
Efficient synthesis of glycerol carbonate by doping metallic copper in palladium-catalyzed glycerol system for carbonylation reaction†
Zhihao
Lv
,
Pengpeng
Huang
,
Pingbo
Zhang
 *,
Mingming
Fan
,
Pingping
Jiang
and
Yan
Leng
*,
Mingming
Fan
,
Pingping
Jiang
and
Yan
Leng
The Key Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, P. R. China. E-mail: pingbozhang@126.com
First published on 24th November 2023
Abstract
M–N–C materials derived from zeolitic imidazolium frameworks have exhibited remarkable efficacy as carriers for catalyzing glycerol carbonylation reactions, attributable to their meticulously modifiable pore size architecture, elevated catalytic selectivity, and commendable stability. In this study, we accomplished the successful integration of Cu2+ ions into the growth kinetics of ZIF-8 through a facile hydrothermal method. Subsequently, the resulting Cu–NC material was calcinated at 950 °C in a nitrogen atmosphere. This derived material served as an optimal substrate for the deposition of palladium nanoparticles, yielding the Pd/Cu–NC catalyst. The catalytic potential of this catalyst was evidenced by its superior efficiency in driving the glycerol carbonate synthesis from glycerol, surpassing the performance of the NC carrier in isolation. Impressively, a 98.3% yield and 99.7% selectivity were achieved within a mere 2-hour reaction span, conducted at 140 °C and 4 MPa. Employing density functional theory simulations, we delved into the intricate mechanisms governing carbonyl formation and ring closure during the oxidative carbonylation of glycerol. The strategic introduction of metallic copper during the initial phase of the transition state prominently underscored the robust interaction between copper and palladium. This interaction engendered a harmonized and systematically orchestrated charge distribution encircling the metallic palladium, thereby facilitating a more stable trajectory for ring formation processes.
Introduction
Biodiesel, derived from renewable biomass resources, serves as a pivotal means to curtail greenhouse gas emissions and air pollutants. Moreover, its potential for blending with conventional diesel holds promise in mitigating reliance on conventional diesel energy sources.1,2 However, the burgeoning biodiesel chemical industry has engendered an escalating surplus of waste glycerol, a consequential by-product, thereby constraining the biodiesel sector.1,3 Consequently, the sustainable valorization of glycerol has emerged as a focal point for burgeoning research endeavors.4 In contemporary times, catalytic conversion strategies have yielded an array of glycerol derivatives, encompassing hydrogen, propylene glycol, dihydroxyacetone, glycerol acid, acrolein, glycerol esters, polyglycerol, and glycerol carbonate.5 Among these, glycerol carbonate (4-hydroxymethyl-2-oxo-1,3-dioxolane) stands out as a molecule harboring both hydroxyl and 2-oxo-1,3-dioxolane moieties, thereby endowing it with versatile reactivity and applications, spanning bio-based solvents, battery electrolyte fluids, organic synthesis, cosmetics, and an array of surfactants.6 The avenues for glycerol carbonate synthesis encompass four principal methodologies: phosgene gas approach, ester exchange technique, urea alcoholysis process, and oxidative carbonylation route.7–10 Of these, oxidative carbonylation has garnered considerable attention as an environmentally benign and sustainable synthetic route, marked by heightened atomic utilization efficiency, cost-effectiveness, and the sole generation of water as a by-product.The crux of the matter revolves around the identification of proficient catalysts for the oxidative carbonylation of glycerol, aimed at synthesizing glycerol carbonate. Among the catalytic systems reported in the literature are PdCl2(phen)/KI, CuCl2, Pd@PQP-NHC, and Pd(OAc)2/Mn(acac)3/KBr.10–13 Nonetheless, many of these homogeneous catalysts remain plagued by challenges encompassing intricate product separation, modest catalytic activity, and limited reusability. Consequently, the emergence of effective heterogeneous catalysts offers promise in overcoming these limitations. Notably, Liu et al. harnessed a zeolite-Y-immobilized Pd complex (PdCl2(phen)@Y) as a retrievable catalyst for glycerol oxidative carbonylation, achieving a commendable turnover frequency (TOF) value of 317 h−1.14 Similarly, Wang et al. reported an impressive glycerol conversion of 82.2% and an elevated TOF value of 900 h−1 with Pd/C, facilitated by NaI assistance.15 These instances underscore the exceptional efficacy of heterogeneous Pd catalysts in glycerol oxidative carbonylation reactions. Consequently, these catalysts offer promising prospects as primary active centers for the formulation of heterogeneous catalysts, yielding enhanced yields and activity.
Zeolite imidazolium framework materials (ZIFs), a subset of metal–organic frameworks (MOFs), uniquely amalgamate the favorable attributes of MOFs, encompassing substantial specific surface areas and modifiable pore structures, while additionally showcasing robust thermal stability and structural integrity. Moreover, ZIFs offer a simpler and more convenient synthesis route compared to conventional MOFs. These materials have garnered extensive application across catalysis, gas storage and adsorption, water treatment, gas separation, and drug delivery arenas.16,17 However, when employed in isolation, catalysts fashioned from ZIF-8 grapple with limitations pertaining to their pore dimensions, thereby hampering their utility in the catalytic conversion of oils and fats. Nevertheless, ZIFs stand as abundant sources of nitrogen. Leveraging the electronegativity and atomic radius of nitrogen, akin to carbon, allows for selective nitrogen doping within sp2 hybridized carbon materials. Nitrogen doping engenders shifts in the electronic characteristics of carbon atoms, thus augmenting catalytic efficiency.18 Ergo, the strategic choice of ZIFs as precursors for fabricating nitrogen–metal–carbon (M–N–C) catalysts emerges as a promising avenue, engendering robust interactions between transition metal atoms and nitrogen moieties. A case in point, Jin et al. harnessed atomically dispersed copper within ZIF-8-derived N-doped carbon matrices to fuel high-performance oxygen electrocatalysis in zinc–air batteries.19 The resultant rechargeable zinc–air batteries exhibited exemplary long-term stability and elevated current efficiency.
In this study, we have successfully employed Cu–NC materials derived from ZIF-8 as carriers, effectively loaded with palladium nanoparticles. This approach has enabled the creation of meticulously dispersed Pd/Cu–NC catalysts, showcasing exceptional yields and selectivity. Furthermore, our investigation delved into the impact of diverse Cu doping levels on the reaction's outcome. Notably, the yield demonstrated a trend akin to a volcano plot, characterized by initial escalation followed by subsequent reduction. To delve deeper, we employed density functional theory to simulate the transition state of glycerol synthesis into glycerol carbonate, focusing on two pivotal steps. Through this approach, we achieved a comprehensive understanding of the reaction process, including the formation of carbonyl groups. Additionally, our theoretical simulations shed light on the role of Cu in the formation of rings within the process.
Preparation of catalyst
Pd/Cu–NC was methodically synthesized as delineated in Fig. 1. The process commenced with the utilization of Cu-ZIF-8, prepared through a hydrothermal approach, as the precursor. Subsequent to this, the Cu–NC carrier was achieved through a meticulous calcination regimen conducted at 950 °C under a nitrogen atmosphere. The ensuing step encompassed the loading of palladium nanoparticles onto the Cu–NC carrier, realized via the impregnation method, leading to the ultimate formation of Pd/Cu–NC. Comprehensive details pertaining to the sequential experimental procedures involving Cu-ZIF-8, Cu–NC, and Pd/Cu–NC can be found in the supporting literature. Additionally, the preparation of Pd/ZIF-8, Pd/NC, Pd/Cu(Im)2, and ball-milled Pd–Cu–NC are elaborated within the ESI.† Noteworthy, the genuine contents of Pd and Cu in the resulting Pd/Cu–NC were quantified via inductively coupled plasma emission spectrometry (ICP-OES) at 10.8 wt% and 2.16 wt%, respectively. | ||
| Fig. 1 Schematic representation of the synthesis procedures of Pd/Cu–N–C. | ||
Activity test
The catalytic assessment was conducted within a 50 mL stainless steel autoclave reactor, with the reaction pathway elucidated in Scheme 1. All reagents are reacted within a 10 mL PTFE liner, strategically positioned within the autoclave reactor to avert any interaction with the metallic enclosure. By this design, the gas mixture occupied a free volume spanning 35–40 mL, as reported previously.15 To ensure distinctive and reproducible peaks, we employed silylation to convert the product into the corresponding silylated derivative, thus lowering its boiling point for subsequent gas-phase analysis.11 The standard curve for the internal standard method and relevant formulas can be found in the ESI.† | ||
| Scheme 1 Catalyst activity test reaction equation. | ||
In a prototypical experiment, a solution of propanetriol (15 mmol) dissolved in dimethylacetamide (DMA) (10 mL), was prepared. This solution was combined with the Pd/Cu0.356–NC catalyst (represented by ‘x’, indicating the Cu to Zn mole ratio) at a quantity of 0.007 g (corresponding to Pd content of 0.011 mmol), and augmented with KI (0.11 mmol). The resulting solution, characterized by its deep black hue, was then introduced into a glass bottle. This assembly was subsequently placed within the autoclave reactor, hermetically sealed, purged, and pressurized with a gaseous mixture of O2 (1.3 MPa) and CO, affording a combined pressure of 4 MPa. The reaction milieu was elevated to 140 °C, whereupon the reaction was sustained for a duration of 2 hours.
Simulation models and methods
For a better study of the formation of carbonyl groups during the reaction process, the process of ring formation, and the promotion of copper in the reaction process, we have utilized density functional simulation for the study, and the methodology and basis set used refer to the supporting literature.Results and discussion
The calcined carriers and catalysts underwent scrutiny through scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Notably, the micrographs in Fig. 2a–d, alongside Fig. S2a,† collectively illustrate the discernible presence of a consistent rhombic dodecahedral architecture within ZIF-8, Cu-ZIF-8, Cu–NC, and Pd/Cu–N–C.20 This unaltered morphology following Cu modification and successive calcination signifies the robust structural integrity of ZIF-8. Conversely, Cu–N–C, characterized by its relatively uneven surface replete with micropores, ostensibly attributed to the volatilization of Zn during pyrolysis, showcases a distinct contrast against Cu–NC and Cu-ZIF-8.18 This distinctive feature is suggestive of an augmented defect density within the geometry of Cu–NC, potentially contributing to its heightened activity as a carrier in comparison to Cu-ZIF-8. In-depth analysis via energy-dispersive X-ray spectroscopy (EDS) energy spectra and elemental distribution maps, as depicted in Fig. 2f–i and S2c,† underscores the homogenous co-doping of Cu and N within the carbon framework. A compelling observation arises in the HAADF-TEM mode, wherein the enhanced brightness of Cu atoms uniformly pervades the darker carbon carriers. This phenomenon arises due to the higher atomic number of Cu relative to that of C and N, consequently manifesting distinct contrasts.21 Which provides a better synergy between copper and palladium. | ||
| Fig. 2 (a, b and d) TEM images of Cu-ZIF-8, Cu–NC and Pd/Cu–NC (c) SEM images of Pd/Cu–NC. (e–i) Elemental mapping of C, N, Cu, and Pd. | ||
The crystalline phases inherent to the samples, in conjunction with the loading outcomes, were meticulously characterized through X-ray diffractometry (XRD) and Fourier infrared spectroscopy (FT-IR). The XRD profiles of NC, Cu–NC, and Pd/Cu–NC notably exhibit a broad diffraction peak cantered around 23.5° (Fig. 3a), signifying correspondence with the (002) diffraction peak ascribed to graphitic carbon, this suggests that the successful synthesis of C skeleton.22 Further insight is gleaned from the FT-IR spectra (Fig. 3b), wherein discernible features emerge: the vibrational peak at 3123 cm−1 pertaining to the C–H bond of 2-methylimidazolylmethyl, the stretching vibration at 1585 cm−1 emblematic of the imidazolium five-membered ring's C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N moiety, and the distinctive stretching vibration at 1401 cm−1 indicative of the C–N bond. Collectively, these observations affirm the transference of the structural attributes from ZIF-8 to the calcined catalyst, as reported elsewhere.23,24 Notably, the peak range spanning 600–800 cm−1 aligns with the metal–ligand vibrational spectrum intrinsic to ZIF-8. Intriguingly, for the last three calcined samples, a conspicuous diminution in zinc ligand vibrational peaks is discerned, an outcome attributed to the volatilization of zinc, precipitated by the rigorous nitrogen atmosphere at 950 °C, surpassing the boiling point of zinc (906 °C).
N moiety, and the distinctive stretching vibration at 1401 cm−1 indicative of the C–N bond. Collectively, these observations affirm the transference of the structural attributes from ZIF-8 to the calcined catalyst, as reported elsewhere.23,24 Notably, the peak range spanning 600–800 cm−1 aligns with the metal–ligand vibrational spectrum intrinsic to ZIF-8. Intriguingly, for the last three calcined samples, a conspicuous diminution in zinc ligand vibrational peaks is discerned, an outcome attributed to the volatilization of zinc, precipitated by the rigorous nitrogen atmosphere at 950 °C, surpassing the boiling point of zinc (906 °C).
 | ||
| Fig. 3 (a) XRD patterns of NC, Cu–NC and Pd/Cu–NC. (b) FT-IR spectrogram of ZIF-8, NC, Cu–NC and Pd/Cu–NC. | ||
Distinctive XRD patterns for Cu–NC (Fig. 3a) underscore the emergence of well-defined diffraction peaks at 43.3° and 50.4°, attributable to the (111) and (200) reflections, respectively, of metallic copper, thereby attesting to the presence of Cu (PDF # 04-0836). Concomitantly, the crafted catalysts reveal two broad diffraction peaks at 40.23° and 46.80°, aligned with the (111) and (200) reflections of Pd metal, thus corroborating the successful palladium loading.25 A remarkable finding unfolds upon Pd loading – the erstwhile discernible Cu peaks remarkably diminish. This intriguing phenomenon is ascribed to the exceedingly dispersed nature of Cu, facilitated by its nominal content, ultimately manifesting as high dispersion within the carrier's scaffold. This unique configuration renders the detection of high-content Pd-induced diffraction peaks for Cu implausible.21
To investigate the valence state of the Cux–NC carrier (where ‘x’ signifies the Cu–Zn molar ratio) as well as the resulting Pd/Cu–NC catalyst, an X-ray photoelectron spectroscopy (XPS) analysis was undertaken. The XPS broad peak spectra (Fig. 4a) substantiate the presence of carbon, nitrogen, and oxygen elements in both the carrier and catalyst. The oxygen elements can be attributed to water molecules adsorbed by the carrier, in concordance with the hydrogen bonding vibrational peaks of water molecules associated with ZIF-8, as established by earlier infrared spectroscopy (Fig. 3b) at 3450 cm−1. The observation of the 2p peak of Zn within the precursor of Cu0.356–NC, Cu0.356-ZIF-8 (Fig. S3a†), further corroborates the almost complete vaporization of calcined Zn ions.26 Specifically, the positions of Zn 2p1/2 and Zn 2p3/2 peaks, located at 1043.88 eV and 1021.81 eV, respectively (Fig. S3b†), substantiate the prevalence of Zn2+ in elemental Zn, primarily in the form of Zn2+.27
 | ||
| Fig. 4 XPS wide spectra for (a) Cu0.356–NC and Pd/Cu0.356–NC. (b–d) XPS spectra of Cu 2p3/2 and Pd 3d for the prepared catalysts. (e) Electron transport sketch map. | ||
Furthermore, the distinct appearance of the Cu 2p3/2 characteristic peak in Fig. 3b validates the effective dispersion of Cu within the Cu–NC carriers.28,29 The characteristic peak at 931.4 eV within the Cu 2p spectrum indicates the presence of Cu(0)/Cu(I) species.21 This observation aligns with the XRD results, whereas the peak at 934.5 eV attests to the presence of Cu(II) species. Notably, in the absence of palladium loading (Fig. 4c), these two peaks shifted by approximately 1.8 eV. We believe that conventional physical loading should not lead to such a result, and that during the experiments, in order to allow the reduction of divalent palladium to palladium nanoparticles to ensure its dispersion on the carrier, it was necessary to pre-mix the carrier with palladium dichloride for two hours prior to the addition of the reductant, which led to the fact that divalent palladium would partially displace the divalent copper in the same way as divalent copper had displace divalent zinc before, which led to the result, and that such a metal displacements had already been reported in a generalized manner.32–34 That attributed to the synergistic interaction between Cu and Pd. Remarkably, the inclusion of palladium chloride potentially led to partial doping of divalent palladium into the carrier backbone, supplanting copper atoms and fostering robust Cu–Pd interactions (Fig. 4e). This intricate interaction was a pivotal factor contributing to the augmented selectivity of the yield. The ensuing characteristic peaks of Pd(0) and Pd(II), represented by Pd 3d3/2 and Pd 3d5/2, respectively, manifest on the Pd/Cu–NC catalyst at 340.07 eV, 341.09 eV, and 334.80 eV, 335.99 eV. This affirms the efficacious loading of palladium nanoparticles and lends credence to the hypothesis of the presence of divalent palladium, corroborating earlier conjectures. Furthermore, the C 1s peak (Fig. 5a) substantiates the dimethylimidazole structure within Cu0.356-ZIF-8, Cu0.356–NC, and Pd/Cu0.356–NC. Examining the N 1s spectrum (Fig. 5b) reveals the presence of two peaks at 397.92 eV and 403 eV, attributing to N–Zn & N–Cu, and N oxides, respectively.
 | ||
| Fig. 5 XPS spectra of C 1s for (a) Cu0.356-ZIF-8, Cu0.356–NC, Pd/Cu0.356–NC. XPS spectra of N 1s for (b) Cu0.356-ZIF-8, Cu0.356–NC, Pd/Cu0.356–NC. | ||
Table 1 presents the specific surface area, average pore diameter, and pore volume parameters of Cu0.356-ZIF-8, Cu0.356–NC, and Pd/Cu0.356–NC. As indicated in the table, the specific surface area of Cu0.356-ZIF-8 was recorded at 1801.82 m2 g−1, accompanied by an average pore diameter of 1.22 nm and a pore volume of 0.6268 cm3 g−1. Notably, the calcination process engendered a discernible transformation in the pore structure of Cu0.356–NC. Despite this change, the specific surface area of Cu0.356–NC remained impressively high at 1393.4 m2 g−1. This augmented surface area bears implications for enhanced molecular interaction, such as that of glycerol, by affording a profusion of reaction sites. Moreover, a comparative analysis with the original Cu0.356-ZIF-8 pore size (1.22 nm) and pore volume (0.6268 cm3 g−1) revealed a significant enhancement in the pore diameter (1.22 nm) and pore volume (0.6268 cm3 g−1) for Cu0.356–NC post-calcination. This augmentation in pore size and volume notably contributes to an augmented reactivity.
| Samples | Surface areaa (m2 g−1) | Average pore diameterb (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|
| a Measured using N2 adsorption with the Brunauer–Emmett–Teller (BET) method. b Pore size in diameter calculated by the desorption data using Barrett–Joyner–Halenda (BJH) method. | |||
| Cu0.356-ZIF-8 | 1801.82 | 1.22 | 0.6268 |
| Cu0.356–NC | 1393.40 | 2.49 | 0.9937 |
| Pd/Cu0.356–NC | 1081.43 | 3.20 | 0.8670 |
The adsorption–desorption isotherms depicted in Fig. 6b for Cu-ZIF-8 conformed to the type I hysteresis loops, indicative of the presence of abundant micropores within the samples. Detailed pore size distribution analysis revealed a prevailing pore size distribution ranging between 1–3 nm for Cu-ZIF-8.30 However, post-calcination (Fig. 6a), a shift away from type II hysteresis loops in the adsorption–desorption isotherms became evident, implying that the catalyst primarily adopted a mesoporous configuration following calcination. This alteration was accompanied by a noteworthy transition in the dominant pore size distribution to the range of 3–5 nm. Intriguingly, upon loading palladium metal nanoparticles, an enlargement of pore diameters was observed. This observation is ascribed to the effects of particle stacking and filling. The aggregation of particles has the potential to reshape and resize the original pores, while the presence of nano-metal particles might fill the pore spaces within the carrier, consequently leading to a reduction in the effective pore volume. This cascade of effects ultimately culminates in larger apparent pore sizes, a phenomenon corroborated by the concomitant decrease in pore volume.31
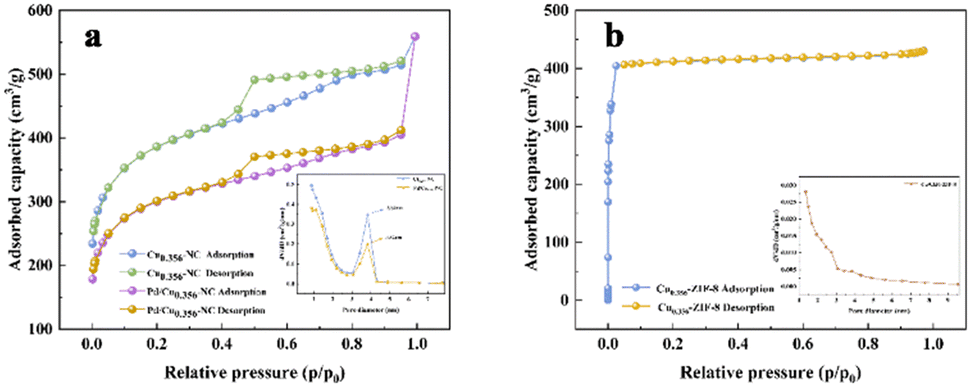 | ||
| Fig. 6 (a and b) N2 adsorption–desorption isotherms and pore diameter distributions diagram of prepared carrier a and catalyst b. | ||
Fig. 7 delineates the interplay between yield and selectivity across diverse catalysts. Notably, in the context of catalysts loaded with uncalcined carriers, Cu-doped catalysts conspicuously outperformed Pd/ZIF-8. This phenomenon can be attributed to the collaborative prowess of bimetallic constituents and structural changes following metal doping. The transformation of the carrier's pore structure from microporous to mesoporous post-calcination, coupled with the inherent catalytic potency of the NC material, culminated in a substantial surge in yield. Additionally, we employed the ball milling method to physically blend palladium chloride, ZIF-8, and copper nitrate, followed by calcination. The resulting catalysts exhibited a markedly inferior performance compared to the Pd/Cu–NC counterparts, serving as a testament to the superior catalytic efficacy of the Cu–NC material as a carrier.
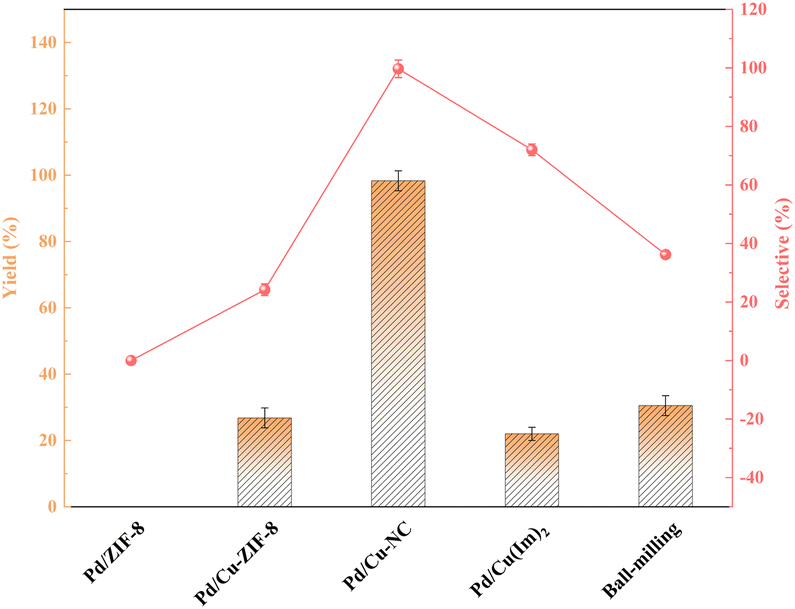 | ||
| Fig. 7 Effect of different catalysts on the reaction. | ||
The optimization outcomes of the experimental investigation are depicted in Fig. 8, illustrating the correlation between reaction yield, selectivity, and key variables, namely time (Fig. 8a), pressure (Fig. 8b), temperature (Fig. 8c), and yield (Fig. 8d). The graphical representation distinctly indicates A near-linear dependence of reaction yield and selectivity on these aforementioned factors. However, noteworthy is the observation that when the temperature surpasses a certain threshold (denoted as temperature 1), there is a pronounced reduction in both yield and selectivity. Specifically, conditions characterized by 140 °C temperature, 4 MPa pressure, and a loading of 17.26% lead to an impressive 91.1% yield and 93.4% selectivity achieved within a mere 1.5-hour duration. Further analysis at identical temperature and pressure conditions showcases an optimal performance, delivering an elevated 98.3% yield and an exceptional 99.7% selectivity in just 2 hours. Evidently, this reaction underscores the synergistic features of high temperature, exceptional selectivity, and commendable yield. The combination of elevated temperature, superior selectivity, and notable yield truly delineates the salient attributes of this reaction.
 | ||
| Fig. 8 Optimization of catalyst with respect to (a) time (b) pressure, (c) temperature, (d) load. | ||
Our investigation delved into the impact of copper metal doping on the reaction, illustrated in Fig. 9. It is evident that copper doping plays a pivotal role in influencing yield. When copper is introduced as a dopant, a discernible trend emerges in yield and selectivity, displaying characteristics reminiscent of a volcano plot. This phenomenon can be ascribed to the interplay of multiple factors. In instances where the metal content is meager, the catalyst's activity might be compromised due to an insufficient number of active metal sites. With an incremental increase in metal content, the catalytic activity tends to rise, presumably owing to an augmentation in active sites, thereby expediting the reaction rate. However, an excessive metal content might foster the adsorption of an excess of reaction intermediates, giving rise to competing reactions or by-product formation (notably, glycerol's tendency to form monomers under divalent copper-catalyzed conditions). This, in turn, hampers the reaction's selectivity. The 82% yield observed in the case of undoped copper could be attributed to metal loss during the direct calcination of ZIF-8 at 950 °C under nitrogen, leading to backbone collapse and fragmentation (Fig. S2b†). The resultant defects are likely responsible for the increased yield and selectivity, which, counterintuitively, impairs catalyst recovery.
 | ||
Fig. 9 The effect of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn mole ratio on the reaction. :Zn mole ratio on the reaction. | ||
The elucidation of the mechanism governing glycerol carbonylation for glycerol carbonate synthesis was initially proposed by Hu et al. in 2010.10 While their work provided an initial framework, the intricate details encompassing the formation of the carbonyl group, the ring-forming process, and the ultimate desorption stages were not comprehensively explored (Fig. 10 procedures 1 and 2). Our research group has extended the mechanistic investigation of this reaction, delving into the intricacies of carbonyl group formation, the subsequent ring-forming process, and the terminal desorption, utilizing density functional theory (DFT). Furthermore, we have elucidated the interactions between copper and palladium via electrostatic potentials.
 | ||
| Fig. 10 The reaction mechanism. | ||
To simplify the workload of simulation calculations, we adopt the transition state in the reaction process to explore the synergistic effect between Cu and Pd. For the initial formation of the carbonyl group (Fig. 11), a sequential process is envisioned. Initially, carbon monoxide adsorbed on metal palladium triggers the rupture of the palladium–oxygen bond. Subsequently, oxygen-negative ions partake in nucleophilic addition to the triple bond of carbon monoxide, culminating in the formation of the carbonyl group. The initial electrostatic potential diagram (Fig. 12a) illustrates a lower electrostatic potential at the palladium–oxygen bond, inducing a more uniform charge distribution that heightens the reactivity of the carbonyl–oxygen bond. Upon an exchange reaction between another hydroxyl hydrogen and an iodide ion anion, a six-membered ring emerges. However, this ring's stability is precarious (Fig. 12b), often leading to its attachment to the carbon and palladium bond. In this region, the electrostatic potential is elevated, indicating an uneven charge distribution that facilitates chemical bond rupture. Consequently, negative carbon ions mount a subsequent attack on the oxygen atom, culminating in the product, the formation of the glycerol carbonate ring, alongside the detachment of palladium. From an electrostatic potential analysis subsequent to the incorporation of copper (depicted in Fig. 12c and d), it becomes conspicuous that the presence of copper exerts a pronounced influence on the palladium-driven reaction pathway.
 | ||
| Fig. 11 Intermediate state potential energy diagram. | ||
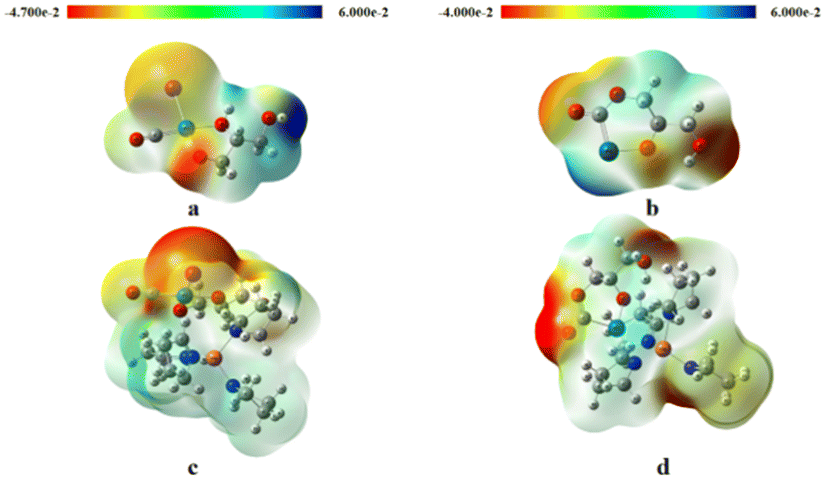 | ||
| Fig. 12 (a and b) Initial state electrostatic potential diagram (c and d) doped copper initial state electrostatic potential diagram. | ||
Possible mechanism of copper–palladium synergistic promoting reaction is shown in Scheme 2. The electrostatic potential profile of the palladium center experiences a diminution in intensity following the introduction of copper, a trend particularly accentuated during the cyclization phase. This phenomenon is highlighted by the reduction of the inherent charge associated with the metal palladium from 0.474 to 0.245 during the cyclization process. Consequently, an augmented degree of uniformity and ordered charge distribution encircles the metal palladium, consequently amplifying the stability quotient within the cyclization process. This augmented stability can be ascribed to the interplay of charge transfer dynamics between copper and palladium, alongside their collaborative synergistic effect. This synergistic interplay engenders an elevation in the overall reactivity of the reaction, thereby contributing to the observed enhancement, this is also corroborated with the catalyst study of copper doping action activity experiments (Fig. 9).
 | ||
| Scheme 2 Possible mechanism of copper–palladium synergistic promoting reaction. | ||
Conclusions
In summary, our investigation has led us to the discovery of a highly efficient catalyst, Pd/Cu0.356–NC, capable of catalyzing the glycerol carbonylation reaction to synthesize glycerol carbonate. Leveraging the advantageous attributes of the zeolite imidazolium framework, such as its high specific surface area and exceptional stability, we have successfully enhanced its catalytic performance through the incorporation of Cu metal subsequent to calcination. Under conditions of 140 °C and 4 MPa, with a loading of 17.26%, a remarkable yield of 91.1% and selectivity of 93.4% was achieved within 1.5 hours. Impressively, an even higher yield of 98.3% along with an outstanding selectivity of 99.7% was attained within 2 hours. Furthermore, the incorporation of copper metal within the zeolite imidazolium framework emerged as a pivotal factor in heightening catalytic activity. Notably, the copper content exhibited a distinctive volcano plot trend, illustrating its influence on the reaction's efficiency. This catalytic system showcases the potential of bimetallic catalysts, warranting further exploration within this domain. Additionally, our study delved into the mechanistic intricacies of the glycerol carbonylation reaction for glycerol carbonate synthesis. Through the utilization of density functional theory, we simulated the crucial processes involving the formation of the carbonyl group and the subsequent five-membered ring formation in glycerol carbonate. This in-depth analysis not only deepened our comprehension of the reaction mechanism but also shed light on the pivotal role of copper metal in this intricate process. This exploration not only enhances our understanding of the reaction system but also establishes a new avenue for future research directions. In essence, our findings contribute to the advancement of catalytic science, presenting a promising catalyst and unravelling mechanistic insights that hold promise for sustainable glycerol carbonate synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial supports from the Natural Science Foundation of China (NSFC) (No. 22378163) and MOE & SAFEA for the 111 Project (B13025) are gratefully acknowledged.References
- I. Ambat, V. Srivastava and M. Sillanpää, Renewable Sustainable Energy Rev., 2018, 90, 356–369 CrossRef CAS.
- A. C. Pinto, L. L. N. Guarieiro, M. J. C. Rezende, N. M. Ribeiro, E. A. Torres, W. A. Lopes, P. A. de P. Pereira and J. B. de Andrade, J. Braz. Chem. Soc., 2005, 16, 1313–1330 CrossRef CAS.
- M. Balat and H. Balat, Appl. Energy, 2010, 87, 1815–1835 CrossRef CAS.
- H. Rastegari, H. Jazini, H. S. Ghaziaskar and M. Yalpani, in Biodiesel: From Production to Combustion, ed. M. Tabatabaei and M. Aghbashlo, Springer International Publishing, Cham, 2019, pp. 101–125 Search PubMed.
- C. H. Zhou, H. Zhao, D. S. Tong, L. M. Wu and W. H. Yu, Catal. Rev.: Sci. Eng., 2013, 55, 369–453 CrossRef CAS.
- G. Galletti, P. Prete, S. Vanzini, R. Cucciniello, A. Fasolini, J. De Maron, F. Cavani and T. Tabanelli, ACS Sustainable Chem. Eng., 2022, 10, 10922–10933 CrossRef.
- S. Franklin, United States, US2446145A, 1948.
- J. Li and T. Wang, J. Chem. Thermodyn., 2011, 43, 731–736 CrossRef CAS.
- L. Wang, Y. Ma, Y. Wang, S. Liu and Y. Deng, Catal. Commun., 2011, 12, 1458–1462 CrossRef CAS.
- J. Hu, J. Li, Y. Gu, Z. Guan, W. Mo, Y. Ni, T. Li and G. Li, Appl. Catal., A, 2010, 386, 188–193 CrossRef CAS.
- M. Casiello, A. Monopoli, P. Cotugno, A. Milella, M. M. Dell'Anna, F. Ciminale and A. Nacci, J. Mol. Catal. A: Chem., 2014, 381, 99–106 CrossRef CAS.
- Y. Lei, G. Lan, M. Fan and G. Li, Catal. Commun., 2020, 140, 106007 CrossRef CAS.
- F. Doro, P. Winnertz, W. Leitner, A. Prokofieva and T. E. Müller, Green Chem., 2011, 13, 292–295 RSC.
- Y. Liu, Z. Li, Q. Yu, Y. Chen, Z. Chai, G. Zhao, S. Liu, W.-C. Cheong, Y. Pan, Q. Zhang, L. Gu, L. Zheng, Y. Wang, Y. Lu, D. Wang, C. Chen, Q. Peng, Y. Liu, L. Liu, J. Chen and Y. Li, J. Am. Chem. Soc., 2019, 141, 9305–9311 CrossRef CAS PubMed.
- L. Wang, Y. Liu, C. Liu, R. Yang and W. Dong, Sci. China: Chem., 2013, 56, 1455–1462 CrossRef CAS.
- R. Chen, J. Yao, Q. Gu, S. Smeets, C. Baerlocher, H. Gu, D. Zhu, W. Morris, O. M. Yaghi and H. Wang, Chem. Commun., 2013, 49, 9500–9502 RSC.
- J. Li, X. Wang, G. Zhao, C. Chen, Z. Chai, A. Alsaedi, T. Hayat and X. Wang, Chem. Soc. Rev., 2018, 47, 2322–2356 RSC.
- W. Zhu, S. Ndayiragije, X. Zuo, X. Zhang, G. Wang and X. Wang, J. Environ. Chem. Eng., 2022, 10, 107758 CrossRef CAS.
- W. Jin, Z. Lu, Q. Wang, Y. Zhu, H. Pan, S. Yao, Z. Fang, X. Huang and X. Chen, JPhys Mater., 2021, 4, 024006 CrossRef CAS.
- S. Sun, Z. Yang, J. Cao, Y. Wang and W. Xiong, J. Solid State Chem., 2020, 285, 121219 CrossRef CAS.
- Y. Yao, H. Yin, Y. Zhang, F. Wei, H. Hu, Y. Tang and S. Wang, Chem. Eng. J., 2021, 426, 131801 CrossRef CAS.
- M. Kuang, Q. Wang, P. Han and G. Zheng, Adv. Energy Mater., 2017, 7, 1700193 CrossRef.
- S. Tanaka and Y. Tanaka, ACS Omega, 2019, 4, 19905–19912 CrossRef CAS PubMed.
- B. Chi, L. Zhang, X. Yang, Y. Zeng, Y. Deng, M. Liu, J. Huo, C. Li, X. Zhang, X. Shi, Y. Shao, L. Gu, L. Zheng, Z. Cui, S. Liao and G. Wu, ACS Catal., 2023, 13, 4221–4230 CrossRef CAS.
- D. A. Bulushev, F. S. Golub, S. V. Trubina, V. V. Zvereva, L. G. Bulusheva, E. Y. Gerasimov, M. Navlani-García, A. D. Krot and H. S. Jena, ACS Appl. Nano Mater., 2022, 5, 12887–12896 CrossRef CAS.
- X. Wang, Y. Zhao, Y. Sun and D. Liu, Molecules, 2022, 27, 4312 CrossRef CAS.
- Q. Guo, C. Chen, L. Zhou, X. Li, Z. Li, D. Yuan, J. Ding, H. Wan and G. Guan, Microporous Mesoporous Mater., 2018, 261, 79–87 CrossRef CAS.
- Z. Li, Y. Sun, J. Xing, Y. Xing and A. Meng, J. Hazard. Mater., 2018, 352, 204–214 CrossRef CAS PubMed.
- M. Shakeel, M. Arif, G. Yasin, B. Li and H. D. Khan, Appl. Catal., B, 2019, 242, 485–498 CrossRef CAS.
- J. Cao, S. Sun, X. Li, Z. Yang, W. Xiong, Y. Wu, M. Jia, Y. Zhou, C. Zhou and Y. Zhang, Chem. Eng. J., 2020, 382, 122802 CrossRef CAS.
- Y. Song and Q. Zheng, Crit. Rev. Solid State Mater. Sci., 2016, 41, 318–346 CrossRef CAS.
- N. Wang, X. Zhao, R. Zhang, S. Yu, Z. H. Levell, C. Wang, S. Ma, P. Zou, L. Han, J. Qin, L. Ma, Y. Liu and H. L. Xin, ACS Catal., 2022, 12, 4156–4164 CrossRef CAS.
- V. Armel, S. Hindocha, F. Salles, S. Bennett, D. Jones and F. Jaouen, J. Am. Chem. Soc., 2017, 139, 453–464 CrossRef CAS PubMed.
- H. Tang, S. Cai, S. Xie, Z. Wang, Y. Tong, M. Pan and X. Lu, Adv. Sci., 2016, 3, 1500265 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Standard curves, GC methods, preparation method and catalyst characterization methods and results. See DOI: https://doi.org/10.1039/d3cy01189e |
| This journal is © The Royal Society of Chemistry 2024 |