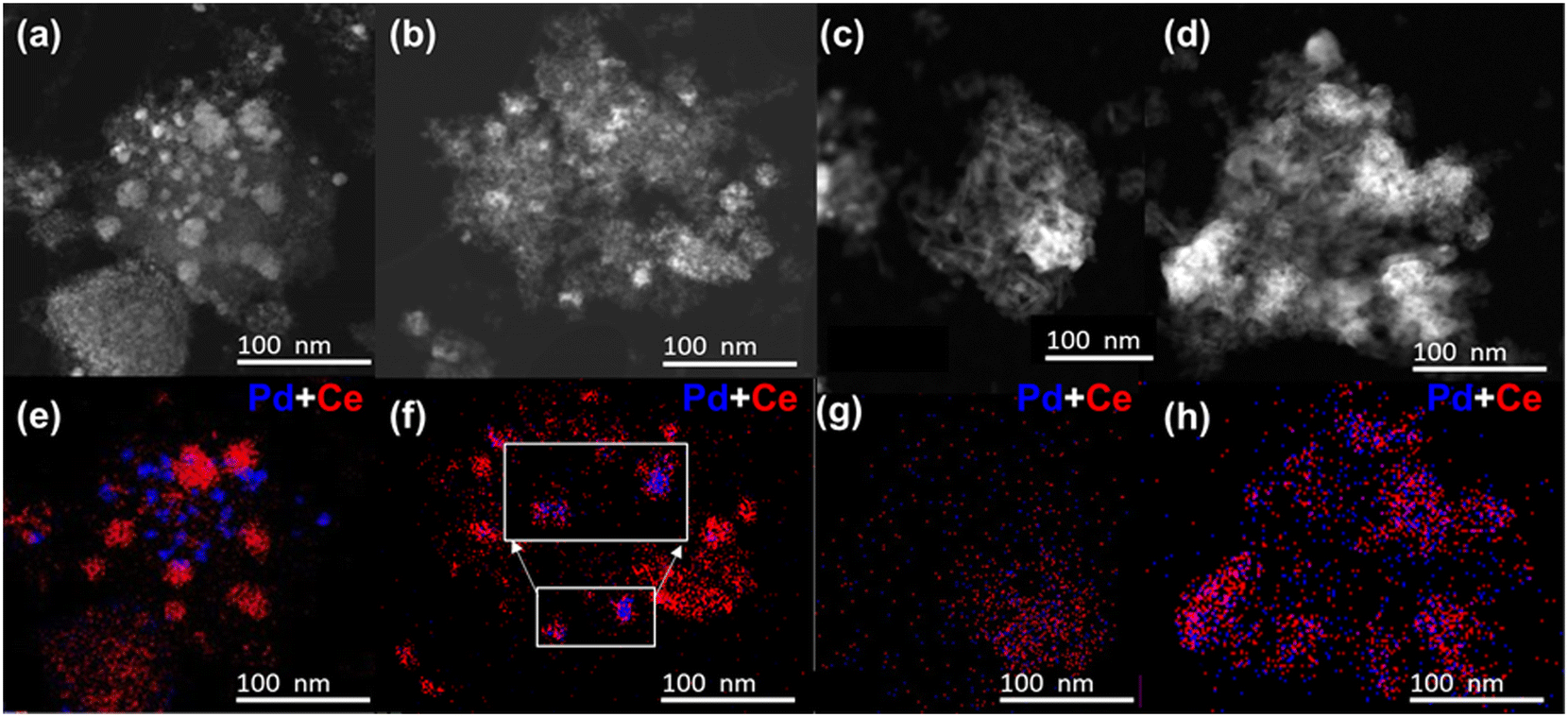 | ||
| Fig. 3 Chemical imaging of the fresh and reduced Pd/CeOx/γ-Al2O3 (Pd/Ce/Al) and CeOx/Pd/γ-Al2O3 (Ce/Pd/Al) catalysts nanostructure. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and X-ray energy dispersive spectroscopy (EDS) elemental maps of Pd/Ce/Al-F (a and e), Ce/Pd/Al-F (b and f), Pd/Ce/Al-R (c and g), and Ce/Pd/Al-R (d and h). Pd and Ce in blue and red, respectively; F = fresh; R = reduced. | ||
Inspired by previous work on core-shell Pd/CeO2 catalysts, for which exposure to a reducing atmosphere caused Pd encapsulation by CeOx,32 we explored the effect of a reduction treatment on the Pd/Ce/Al and Ce/Pd/Al catalysts, with the aim to modify the interaction of Pd and Ce, the nanostructure of the catalysts, and therefore their activity. A temperature of reduction of 500 °C was chosen based on temperature programmed reduction (TPR) results (see Fig. S1, Table S1,† for details). After in situ reduction at 500 °C with hydrogen, the Pd/Ce/Al-R and Ce/Pd/Al-R catalysts (R for reduced) were tested for methane oxidation reaction, to determine the effect of reduction on catalytic performance (Fig. 2).
The reduced catalysts displayed better activities compared to the fresh calcined catalysts, and the effect of reduction was more perceptible at 330–390 °C for both catalysts. The initial reaction rate (rCH4) and turnover frequency (TOF) of methane oxidation reaction with the reduced catalysts also increased by about 3.0–3.5 times compared to the values with the fresh catalysts (Table 1).23,32 The promotion effect was more pronounced for the reduced Ce/Pd/Al catalyst, with a sharp rise in the activity (Fig. 2A). However, the activation energies of the fresh and reduced Ce/Pd/Al catalysts were comparable (Fig. 2B). A higher activation energy value was instead observed for the Pd/Ce/Al-R catalyst. This could be due to changes in the Pd–Ce interface and higher activation barrier due to calcination at a high temperature.22,29,32–35 Reduction may be effective to make the PdO particles more accessible for catalysis through restructuring, changes in the surface and nature of the adsorbed intermediate species as discussed further in the later sections.23,29,32–37
The stability of the catalysts was tested for 50 h during time-on-stream operation at 350–390 °C (Fig. 2C), which shows an overall stable performance for all the catalyst materials in the low temperature kinetic regions of the methane oxidation reaction.20,22
To investigate the possibility of hysteresis38 between the ignition and extinction temperatures, we performed three consecutive ignition (heating)–extinction (cooling) experiments with each reduced catalyst (Pd/Ce/Al-R and Ce/Pd/Al-R) to obtain the ignition–extinction curves (Fig. S2 and S3†). The ignition and extinction temperatures for methane oxidation reaction with the reduced (Pd/Ce/Al-R and Ce/Pd/Al-R) catalysts were 280 °C and 250 °C, respectively showing negligible hysteresis (Table S2†). The methane conversions remained very high (90–99%) at 500–600 °C after three consecutive ignition–extinction cycles spanning over 48–50 h, thus confirming the high stability of both reduced catalysts after recycling (Fig. S2 and S3†).
To understand the observed trends in catalytic activity and investigate the effect of reduction on the morphology and distribution of PdO particles in the Pd/Ce/Al and Ce/Pd/Al catalysts, we performed EDS and HAADF-STEM characterization (Fig. 3). The HAADF-STEM images of Pd/Ce/Al-F catalyst (Fig. 3a) displayed PdO nanoparticles dispersed on the surface of the aluminum oxide. The EDS mappings of Pd/Ce/Al-F catalyst (Fig. 3e) indicated that Pd was mostly located on the aluminum oxide support and not on CeOx. In contrast, the EDS signals in Ce/Pd/Al-F catalyst overlapped between Pd and Ce, indicating the PdO particles were in close contact with CeOx (Fig. 3f) due to strong Pd–Ce interactions.19,23,24,27,29,35,39 Such nanostructure made PdOx particles less exposed to the surface possibly due to partial embedment on the cerium oxide support,24,35,39 and therefore less able to catalyze the methane oxidation, potentially accounting for the lower activity of the Ce/Pd/Al F catalyst compared to Pd/Ce/Al-F.
The HAADF-STEM images and EDS mappings (Fig. 3c, d, g and h) of the reduced Pd/Ce/Al-R and Ce/Pd/Al-R catalysts clearly showed that reduction brought Pd and Ce in close contact in both catalysts. After reduction, the EDS signals of Pd and Ce were correlated in both the Pd/Ce/Al-R and the Ce/Pd/Al-R catalyst, suggesting that Pd migrated to CeOx domains (Fig. 3g and h).19,22,23,29,32–35 The EDS chemical imaging (Fig. 3g and h) also indicated that reduction restructured the catalyst surface by dispersing the cerium oxide particles on the support surface and anchoring/encapsulating the Pd particles around cerium oxide. Reduction may be effective to make the PdO particles more accessible for catalysis through restructuring, changes in the surface and nature of the adsorbed intermediate species as discussed further in the later sections.23,29,32–37,40 This observation explains why the reduced catalysts have more similar activity, and indicates that strong metal support interactions between Pd nanoparticles and CeOx in the support play a major role in the catalytic activity of the reduced catalysts.
The tendency for Pd and ceria to redistribute and come together can be explained by atom trapping, commonly observed for CeO2 and exploited to form single atom catalysts. Atom trapping is known for Pt especially on CeO226,28,41,42 and was also observed for Pd.26–28 In our case, we can hypothesize that Pd tends to anchor on CeO2, and the particles migrate or form as aggregation of single atoms trapped on CeO2.
To quantify the exposed Pd surface we performed pulsed CO chemisorption experiments (Table 2). Since CO chemisorption on Pd could be linear (Pd:CO = 1:1) or bridged (Pd:CO = 2:1)43 as seen in DRIFTS experiments (described later), we calculated the dispersion and particle size values assuming both stoichiometries (Table 2). The average particle size range includes the size based on these two stoichiometries. Pd/Ce/Al-F catalyst had smaller apparent Pd nanoparticles (average size of 9–17 nm) compared to the Ce/Pd/Al-F catalyst (average size of 12–25 nm). The increase in apparent average particle size of the Ce/Pd/Al-F catalyst is consistent with agglomeration and/or lower accessibility of the Pd active phase.25 Both accessibility and nanoparticle size help explain the higher activity of the Pd/Ce/Al-F catalyst, despite the limited contact between Pd and Ce observed by STEM. The higher activity of the Pd/Ce/Al-F catalyst is consistent with the facilitated methane activation on smaller nanoparticles because of the structure sensitivity of σ bond activation.29,32 Notably, the apparent dispersion and particle size of Pd did not change after reduction for both catalysts (Table 2) even though the Pd and Ce particles were brought closer after reduction (Fig. 3g and h). This indicates that better catalytic activities by the reduced catalysts could be due to factors other than particle size, including higher reactivity of PdOx (x < 1) species and changes in the morphology, including stronger Pd–Ce interactions in the reduced catalysts. The dispersion of PdO particles in both fresh/spent and reduced/spent catalysts increased (Table 2) suggesting restructuring of the catalytic surface during methane oxidation reaction.33,34
To elucidate the chemical state of the Pd active phases, we performed XRD and ex situ XPS analysis of the fresh and reduced Pd/Ce/Al and Ce/Pd/Al catalysts. The XRD patterns of the catalysts are given in Fig. 4. For the Pd/Ce/Al-F and Ce/Pd/Al-F catalysts, the diffraction peaks corresponding to cubic CeO2 (COD 7217887), γ-Al2O3 (ICCD 00-004-0858), tetragonal PdO (COD 414668), and Pd (COD 1011105) were identified. For the CeO2, the major peaks appeared at 28.8°, 33.3°, 47.9°, and 56.9° assigned to (111), (200), (220) and (311) crystalline planes, respectively. For γ-Al2O3, peaks at 46° and 68° corresponding to the (400) and (440) planes were observed. For PdO, major peaks at 34°, 45°, 55°, and 61° assigned to (101), (012), (112), and (200) planes appeared for the catalysts calcined at 850 °C indicating larger PdO crystallite sizes, which correlates with the particle size obtained by CO chemisorption (Table 2).22,24,44,45 After reduction, the PdO diffraction peaks disappeared, and a new peak at 40° assigned to Pd0 (111) appeared indicating the formation of metallic Pd particles after reduction. The Pd/Ce/Al-R catalyst showed a sharper Pd0 peak compared to the Ce/Pd/Al-R catalyst, indicating a larger average crystallite size of Pd, and yet showed better dispersion (Table 2). This seemingly contradictory result indicates that Pd0 was still partially embedded by CeOx and was less accessible in the Ce/Pd/Al-R catalyst.
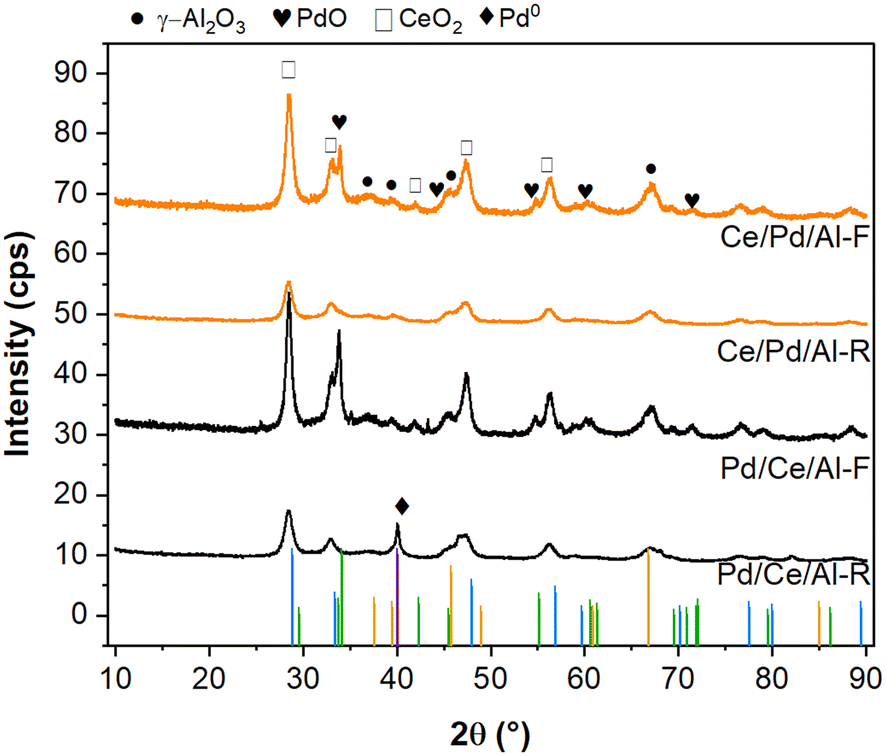 | ||
| Fig. 4 X-ray diffraction (XRD) patterns of fresh and reduced Pd/CeOx/γAl2O3 (Pd/Ce/Al) and CeOx/Pd/γAl2O3 (Ce/Pd/Al) catalysts. F = fresh; R = reduced. Reference patterns: γ-Al2O3 (ICCD 00-004-0858) in yellow, tetragonal PdO (COD 414668) in green, CeO2 (COD 7217887) in blue, Pd (COD 1011105) in purple. | ||
Fig. 5 shows the ex situ XPS spectra in the Pd 3d region of the fresh, reduced and spent Pd/Ce/Al and Ce/Pd/Al catalysts, together with the fitting results of the signals (Table S3†). The 3d5/2 peak in the Pd 3d spectrum was deconvoluted into 3 peaks at 335.1, 336.5, and 338 eV, corresponding to the binding energies (BE) of Pd0, PdO and PdOx (where x > 1 or Pdδ+ where δ > 2), respectively.14,46,47 PdO (80–90 at%) was the major phase in the fresh Pd/Ce/Al and Ce/Pd/Al catalysts (Table S3†). The lower intensity observed for Pd0 signals in the reduced Ce/Pd/Al-R catalyst compared to all the other catalytic materials indicate that Pd was less available to the surface, in agreement with CO chemisorption results. The Pd/Ce/Al-R-S (R-S indicates reduced-spent) catalyst contained 4 at% Pd0, 79 at% PdO and 15 at% PdOx (Table S3†). However, the % methane conversion was similar with both reduced catalysts after methane oxidation reaction at 500 °C for 1 h (Fig. 2A), suggesting that PdO and not Pd0 was the active phase under oxidizing conditions.24,27,44,45 The XPS data on the at% of Ce+3 and Ce+4 in the fresh and reduced catalysts are given in Table S4.† The fresh catalysts contained about 30 at% Ce+3 and 70 at% Ce+4 and the Ce+3 content increased by about 10–13 at% in the reduced catalysts indicating the role of reduction in the redox chemistry of these catalysts.22,23,27,45
 | ||
| Fig. 5 X-ray photoelectron spectroscopy (XPS) results in the Pd 3d region, with the fitting of Pd 3d peaks.(A) Pd/CeOx/γAl2O3 (Pd/Ce/Al); (B) CeOx/Pd/γ-Al2O3 (Ce/Pd/Al); R = reduced. Due to significant noise and low intensity in the XPS of Ce/Pd/Al-R and Ce/Pd/Al-R-S catalysts, deconvolution and peak fitting were not performed for these catalysts. | ||
To gain insights into the reaction mechanisms on the synthesized catalyst materials, we performed operando DRIFTS experiments under CH4 oxidation reaction conditions over the fresh and reduced Pd/Ce/Al and Ce/Pd/Al catalysts (Fig. 6A and B). The fresh catalysts did not show reaction intermediate accumulating on the surface, possibly indicating that the rate determining step was the CH4 dissociative adsorption. On the other hand, on pre-reduced Pd/Ce/Al-R and Ce/Pd/Al-R catalysts, adsorbed CO species were observed in the 2200–1800 cm−1 region. Four bands were observed, at 2133, 2081–2088, 1988 and 1936 cm−1, which were assigned respectively to CO/PdOx species, linear CO/Pd, bridged CO/Pd and multifold CO/Pd.48,49 The bands resemble the previously reported 1922, 1966, and 2088 cm−1 peaks for Pd/Al2O3 catalysts, assigned to μ2 bridge-bonded CO on Pd(111) facets, μ2 bridge-bonded CO on the Pd NPs edges/corners, and linear CO on HCSs, respectively.50 Pd/Ce/Al showed higher adsorbed CO signal intensity compared to Ce/Pd/Al, indicating lower Pd availability in the latter, in agreement with XPS and CO chemisorption results. Moreover, the Ce/Pd/Al-R catalysts presented a less pronounced signal at 1936 cm−1 when compared to Pd/Ce/Al-R, while the 1988 cm−1 signal intensity is more comparable between the two catalysts. This indicates that CeOx preferentially decorated the facets of Pd NPs vs. the edge of the particles in the Ce/Pd/Al catalyst, according to the analysis of Wang et al.50 Notably, regardless of the catalyst composition, the CO species were loosely bound, and were removed already at 150 °C.
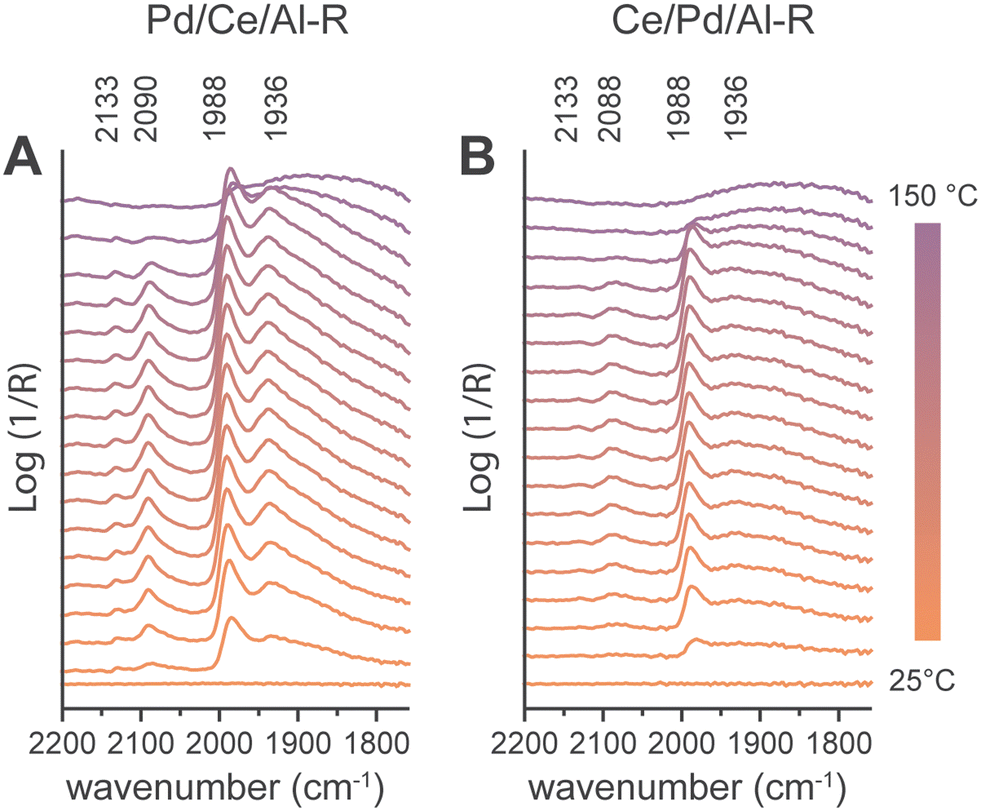 | ||
| Fig. 6 Operando Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS) spectra under CH4 oxidation reaction conditions, for (A) Pd/Ce/Al-R and (B) Ce/Pd/Al-R catalysts after calcination at 850 °C and in situ reduction at 450 °C in 50 vol% H2/N2. Conditions: CH4 1 vol%, O2 4 vol/%, N2 balance, 50 mL min−1 total flow. R = reduced. | ||
Since aging at high temperature (>700 °C) could affect the activity and thermal stability due to deterioration of the support structure, reduced specific surface area, and/or sintering,20,25,27,35 we tested the fresh and reduced Pd/Ce/Al and Ce/Pd/Al catalysts for methane oxidation after a treatment at 850 °C for 5 h under methane oxidation reaction conditions. In general, aging resulted in partial loss of catalytic activity for all catalysts (Fig. 7A), but the pre-reduced catalysts showed more resistance against aging (Fig. 7A, Table S5†). A methane conversion higher than 90% was achieved at 500–850 °C for the Ce/Pd/Al-R-a catalyst and above 700 °C for the Pd/Ce/Al-R-a catalyst (Table S5†). The Pd/Ce/Al-R-a catalyst showed slightly better activity at lower temperatures but the Ce/Pd/Al-R-a catalyst outperformed in the higher temperature range of 500–850 °C (Fig. 7B).
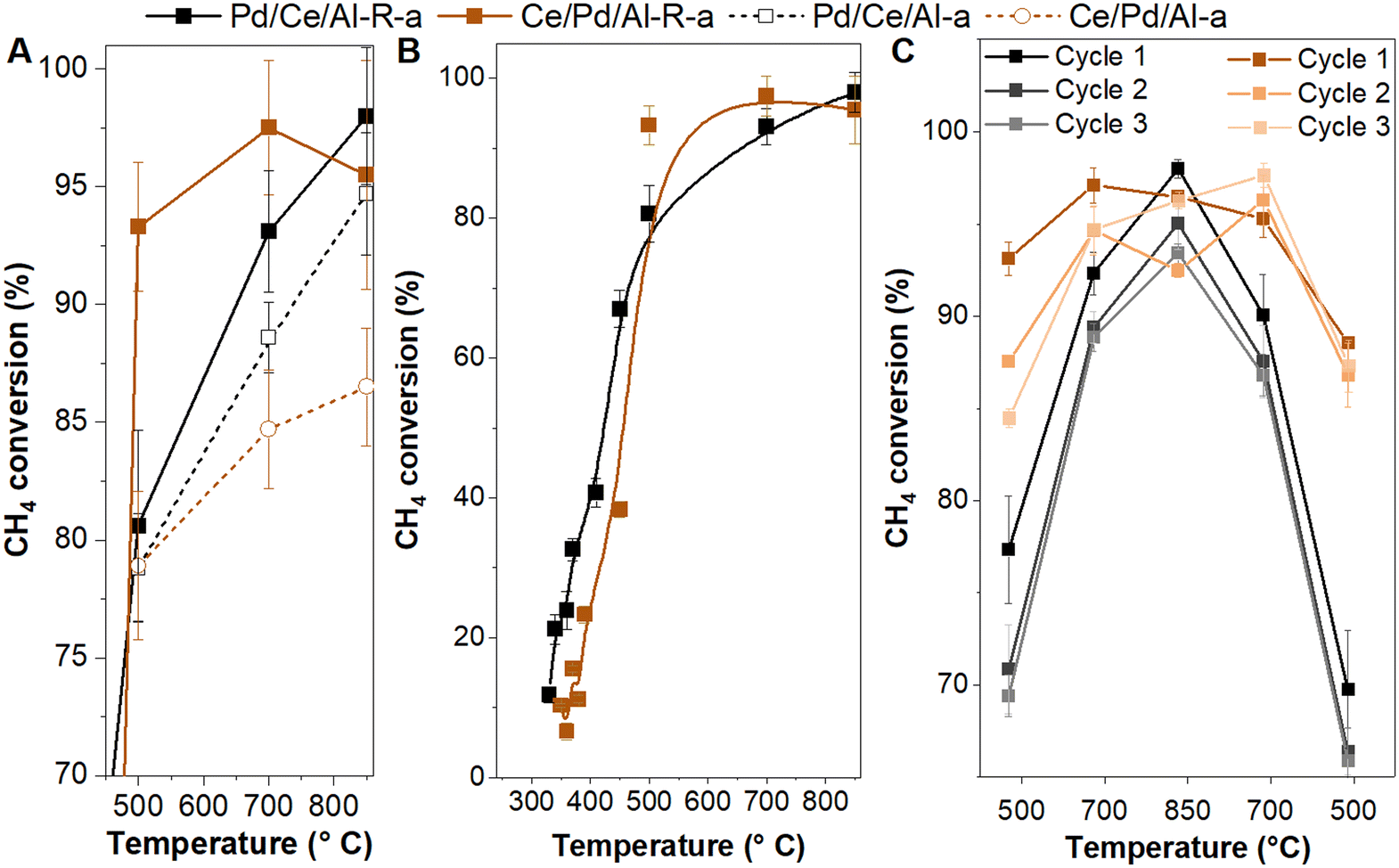 | ||
| Fig. 7 (A and B) CH4 conversion as function of temperature, and (C) catalytic cycles for Pd/CeOx/γAl2O3 (Pd/Ce/Al) and CeOx/Pd/γAl2O3 (Ce/Pd/Al) catalysts. Conditions: CH4 1 vol%, O2 4 vol/%, N2 balance; GHSV 90000 h−1; (A and C) the Weisz-Prater (Cw-p) values for these catalysts were between 6–9 at 500–850 °C indicating internal MTL, Mear's criteria indicates the absence of external MTL; R = reduced; a = aged. | ||
The HDAAF-STEM and EDS images (Fig. 8) of the reduced and aged (Pd/Ce/Al-R-a and Ce/Pd/Al-R-a; R-a indicates reduced and aged) catalysts showed that the Pd/PdO nanoparticles were still anchored by cerium oxide even after aging of the reduced catalysts with a methane gas mix (1% CH4, 4% O2, balance N2) for 5 h at 850 °C. The anchoring effect may explain how a reduction pretreatment could restore and maintain stability of the catalysts during methane oxidation reaction at high temperatures.
 | ||
| Fig. 8 Nanostructure of the pre-reduced aged catalysts. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and X-ray energy dispersive spectroscopy (EDS) elemental maps of Pd/Ce/Al-R-a (a and b); and Ce/Pd/Al-R-a (c and d). Pd and Ce in blue and red, respectively; R-a = reduced and aged. | ||
The high temperature stability of the reduced and aged (R-a) catalysts was further tested by performing catalytic cycles between 500 and 850 °C (Fig. 7C). The methane conversion over Ce/Pd/Al-R-a decreased over time but remained higher than 85% at 500 °C after three catalytic cycles involving heating and cooling over a span of about 24 h (Fig. 7C). On the other hand, Pd/Ce/Al-R-a showed a gradual deactivation to methane conversion values of around 70% at 500 °C. A similar observation was also made by Colussi et al.51 and they suggested that the Pd particles in contact with the Al2O3 support did not get re-oxidized completely to PdO/PdOx during the cooling cycles. These results show that the reduced Ce/Pd/Al-R was the most thermally stable catalyst, despite having a lower starting activity as fresh catalyst (Fig. 2). We interpret this as an effect of better encapsulation, as shown by reduced Pd accessibility in DRIFTS, CO chemisorption and XPS.
In Fig. 7A and C, with temperature increase from 500 to 850 °C the methane conversion increased by only 5–20% and did not reach 100%, despite the high temperature, which indicates the presence of mass transfer limitations (MTL). This was verified by means of the Weisz-Prater and Mear's criteria for internal and external mass transfer limitations, respectively.52,53 The MTL data and calculations for the reduced/aged (Pd/Ce/Al-R-a and Ce/Pd/Al-R-a) and aged (Pd/Ce/Al-a and Ce/Pd/Al-a) catalysts are given in the ESI.† The Weisz-Prater (Cw-p) values for these catalysts were between 6–9 at 500–850 °C (Table S6†). The results indicate that at high temperatures the methane oxidation reaction with the reduced/aged catalysts was limited by internal mass transfer. Contrary to this, values between 1.1 × 10−2 and 8.1 × 10−3 were estimated for the Mear's criteria which indicates the absence of external mass transfer limitations (Table S6†). The internal mass transfer limitations (MTL) started above 340 °C (Cw-p > 1) for the reduced/aged catalysts (Fig. 7B). The trends in activity and deactivation in Fig. 7C can thus be interpreted in terms of loss of surface area and porosity in the catalyst, due to thermal aging at a high temperature.
4. Conclusions
The goal of this work was to synthesize active and stable Pd-based methane oxidation catalysts on a support material consisting of cerium oxide and aluminum oxide by sequential impregnation and calcination at a high temperature of 850 °C for methane oxidation. The CeOx–Al2O3 support material provided an increased stability to CeOx through the high dispersion of Al2O3 and support-support interactions. The PdO/CeOx–Al2O3 (Pd/Ce/Al) catalyst synthesized by impregnating palladium nitrate on CeOx–Al2O3 showed high methane oxidation activity due to presence of small PdO/PdOx nanoparticles mostly on the Al2O3 support. In contrast, a reference CeOx/PdO–Al2O3 (Ce/Pd/Al) catalyst material, prepared by impregnating cerium nitrate on PdO/Al2O3, exhibited low activity due to partial embedment of PdO on CeOx in the support material. This resulted in less availability of PdO/PdOx active sites, as confirmed by DRIFTS and CO chemisorption measurements. To improve the catalytic activity and stability, the catalysts were pretreated by reduction with hydrogen at 500 °C. Both reduced catalysts showed improved activity and stability against thermal aging and cycling to 850 °C with respect to their calcined counterparts. The EDS/STEM data of reduced Pd/Ce/Al and Ce/Pd/Al catalysts clearly showed that reduction brought Pd and Ce in contact regardless of how the catalyst was prepared. The STEM/EDS chemical imaging (Fig. 3) also indicated that reduction restructured the catalyst surface by dispersing the cerium oxide particles and anchoring the Pd particles around cerium oxide. The morphological changes induced by reduction pretreatment made the Pd particles more accessible for methane oxidation reaction. The results showed that simple reduction pretreatments could improve the catalytic activity and stability of the catalysts at both low and high reaction temperatures through changing Pd–Ce interactions, restructuring of the surface and changes in the nature and types of adsorbed species, as shown by HRTEM, EDS and operando DRIFTS. We suggest that the proposed strategy can be used as a scalable alternative to embedding of Pd NPs by CeO2 in core-shell systems for enhanced thermal stability in exhaust after treatment applications.Author contributions
Anil Banerjee: conceptualization, methodology, visualization, formal analysis, validation, formal writing – original draft, writing – review & editing, supervision, project management; Laura Proaño: investigation, methodology, visualization, formal analysis, validation, writing – review & editing; Alexis Alvarez: investigation, methodology, validation; Imani Rogers: investigation, methodology, validation; Jihyeon Park: investigation, methodology, validation; Maddison Montgomery: investigation, visualization, writing – review & editing; Mehmet Z. Billor: investigation, methodology, visualization; Bert M. Weckhuysen: investigation, writing – review & editing; Matteo Monai: conceptualization, methodology, investigation, visualization, formal analysis, validation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Logistical support and internal research grants for this project, and a graduate teaching assistantship to J. P. provided by Columbus State University; Fulbright Colombia and Ministerio de Ciencia, Tecnología e innovación for the Fulbright-Minciencias cohort 2020 scholarship to L. P. for doctorate studies; the Advanced Research Center Chemical Building Blocks Consortium (ARC CBBC) funding to B. M. W. and M. M.; Prof. Yong Ding, School of Materials Science and Engineering, Georgia Institute of Technology for STEM/EDS characterization.References
- P. Gélin and M. Primet, Appl. Catal., B, 2002, 39, 1–37 CrossRef.
- J. Chen, H. Arandiyan, X. Gao and J. Li, Catal. Surv. Asia, 2015, 19, 140–171 CrossRef CAS.
- M. Monai, T. Montini, R. J. Gorte and P. Fornasiero, Eur. J. Inorg. Chem., 2018, 2884–2893 CrossRef CAS.
- F. Nkinahamira, R. Yang, R. Zhu, J. Zhang, Z. Ren, S. Sun, H. Xiong and Z. Zeng, Adv. Sci., 2023, 10, 1–53 Search PubMed.
- Z. Tang, T. Zhang, D. Luo, Y. Wang, Z. Hu and R. T. Yang, ACS Catal., 2022, 12, 13457–13474 CrossRef CAS.
- C. Huang, W. Shan, Z. Lian, Y. Zhang and H. He, Catal. Sci. Technol., 2020, 10, 6407–6419 RSC.
- R. Farrauto, Science, 2012, 337, 659–661 CrossRef CAS PubMed.
- J. B. Miller and M. Malatpure, Appl. Catal., A, 2015, 495, 54–62 CrossRef CAS.
- J. J. Willis, A. Gallo, D. Sokaras, H. Aljama, S. H. Nowak, E. D. Goodman, L. Wu, C. J. Tassone, T. F. Jaramillo, F. Abild-Pedersen and M. Cargnello, ACS Catal., 2017, 7, 7810–7821 CrossRef CAS.
- A. C. Banerjee, J. M. McGuire, O. Lawnick and M. J. Bozack, Catalysts, 2018, 8, 266–283 CrossRef.
- L. M. T. Simplício, S. T. Brandão, E. A. Sales, L. Lietti and F. Bozon-Verduraz, Appl. Catal., B, 2006, 63, 9–14 CrossRef.
- K. Murata, Y. Mahara, J. Ohyama, Y. Yamamoto, S. Arai and A. Satsuma, Angew. Chem., Int. Ed., 2017, 56, 15993–15997 CrossRef CAS.
- R. J. Farrauto, M. C. Hobson, T. Kennelly and E. M. Waterman, Appl. Catal., A, 1992, 81, 227–237 CrossRef CAS.
- D. R. Fertal, M. Monai, L. Proaño, M. P. Bukhovko, J. Park, Y. Ding, B. M. Weckhuysen and A. C. Banerjee, Catal. Today, 2021, 382, 120–129 CrossRef CAS.
- M. Danielis, L. E. Betancourt, I. Orozco, N. J. Divins, J. Llorca, J. A. Rodríguez, S. D. Senanayake, S. Colussi and A. Trovarelli, Appl. Catal., B, 2021, 282, 119567 CrossRef CAS.
- S. Colussi, A. Gayen, M. F. Camellone, M. Boaro, J. Llorca, S. Fabris and A. Trovarelli, Angew. Chem., Int. Ed., 2009, 48, 8481–8484 CrossRef CAS.
- T. Guo, J. Du, J. Wu, S. Wang and J. Li, Chem. Eng. J., 2016, 306, 745–753 CrossRef CAS.
- R. Ramírez-López, I. Elizalde-Martinez and L. Balderas-Tapia, Catal. Today, 2010, 150, 358–362 CrossRef.
- M. Cargnello, J. J. D. Jaén, J. C. H. Garrido, K. Bakhmutsky, T. Montini, J. J. C. Gámez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–718 CrossRef CAS PubMed.
- X. Liu, J. Liu, F. Geng, Z. Li, P. Li and W. Gong, Front. Chem. Sci. Eng., 2012, 6, 34–37 CrossRef CAS.
- X. Feng, W. Li, D. Liu, Z. Zhang, Y. Duan and Y. Zhang, Small, 2017, 13, 1–8 Search PubMed.
- X. Yang, C. Du, Y. Guo, Y. Guo, L. Wang, Y. Wang and W. Zhan, J. Rare Earths, 2019, 37, 714–719 CrossRef CAS.
- M. Wu, W. Li, X. Zhang, F. Xue, T. Yang, L. Yuan, L. Y. M. Wu, W. Li, X. Zhang, F. Xue and T. Yang, ChemCatChem, 2021, 13, 3490–3500 CrossRef CAS.
- D. R. Fertal, M. P. Bukhovko, Y. Ding, M. Z. Billor and A. C. Banerjee, Catalysts, 2020, 10, 1–16 CrossRef.
- Y. Liu, C. Hu, K. Xiong, Y. Sun and L. Bian, Chem. Phys. Lett., 2020, 761, 138064 CrossRef CAS.
- A. K. Datye and H. Guo, Nat. Commun., 2020, 11, 2133 CrossRef.
- G. Spezzati, Y. Su, J. P. Hofmann, A. D. Benavidez, A. T. DeLaRiva, J. McCabe, A. K. Datye and E. J. M. Hensen, ACS Catal., 2017, 7, 6887–6891 CrossRef CAS.
- R. Alcala, A. DeLaRiva, E. J. Peterson, A. Benavidez, C. E. Garcia-Vargas, D. Jiang, X. I. Pereira-Hernández, H. H. Brongersma, R. ter Veen, J. Staněk, J. T. Miller, Y. Wang and A. Datye, Appl. Catal., B, 2021, 284, 119722 CrossRef CAS.
- J. Chen, Y. Wu, W. Hu, P. Qu, G. Zhang, P. Granger, L. Zhong and Y. Chen, Appl. Catal., B, 2020, 264, 118475 CrossRef.
- J. Chen, J. Zhong, Y. Wu, W. Hu, P. Qu, X. Xiao, G. Zhang, X. Liu, Y. Jiao, L. Zhong and Y. Chen, ACS Catal., 2020, 10, 10339–10349 CrossRef CAS.
- J. Ma, Y. Lou, Y. Cai, Z. Zhao, L. Wang, W. Zhan, Y. Guo and Y. Guo, Catal. Sci. Technol., 2018, 8, 2567–2577 RSC.
- N. L. Wieder, M. Cargnello, K. Bakhmutsky, T. Montini, P. Fornasiero and R. J. Gorte, J. Phys. Chem. C, 2011, 115, 915–919 CrossRef CAS.
- C. Chen, J. Cao, M. Cargnello, P. Fornasiero and R. J. Gorte, J. Catal., 2013, 306, 109–115 CrossRef CAS.
- R. Burch and F. J. Urbano, Appl. Catal., A, 1995, 124, 121–138 CrossRef CAS.
- P. Lott, P. Dolcet, M. Casapu, J. D. Grunwaldt and O. Deutschmann, Ind. Eng. Chem. Res., 2019, 58, 12561–12570 CrossRef CAS.
- S. Xie, Z. Wang, W. Tan, Y. Zhu, S. Collier, L. Ma, S. N. Ehrlich, P. Xu, Y. Yan, T. Xu, J. Deng and F. Liu, Environ. Sci. Technol., 2021, 55, 7624–7633 CrossRef CAS PubMed.
- K. A. Ledwa, L. Kępiński and M. Pawlyta, Mater. Res. Bull., 2021, 141, 111357 CrossRef CAS.
- R. M. Al Soubaihi, K. M. Saoud, F. Ye, M. T. Zar Myint, S. Saeed and J. Dutta, Microporous Mesoporous Mater., 2020, 292, 109758 CrossRef CAS.
- L. Piccolo, Catal. Today, 2020, 373, 1–18 Search PubMed.
- S. W. Chee, J. M. Arce-Ramos, W. Li, A. Genest and U. Mirsaidov, Nat. Commun., 2020, 11, 2133 CrossRef CAS.
- H. N. Pham, A. Delariva, E. J. Peterson, R. Alcala, K. Khivantsev, J. Szanyi, X. S. Li, D. Jiang, W. Huang, Y. Sun, P. Tran, Q. Do, C. L. Dimaggio, Y. Wang and A. K. Datye, ACS Sustainable Chem. Eng., 2022, 10, 7603–7612 CrossRef CAS.
- J. Jones, I. B. Perry, G. Lu, P. Liu and S. L. Buchwald, Science, 2016, 353, 150–154 CrossRef CAS.
- P. Canton, G. Fagherazzi, M. Battagliarin, F. Menegazzo, F. Pinna and N. Pernicone, Langmuir, 2002, 18, 6530–6535 CrossRef CAS.
- S. Colussi, A. Trovarelli, E. Vesselli, A. Baraldi, G. Comelli, G. Groppi and J. Llorca, Appl. Catal., A, 2010, 390, 1–10 CrossRef CAS.
- L. M. T. S. Rodrigues, R. B. Silva, M. G. C. Rocha, P. Bargiela, F. B. Noronha and S. T. Brandão, Catal. Today, 2012, 197, 137–143 CrossRef CAS.
- B. V. Christ, Handbook of Monochromatic XPS Spectra. The Elements of Native Oxides, Wiley, 2000 Search PubMed.
- B. Van Devener, S. L. Anderson, T. Shimizu, H. Wang, J. Nabity, J. Engel, J. Yu, D. Wickham and S. Williams, J. Phys. Chem. C, 2009, 113, 20632–20639 CrossRef CAS.
- K. Murata, E. Eleeda, J. Ohyama, Y. Yamamoto, S. Arai and A. Satsuma, Phys. Chem. Chem. Phys., 2019, 21, 18128–18137 RSC.
- S. D. Ebbesen, B. L. Mojet and L. Lefferts, Phys. Chem. Chem. Phys., 2009, 11, 641–649 RSC.
- H. Wang, X.-K. Gu, X. Zheng, H. Pan, J. Zhu, S. Chen, L. Cao, W.-X. Li and J. Lu, Adv., 2019, 5, 1–8 CAS.
- S. Colussi, A. Trovarelli, G. Groppi and J. Llorca, Catal. Commun., 2007, 8, 1263–1266 CrossRef CAS.
- S. J. Park, M. P. Bukhovko and C. W. Jones, Chem. Eng. J., 2020, 420, 130369 CrossRef.
- J. T. García-Sánchez, R. Valderrama-Zapata, L. F. Acevedo-Córdoba, D. Pérez-Martínez, S. Rincón-Ortiz and V. G. Baldovino-Medrano, ACS Catal., 2023, 13, 6905–6918 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Hydrogen temperature programmed reduction of Pd/Ce/Al-F and Ce/Pd/Al-F catalysts (Fig. S1), calculated hydrogen consumption during hydrogen temperature programmed reduction (Table S1), ignition-extinction curves of Pd/Ce/Al-R catalyst (Fig. S2), ignition-extinction curves of Ce/Pd/Al-R catalyst (Fig. S3), ignition-extinction profile of Pd/Ce/Al-R and Ce/Pd/Al-R catalysts (Table S2), at% Pd, PdO, PdOx in Pd 3d XPS spectra of Pd/Ce/Al and Ce/Pd/Al catalysts (Table S3), at% Ce(III) and Ce(IV) in Ce 3d XPS spectra of Pd/Ce/Al and Ce/Pd/Al catalysts (Table S4), effect of aging and reduction on stability of the as-prepared catalysts (Table S5), mass transfer limitations data for Pd/Ce/Al and Ce/Pd/Al catalysts (Tables S6 and S7), mass transfer limitations calculations. See DOI: https://doi.org/10.1039/d3cy01193c |
| This journal is © The Royal Society of Chemistry 2024 |