
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMeasuring tryptophan dynamics using fast scan cyclic voltammetry at carbon fiber microelectrodes with improved sensitivity and selectivity†
Isabella Schapiraa,
Margaret R. O'Neilla,
Lillian Russo-Savageb,
Terdha Narlac,
Kathryn A. Lapraded,
James M. Staffordbd and
Yangguang Ou *ab
*ab
aDepartment of Chemistry, University of Vermont, USA. E-mail: you@uvm.edu
bNeuroscience Graduate Program, University of Vermont, USA
cDepartment of Pharmacology, University of Vermont, USA
dDepartment of Neurological Sciences, University of Vermont, USA
First published on 4th September 2023
Abstract
Despite the fact that tryptophan (Trp) is an essential amino acid that humans typically obtain through diet, there are several interesting tryptophan dynamics at play in the body. Quantifying and understanding these dynamics are crucial in studies of depression, autism spectrum disorder, and other disorders that involve neurotransmitters directly synthesized from tryptophan. Here we detail the optimization of waveform parameters in fast scan cyclic voltammetry at carbon fiber microelectrodes to yield four-fold higher sensitivity and six-fold higher selectivity compared to previously reported methods. We demonstrate the utility of our method in measuring (1) exogenous Trp dynamics from administration of Trp to PC-12 cells with and without overexpression of tryptophan hydroxylase-2 and (2) endogenous Trp dynamics in pinealocyte cultures with and without stimulation via norepinephrine. We observed interesting differences in Trp dynamics in both model systems, which demonstrate that our method is indeed sensitive to Trp dynamics in different applications.
Introduction
Serotonin is an important neurotransmitter in the brain and dysregulation of serotonin levels has been associated with various disorders, including depression1–4 and autism spectrum disorder (ASD).5,6 Serotonin itself has been hypothesized to not cross the blood–brain barrier.7–9 There is growing evidence that suggests the gut microbiome communicates with the brain in a bidirectional manner through the vagal nerve and through biomarkers that can cross the blood–brain barrier (BBB). One of these biomarkers that can cross is the amino acid tryptophan (Trp), the precursor to serotonin.Trp is one of the naturally occurring amino acids that is essential for protein synthesis and plays a fundamental role in nutrition and health.10 Once ingested, Trp is distributed throughout the body via the circulatory system.11 It is also the precursor to several brain neurotransmitters (i.e. serotonin, melatonin)9,10 and other biomarkers that participate in the crosstalk in the gut–brain axis (i.e. kynurenine, vitamin B6).12,13 As a result, it has been hypothesized that Trp itself plays crucial roles in the brain, the gut, and the various disorders that involve the gut–brain axis, including depression14–20 and autism spectrum disorder.21–25
Humans obtain most of necessary Trp from diet, but Candida, Streptococcus, Escherichia, Enterococus, and other microorganisms in the gut microbiome express Trp synthase that catalyzes the Trp biosynthesis from serine and indole-3-glycerol-phosphate.26 The gut microbiome also plays a major role in regulating the available amount of circulating Trp by modulating enzymes in various Trp metabolism pathways: serotonin, kynurenine, and indole.26,27 These studies demonstrate that while Trp is not synthesized by mammalian cells, there are still biosynthetic and degradation pathways that alter Trp levels.
In addition to contribution from the microbiome, there are other factors that can influence Trp dynamics. Approximately 75–85% (and as high as 95%) of Trp in the body is bound to albumin protein.28 While only ∼5–10% is suspected to be free under normal conditions,29 because Trp has a higher affinity for the BBB transporter than albumin,28,30–36 it has been estimated that as much as 75% of free Trp can cross the BBB.37 And while total Trp usually does not change much, free Trp can change drastically. For example, changes in peripheral Trp can cause changes in free Trp in the central nervous system.38,39 Endogenous free Trp levels can also change due to various factors. For example, in a study done by Chaouloff et al.,40 researchers trained rats to run on a treadmill for 2 h. Even though total Trp was not altered, the amount of free Trp increased by 70% post-run in the plasma and by 80% in the brain. These studies highlight that there is indeed interesting endogenous Trp dynamics in the brain and periphery and these can be altered by various factors.
Common practice for Trp monitoring in foods, bodily fluids, and tissue often utilize chromatography methods.41–47 For free Trp measurements, researchers often rely on colorimetric methods, near-infrared reflectance spectroscopy, optical sensors, optimized high performance liquid chromatography with mass spectrometry detection, and isotopic labelling (reviewed in Friedman10 and Khan et al.48). While these methods are quantitative and can provide spatial and/or temporal information of Trp, they are limited by sample preparation and/or need for separation prior to detection. There have been efforts to measure free Trp in situ using microdialysis coupled to high performance liquid chromatography and UV-vis detection. While powerful, this technique does have some limitations. Firstly, the time resolution of microdialysis-HPLC-UV-vis is limited by collection and separation times. While separations as fast as under a min has been reported for catecholamines, all of the prior free Trp measurements have 20 min time resolutions.38–40 Interestingly, these time-resolved data show an immediate change in free Trp within the first 20 min but these dynamics were too rapid to be resolved by conventional methods. Secondly, the large size of the microdialysis probe relative to the intercapillary distance of the vasculature has been shown by others to cause foreign body response in the surrounding tissue.49,50 While all of these methods have provided a plethora of valuable information, they are either performed ex situ, require a specialist to operate the instrumentation, involve sample treatment protocols before detection (including removing interfering species), has poor spatiotemporal resolution, and/or causes foreign body response (and therefore may result in signal artifacts).
Thus, there is still a need for a sensitive and selective Trp quantitation technique for in situ monitoring in cells, tissues, and in vivo. Moreover, release dynamics of Trp metabolites in the brain occur on the order of subseconds,51,52 so it is possible Trp dynamics occurs on comparable scales as well. Furthermore, Trp concentrations are hypothesized to vary between different cell types and brain regions53,54 so micron-scale spatial resolution would be advantageous. A variety of electrochemical methods and electrode platforms have been developed for the quantitation of Trp (reviewed in Khan et al.48). A promising technique capable of making measurements at the appropriate spatiotemporal scale is fast scan cyclic voltammetry (FSCV) at carbon fiber microelectrodes (CFMs). FSCV provides 100 ms temporal resolution for continuous recordings. CFMs are small (cause less damage),55 biocompatible (reviewed in Hejazi et al.56), and provide a versatile surface for different modifications (reviewed in Ou et al.57 and Rafi et al.58). The Venton lab first demonstrated the utility of using FSCV at CFMs to measure gonadotropin-releasing hormone, which contains Trp and Tyr.59 The Ross lab demonstrated the utility of FSCV at CFMs (via the application of the dopamine waveform) to measure electroactive free amino acids, including Trp, with 100 ms timescale and micron spatial resolution.60 However, their paper did not demonstrate selective measurement of Trp over dominant interfering species such as Tyr, which has overlapping oxidation peaks. This is an important issue to address as there are significant amounts of dipeptides containing Tyr such as Tyr–Tyr, Tyr–Gly, Tyr–Glu in the brain and there are significant dynamics in these dipeptide levels.61 One of these regions is the hippocampus, a primary area of interest for studying depression and autism spectrum disorder. Since these dipeptides also contain Tyr, they also have overlapping oxidation peaks as Trp.
In this work, we utilize a sawhorse waveform in FSCV recordings on CFMs and optimize the parameters of said waveform to gain significant selectivity of Trp detection over that for Tyr. In fact, we demonstrate that our optimized conditions permit high selectivity for Trp detection over that for Tyr while maintaining high sensitivity for Trp. We also demonstrate the utility of our method in a real sample by measuring exogenous and endogenous Trp dynamics in cultured PC-12 cells and pinealocytes.
Experimental
Chemicals and instrumentation
The chemicals used were analytical grade and were purchased from MilliporeSigma (St. Louis, MO), unless otherwise stated. Phosphate buffered saline (PBS) tablets were purchased from Fisher Bioreagents (Waltham, MA, USA). 1 × PBS (pH 7.40) was prepared by dissolving five PBS tablets in 1.0 L nanopure water (Milli-Q IQ7000, MilliporeSigma, St. Louis, MO). 0.5 mM Trp and Tyr stock solutions were prepared in PBS and frozen into small aliquots until use. On the day of the experiment, stock solutions were thawed at room temperature and standard solutions of 0.5 μM, 1 μM, 3 μM, and 5 μM tryptophan and tyrosine solutions were prepared from the stock. Dopamine and serotonin stocks (100 μM each) were made in PBS and frozen into small aliquots until use. On the day of the experiment, stock solutions were thawed at room temperature and 1 μM dopamine (FisherSci, Hampton, NH) and serotonin (Krackeler, Albany, NY) standards were made via dilution using PBS. As these compounds are sensitive to air oxidation, we used solutions within the hr.Electrode fabrication and fast scan cyclic voltammetry
A single carbon fiber (GoodFellow, Pittsburgh, PA, USA) of 7 μm diameter was aspirated into a 0.5 mm inner diameter (1.00 mm outer) borosilicate glass capillary (A-M Systems, Inc., Carlsberg, WA, USA) using a vacuum pump (Gast). These filled capillary tubes were subsequently pulled using a vertical puller (PC-100 or PE-22, Narishige Group, Japan) to create a tapered seal insulating the carbon fiber. While observing the electrode under a microscope, the protruding carbon fiber was then cut to a length between 130 μm to 160 μm. All the results below are reported as current densities, in which the measured current is normalized to the geometric surface area of the electrode and reported in unit of pA μm−2. A nichrome wire with a soldered pin (Phoenix Enterprises/PE Connectors, Chatsworth, CA, USA) was then coated with silver epoxy paint (GC Electronics, Rockford, IL, USA) and inserted into the distal end of the fiber-filled capillary. A polyolefin heat shrink tubing (Mouser, Mansfield, TX, USA) then is heat-sealed around the electrode/nichrome wire junction to secure them in place.We employed a 2-electrode system comprised of a pseudo-Ag/AgCl reference (made in-house) and the CFM working electrode. Fast scan cyclic voltammetry (FSCV) was performed using a custom-built integration unit that includes a Dagan Chem-Clamp (Dagan Corporation, Minneapolis, MN, USA), National Instrument data acquisition cards, Pine Instrument Dagan-compatible headstage, and WCCV 3.6 software (Knowmad Technologies LLC, Tucson, AZ, USA). All experiments were performed in a Faraday cage (AutoMate, Berkeley, CA, USA). Electrodes were cycled in each solution for 10 min prior to any data acquisition. A sawhorse waveform was applied at a frequency of 10 Hz for all experiments. More details on the waveform are presented later in Results and discussion.
For FSCV recordings, cells were removed from the incubator and transported to the Faraday cages. Manual micromanipulators (Narashige Group, Japan) were used to submerge the CFMs into the media and approach the cells as close as possible without breaking. For PC-12 measurements, we measured ∼2–3 seconds of FSCV background before using a micropipette to exogenously apply 5 μM Trp into the media near the CFM while continuously recording for up to 60 seconds. This was repeated 4 more times at different locations in the well plate as well as in different culture plates for PC-12 cells. We performed these measurements 6 times for PC-12 cells with overexpression of TPH2. For pinealocytes, we measured baseline for 5 min (30 s files, 30 s in between), then added either media or norepinephrine, and continued to measure for 30 min.
Flow injection analysis
A continuous flow of PBS (buffer) was pumped into a flow cell (built in-house, Cole-Parmer, Vernon Hills, IL) at 1.81 mL min−1 using a syringe pump (Fisher Scientific, Waltham, MA, USA). Reference and working electrodes were attached to the flow cell accordingly. A 5 s bolus of Trp, Tyr, or both was introduced to the electrodes by using a 6-port injection valve (VICI, Houston, TX, USA).Data collection
Custom software WCCV 3.6 software (Knowmad Technologies LLC, Tucson, AZ, USA) was employed for the collection and initial analysis of data, which includes background subtraction, signal averaging, and digital filtering. Data was filtered using a fourth order Butterworth low pass filter. The cyclic voltammograms (CVs) and i–t (current vs. time) plots were stacked over time to create a 2D color plot. The x-axis is time (30 s files unless otherwise stated), the y-axis is potential applied (V), and the color indicates current. The CVs and i–t plots were exported as text files and analyzed in Excel and GraphPad Prism 9. The max currents at the oxidation peak corresponding to Trp oxidation (∼1.0 V vs. pseudo Ag/AgCl) were normalized to the geometric surface area of each electrode. Grubbs test was used to remove any outliers and errors were propagated appropriately to combine the different trials from different electrodes. All statistical tests, including Grubbs', t-tests, and one-way ANOVA Dunnett T3 multiple comparison test, as well as linear regression were conducted in GraphPad Prism 9. Experimental alpha was set at 0.05.Cell culture
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well with pinealocyte culture media. Cells were maintained for ∼2 weeks in a 37 °C humidified incubator with 5% CO2 until FSCV measurements were ready to be performed.
000 cells per well with pinealocyte culture media. Cells were maintained for ∼2 weeks in a 37 °C humidified incubator with 5% CO2 until FSCV measurements were ready to be performed.Generation of TPH2 overexpressing PC-12
Results & discussion
Trp is one of the 20 naturally occurring aromatic amino acids and is electroactive, meaning it can be readily oxidized or reduced. Nguyen et al.63 demonstrated that Trp undergoes an initial 2-electron, 2-proton oxidation, a step that was attributed to the observed anodic peak at 0.9 V on a pyrolytic graphite electrode vs. SCE. Others64 have attributed the loss of the pyrrolic double bond and addition of a ketone adjacent to the secondary amine as the primary oxidation event observed on glassy carbon (vs. Ag/AgCl (3 M KCl)). Trp oxidation was observed using FSCV at CFM by Ross and colleagues60 at 1.06 V, and we observe a similar oxidation peak at 1.0 V on the forward scan (Fig. 1).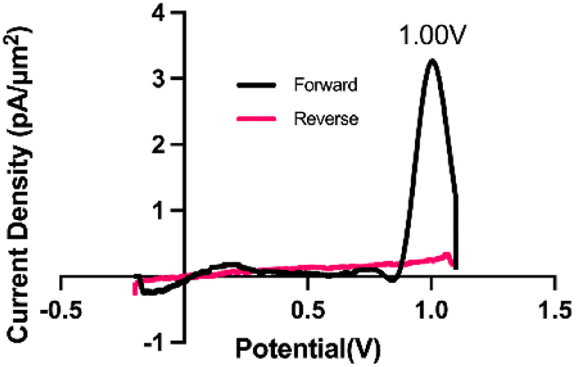 | ||
| Fig. 1 Representative cyclic voltammogram for Trp oxidation on carbon fiber microelectrode using the multi-scan rate waveform. Black trace indicates the forward scan while the red trace indicates the reverse scan. The primary oxidation peak occurs at 1.00 V. | ||
The waveform parameters (holding potential, transition potential, switching potential, switching potential duration, and scan rate) of a multi-scan rate sawhorse waveform (MSW) were optimized for the sensitive and selective detection of Trp over Tyr. The original MSW (Fig. 2a) was derived from work published by Sombers and colleagues for the detection of enkephalin using FSCV at CFMs.65,66 The first scan rate between the holding potential (A, Fig. 2a) and transition potential (B, Fig. 2a) was held constant at 100 V s−1 because there are no oxidation or reduction peaks of interest occurring in those potentials. The primary oxidation peak occurs between the transition potential (B, Fig. 2a) and the switching potential (C, Fig. 2a). The scan rate in this potential range was varied (discussed below).
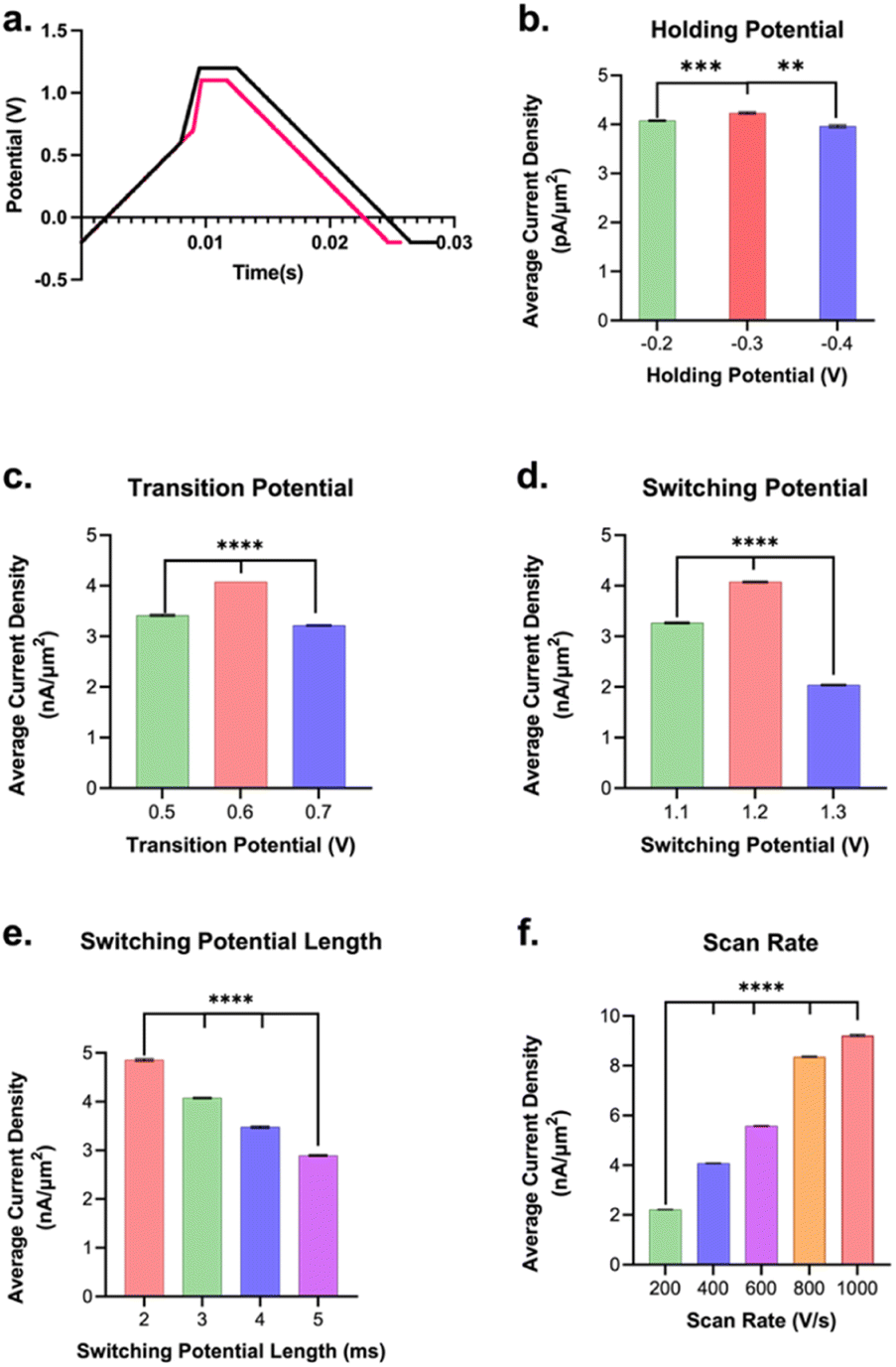 | ||
| Fig. 2 Summary showing the average peak current density (pA μm−2) and SEM (error bars) for 1 μM Trp bolus injected into the flow injection analysis system and how varying each respective parameter affected the observed current density for Trp oxidation. The average current densities were calculated by averaging the points in the bolus region of the IT curves for each electrode and then averaging these values together between electrodes (and propagating the appropriate SEMs). (a) is an overlay of the original and optimized waveforms. Point A indicates holding potential, B is the transition potential, and C is the switching potential. (b) Current density was greatest for a holding potential of −0.3 V over those at −0.2 V and −0.4 V. (c) The largest signal for Trp was found to be with a transition potential of +0.6 V over those of +0.5 V and +0.7 V. (d) The average current density was highest for switching potential of +1.2 V when compared to those at +1.1 V and +1.3 V. (e) The switching potential duration of 2 ms yielded the largest average current density when compared to longer hold times (3, 4, or 5 ms). (f) The scan rate that yielded the largest signal was 1000 V s−1 **p < 0.01, ****p < 0.0001. | ||
Each parameter in the MSW (vide supra) was varied systematically. The assumption is that these parameters behave independently from each other, and, from our experience, there has been no evidence indicating otherwise. Fig. 2b–f summarizes the results from the optimization of MSW waveform parameters for maximal sensitivity towards Trp detection. Maximum sensitivity towards Trp detection was observed at intermediate values that we tested for the holding potential, transition potential, and switching potentials. The utility of the holding potential is to: (1) oxidize interfering species such that their anodic currents become part of the background signal and can be subtracted out – this applies for serotonin detection, for example, due to the positive holding potential;67,68 and/or (2) preconcentrate analytes of interest through electrostatic interactions – this applies for dopamine, for example.69 Here, a (−)0.3 V holding potential yielded the maximal sensitivity, which makes sense as at pH 7.40, the majority of the Trp molecules have a positive charge on the amino terminal group, so a negative surface potential on the electrode would preconcentrate it onto the electrode surface. It is possible that scanning the holding potential to even more negative values would preconcentrate interfering species, thereby lowering the sensitivity for Trp. The optimal transition potential was +0.6 V. The transition potential is the point where the scan rate switches from 100 V s−1 to higher scan rates and should precede the potential at which we expect our primary oxidation peak. At potentials between the holding and transition potential, the resulting currents were significantly lower due to the slower scan rate (i.e. 100 V s−1). Thus, the smaller oxidation peak in Trp observed by others (∼0.43 V in Weese-Myers et al.60) was not apparent in our CVs. Others have observed similar phenomenon using the multi-scan rate sawhorse waveform.65,66
The switching potential is the upper limit potential and serves to: (1) over-oxidize the surface groups to increase adsorption; and/or (2) renew the electrode surface by stripping the topmost layer of carbon. These phenomena have been observed and discussed by others prior.66,70,71 The optimal switching potential that gave the best signal was 1.2 V. Moreover, maximal sensitivity for Trp was observed when the switching potential (or amperometric potential as referred to by Calhoun et al.65) was held for 2 ms (Fig. 2e). Longer duration displays a scenario of diminishing returns. While having a switching potential just below the stripping limit (1.3 V) and a relatively shorter amperometric hold time yield the highest sensitivity for Trp sensing may initially seem counterintuitive, we hypothesize that these results can be explained via surface polymerization. It is possible that Trp polymerization on the carbon surface is enhancing the signal for Trp measurement. This is in accordance with work done by others72 that demonstrated glutamate polymerization on CFMs yielding greater sensitivity of the electrode to dopamine detection compared to pre-polymerization. We believe that a similar phenomenon is occurring with Trp. This could explain why the highest sensitivity observed was for a switching potential just below stripping and also for the lowest amperometric holding duration of 2 ms.
Lastly, the maximal sensitivity was observed for the 1000 V s−1 scan rate (highest scan rate tested) over the lower values (Fig. 2f). This makes sense as there is an expected increase in signal over background with an increase in scan rate, in agreement with those reported previously.73
We then assessed how the different waveform parameters above can be utilized to optimize for selectivity of Trp over Tyr signal. This is of particular interest as Trp and Tyr has a maximum oxidation peak at the same potential (∼1.0 V) in an FSCV experiment on a CFM vs. Ag/AgCl. We injected boluses of 1 μM Trp and, separately, boluses of 1 μM Tyr into the FIA system and evaluated their respective current densities at 1.0 V vs. Ag/AgCl. Unless otherwise stated we tested 3–5 electrodes per condition. Then the current density from Trp oxidation peak was divided by that for the Tyr oxidation peak from the same conditions to yield a Trp:Tyr selectivity ratio. We observed that a holding potential of −0.2 V yielded the highest selectivity ratio for Trp:Tyr (Fig. 3a). Now this optimal condition for selectivity differed from that for optimal holding potential for sensitivity (vide supra). Because the loss in selectivity (−50%) going from −0.2 V to −0.3 V holding potential was significantly greater than the gain in sensitivity going from −0.2 V to −0.3 V (+3.8%), we decided to select −0.2 V as the optimal holding potential overall. Moreover, we observed that a transition potential of +0.7 V yielded the highest selectivity ratio compared to those of smaller values (Fig. 3b). The optimal transition potential for sensitivity was determined to be +0.6 V (see above). The loss in selectivity (−87%) was significantly greater going from +0.7 to +0.6 V than the gain in sensitivity from +0.7 to +0.6 V (+27%). Thus, the optimal transition potential we selected was +0.7 V.
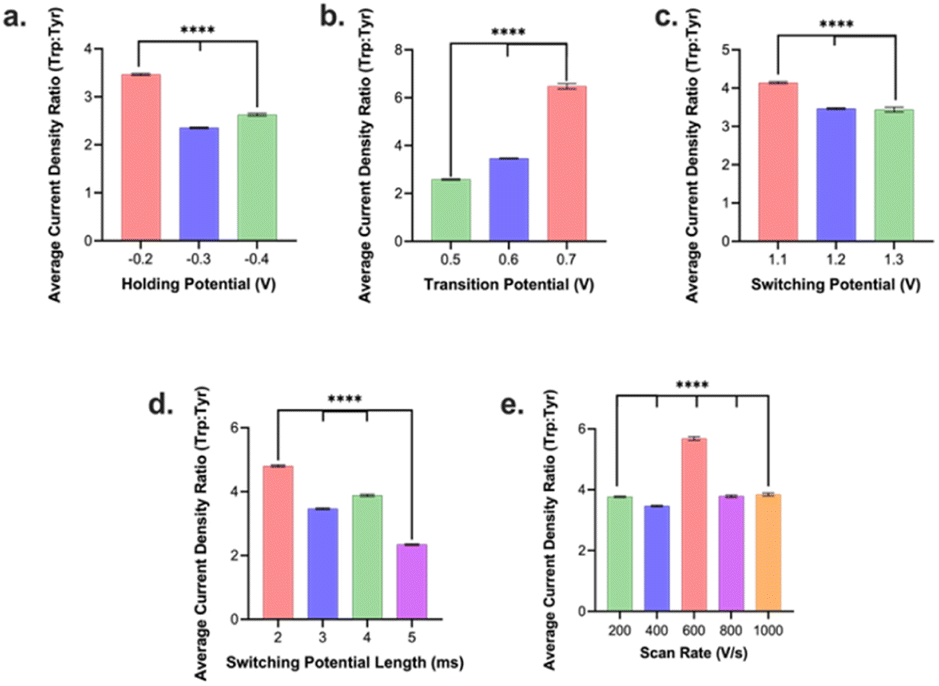 | ||
| Fig. 3 Plot of ratios of current densities of Trp to Tyr as an indicator of selectivity for the different optimized parameters: (a) holding potential, (b) transition potential, (c) switching potential, (d) duration of switching potential, and (e) scan rate. Error bars are ±SEM. ****p < 0.0001. | ||
For switching potential, we discovered that a switching potential of +1.1 V yielded the highest selectivity ratio (Fig. 3c). This contrasted with the +1.2 V switching potential determined to yield the maximum sensitivity. Here, the relative loss of selectivity/gain of sensitivity were relatively comparable (∼20–25%) so we opted to go with +1.1 V in favor of slightly higher selectivity for a slight loss in sensitivity. For switching potential duration, 2 ms was the optimal condition that yielded the maximum sensitivity and selectivity (Fig. 3d). In contrast to sensitivity results for scan rate, 600 V s−1 yielded the highest selectivity for Trp signals over Tyr and we decided to favor selectivity at the slight sacrifice of reduced sensitivity.
We have demonstrated above a significant improvement in selectivity for Trp oxidation over that for Tyr with the optimized MSW (Fig. 3, −0.2 V holding, +0.7 V transition, +1.1 V switching, 2 ms duration of switching potential, 600 V s−1 scan rate between the transition and switching potentials). We then wanted to assess whether the presence of Tyr in the same solution would affect the Trp signal in any way. Thus, we performed injections of a mixture of 1 μM Trp + 1 μM Tyr into the FIA and compared/contrasted that current density with those of Trp and Tyr alone (Fig. 4a). We observed that while there is a significant drop in Trp current density as a result of the presence of an equivalence concentration of Tyr, the current density is still significantly higher (4-fold) than that for Tyr alone. We hypothesize that this decrease is not due to the oxidation of Tyr at the electrode surface (since it's relatively small) but merely the shielding of Trp from interacting with the electrode by the presence of Tyr.
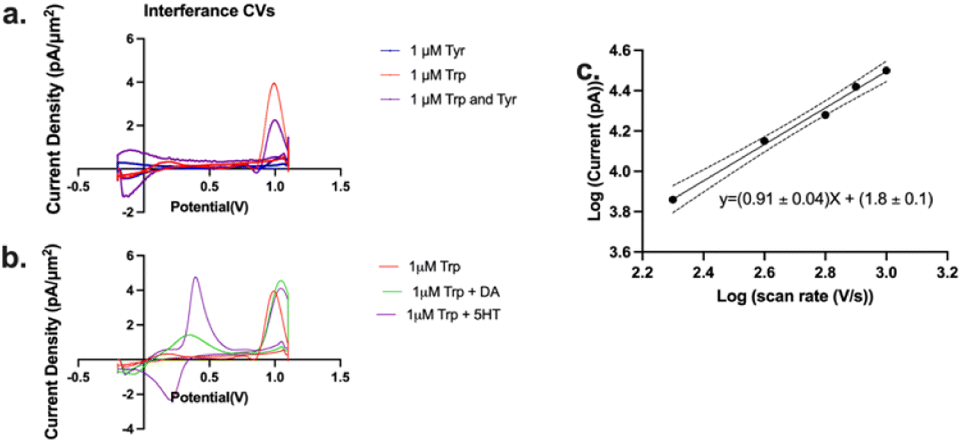 | ||
| Fig. 4 (a) Representative cyclic voltammograms of injections of 1 μM Trp (red), 1 μM Tyr (blue), and a mixture of the two (purple) onto a flow injection analysis system. (b) Representative cyclic voltammograms of injections of 1 μM Trp (red), 1 μM Trp with 1 μM dopamine (DA), and 1 μM Trp with 1 μM serotonin (5HT). (c) Log–log plot of the current vs. scan rate for oxidation of Trp. The dashes lines indicate 95% confidence interval for linear regression analysis. Slope of the regression line is 0.91 ± 0.04, which suggests an adsorption-limited mechanism of electron transfer. | ||
Next, we tested whether other common interferents such as dopamine and serotonin would affect the Trp signal. As seen in Fig. 4b, while the presence of dopamine and serotonin did shift the Trp peak slightly, dopamine and serotonin oxidation were observed at a much lower potential than that for Trp oxidation so interference was negligible.
We also obtained a slope of 0.91 ± 0.04 in a log–log plot of the current vs. scan rate (Fig. 4c). Theory predicts that a slope of 1 is indicative of an entirely adsorption-driven process while a slope of 0.5 indicates a solely diffusion-driven process.42,74,75 Our slope indicates that the signal we obtained is primarily adsorption-driven with some contribution from diffusion and this is in agreement with those reported by others.60,75
As summarized in Fig. 5a and b, the original MSW did yield significantly lower current density than the dopamine waveform at all concentrations (****p < 0.0001). The optimized MSW, however, yielded significantly higher sensitivity for Trp detection than the original MSW at all four concentrations that were evaluated (****p < 0.0001). The optimized MSW yielded higher current density for Trp than the dopamine waveform at all concentrations evaluated (****p < 0.0001).
 | ||
| Fig. 5 (a) Calibration curves for Trp using three different waveforms (optimized MSW (green triangle), dopamine (magenta circle), and original MSW (blue square)). Error bars (±SEM) are too small to be visible. Only the t-test results comparing optimized vs. original waveforms are denoted in the graph. (b) Zoomed-in plot of the 0.5 μM Trp showing how the optimized MSWs yielded higher current density than the dopamine and original waveform. ****p < 0.0001. | ||
These conclusions were further validated by the calculated limit of detection (LOD) and quantitation (LOQ) (Table 1). LOD and LOQ were calculated from standard deviation of the baseline noise in the IT curves and the corresponding calibration curve for each waveform (n = 5 for each category). There is no statistical difference between those reported for the dopamine and original MSW (p = 0.0588). Measurements using the optimized MSW, however, did show significantly lower LOD and LOQ than those for the dopamine (p < 0.01) and original MSW (p < 0.05). We conclude, therefore, that the optimized MSW provided conditions for the most sensitive detection of Trp of the three waveforms. In fact, based on these data, we can report a 4-fold improvement in the limit of detection using the optimized waveform compared to the dopamine waveform and a 20-fold better sensitivity compared to the original MSW.
| Dopamine waveform | Original MSW | Optimized MSW | |
|---|---|---|---|
| LOD (nM) | 41 ± 20 | 190 ± 150 | 9.0 ± 0.6 |
| LOQ (nM) | 100 ± 30 | 290 ± 190 | 34 ± 6 |
Next, we investigated fouling on the electrode surface as a function of multiple repeated injections of Trp using the optimized waveform (Fig. 6). A one-way ANOVA Dunnett's T3 multiple comparison test revealed that there is no statistical difference between any of the 25 consecutive injections. Thus, we conclude that with the optimized waveform, multiple consecutive injections of at least 25 should not cause any fouling on the electrode surface.
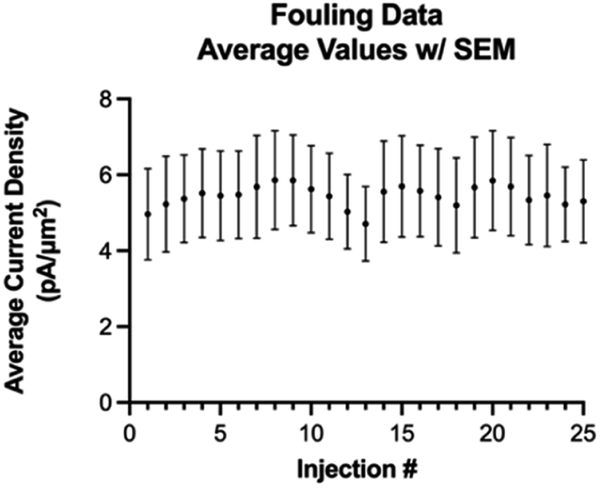 | ||
| Fig. 6 Average current density over 25 consecutive injections (error bars = SEM). | ||
Lastly, we performed Trp measurements in cultured PC-12 cells and pinealocytes as proof-of-principle to demonstrate our ability to measure different Trp dynamics (both exogenous and endogenous changes). While we may be the first to use FSCV at CFMs to measure Trp in cell cultures, we are not the first to use FSCV to measure Trp-containing compounds in cell cultures. In fact, the Venton lab first demonstrated the utility of using FSCV at CFMs to measure gonadotropin-releasing hormone, which contains Trp and Tyr.59 Moreover, Kennedy and colleagues utilized FSCV at CFMs to measure α-melanocyte stimulating hormone and other Trp- and Tyr-containing oligopeptides.75 The authors reported comparable temporal resolution as our method reported here but utilized a slightly larger fiber (9 μm in diameter) beveled at 45° into a disc electrode. Assuming an elliptical shape of the electrode, this would translate to a current density of ∼4 pA μm−2 for the authors' highest observed Trp or Trp-containing peptide. In contrast, with our method, we observe a nearly 2-fold higher current density under comparable conditions. Moreover, we perform further optimization for selectivity here.
First, we cultured two types of cells: PC-12 and PC-12 with an overexpression of tryptophan hydroxylase 2 (TPH2). TPH is the rate limiting enzyme in the synthesis of serotonin from Trp and is localized intracellularly. While TPH1 is expressed primarily in the pineal gland and the periphery, TPH2 is expressed mainly in serotonergic neurons in the brain.76,77 We positioned a CFM in the extracellular media surrounding the cells (with a pseudo-Ag/AgCl in the same well). We then exogenously added Trp We observed a large range of maximum current densities in the individual recordings even within the same well plate of cells. This can be rationalized by the fact that the current densities depend on: (1) how close the CFMs are positioned near the cell membranes; and (2) how close the Trp is applied near the CFMs. In order to quantitatively compare between the different recordings, we normalized each current density vs. time profile to the maximum signal from the exogenously added Trp (excluding higher currents that were due to mechanical noise of adding the Trp itself, usually present at the time of addition). As seen in Fig. 7, the normalized data revealed two distinct profiles. The general shape of the time profiles is similar for both types of PC-12 cells: there is an initial increase due to the application of the exogenous Trp and then a subsequent decay. Both decay curves should include contribution from mass transport (i.e. diffusion) and possibly from the activity of any membrane-bound transporters. Our data shows a slower decay curve for those Trp measurements done with PC-12 cells with TPH2(+) than their wildtype counterpart. Assuming that the Trp decay curves in both should experience similar contributions from mass transport, then the discrepancy between the two could indicate differences in transport activity that internalize amino acids such as Trp into the cells. In fact, these data are incredibly reproducible, as indicated by the relatively small error bars (±SEM) at each time point (pale blue for PC-12 with TPH2(+), pale green for PC-12 wildtype). Wilcoxon t-test results revealed a significant difference between the two decay profiles (****p < 0.0001). A speculative hypothesis to explain these data is that the PC-12 cells with TPH2(+) may have lower expression of amino acid transporters.78,79 This hypothesis is supported by the following evidence. First, PC-12 cells are suspended cells and are presumably more rapidly dividing than PC-12 with TPH2(+), which are adherent cells. LAT1, L-type amino acid transporter 1, is the transporter protein responsible for shuttling Trp across the membrane and has been linked to cell proliferation.80,81 It is possible that PC-12 with TPH2(+) has lower LAT1 activity than PC-12 wildtype since the former are adherent. More experiments will be needed to evaluate and test this hypothesis. Nevertheless, these recordings demonstrate the utility of our optimized method for measuring and differentiating Trp dynamics (from an exogenous source) between two cell lines with good spatiotemporal resolution.
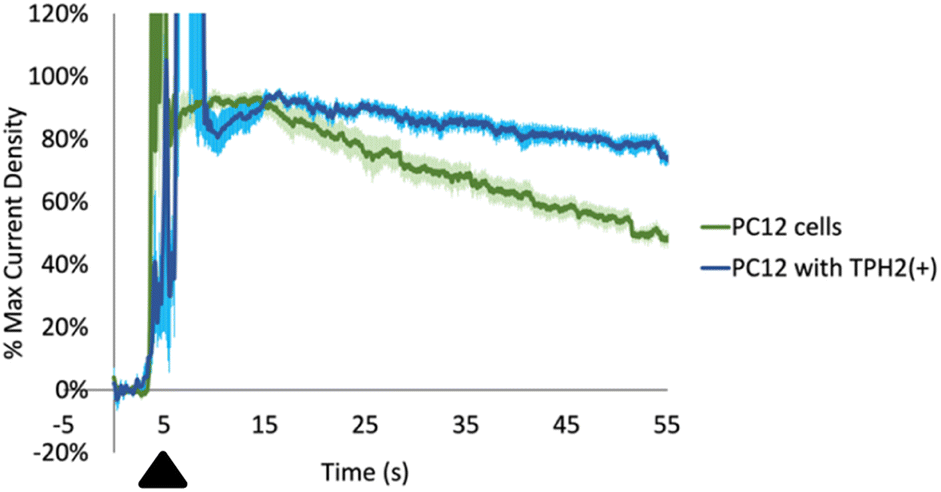 | ||
| Fig. 7 Normalized current density (to the maximum current in each curve, excluding those signals called by mechanical noise from the injection itself) for exogenously applied 10 μL of 5 μM Trp (black triangle) to cultured PC-12 cells (green, n = 5 measurements) and PC-12 cells with overexpressed tryptophan hydroxylase 2 (TPH2(+), n = 6 measurements). Pale blue and pale green error bars indicate SEM. ****p < 0.0001. Time resolution was 100 ms. | ||
We also cultured pinealocytes, which were harvested from pineal gland of rats. Upon exposure to norepinephrine, these cells produce melatonin from Trp in the surrounding media. In this protocol, baseline recordings were done for 5 min, then either 5 μL water or 5 μL water with 1 mM norepinephrine (NE) were added to the pinealocytes near the electrode. The current densities corresponding to Trp oxidation was then plotted against time for control group (water injection) and experimental group (NE). The binned data with 1 min time resolution is shown in Fig. S2.† Since the Trp dynamics in this particular system was slower than our time resolution of 100 ms, we binned the data further (7 min bins) to show the dynamics more clearly (Fig. S3†). We calculated the difference in current density at each time point between the control and NE group and then normalized as % control (water). As seen in Fig. 8, there is a significant decrease in Trp at time points 28 min (p = 0.0153) and 35 min (p = 0.0070) compared to the beginning of the experiment. We performed linear regression to demonstrate these trends in a more quantitative manner. The linear regression for control group (blue) was y = (0.0010 ± 0.0009) × X + (0.05 ± 0.02), with a positive slope. The linear regression for the NE group (pink) was y = (−0.0007593 ± 0.0009) × X + (0.08 ± 0.02), with an overall negative slope. These results makes sense as the cells exposed to NE were stimulated to make melatonin from Trp, thereby decreasing the amount of Trp in the media in the process. It also makes sense that this process takes some time as the Trp would need to transported into the cells via transporters before melatonin production can begin. This is also on par with previous blood sampling data of melatonin levels in humans,82 in which the data shows an increase in melatonin ∼30–60 min after dark cycle begins (when norepinephrine would be presumably released, assuming that sunset is around 20:00 h). Collectively, these results demonstrate that our method was capable of measuring endogenous Trp dynamics as well.
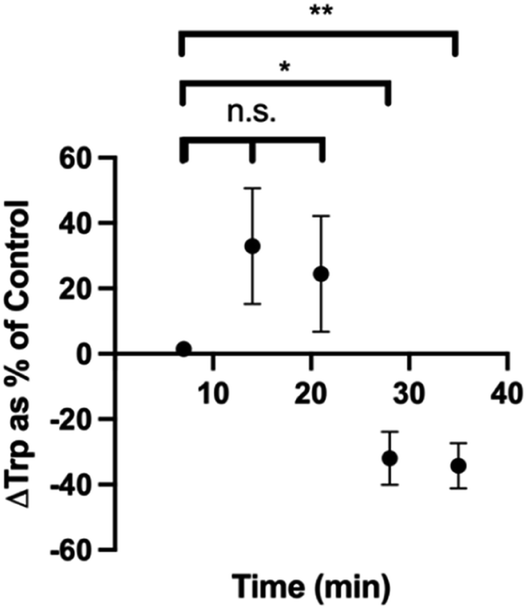 | ||
| Fig. 8 Plot of changes in Trp levels reported as % control over time. Control data is the current density plot over time for water injection. *p < 0.05. **p < 0.01. n.s. = not significant. | ||
Conclusions
We developed an improved method for sensitive and selective Trp sensing via the application of an optimized multi-scan rate sawhorse waveform using FSCV at CFMs. We demonstrated four-fold higher sensitivity for Trp sensing over previous methods and a six-fold higher current density for Trp signals than those for Tyr, one of the primary interfering species that has similar oxidation potentials. We utilized the optimized method and measured exogenously applied extracellular Trp in PC-12 and PC-12 with overexpression of TPH2. The time-dependent measurements revealed similar increase, but interesting differences in the decay curves. We also demonstrated the utility of the method in measuring time resolved endogenous Trp dynamics in pinealocytes either exposed to water or water with norepinephrine. There were clear differences between the two groups. Altogether, our optimized method has the spatiotemporal resolution, sensitivity, and selectivity to monitor exogenously added and endogenously present Trp dynamics in different cell cultures. These proof-of-concept studies are promising for anticipated future work in vivo.Author contributions
I. S. – conceptualization, formal analysis, investigation, methodology, validation, visualization, writing – review & editing; M. O. – conceptualization, formal analysis, investigation, validation, writing – review & editing; L. R-S. – formal analysis, investigation, methodology, visualization, writing – review & editing; T. N. – formal analysis, methodology, visualization, writing – review & editing; K. L. – resources, methodology; J. S. – resources, formal analysis, funding acquisition, project administration, writing – review & editing; Y. O. – conceptualization, project administration, formal analysis, funding acquisition, writing – original draft, writing – review & editing.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
TPH2 overexpressing cell line creation and pinealocyte culture method development was supported under Defense Advanced Research Projects Agency (DARPA) agreement number FA8650-21-2-7120. This material is based on research sponsored by 711 Human Performance Wing (HPW) and DARPA under agreement number FA8650-21-2-7120. The U.S. Government is authorized to reproduce and distribute reprints for Governmental purposes notwithstanding any copyright notation thereon. The views and conclusions contained herein are those of the authors and should not be interpreted as necessarily representing the official policies or endorsements, either expressed or implied, of 711 HPW and DARPA or the U.S. Government. The authors would also like to thank the University of Vermont for funding this work (YO, IS, LRS, TN, MO), Prof. David Punihaole for useful discussions relating to this manuscript, and UVM-LCOM Flow Cytometry and Cell Sorting Facility (RRID:SCR_022147) for sorting cell lines used in this study.Notes and references
- A. Coppen, Br. J. Psychiatry, 1967, 113, 1237–1264 CrossRef CAS PubMed.
- D. Healy, 2015, 350, p. h1771.
- B. J. Sadock, V. A. Sadock and P. Ruiz, Kaplan & Sadock's Comprehensive Textbook of Psychiatry, Wolters Kluwer, Philadelphia, 2017 Search PubMed.
- Psychiatric Times, https://www.psychiatrictimes.com/view/psychiatrys-new-brain-mind-and-legend-chemical-imbalance, (accessed June 2023).
- R. Marotta, M. C. Risoleo, G. Messina, L. Parisi, M. Carotenuto, L. Vetri and M. Roccella, Brain Sci., 2020, 10, 163 CrossRef CAS PubMed.
- D. C. Chugani, Mol. Psychiatry, 2002, 7, S16–S17 CrossRef PubMed.
- C. Jonnakuty and C. Gragnoli, J. Cell. Physiol., 2008, 217, 301–306 CrossRef CAS PubMed.
- R. El-Merahbi, M. Löffler, A. Mayer and G. Sumara, FEBS Lett., 2015, 589, 1728–1734 CrossRef CAS PubMed.
- A. Bektaş, H. Erdal, M. Ulusoy and İ. T. Uzbay, Turk. J. Gastroenterol., 2020, 31, 721 CrossRef PubMed.
- M. Friedman, Int. J. Tryptophan Res., 2018, 11, 1178646918802282 Search PubMed.
- D. M. Richard, M. A. Dawes, C. W. Mathias, A. Acheson, N. Hill-Kapturczak and D. M. Dougherty, Int. J. Tryptophan Res., 2009, 2, IJTR–S2129 Search PubMed.
- P. J. Kennedy, J. F. Cryan, T. G. Dinan and G. Clarke, Neuropharmacology, 2017, 112, 399–412 CrossRef CAS PubMed.
- Y. Li, Z.-Y. Luo, Y.-Y. Hu, Y.-W. Bi, J.-M. Yang, W.-J. Zou, Y.-L. Song, S. Li, T. Shen and S.-J. Li, Microbiome, 2020, 8, 1–23 CrossRef PubMed.
- L. Dell'Osso, C. Carmassi, F. Mucci and D. Marazziti, Curr. Pharm. Des., 2016, 22, 949–954 CrossRef PubMed.
- A. S. Correia and N. Vale, Int. J. Mol. Sci., 2022, 23, 8493 CrossRef CAS PubMed.
- C. Chojnacki, T. Popławski, J. Chojnacki, M. Fila, P. Konrad and J. Blasiak, Nutrients, 2020, 12, 3183 CrossRef CAS PubMed.
- L. Lanser, P. Kink, E. M. Egger, W. Willenbacher, D. Fuchs, G. Weiss and K. Kurz, Front. Immunol., 2020, 11, 249 CrossRef CAS PubMed.
- H. Liaqat, A. Parveen and S. Y. Kim, Nutrients, 2022, 14, 3270 CrossRef CAS PubMed.
- M. Reuter, V. Zamoscik, T. Plieger, R. Bravo, L. Ugartemendia, A. B. Rodriguez and P. Kirsch, Nutr. Res., 2021, 85, 14–20 CrossRef CAS PubMed.
- I. Lukić, D. Getselter, O. Koren and E. Elliott, Front. Behav. Neurosci., 2019, 13, 123 CrossRef PubMed.
- L. Boccuto, C.-F. Chen, A. R. Pittman, C. D. Skinner, H. J. McCartney, K. Jones, B. R. Bochner, R. E. Stevenson and C. E. Schwartz, Mol. Autism, 2013, 4, 1–10 CrossRef PubMed.
- N. Israelyan and K. G. Margolis, Pharmacol. Res., 2019, 140, 115–120 CrossRef PubMed.
- L. Xiao, J. Yan, T. Yang, J. Zhu, T. Li, H. Wei and J. Chen, Msystems, 2021, 6, 1–17 Search PubMed.
- J. Kałużna-Czaplińska, J. Jóźwik-Pruska, S. Chirumbolo and G. Bjørklund, Metab. Brain Dis., 2017, 32, 1585–1593 CrossRef PubMed.
- E. Daly, C. Ecker, B. Hallahan, Q. Deeley, M. Craig, C. Murphy, P. Johnston, D. Spain, N. Gillan and M. Gudbrandsen, Brain, 2014, 137, 2600–2610 CrossRef PubMed.
- M. Berger, J. A. Gray and B. L. Roth, Annu. Rev. Med., 2009, 60, 355–366 CrossRef CAS PubMed.
- R. Valladares, L. Bojilova, A. H. Potts, E. Cameron, C. Gardner, G. Lorca and C. F. Gonzalez, Faseb. J., 2013, 27, 1711–1720 CrossRef CAS PubMed.
- S. D. Hood, C. J. Bell and D. J. Nutt, Aust. N. Z. J. Psychiatry, 2005, 39, 558–564 CrossRef PubMed.
- A. A. B. Badawy and G. Guillemin, Int. J. Tryptophan Res., 2019, 12, 1178646919868978 Search PubMed.
- R. J. Wurtman, F. Hefti and E. Melamed, Pharmacol. Rev., 1980, 32, 315–335 CAS.
- E. L. Sainio, K. Pulkki and S. N. Young, Amino Acids, 1996, 10, 21–47 CrossRef CAS PubMed.
- J. D. Fernstrom, Physiol. Rev., 1983, 63, 484–546 CrossRef CAS PubMed.
- P. Etienne, S. N. Young and T. L. Sourkes, Nature, 1976, 262, 144–145 CrossRef CAS PubMed.
- A. Yuwiler, W. H. Oldendorf, E. Geller and L. Braun, J. Neurochem., 1977, 28, 1015–1023 CrossRef CAS PubMed.
- W. M. Pardridge, J. Neural Transm., Suppl., 1979, 15, 43–54 CAS.
- R. J. Wurtman and W. M. Pardridge, presented in part at the Proceedings of the International Symposium, Prilly/Lausanne, 1978 Search PubMed.
- R. J. Geor and P. A. Harris, in Equine Sports Medicine and Surgery, ed. K. W. Hinchcliff, A. J. Kaneps and R. J. Geor, W.B. Saunders, 2nd edn, 2014, pp. 819–834, DOI:10.1016/B978-0-7020-4771-8.00037-5.
- P. H. Hutson, G. S. Sarna, B. D. Kantamaneni and G. Curzon, J. Neurochem., 1985, 44, 1266–1273 CrossRef CAS PubMed.
- G. S. Sarna, P. H. Hutson, M. T. O'Connell and G. Curzon, J. Neurochem., 1991, 56, 1564–1568 CrossRef CAS PubMed.
- F. Chaouloff, G. A. Kennett, B. Serrurrier, D. Merino and G. Curzon, J. Neurochem., 1986, 46, 1647–1650 CrossRef CAS PubMed.
- M. K. Gaitonde and T. Dovey, Biochem. J., 1970, 117, 907–911 CrossRef CAS PubMed.
- J. C. Zahnley and J. G. Davis, Biochem. J., 1973, 135, 59–61 CrossRef CAS PubMed.
- M. Friedman, J. Agric. Food Chem., 2004, 52, 385–406 CrossRef CAS PubMed.
- M. Friedman, C. E. Levin, A. T. Noma, W. C. Montague and J. C. Zahnley, in Progress in tryptophan and serotonin research, De Gruyter, 2019, pp. 119–124 Search PubMed.
- A. S. Inglis, in Methods in Enzymology, Elsevier, 1983, vol. 91, pp. 26–36 Search PubMed.
- H. Yamada, H. Moriya and A. Tsugita, Anal. Biochem., 1991, 198, 1–5 CrossRef CAS PubMed.
- G. A. Bubenik, R. O. Ball and S. F. Pang, J. Pineal Res., 1992, 12, 7–16 CrossRef CAS PubMed.
- Z. A. Khan, P. J.-S. Hong, C. H. Lee and Y. Hong, Int. J. Nanomed., 2021, 16, 6861–6888 CrossRef CAS PubMed.
- A. Jaquins-Gerstl and A. C. Michael, J. Neurosci. Methods, 2009, 183, 127–135 CrossRef PubMed.
- A. Jaquins-Gerstl and A. C. Michael, Analyst, 2015, 140, 3696–3708 RSC.
- E. C. Dankoski and R. M. Wightman, Front. Integr. Neurosci., 2013, 7, 44 CAS.
- A. L. Hensley, A. R. Colley and A. E. Ross, Anal. Chem., 2018, 90, 8642–8650 CrossRef CAS PubMed.
- Y. Murakami and K. Saito, Int. J. Tryptophan Res., 2013, 6, IJTR–S11558 Search PubMed.
- E. Höglund, Ø. Øverli and S. Winberg, Front. Endocrinol., 2019, 10, 158 CrossRef PubMed.
- T. D. Y. Kozai, A. S. Jaquins-Gerstl, A. L. Vazquez, A. C. Michael and X. T. Cui, ACS Chem. Neurosci., 2015, 6, 48–67 CrossRef CAS PubMed.
- M. Hejazi, W. Tong, M. R. Ibbotson, S. Prawer and D. J. Garrett, Front. Neurosci., 2021, 15, 658703 CrossRef PubMed.
- Y. Ou, A. M. Buchanan, C. E. Witt and P. Hashemi, Anal. Methods, 2019, 11, 2738–2755 RSC.
- H. Rafi and A. G. Zestos, J. Electrochem. Soc., 2021, 168, 057520 CrossRef CAS PubMed.
- K. M. Glanowska, B. J. Venton and S. M. Moenter, J. Neurosci., 2012, 32, 14664–14669 CrossRef CAS PubMed.
- M. E. Weese-Myers and A. E. Ross, J. Electrochem. Soc., 2021, 168, 126524 CrossRef CAS.
- T. Vallianatou, N. B. Bechet, M. S. P. Correia, I. Lundgaard and D. Globisch, Metabolites, 2021, 12, 21 CrossRef PubMed.
- J. M. Stafford, C.-H. Lee, P. Voigt, N. Descostes, R. Saldaña-Meyer, J.-R. Yu, G. Leroy, O. Oksuz, J. R. Chapman and F. Suarez, Sci. Adv., 2018, 4, eaau5935 CrossRef CAS PubMed.
- N. T. Nguyen, M. Z. Wrona and G. Dryhurst, J. Electroanal. Chem. Interfacial Electrochem., 1986, 199, 101–126 CrossRef CAS.
- T. A. Enache and A. M. Oliveira-Brett, Electroanalysis, 2011, 23, 1337–1344 CrossRef CAS.
- S. E. Calhoun, C. J. Meunier, C. A. Lee, G. S. McCarty and L. A. Sombers, ACS Chem. Neurosci., 2018, 10, 2022–2032 CrossRef PubMed.
- A. C. Schmidt, L. E. Dunaway, J. G. Roberts, G. S. McCarty and L. A. Sombers, Anal. Chem., 2014, 86, 7806–7812 CrossRef CAS PubMed.
- B. P. Jackson, S. M. Dietz and R. M. Wightman, Anal. Chem., 1995, 67, 1115–1120 CrossRef CAS PubMed.
- P. Hashemi, E. C. Dankoski, J. Petrovic, R. B. Keithley and R. M. Wightman, Anal. Chem., 2009, 81, 9462–9471 CrossRef CAS PubMed.
- D. L. Robinson, B. J. Venton, M. L. A. V. Heien and R. M. Wightman, Clin. Chem., 2003, 49, 1763–1773 CrossRef CAS PubMed.
- P. Takmakov, M. K. Zachek, R. B. Keithley, P. L. Walsh, C. Donley, G. S. McCarty and R. M. Wightman, Anal. Chem., 2010, 82, 2020–2028 CrossRef CAS PubMed.
- M. L. A. V. Heien, P. E. M. Phillips, G. D. Stuber, A. T. Seipel and R. M. Wightman, Analyst, 2003, 128, 1413–1419 RSC.
- J. Holmes, C. E. Witt, D. Keen, A. M. Buchanan, L. Batey, M. Hersey and P. Hashemi, Anal. Chem., 2021, 93, 10762–10771 CrossRef CAS PubMed.
- D. O. Wipf, E. W. Kristensen, M. R. Deakin and R. M. Wightman, Anal. Chem., 1988, 60, 306–310 CrossRef CAS.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical methods: fundamentals and applications, John Wiley & Sons, 2022 Search PubMed.
- C. D. Paras and R. T. Kennedy, Electroanalysis, 1997, 9, 203–208 CrossRef CAS.
- D. J. Walther, J.-U. Peter, S. Bashammakh, H. Hortnagl, M. Voits, H. Fink and M. Bader, Science, 2003, 299, 76–76 CrossRef CAS PubMed.
- K. W. Ottenhof, M. Sild, M. L. Lévesque, H. G. Ruhé and L. Booij, Neurosci. Biobehav. Rev., 2018, 92, 29–42 CrossRef CAS PubMed.
- T. Nemoto, S. Horie, Y. Okuma, Y. Nomura and T. Murayama, Neurosci. Lett., 2001, 311, 117–120 CrossRef CAS PubMed.
- T. Nemoto, N. Shimma, S. Horie, T. Saito, Y. Okuma, Y. Nomura and T. Murayama, Eur. J. Pharmacol., 2003, 458, 17–24 CrossRef CAS PubMed.
- L. Quan, R. Ohgaki, S. Hara, S. Okuda, L. Wei, H. Okanishi, S. Nagamori, H. Endou and Y. Kanai, J. Exp. Clin. Cancer Res., 2020, 39, 1–17 CrossRef PubMed.
- W. Luo, H. Zhang, Y. Zhang, P. Liang, X. Wang, J. Ma, D. Tan, Y. Tan, J. Song and P. Ji, Am. J. Transl. Res., 2020, 12, 6665 CAS.
- S. Benloucif, H. J. Burgess, E. B. Klerman, A. J. Lewy, B. Middleton, P. J. Murphy, B. L. Parry and V. L. Revell, J. Clin. Sleep. Med, 2008, 4, 66–69 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04551j |
| This journal is © The Royal Society of Chemistry 2023 |