
Site specifically probing the unfolding process of human telomere i-motif DNA using vibrationally enhanced alkynyl stretch†
Tiantian
Dong
ab,
Pengyun
Yu
 ab,
Juan
Zhao
ab and
Jianping
Wang
*ab
ab,
Juan
Zhao
ab and
Jianping
Wang
*ab
aBeijing National Laboratory for Molecular Sciences, Molecular Reaction Dynamics Laboratory, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: jwang@iccas.ac.cn; Fax: (+86)-010-62563167; Tel: (+86)-010-62656806
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 4th January 2024
Abstract
The microscopic unfolding process of a cytosine-rich DNA forming i-motif by hemi-protonated base pairs is related to gene regulation. However, the detailed thermal unfolding mechanism and the protonation/deprotonation status of site-specific cytosine in DNA in a physiological environment are still obscure. To address this issue, a vibration-enhanced C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C probe tagged on 5′E terminal cytosine of human telomere i-motif DNA was examined using linear and nonlinear infrared (IR) spectroscopies and quantum-chemistry calculations. The CC probe extended into the major groove of the i-motif was found using nonlinear IR results only to introduce a minor steric effect on both steady-state structure and local structure dynamics; however, its IR absorption profile effectively reports the cleavage of the hemi-protonated base pair of C1–C13 upon the unfolding with C1 remaining protonated. The temperature mid-point (Tm) of the local transition reported using the CC tag was slightly lower than the Tm of global transition, and the enthalpy of the former exceeds 60% of the global transition. It is shown that the base-pair unraveling is noncooperative, with outer base pairs breaking first and being likely the rate limiting step. Our results offered an in-depth understanding of the macroscopic unfolding characteristics of the i-motif DNA and provided a nonlinear IR approach to monitoring the local structural transition and dynamics of DNA and its complexes.
C probe tagged on 5′E terminal cytosine of human telomere i-motif DNA was examined using linear and nonlinear infrared (IR) spectroscopies and quantum-chemistry calculations. The CC probe extended into the major groove of the i-motif was found using nonlinear IR results only to introduce a minor steric effect on both steady-state structure and local structure dynamics; however, its IR absorption profile effectively reports the cleavage of the hemi-protonated base pair of C1–C13 upon the unfolding with C1 remaining protonated. The temperature mid-point (Tm) of the local transition reported using the CC tag was slightly lower than the Tm of global transition, and the enthalpy of the former exceeds 60% of the global transition. It is shown that the base-pair unraveling is noncooperative, with outer base pairs breaking first and being likely the rate limiting step. Our results offered an in-depth understanding of the macroscopic unfolding characteristics of the i-motif DNA and provided a nonlinear IR approach to monitoring the local structural transition and dynamics of DNA and its complexes.
1. Introduction
Cytosine-rich sequences can fold into an intercalated four strand i-motif structure via hemi-protonated base pairs ([C–H–C]+) under weak acidic or even neutral conditions and unfold into a hairpin-like or a random-coil structure under basic conditions or at high temperature. Both the conformation transformation process and structure stability of i-motif DNA are related to the transcription, gene regulation and genomic function in biological systems.1–5 The i-motif structure has also been widely used in biotechnology, drug delivery, molecular switching and nanostructure construction because of its unique proton-induced folding/unfolding properties.6,7 The structural stability and the conformational dynamics of the folding/unfolding of the i-motif DNA are of great significance for gene regulation8,9 as well as for molecular device fabrication.6,7,10,11 The folding and unfolding process of the i-motif is known to be both pH and temperature dependent, and reversible.12 The kinetics of the folding/unfolding of the i-motif DNA has been an important research topic, and has been studied by using several methods, such as nuclear magnetic resonance (NMR) spectroscopy, circular dichroism (CD) spectroscopy, fluorescence resonance energy transfer (FRET) and surface plasmon resonance (SPR) spectroscopy.13–18 Time-resolved spectroscopy and molecular dynamics simulations have been recently used to explore the detailed mechanism steps of the folding/unfolding and the asymmetric structures of hemi-protonated base pairs, as well as the characteristics of protonation–deprotonation of related bases.14,16,19–21Using stopped-flow circular dichroism (SFCD) spectroscopic measurements, a pH induced unfolding model involving two steps of neutralization of protons was proposed, in which the rate-limiting step was found to be preferably the cooperative neutralization of two of the totally six protons.16 This work was supported by all-atom molecular dynamics simulations.20 Although there have been extensive works on the pH-induced folding/unfolding pathways of the i-motif structure, temperature-induced folding/unfolding dynamics are much less studied. Molecular dynamics simulations were carried out at higher temperature to reveal the conformation of stable intermediates in the deprotonated state during the thermodynamic unfolding period of the i-motif DNA.21 Recently, it was shown by temperature-dependent infrared measurements that for thermal unfolding, the outer hydrogen bonds involving C(2)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(2) and N(4)H2 amino groups break first, subsequently followed by the breaking of the hydrogen bond involving N(3)+.22 However, the microscopic mechanism and pathway of the unfolding of the i-motif DNA, particularly the site-specific protonation/deprotonation status of a given nucleobase is still under debate.
O(2) and N(4)H2 amino groups break first, subsequently followed by the breaking of the hydrogen bond involving N(3)+.22 However, the microscopic mechanism and pathway of the unfolding of the i-motif DNA, particularly the site-specific protonation/deprotonation status of a given nucleobase is still under debate.
Vibrational spectroscopy has shown excellent sensitivity to biomolecular conformation and has been a suitable tool to study protein and DNA structural transition dynamics.23–27 An external vibrational probe tagged on a biomolecule with its vibrational absorption outside the major absorption regions of the biomolecules has become a very useful method for site-specific monitoring.28–31 Recently, phenylacetylene was used as a Raman tag to report the protonation/deprotonation status of a nucleobase in the folded- and unfolded-states of the i-motif DNA.32 In our recent work, we have explored a novel vibrationally enhanced alkynyl probe with a single absorption band that has excellent sensitivity for the local structure of nucleobases and their solvent environment, as well as the hydrogen-bonding interaction between the nucleobases and solvent.33 Since no new polar short-range interaction is introduced by the alkynyl group, the interpretation of the observed IR spectra can be easily carried out. Because of this, it is advantageous to be used for monitoring site-specific and local structures and dynamics of DNA as well as tracking the forming and breaking events of the nucleobase pairs, and the protonation/deprotonation status of the nucleobases. To this end, the extent of perturbations of the extrinsically tagged IR probe to the local structure is an important and complicated issue that determines the applicability of the probe and the reasonability of the obtained experimental results. As will be seen from the FTIR spectral results in this work, the externally tagged probe shows little effect on the equilibrium structure of the i-motif DNA. However, several studies pointed out that an apolar methyl group or polar hydroxymethyl group tagged on cytosine may alter its local hydration and base pairing dynamics, as well as structure flexibility.34–36
In this work, we use infrared spectroscopy employing a vibrationally enhanced alkynyl probe tagged on the site-specific nucleobase of hTelo i-motif DNA (PDB:1EL2, as illustrated in Fig. 1a) to monitor the thermally unfolding process and the protonation–deprotonation properties of the selected nucleobase. Herein, the alkynyl probe is triisopropylsilylacetylene (TIPSE) and the alkynyl probe modified deoxycytidine is TIPSEdC.33 The CC probe was tagged on the 5′E terminal cytosine (C1) by the conventional solid-phase DNA synthesis method37 to track the status of the C1–C13 nucleobase pair during the unfolding process of the i-motif DNA.
 | ||
| Fig. 1 (a) A scheme of the intercalated, four-stranded hTelo i-motif DNA structure. The sequence is marked with letter C (cytosine), T (thymine), A (adenine) and number subscript. (b) Illustration of the CC probe (red marker) labeled C1 nucleobase of the hTelo i-motif DNA strand in the folded structure. Straight-line arrows indicate the major and minor grooves of the i-motif structure. The i-motif structure frame is showed in the residue colored tube model. Hemi-protonated base pair of C1–C13 is highlighted in blue and hydrogen bonds are displayed with a colored dashed line. (c) Chemical structure of the CC probe labeled C1 nucleobase. | ||
The structure of the TIPSE-labeled (named as CC-labeled) i-motif strand is displayed in Fig. 1b, and the chemical structure of the IR probe is shown in Fig. 1c. We carried out temperature-dependent FTIR experiment for both unlabeled and CC-labeled strands. The transition temperature (Tm) derived from the CC stretch for the local transition was found to be slightly lower than the global transition temperature, indicating that the cleavage of outer at least the terminal base pair involving the C1–C13 base pair is likely to be one of the initial steps of the unfolding and may also be the rate-limiting step. For C1, it remains to be protonated during the unfolding of the i-motif DNA. Detailed analysis is given in this work.
In addition, the extent of perturbations to the local structure and solvation dynamics from the externally tagged CC probe was explored in both the unlabeled and CC-labeled strands using ultrafast nonlinear IR spectroscopy including two-dimensional IR (2D IR) spectroscopy. The extrinsic CC probe tagged to C1 was found to have a minor perturbative effect on the local structure dynamics, solvent dynamics, and structural flexibility of the i-motif DNA.
2. Experimental and methods
2.1. Sample preparation
The hTelo i-motif DNA is a 22 (nucleotides, nt) oligomer with the sequence of 5′-CCCTAACCCTAACCCTAACCCT-3′,38 however, U7 of 1EL2 was replaced by C7 in this work. The CC labeled C1 strand is 5′-TIPSECCCTAACCCTAACCCTAACCT-3′. Both unlabeled and CC-labeled strands were synthesized by the Suzhou Biosyntech Co., Ltd. The TIPSE modified deoxycytidine phosphoramidite (TIPSEdC phosphoramidite) was prepared in this work using the conventional phosphosamidite synthesized method.37 The synthetic details and NMR characterizations are shown in Scheme S1 and Fig. S1–S5, ESI.† The MS spectrum of the CC labeled strand is shown in Fig. S6 (ESI†) to show that the CC group has been labeled on the i-motif strand. Powdered samples were at default purity and centrifuged twice at a speed of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 minutes. Then all labile protons of the two samples were exchanged in deuterium oxide (D2O) three times. The prepared buffer D2O solution is pH* 7.6 Dulbecco's phosphate-buffered saline (DPBS buffer solution) with the pH* value adjusted by deuterium chloride (DCl). pH* refers to the uncorrected (for D2O) pH-meter reading value at 25 °C. Both unlabeled and CC-labeled strands were dissolved in pH* 4.5 DPBS buffer solution at a concentration of 10 mM for temperature-dependent FTIR measurement and ultrafast nonlinear infrared spectroscopic measurement.
000 rpm for 10 minutes. Then all labile protons of the two samples were exchanged in deuterium oxide (D2O) three times. The prepared buffer D2O solution is pH* 7.6 Dulbecco's phosphate-buffered saline (DPBS buffer solution) with the pH* value adjusted by deuterium chloride (DCl). pH* refers to the uncorrected (for D2O) pH-meter reading value at 25 °C. Both unlabeled and CC-labeled strands were dissolved in pH* 4.5 DPBS buffer solution at a concentration of 10 mM for temperature-dependent FTIR measurement and ultrafast nonlinear infrared spectroscopic measurement.
2.2. Methods
Temperature-dependent FTIR spectra were recorded using a commercial Nicolet 6700 FTIR spectrometer with an attached homemade temperature-controlling device for samples. Sample solutions were held in a double-sample-cell holder with a half-cell sample solution and a half-cell solvent (DPBS buffer solution). The FTIR spectra of the two samples were collected from 25 °C to 90 °C, with a step of 5 °C. Each spectrum is solvent background subtracted.Ultrafast nonlinear IR spectroscopy was used to characterize the structure dynamics of the unlabeled and CC-labeled strands in the 6-μm wavelength region. 2D IR spectra of the two samples were collected using a pulse-shaped 2D IR apparatus.39,40 Briefly a laser of 800-nm center wavelength, 3-mJ pulse energy, 25-fs pulse width, and 1-kHz repetition rate generated using a commercial Ti-sapphire laser amplifier was modulated into the needed mid-infrared pulse (center frequency of 1740 cm−1) through an optical parametric amplifier (OPA) and a difference frequency generator (DFG). The out pulse is split into two pulses, one is called the pump pulse, passing a series of IR optics, entering the pulse shaper, and generating a pair of delay time (i.e., the coherence time, τ) adjustable pump pulses, the other is called the probe pulse, passing the IR polarizer and half-wave plate and entering the IR monochromator along the same direction as the generated nonlinear IR signal. Detection frequency (ωt) is defined by the IR monochromator and grating. The delay time between the paired pump pulses and the probe pulse is called the waiting time (Tw). The 2D IR signal was collected in the pump–probe geometry at each of a series of Tw. Additionally, the magic-angle IR pump–probe spectroscopic measurement was carried out using the same experimental setup with the 54.7 degree between the laser electric-field directions of the pump pulse pair (at τ = 0) and the probe pulse.
Quantum chemistry calculations were performed using the density functional theory (DFT + D3) at the level of B3LYP by Gaussian 09,41 in which the D3 dispersion correction scheme calculates the inter- and intramolecular dispersion interactions that are useful for large (supramolecular) systems by employing the given system coordinates (and atomic numbers). The 6-311+G** basis set was used for geometry optimization of the protonated/non-protonated cytosine and nucleobase pair structures, followed by IR spectral simulations. Even though the size of the model dimer systems (≤91 atoms) considered in this work is not too large, the dispersion correction on DFT is still considered in our computation.
3. Results and discussion
3.1. Temperature-dependent FTIR spectroscopy
Both the unlabeled and CC-labeled strands are in the folded state (i-motif structure) when dissolved in pH* 4.5 DPBS buffer solution at room temperature (25 °C) as illustrated using the CD spectra (shown in Fig. S7, ESI†). Temperature-dependent FTIR spectra of the unlabeled and CC-labeled strands are shown in Fig. 2. In the 6-μm wavelength region, the vibrational absorption is mainly from three nucleobase's vibration modes which could reflect the global folding/unfolding process of the i-motif DNA. The spectral evolution of the CC-labeled strand is consistent with that of the unlabeled strand. Each band is assigned with colored shading and marked with a combined letter-number in Fig. 2a and b, among which, C is cytosine, T is thymine, and A is adenine. With increasing temperature, the C1 band (1665–1655 cm−1) frequency gradually red shifts and intensity gradually increases. The A1 band (1625 cm−1) behaves similarly. In the 1700–1750 cm−1 region, the C+1 (C+ is the protonated cytosine) band intensity decreases whereas the C+1′ band intensity increases, in which one isosbestic point appears at 1707 cm−1 (marked by a black star). In a thermal transition, an isosbestic point may appear between a new band and a vanishing old band, which can be used as an indicator of phase transition and can reveal the transition temperature (mid-point of the structure transition).42–46 Therefore, both the unlabeled and CC-labeled strands have changed from the folded state (i-motif structure) to the unfolded state (random coil) at the temperature ranging from 25 °C to 90 °C, as indicated by the temperature-dependent FTIR spectra. This process seems like a typical two-state transition. As seen in Fig. 2a and b, there seems to be a transition point around 60 °C (drawn in a colored dotted line), at which all band frequencies or/and intensities especially the C1 band and pair of C+1/C+1′ peaks change most drastically.
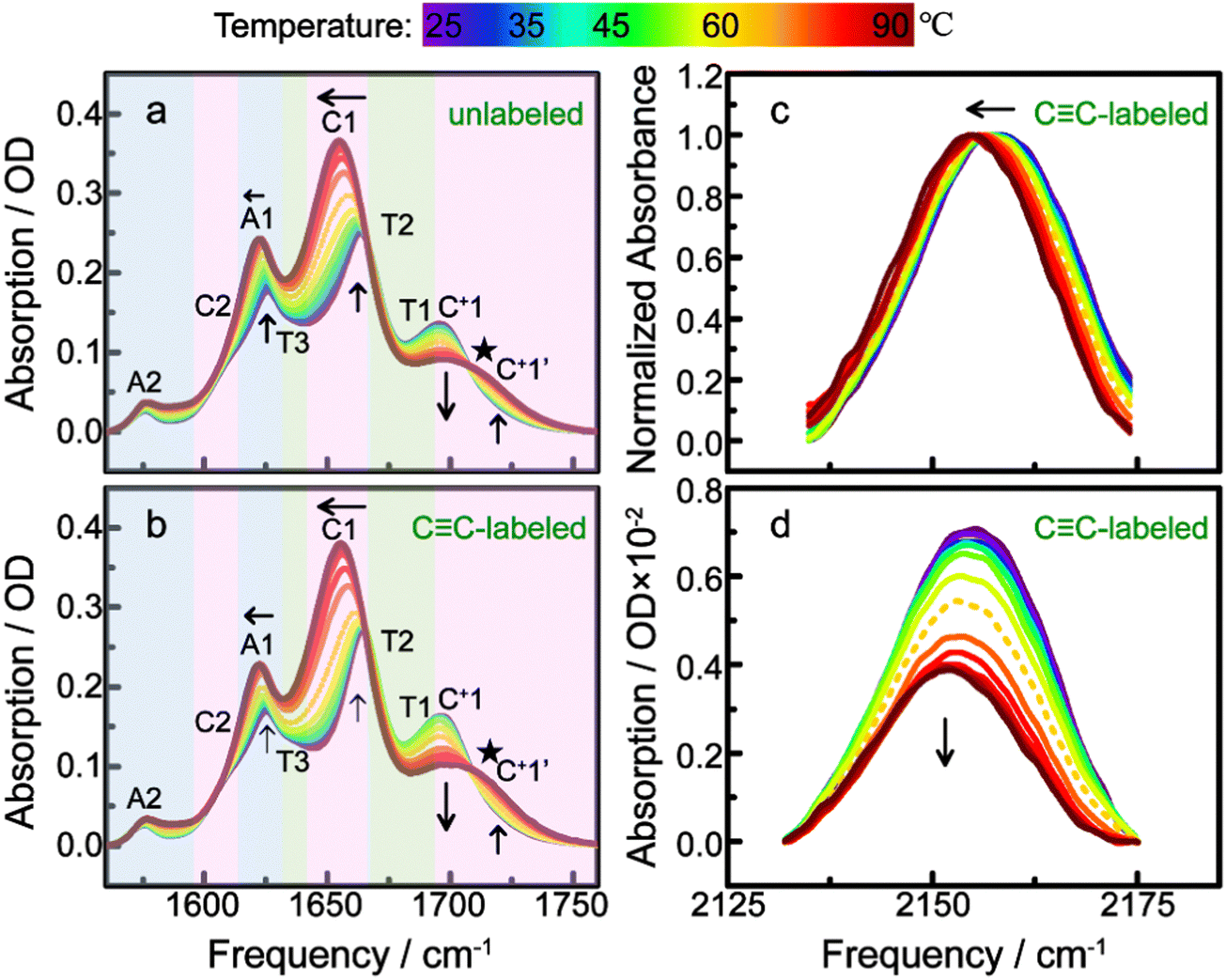 | ||
| Fig. 2 Temperature-dependence of the FTIR spectra of the unlabeled (a) and CC-labeled (b) h-Telo i-motif DNA strand in pH* 4.5 DPBS buffer solution in the 6 μm wavelength region collected from 25 to 90 °C. The CC stretching absorption region of the intensity normalized (c) and baseline corrected (d) spectra of the CC-labeled strand. Colored shadings indicate the contribution of three nucleobases to the spectra and each peak is marked with the combined letter-number in (a) and (b). Arrows indicate the directions of changes of peak intensity gain/loss and frequency shift with increasing temperature. The dotted line of the 60 °C spectrum in each panel indicates the case near thermal unfolding transition temperature. Black star in (a) and (b) indicates the isosbestic point. | ||
Comparing the unlabeled and CC-labeled strands, the characteristics of key spectral changes and their temperature dependencies are quite similar for the two samples. This indicates that the external CC probe tagged on C1 has little effect on the steady-state i-motif structures and transformation between structures, even though the label has a relatively large terminal group (TIPS). This is probably because the probe extends toward into the major groove of the i-motif structure and TIPS is far enough from the main bulk structure (Fig. 1b). This point has been reported by our recent work of the CC probe.33 Hence the TIPS group yields only a minor steric effect on the local structure of the i-motif DNA studied here.
The stretching vibration peak of CC tagged on C1 also changed gradually as displayed in Fig. 2c and d. The peak position red shifts, and intensity decreases as a function of temperature, indicating that this label can sense the unfolding process of the i-motif DNA, in particular the C1–C13 base pair cleavage process as indicated in Fig. 1b. The CC spectral change also gave a transition mid-point temperature of around 60 °C, as shown by the dotted spectral line in both Fig. 2c and d. Therefore, the temperature dependence of the CC band could reveal the thermally unfolding process of the i-motif DNA and the local structure transition involving C1–C13 base pair cleavage. However, two issues should be pointed out. The first is the components of the CC band. From the folded-state to the unfolded-sate, the CC band red shifted about 4 cm−1. It is feasible but unnecessary to split the CC band into two or more peaks representing the absorption components for different states, because this would complicate spectral interpretation. Instead, the CC band could be treated as a whole in which as the unfolded component increases, the absorption of the lower-frequency component increases and the overall CC band red shifts. The second is the incompleteness of the CC band line shape shown in both Fig. 2c and d, which is due to the presence of both OD stretching vibration and bending-libration combination absorption bands of D2O that interacts with the higher-frequency peak wings of the CC band. Baseline corrected spectra are shown in Fig. 2d.
The IR absorption peaks in the 6-μm region were further used to characterize the unfolding process especially for making a distinction between the global transition and local transition processes. We collected the IR spectra of three nucleosides (dA, dT and dC) and protonated dC (dC+) at room temperature, which are shown in Fig. S8a (ESI†). The vibrational absorption band of each base of the unfolded i-motif structure (random coil) shown in Fig. 2a and b is very close to that of the free nucleoside monomer. The superposed spectrum of nucleosides' monomer spectra with the ratio (4:6:6:6) shown in Fig. S8b (ESI†) nearly coincided to the spectra of the unfolded i-motif strand shown in Fig. 2a. The 6-μm bands of i-motif DNA strand were further assigned based on the assignment of nucleoside monomers, which are marked similarly in the two figures. Peak assignment of the protonated/deprotonated cytosine in both the folded and unfolded forms is crucial, because the hemi-protonated base pair breaking involves cytosine, whose IR signature changes dramatically whereas those of the A and T bases only change slightly during the unfolding of the i-motif DNA.
From the folded to unfolded (random coil) structure of the i-motif, the CO stretching vibrational absorption frequency of cytosine (band C1) shifted from 1664 cm−1 to 1655 cm−1 as base pairs break and the π–π base-pair stacking degrades. The change of the C1 band is directly related to the structural transformation and reflects the global unfolding process. Protonated cytosine (dC+) of the folded-state has an IR peak located in 1709 cm−1, which is shown in Fig. S8a (ESI†). As temperature increases, the concentration of the folded component decreases whereas the unfolded component increases. This is reflected by the intensity of the C+1 band at 1696 cm−1 (folded state) that decreases, and by that of the C+1′ band at 1720 cm−1 (unfolded state) that increases.
The frequency assignment of these two bands is consistent with previous reports.26,47 Specifically, Wieser's group indicated that a new absorption band at 1694-cm−1 is the unique feature for the intercalated structure forming,47 whereas a recent work of the Tokmakoff group pointed out that the growth of the 1720 cm−1 band indicates a gain of the protonated cytosine in the random-coil state.26 Therefore, the changes in the paired C+1/C+1′ bands can be used to reflect the global unfolding process in this work. In addition, two shoulder bands appeared near 1689 cm−1 and 1667 cm−1 corresponding to thymine carbonyl stretch (T1 and T2 in Fig. 2a and b) which are consistent with dT's absorption shown in Fig. S8a (ESI†). A shoulder band near 1637 cm−1 corresponds to the thymine ring vibration (T3 in Fig. 2a and b), while the shoulder near 1609 cm−1 corresponds to the ring mode of cytosine (C2 in Fig. 2a and b). The 1625-cm−1 and 1575-cm−1 peaks correspond to the ring modes of adenine (A1 and A2 in Fig. 2a and b). These band assignments are listed in Table S1 (ESI†).
3.2. Two-state model
To determine the transition temperature (Tm), the temperature dependence of selected spectral parameters (band frequency and intensity) is fitted with a sigmoidal function derived from the two-state model:22,48 | (1) |
The C1 band frequency andintensity, C+1′ band intensity, and CC band frequency and intensity were fitted to the two-state model, and fitting curves are shown in Fig. 3. For the C1 band intensity, the intensity growth of 1653 cm−1 (unfolded state) in the difference spectra (shown in Fig. S9, ESI†) was used, in which each spectrum was subtracted from that at 25 °C. Fitting the C1 band frequency (Fig. 3a) yielded a Tm value of 62.7 °C and 63.2 °C for the unlabeled and CC-labeled strands, respectively. Fitting the C1 band intensity yielded a Tm value of 62.9 °C and 63.0 °C, respectively, for both strands as shown in Fig. 3b. The growth of the C+1′ band was also fitted to give the Tm values of 62.8 °C and 63.0 °C, respectively, for both strands as shown in Fig. 3c. The red shift of the C1 band, the growth of the 1653-cm−1 band and that of the C+1′ band are all related to the six hemi-protonated base pair cleavage process which is linearly correlated with the concentration of the random coil structure. The global transition mid-point of CC-labeled strand (i.e., 63.1 °C on average) is found to be only 0.3 °C higher than that of the unlabeled strand (i.e., 62.8 °C on average). The transition temperature region of the CC-labeled strand is slightly narrower than that of the unlabeled strand as indicated in Fig. 3a–c. These results illustrate that the CC-labeled strand is slightly more stable than the unlabeled strand, indicating that the external probe has little perturbation in the steady-state structure of the i-motif DNA including the folded, partially folded and unfolded structures. The global Tm determined in this work is slightly higher than previously reported Tm from a 21 nucleotide (nt) strand (i.e., 59.5 °C) by CD spectroscopy32 and is slightly lower than previously reported Tm from a 24 nt strand (i.e., 66.2 °C)52 under similar conditions.
 | ||
| Fig. 3 Thermal unfolding curves of the unlabeled strand (wine line) and the CC-labeled strand (navy line) monitored by linear IR spectroscopy at (a) 1664 cm−1 (C1 band) frequency shift, (b) 1653 cm−1 (C1 band) intensity increase from the difference spectrum, and (c) 1720 cm−1 (C+1′ band) intensity increase. (d) 2155 cm−1 (CC stretch) frequency shift (black line) and intensity decrease (green line) for the CC-labeled strand. Solid line represents least-square fitting to the two-state unfolding transition. | ||
The temperature dependence of CC band frequency was also fitted with the two-state model and yielded a Tm value of 61.2 °C (shown in Fig. 3d with a black curve). Moreover, fitting temperature dependence of the CC band intensity yielded a Tm value of 61.5 °C (shown in Fig. 3d with a green curve). The relationship between the CC band's red shift and its intensity loss appears to be linear (Fig. S10, ESI†), suggesting that both of them are related to the C1–C13 base pair breaking and the local site denaturation. The Tm derived from the CC stretch presents the local transition mid-point. Significantly, the local transition mid-point given by the CC stretch (ca. 61.4 °C on average) is smaller than that of the global transition mid-point (ca. 63.1 °C on average). The difference is ca. 1.7 °C. Hence it is believed that the hemi-protonated base pairs of C1–C13 breaking precede the global denaturation, and are perhaps among the initial steps of base pair breaking during the thermal unfolding.16 The unfolding pathway may include an initial transition phase that is associated with the partial unfolding of the base pairs or possibly one of the outer/terminal base pairs before the remaining structure unfolding.
The non-cooperativity of base pairs results in a broad distribution of segments with different cleavage of base pairs, which may tilt the initial denaturation curves in the lower-temperature region (as illustrated in Fig. 3a–c), but does not form a new sigmoidal transition yet.53 As shown in Fig. 3d (black dotted line), at the initial transition stage, the CC stretching frequency shifts on the basis of a large slope which significantly deviates from the two-state model. This is reasonable because the CC band would shift significantly only when the hemi-protonated base pair C1–C13 breaks. The linear shift of the initial transition stage is probably due to the C1–C13 base pair weakening along with the degradation of the i-motif structure as temperature increases. At the ending stage of the transition, the red shift of the CC band is not completely linear but fluctuates, which is probably due to the temperature effect.
To examine the temperature effect on the CC stretch, we carried out T-dependent FTIR spectroscopy of CC-labeled protonated and non-protonated cytosine dissolved in the DPBS buffer solution, respectively. Because of the low solubility of TIPSEdC dissolved in DPBS buffer solution, TMSEdC instead of TIPSEdC to be used for FTIR measurements here. The T-dependent spectral parameters of the 6-μm region and CC stretching region of TMSEdC and TMSEdC+ are shown in Fig. S11 (ESI†). As illustrated in Fig. S11c (ESI†), the CC stretch of TMSEdC had a linear red-shift with a total shifted wavenumber of 3 cm−1. As illustrated in Fig. S11d (ESI†), the CC stretch of protonated TMSEdC+ had a fluctuated red-shift with a total shifted wavenumber of 2 cm−1. As illustrated in Fig. S11b (ESI†), the 1714 cm−1 band of protonated TMSEdC+ had a fluctuated blue-shift with a total shifted wavenumber of 1 cm−1. It can be seen that the temperature effect does induce the 6-μm region or CC band shifting with a small range of wavenumber but not a two-state model. Given the experimental uncertainty, it is not clear that the temperature induced frequency shift is meaningful or not. Therefore, the temperature effect is ignored in this work. The changes of CC stretch and the 6-μm region with increasing temperature mainly reflected the structure transformation of i-motif DNA. To further examine the CC stretch of the protonated or non-protonated state, the pH-dependent spectra of the CC stretch of TMSEdC was collected and shown in Fig. S11a (ESI†). The CC stretch of protonated TMSEdC+ blue shifted by 10 cm−1 and was much weaker than that of non-protonated. In addition, the CC stretch red-shifting by a wavenumber of 5 cm−1 induced by i-motif structure unfolding in this work is very nearly to that amount of TMSEdC induced by the hydrogen-bonding interaction of a protonic solvent.33 This further illustrated that the changes of the CC stretch mainly reflected the hydrogen bonding forming/breaking process of the C1–C13 base pair. The temperature dependent frequency change of the monomer shown in Fig. S11d (ESI†) is a purely temperature-induced frequency redshift of the CC stretching mode, which could be another source of the tilted frequency change observed in Fig. 3d.
3.3. Thermodynamic parameters
To obtain the thermodynamic parameters (enthalpy ΔHvHm, entropy ΔSm and free energy ΔGm) at Tm for global transition and local site transition from the equilibrium melting dynamics, we used the van’t Hoff approach to plot ln K vs. 1/T as follows:| lnK = ΔHvHm/RT + ΔSm/R, | (2) |
| ΔGm = ΔHvHm − TΔSm | (3) |
| K = [single strand]/[i-modif] = (Fraction single strand)/(1-Fraction single strand), | (4) |
C-labeled cases. And that of local unfolding is extracted from the frequency shift of the CC stretching mode:| Fraction single strand = (IRT − IR25°C)/(IR90°C − IR25°C), | (5) |
°C is that at 25 °C, and IR90°C is that at 90 °C. The van’t Hoff fitting curves for three cases are shown in Fig. 4. The fitting thermodynamic parameters are very close to the previously reported results52,54,55 and are all collected in Table S2 (ESI†). The difference between the reported results and our results is mainly due to the sequence difference of i-motif and may also be due to difference of experimental conditions including ionic strength and pH.
 | ||
| Fig. 4 The van’t Hoff plot derived from the C1 band intensity at 1655 cm−1 for the unlabeled strand (a) and the CC-labeled strand (b), and the CC stretch band frequency shift for the CC-labeled strand (c). | ||
The values of ΔHvHm, ΔSm and ΔGm for the local-site transition (18.30 kcal mol−1, 0.055 kcal mol−1 and −0.12 kcal mol−1) determined using the CC probe were smaller than those for the global unfolding (28.75 kcal mol−1, 0.087 kcal mol−1 and −0.37 kcal mol−1). However, each thermodynamic parameter for the local-site transition accounts for 30–60% of the global unfolding transition for the CC-labeled strand.
The comparison of thermodynamic parameters between the global and local unfolding processes is very important. According to Wadkins work,54 the melting enthalpy of one C–C+ breaking in the i-motif DNA was approximately 5.0 kcal mol−1 while the melting enthalpy of the local site involving C1–C13 breaking was approximately 18.3 kcal mol−1 based on the van’t Hoff fitting results of the present work. Even taking into account the experimental error, the melting enthalpy of the C1–C13 pair breaking is still much larger than that of an usual C–C+ breaking in the i-motif DNA and exceeds 60% (=18.30/28.75) of total melting enthalpy. The free energy ΔGm of the local site C1–C13 breaking accounts 30% of the total melting free energy for the CC-labeled strand at Tm. These results suggest that the initial step of the C1–C13 breaking is very likely to be in the rate-liming step as it has a larger energy barrier than the rest of the base pairs to be broken when there are two or more steps of unfolding.56,57 It should be pointed out that the local site transition enthalpy derived from the CC probe does not present explicitly the melting enthalpy of C1–C13 breaking but may correspond to the enthalpy change involved in the C1–C13 breaking under the global unfolding process. Thus, whether the C1–C13 breaking is in the rate-liming step or not still requires more experimental evidence such as the unfolding rate obtained from time-resolved temperature-jump spectroscopy.57
The global transitions (Fig. 3a–c) and the local site transition (Fig. 3d) fitted by the two-state model yielded a slightly lower Tm but a quite larger enthalpy for the former, suggesting that the local site involving C1–C13 breaks first, followed by the outer base pair breaking, or that the rate-limiting step includes at least the C1–C13 breaking.
The global transition of six base-pair breaking is found to be not completely cooperative but rather shows a two-state transition behavior. According to the above results, we proposed a thermal unfolding model for the i-motif DNA:
 | (6) |
This model is mechanically consistent with the reported pH-induced unfolding model16 and the results from high-temperature molecular dynamics simulations.21 Although there is no comparative experiments on the CC probe tagged on other nucleobase sites, we speculate that the local transition Tm of other sites is unlikely to be smaller than that of the terminal site (C1–C13) involved this work, and the local transition ΔHvHm of other sites is unlikely to exceed 50% of the total. This suggests that the breakings of these base pairs are non-cooperative. Based on this model, we believe that the initial step of the C1–C13 breaking is very likely to be in the rate-liming step.
3.4. Simulated IR spectra
To further explore the mechanism of the peak frequency and intensity change in the CC stretching mode induced by the C1–C13 breaking, and to understand the asymmetric feature of the hemi-protonated base pairs under the thermal unfolding transition, we carried out DFT calculations on the CC stretching mode of different cases involving the CC-labeled hemi-protonated dimer and protonated/non-protonated deoxycytidine monomer. The optimized structures and the corresponding simulated IR spectra of the CC stretching mode are shown in Fig. 5. Our recent work showed that a limited solvent effect on the CC stretching frequency of TMSEdC,33 and considering the location of the CC moiety in the labeled bases, we carried out frequency calculations of the CC stretching mode in the gas phase.
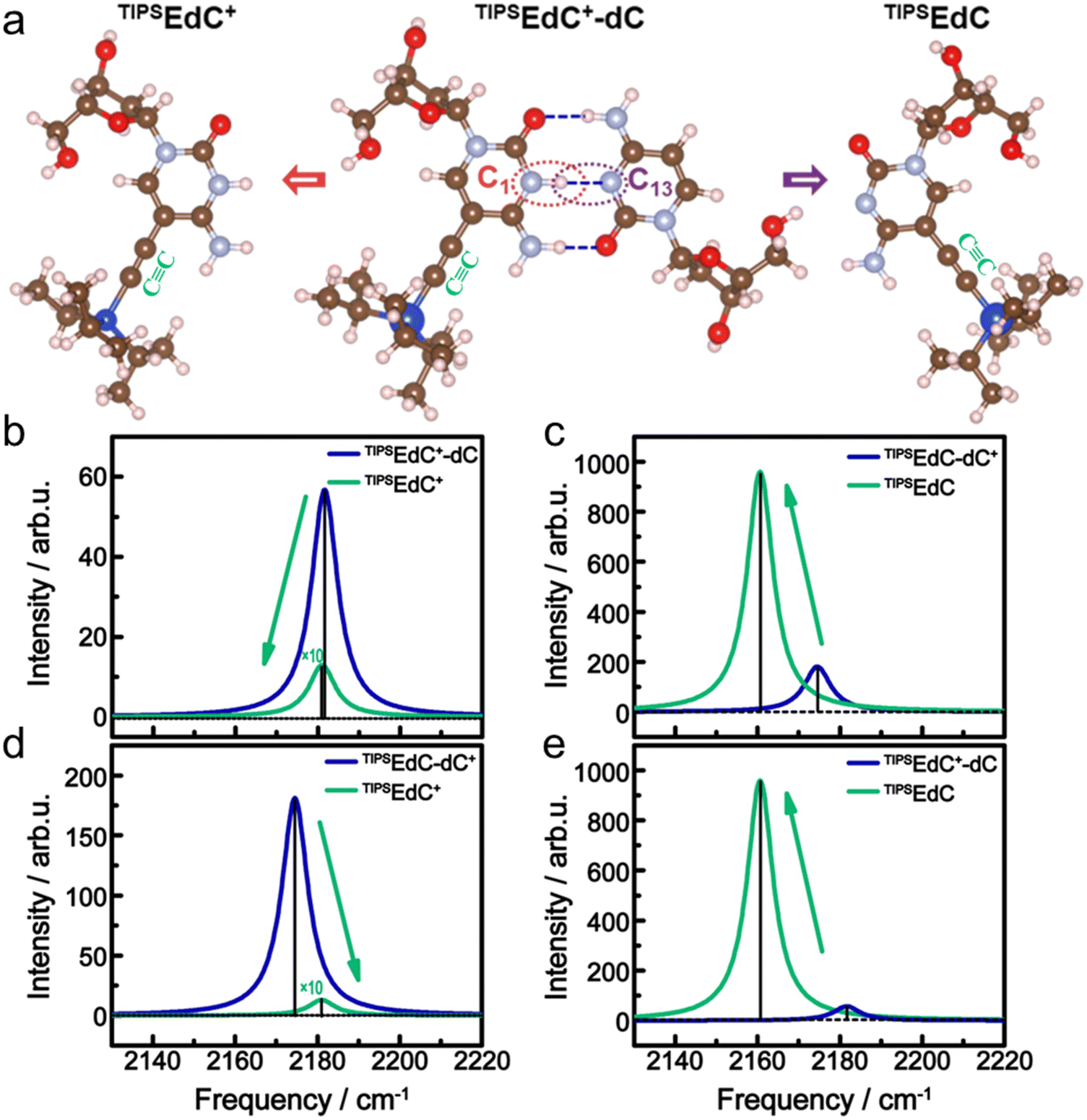 | ||
| Fig. 5 (a) Illustration of the hemi-protonated base pair of the folded state (middle), and the protonated monomer (left) and non-protonated monomer (right) of the unfolded state. Simulated IR spectra of TIPSEdC+-dC vs.TIPSEdC+ (b), TIPSEdC-dC+vs.TIPSEdC (c), TIPSEdC-dC+vs.TIPSEdC+ (d) and TIPSEdC+-dC vs.TIPSEdC (e). The intensity is enlarged by ten times for TIPSEdC+ in (b) and one hundred times in (d). | ||
The calculated frequencies were scaled by a factor of 0.967.59 Here, dC+ is protonated dC and TIPSEdC+ is protonated TIPSEdC. Two CC-labeled hemi-protonated dimers are considered: TIPSEdC+-dC and TIPSEdC-dC+. From Fig. 5b–e, one sees that only the case of changing from TIPSEdC+-dC to TIPSEdC+ (Fig. 5b) shows certain agreement with the experimental results (Fig. 3c and d) in both the frequency red shift and the intensity decrease of the CC stretching mode. In contrast, the calculated CC stretching intensity greatly increased from TIPSEdC+-dC to TIPSEdC (Fig. 5e), which is not what was observed experimentally. Moreover, the CC stretching band may red shift significantly (about 10 cm−1) when TMSEdC+ is deprotonated as illustrated in Fig. S11a (ESI†). Hence, the observed red shift of the CC stretching band (ca. 10 cm−1) induced by the deprotonation of cytosine is much larger than that induced by the unfolding transformation (about 5 cm−1). This infers that base C1 is unlikely to be deprotonated under the thermal unfolding transition of the i-motif.
The CC band red shifts as the hemi-protonated base pair C1–C13 breaks. The CC band intensity is quite low in the protonated cytosine (in the unfolded state) as indicated by the IR spectrum of TMSEdC+ (not shown), but is slightly larger when the hemi-protonated base pair of C1–C13 is formed (in the folded state). The vibrational intensity of the CC stretch of both unfolded and folded but protonated C1 is weaker than that of the non-protonated C1. In addition, as illustrated in Fig. 5c and d, the CC stretching mode is either intensity increased or frequency blue-shifted from TIPSEdC-dC+ to TIPSEdC or to TIPSEdC+, which does not agree with the experimental results. This infers that the proton localization may more likely occur at residue C1 than C13 in the hemi-protonated base pair of C1–C13 in which a delocalized proton is known to oscillate between the two wells.19 However, there has been no proof about the tendency of proton delocalization in C1–C13.19 Thus, the CC probe used here has successfully tracked the protonation status of the C1–C13 pair during the unfolding process of the i-motif DNA.
It should be noted that the chemical environment of the CC-labeled C1–C13 hemi-protonated base pair located on the i-motif structure in the DPBS buffer solution is more complicated than that of a single nucleoside or paired nucleosides. The labeling group faces outward the base-pairing region of the i-motif DNA, being a few covalent bonds away from the hydrogen bonds, so that the hydrogen-bonding effect on the CC stretching frequency is indirect and thus not significant. To examine the effect of non-covalent interactions, including mainly the van der Waals interaction and hydrophobic interaction, between the CC-labeled C1–C13 and nearby bases and associated chemical groups, we visualize the PDB structure of hTelo i-motif DNA38 in Fig. S13 (ESI†). It is shown that the estimated distance between the CC-label and C2 base, or between that and the A12 base, is 6.06 Å or 13.57 Å. Thus, the non-covalent interactions between the CC probe and these two nearby groups are considered to be weak and are not considered specifically in our model calculations shown in Fig. 5. Other interactions, such as the possible π–π stacking effect, may also influence the CC stretching frequency to some extent, but are considered specifically in the calculation of our work.
3.5. Probe perturbations
The extent of perturbations of the extrinsically tagged probe in the local structure and solvation dynamics is examined using 2D IR and pump–probe spectroscopies and the results are shown in Fig. 6. | ||
| Fig. 6 (a) Waiting-time dependent 2D IR spectra of the folded structure of the unlabeled (top row) and the CC-labeled i-motif (bottom row). Dashed lines indicate the values of ωτ for three prominent diagonal peaks of A1, C1, T1 and that for a shoulder peak C2, peaked at 1625 cm−1, 1665 cm−1, 1689 cm−1 and 1609 cm−1, respectively. Purple rectangles indicate the off-diagonal cross peaks between C1 and T1 modes. (b) CLS of the A1 band (0 → 1 transition) for the unlabeled case (wine) and CC-labeled case (navy). (c) Selected delay-time dependent dynamics for the unlabeled case (wine) and the CC-labeled case (navy). Probing ωτ positions are 1665 cm−1 for C1 (0 → 1 transition), 1603 cm−1 for C2 (1 → 2 transition), and 1625 cm−1 for A1 (0 → 1 transition). The peak labeling is described in Fig. 2. In (b), solid curves represent fittings with single exponential function. In (c), solid curves represent fittings with di-exponential function. | ||
Time-dependent and purely absorptive 2D IR spectra of the folded-state with unlabeled strands (top row) and those of the CC-labeled strands (bottom row) are shown in Fig. 6a. Paired diagonal peaks, which are composed of positive peaks (red) coming from the v = 0 → v = 1 transitions (v is vibrational quantum number) and negative peaks (blue) coming from the corresponding 1 → 2 transitions are shown. Off-diagonal cross peaks (coming from vibration mode coupling or energy transfer) are also shown, for example between C1 and T1 modes (Fig. 6a). Three prominent diagonal peaks in the 2D IR spectra are observed at 1625 cm−1, 1665 cm−1 and 1689/1696 cm−1, which are attributed to the A1, C1 and T1/C+1, respectively, as shown in the FTIR spectra (Fig. 2). There is almost no difference in the 2D IR line shape between the CC-labeled strand (Fig. 6a bottom row) and the unlabeled strand (Fig. 6a top row), which is so at varying waiting times. However, it should be noted that the 2D line shape changes as a function of the waiting time (from left to right panels of Fig. 6a), indicating the evolution of vibrational frequency (structural dynamics).
2D IR spectral line shape of the diagonal peaks changing with Tw is a process called spectral diffusion,33,60–62 which provides a direct measurement of the inhomogeneity of a given vibration mode caused by the electric field involving both the solvent and local structure. The spectral diffusion dynamics can be extracted from the time-dependent 2D IR diagonal signal using inverse center-line slope (CLS) method that is defined as δωτ/δωt. The spectral diffusion manifests the dynamics of the well-known frequency-frequency correlation function (FFCF).62 The center-line was extracted for the 0 → 1 transition peaks of A1 (shown in Fig. 6a, marked with a blue dotted line at 1624–1630 cm−1) which presents the local structure dynamics including solvent dynamics of the six adenines on average.
As shown in Fig. 6b, the CLS dynamics of two samples have almost the same initial amplitude value and the same unrelaxed residual value, indicating that the two samples have very similar inhomogeneity for the six adenines in a solvent environment as well as a local chemical environment, and also have similar local structural rigidity involving the six adenines. The CLS of two samples were fitted with a single exponential function with almost the same relaxation time constant (1.6 ps). This time constant reflects the averaged time scale of the local structural fluctuations (including solvent) around the six adenines. These adenine bases are evenly located on the three loops of the folded i-motif structure and are mainly solvent exposed (see Fig. S12, ESI†), hence the results suggest a relatively slower local dynamics than solvent water that typically has a time constant of 1.4–1.7 ps in bulk-like D2O or H2O.63–67 The static component is related to the six adenines' structural fluctuations whose time constants are too long to measure in the experimental time window shown here. The time scale of fluctuations of the entire i-motif DNA structure is much longer than that of solvent water exchange dynamics when DNA is dissolved in aqueous solution. This static component would be observed if the time window is up to ∼100 ps.67,68 In summary, spectral diffusion dynamics shown in Fig. 6b indicates that there is no difference in the local structure dynamics of six adenines on average and solvent dynamics between the unlabeled and CC-labeled strands at the folded state.
From the magic-angle pump–probe spectra (Fig. S14, ESI†), population relaxation of the CO stretching modes in these bases can be examined. For example, two vibrational relaxation dynamics of 0 → 1 transition peaks of C1 and 1 → 2 transition peaks of C2 are shown in Fig. 6c. The selected two transitions dynamics were fitted with a bi-exponential function and gave similar relaxation time constants. The dynamics fitting parameters are shown in Table S3 (ESI†). It was found that these dynamics show no much difference between the unlabeled and CC-labeled strands. These results indicate that the vibrational energy dissipation pathways of the vibrationally excited CO modes in the i-motif DNA not affected by the CC-labeling folded strand are almost the same at each mode. Quite similar vibrational relaxations between the unlabeled and labeled i-motif DNA were also observed in other probing frequencies (data not shown).
To sum up, the extrinsic CC probe tagged on C1 was found to have a negligible perturbation in the equilibrium structural transition process, local structural solvent dynamics and structural flexibility of the i-motif DNA. To further examine the extent of perturbations in the local structural dynamics from the CC probe, ultrafast infrared spectra of the partially folded-state structure of two strands dissolved in pH* 7.6 DPBS buffer solution were also collected. The spectra and related dynamics are shown in Fig. S15 (ESI†) and the fitting parameters for the dynamics are collected in Table S3 (ESI†). There was also no difference in all the corresponding dynamics between the unlabeled and CC-labeled strands. Thus, it is believed that the CC probe has a negligible perturbation in the local structure/solvent dynamics of the i-motif DNA even though the CC probe seems to have a large steric hindrance effect and hydrophobic effect. It appears to be more optimistic when a single CC probe is tagged on the major grooves and extends outward edge (less crowed area) of a DNA double helix or a highly ordered helical structure such as the i-motif DNA. Of course, our infrared spectroscopy has certain limitations and more sophisticated experiments should be carried out to further explore the perturbation induced by the CC probe.69 Here, since the unfolding event occurred particularly on the local site probed by the CC label is found to be unlikely influenced by the structural perturbations from the probe itself, such a label is believed to have certain applicability and practicability. It should be noted that in a recent work of Tokmakoff and coworkers,26 2D IR spectroscopy in combination with ns T-jump was used to probe the unfolding dynamics of another i-motif DNA sequence 5′-CCCTTTCCCTTTCCCTTTCCC-3′ (C3T3) on the ms to second time scale. In their work, the CO stretching mode (and ring thymine mode) can be used directly as the structural probe, which allows either global or local structural dynamics to be followed by 2D IR spectroscopy. In our work reported here, a site-specific, extrinsic IR probe (i.e., the CC probe) was added so that local dynamics can be followed in a spectral window that does not have any intrinsic molecular vibration of DNA.
4. Concluding remarks
In this work, a site-specific thermal unfolding process of the i-motif DNA labeled by a CC probe on terminal cytosine (C1) has been examined using temperature-dependent FTIR spectroscopy, in combination with quantum chemistry calculations. Temperature-dependent IR results indicate that the spectral features in both CO/CC stretching and the labeled CC stretching modes can be understood using a typical two-state model of the conformational transformation of DNA.
The extrinsic CC probe can be used to monitor the unfolding process of the i-motif DNA. The change of the CC stretching frequency (and intensity) is directly related to the hemi-protonated base pair C1–C13 breaking process and the protonated/non-protonated status of C1 under its thermal unfolding process. The local Tm derived from the CC stretch (ca. 61.4 °C on average) is found to be slightly lower than that of the global Tm (ca. 63.1 °C on average), indicating that the C1–C13 breaking process may be in the initial step of the denaturation. In addition, the local-site transition ΔHvHm exceeds 60% of that of the global transition, which implies that the C1–C13 breaking process may be in the rate-limiting step of the denaturation and the remaining base pairs' break quickly follows.
Simulated IR spectra of the CC stretch of the CC-labeled hemi-protonated dimer and protonated/non-protonated monomer demonstrate that protons are more likely located on the C1 side of the hemi-protonated base pair of C1–C13, so that C1 remains to be protonated upon the breakage of the C1–C13 link. The CC probe can successfully report the protonation status of site-specific nucleobase during the unfolding process of the i-motif DNA; the forming and breaking processes of the hemi-protonated base pair C1–C13 lead to blue and red shifts in the CC stretching frequency, while the protonation and deprotonation processes of C1 lead to a transition intensity decrease and increase, respectively, in the CC stretching mode.
Our ultrafast infrared spectroscopic results indicate that the extrinsic CC probe tagged on the i-motif DNA has little perturbation in the steady-state structural formation and transformation, and has little perturbation in the local structural dynamics, solvent dynamics and structural flexibility for the folded and partially folded i-motif DNAs. This may be because that the CC probe extends toward the major groove or the outside of the i-motif structure and avoids steric hindrance. Since there is no new short-range polar interaction (such as hydrogen-bonding interaction) introduced by the CC probe, it may become an advantageous structural and dynamics reporter for DNA. However, further experiments of the CC group labeling on other nucleobase sites and direct monitoring of the associated unfolding events are needed to verify the hypothesis. In addition, the proposed thermal unfolding model of the i-motif DNA from this work appears to be reasonable and generally agrees with that of pH-induced unfolding reported previously. Thus, our work demonstrated that the CC probe can be successfully used to monitor the site-specific unfolding process of the i-motif DNA for the first time. It should be generally applicable to the study of the thermal stability of DNA chains using infrared spectroscopy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21973102, 21573243 and 21327802 to J. W.) is greatly acknowledged.References
- H. A. Day, P. Pavlou and Z. A. Waller, Bioorg. Med. Chem., 2014, 22, 4407–4418 CrossRef CAS.
- T. A. Brooks, S. Kendrick and L. Hurley, FEBS J., 2010, 277, 3459–3469 CrossRef CAS PubMed.
- M. Debnath, S. Ghosh, A. Chauhan, R. Paul, K. Bhattacharyya and J. Dash, Chem. Sci., 2017, 8, 7448–7456 RSC.
- H. Abou Assi, M. Garavis, C. Gonzalez and M. J. Damha, Nucleic Acids Res., 2018, 46, 8038–8056 CrossRef PubMed.
- M. Zeraati, D. B. Langley, P. Schofield, A. L. Moye, R. Rouet, W. E. Hughes, T. M. Bryan, M. E. Dinger and D. Christ, Nat. Chem., 2018, 10, 631–637 CrossRef CAS PubMed.
- S. Modi, M. G. Swetha, D. Goswami, G. D. Gupta, S. Mayor and Y. Krishnan, Nat. Nanotechnol., 2009, 4, 325–330 CrossRef CAS PubMed.
- H. Liu and D. Liu, Chem. Commun., 2009, 2625–2636, 10.1039/b822719e.
- J. A. Kretzmann, K. L. Irving, N. M. Smith and C. W. Evans, NAR Cancer, 2021, 3, zcab048 CrossRef.
- S. L. Brown and S. Kendrick, Pharmaceuticals, 2021, 14, 96 CrossRef CAS.
- C. Wang, Z. Huang, Y. Lin, J. Ren and X. Qu, Adv. Mater., 2010, 22, 2792–2798 CrossRef CAS PubMed.
- D. Liu, E. Cheng and Z. Yang, NPG Asia Mater., 2011, 3, 109–114 CrossRef.
- M. Cheng, J. Chen, H. Ju, J. Zhou and J. L. Mergny, J. Am. Chem. Soc., 2021, 143, 7792–7807 CrossRef CAS.
- J. L. Leroy, Nucleic Acids Res., 2009, 37, 4127–4134 CrossRef CAS.
- A. L. Lieblein, J. Buck, K. Schlepckow, B. Furtig and H. Schwalbe, Angew. Chem., Int. Ed., 2012, 51, 250–253 CrossRef CAS PubMed.
- J. Choi, S. Kim, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2011, 133, 16146–16153 CrossRef CAS.
- C. Chen, M. Li, Y. Xing, Y. Li, C. C. Joedecke, J. Jin, Z. Yang and D. Liu, Langmuir, 2012, 28, 17743–17748 CrossRef CAS PubMed.
- G. Mata and N. W. Luedtke, J. Am. Chem. Soc., 2015, 137, 699–707 CrossRef CAS PubMed.
- Y. Mao and J. Zhang, J. Am. Chem. Soc., 2012, 134, 631–639 CrossRef CAS PubMed.
- A. L. Lieblein, M. Kramer, A. Dreuw, B. Furtig and H. Schwalbe, Angew. Chem., Int. Ed., 2012, 51, 4067–4070 CrossRef CAS.
- J. Smiatek and A. Heuer, RSC Adv., 2014, 4, 17110–17113 RSC.
- J. Smiatek, C. Chen, D. Liu and A. Heuer, J. Phys. Chem. B, 2011, 115, 13788–13795 CrossRef CAS.
- J. Somkuti, O. R. Molnar and L. Smeller, Phys. Chem. Chem. Phys., 2020, 22, 23816–23823 RSC.
- A. Ghosh, J. S. Ostrander and M. T. Zanni, Chem. Rev., 2017, 117, 10726–10759 CrossRef CAS.
- P. J. Sanstead, P. Stevenson and A. Tokmakoff, J. Am. Chem. Soc., 2016, 138, 11792–11801 CrossRef CAS PubMed.
- Y. Xu, R. Oyola and F. Gai, J. Am. Chem. Soc., 2003, 125, 15388–15394 CrossRef CAS.
- B. Ashwood, N. H. C. Lewis, P. J. Sanstead and A. Tokmakoff, J. Phys. Chem. B, 2020, 124, 8665–8677 CrossRef CAS PubMed.
- M. S. Jones, B. Ashwood, A. Tokmakoff and A. L. Ferguson, J. Am. Chem. Soc., 2021, 143, 17395–17411 CrossRef CAS.
- H. Taskent-Sezgin, J. Chung, P. S. Banerjee, S. Nagarajan, R. B. Dyer, I. Carrico and D. P. Raleigh, Angew. Chem., Int. Ed., 2010, 122, 7635–7637 CrossRef.
- H. Kim and M. Cho, Chem. Rev., 2013, 113, 5817–5847 CrossRef CAS.
- J. Ma, I. M. Pazos, W. Zhang, R. M. Culik and F. Gai, Annu. Rev. Phys. Chem., 2015, 66, 357–377 CrossRef CAS.
- R. Adhikary, J. Zimmermann and F. E. Romesberg, Chem. Rev., 2017, 117, 1927–1969 CrossRef CAS PubMed.
- R. Itaya, W. Idei, T. Nakamura, T. Nishihara, R. Kurihara, A. Okamoto and K. Tanabe, ACS Omega, 2021, 6, 31595–31604 CrossRef CAS PubMed.
- T. Dong, P. Yu, J. Zhao and J. Wang, Phys. Chem. Chem. Phys., 2022, 24, 29988–29998 RSC.
- S. Carson, J. Wilson, A. Aksimentiev, P. R. Weigele and M. Wanunu, Nucleic Acids Res., 2016, 44, 2085–2092 CrossRef CAS PubMed.
- T. T. Ngo, J. Yoo, Q. Dai, Q. Zhang, C. He, A. Aksimentiev and T. Ha, Nat. Commun., 2016, 7, 10813 CrossRef CAS PubMed.
- X. Teng and W. Hwang, Biophys. J., 2018, 114, 1791–1803 CrossRef CAS PubMed.
- S. A. Ingale, H. Mei, P. Leonard and F. Seela, J. Org. Chem., 2013, 78, 11271–11282 CrossRef CAS PubMed.
- A. T. Phan, M. Guéron and J.-L. Leroy, J. Mol. Biol., 2000, 299, 123–144 CrossRef CAS PubMed.
- D. B. Strasfeld, S. H. Shim and M. T. Zanni, Phys. Rev. Lett., 2007, 99, 038102 CrossRef PubMed.
- S. H. Shim and M. T. Zanni, Phys. Chem. Chem. Phys., 2009, 11, 748–761 RSC.
- M. J. Frish, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Paterson, Gaussian 09, Revision A. 02, Gaussian, Inc., Pittsburgh PA, 2009 Search PubMed.
- D. Reinstädler, H. Fabian, J. Backmann and D. Naumann, Biochemistry, 1996, 35, 15822–15830 CrossRef.
- P. Renati, Z. Kovacs, A. De Ninno and R. Tsenkova, J. Mol. Liq., 2019, 292, 111449 CrossRef CAS.
- M. Bukleski, S. Dimitrovska-Lazova, V. Makrievski and S. Aleksovska, Spectrochim. Acta, Part A, 2020, 231, 118118 CrossRef CAS PubMed.
- M. Bukleski, S. Dimitrovska-Lazova and S. Aleksovska, Spectrochim. Acta, Part A, 2022, 266, 120462 CrossRef CAS.
- Z. A. Piskulich and W. H. Thompson, J. Phys. Chem. Lett., 2020, 11, 7762–7768 CrossRef CAS PubMed.
- D. Tsankov, M. Krasteva, V. Andrushchenko, J. Van De Sande and H. Wieser, Biophys. Chem., 2006, 119, 1–6 CrossRef CAS PubMed.
- J. Somkuti, M. Houska and L. Smeller, Eur. Biophys. J., 2011, 40, 143–151 CrossRef CAS PubMed.
- M. H. Brumano, E. Rogana and H. E. Swaisgood, Arch. Biochem. Biophys., 2000, 382, 57–62 CrossRef CAS.
- D. Du, M. J. Tucker and F. Gai, Biochemistry, 2006, 45, 2668–2678 CrossRef CAS PubMed.
- M. R. Bunagan, J. Gao, J. W. Kelly and F. Gai, J. Am. Chem. Soc., 2009, 131, 7470–7476 CrossRef CAS PubMed.
- M. McKim, A. Buxton, C. Johnson, A. Metz and R. D. Sheardy, J. Phys. Chem. B, 2016, 120, 7652–7661 CrossRef CAS.
- S. D. Stelea, P. Pancoska, A. S. Benight and T. A. Keiderling, Protein Sci., 2001, 10, 970–978 CrossRef CAS PubMed.
- S. M. Reilly, R. K. Morgan, T. A. Brooks and R. M. Wadkins, Biochemistry, 2015, 54, 1364–1370 CrossRef CAS PubMed.
- M. Kaushik, N. Suehl and L. A. Marky, Biophys. Chem., 2007, 126, 154–164 CrossRef CAS.
- D. T. Leeson, F. Gai, H. M. Rodriguez, L. M. Gregoret and R. B. Dyer, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2527–2532 CrossRef CAS PubMed.
- D. Du, Y. Zhu, C.-Y. Huang and F. Gai, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15915–15920 CrossRef CAS PubMed.
- H. Rahaman, M. K. A. Khan, M. I. Hassan, A. Islam, A. A. Moosavi-Movahedi and F. Ahmad, J. Chem. Thermodyn., 2013, 58, 351–358 CrossRef CAS.
- R. D. Johnson III and E. NIST, Computational chemistry comparison and benchmark database, 2013.
- H. Ishikawa, S. Kim, K. Kwak, K. Wakasugi and M. D. Fayer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19309–19314 CrossRef.
- K. Kwak, D. E. Rosenfeld and M. D. Fayer, J. Chem. Phys., 2008, 128, 204505 CrossRef PubMed.
- D. Li, F. Yang, C. Han, J. Zhao and J. Wang, J. Phys. Chem. Lett., 2012, 3, 3665–3670 CrossRef CAS PubMed.
- S. Park, K. Kwak and M. D. Fayer, Laser Phys. Lett., 2007, 4, 704–718 CrossRef CAS.
- R. Yuan, C. Yan, A. Tamimi and M. D. Fayer, J. Phys. Chem. B, 2015, 119, 13407–13415 CrossRef CAS PubMed.
- S. Park and M. D. Fayer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16731–16738 CrossRef CAS.
- S. A. Yamada, W. H. Thompson and M. D. Fayer, J. Chem. Phys., 2017, 146, 234501 CrossRef PubMed.
- J. T. King and K. J. Kubarych, J. Am. Chem. Soc., 2012, 134, 18705–18712 CrossRef CAS PubMed.
- J. K. Chung, M. C. Thielges, S. E. Bowman, K. L. Bren and M. D. Fayer, J. Am. Chem. Soc., 2011, 133, 6681–6691 CrossRef CAS PubMed.
- B. Ashwood, M. S. Jones, A. L. Ferguson and A. Tokmakoff, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2219124120 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of TIPSEdC phosphoramidite; MS characterization of the CC-labeled DNA strand; CD spectra of the unlabeled and CC-labeled DNA strands; FTIR spectra of nucleosides with band assignment; temperature-dependent FTIR difference spectra; plot of the CC band frequency position vs intensity; temperature-dependent FTIR spectra of TMSEdC and TMSEdC+; fitting results of van’t Hoff function; location of the six adenines in the i-motif DNA; comparison of the unlabeled and CC-labeled strands monitored by the pump–probe spectra; nonlinear IR results of the unlabeled and CC-labeled strands at pH* 7.6; and the fitting results of the population relaxation dynamics. See DOI: https://doi.org/10.1039/d3cp05328h |
| This journal is © the Owner Societies 2024 |