
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign strategies of Pt-based electrocatalysts and tolerance strategies in fuel cells: a review
Wenlei Luo
a,
Yitian Jiang
b,
Mengwei Wang
b,
Dan Lu
b,
Xiaohui Sun
b and
Huahui Zhang
 *b
*b
aNational Innovation Institute of Defense Technology, Academy of Military Science, Beijing 100071, China
bState Key Laboratory of Space Power-sources Technology, Shanghai Institute of Space Power-Sources, 2965 Dongchuan Road, Shanghai 200245, China. E-mail: hhzhang2022@126.com
First published on 7th February 2023
Abstract
As highly efficient conversion devices, proton-exchange-membrane fuel cells (PEMFCs) can directly convert chemical energy to electrical energy with high efficiencies and lower or even zero emissions compared to combustion engines. However, the practical applications of PEMFCs have been seriously hindered by the intermediates (especially CO) poisoning of anodic Pt catalysts. Hence, how to improve the CO tolerance of the needed Pt catalysts and reveal their anti-CO poisoning mechanism are the key points to developing novel anti-toxic Pt-based electrocatalysts. To date, two main strategies have received increasing attention in improving the CO tolerance of Pt-based electrocatalysts, including alloying Pt with a second element and fabricating composites with geometry and interface engineering. Herein, we will first discuss the latest developments of Pt-based alloys and their anti-CO poisoning mechanism. Subsequently, a detailed description of Pt-based composites with enhanced CO tolerance by utilizing the synergistic effect between Pt and carriers is introduced. Finally, a brief perspective and new insights on the design of Pt-based electrocatalysts to inhibit CO poisoning in PEMFCs are also presented.
 Wenlei Luo | Luo Wenlei, graduated from the National University of Defense Technology with a master's degree, majored in aerospace science and technology. He received his doctor's degree in chemistry and process engineering from University of Leeds, the United Kingdom in 2015. He is a researcher in the energy and power. He is currently a member of research group “organic liquid hydrogen storage fuel cell”, led the research on fuel cell technology. |
 Yitian Jiang | Yitian Jiang is a senior engineer at Shanghai Institute of Space Power-sources. He received his master degree from Harbin Institute of Technology in 2012. His current research is focused on the construction and application of electrocatalysts for fuel cells. |
 Mengwei Wang | Wang Mengwei received a MS degree in Inorganic Non-metallic Materials Engineering from University of Science and Technology Beijing (USTB) in 2009. She is now a researcher in fuel cell research group in Shanghai Institute of Space Power-Sources (SISP). Her research interests include the influence of surface chemistry of metal nanoparticles on catalytic activities and stability for fuel cells. |
 Dan Lu | Lu Dan obtained a master's degree from Shanghai University in 2017. She mainly participated in the research on the preparation of fuel cell catalysts and electrodes. She has a strong interest in fuel cell-related technology research and engineering applications. She actively participated in the construction of flexible product production lines of fuel cell reactor collection systems, and carried out extensive and valuable research. |
 Xiaohui Sun | Sun Xiaohui, a senior engineer, received her bachelor's degree in applied chemistry from Tianjin University. She was engaged in the design, research and development, engineering application and quality management of chemical power supply in Shanghai Space Power Supply Research Institute. She participated in the exploration of improving the atomic utilization rate and reactivity of fuel cell catalysts, and also carried out targeted quality management research in the research. |
 Huahui Zhang | Zhang Huahui received her doctor's degree in material processing engineering from Beijing University of Aeronautics and Astronautics (BUAA) in September 2009. She is a researcher in the fields of development of Materials for chemical power. She is a member of group “organic liquid hydrogen storage fuel cell”, and she participated in the research of fuel cell catalysts. Her ambition is to conduct research to improve the performance of chemical power sources. |
1. Introduction
The increasing environmental issues and energy crisis severely threaten sustainable human society due to traditional energy consumption and declining fossil fuels. How to develop efficient storage technologies or conversion devices that can fully utilize sustainable and clean energy has drawn extensive attention.1,2 Based on the guidance, many efforts have focused on developing highly efficient conversion devices, e.g., fuel cells, which can effectively reduce traditional fossil energy consumption and its emissions. Proton exchange membrane fuel cells (PEMFCs) as highly efficient conversion devices have received more attention due to their high-power generation efficiency and environmentally sustainable regeneration.3–5 In PEMFCs, fuel molecules and oxidants can be converted into ions by the utilization of catalysts and migrated continuously in the electrolyte, thus continuously converting chemical energy into electrical energy. Despite the advantages, the anodizing process is very complicated, which involves the sluggish kinetics and formation of intermediates (especially CO) that can occupy the active sites of electrocatalysts, thus leading to decreasing its catalytic performance. Therefore, it is urgently needed to design high-efficiency electrocatalysts for the complicated anodizing process.6–8As a state-of-the-art catalyst, platinum (Pt) can absorb many charged species and then accelerate the formation of active species on its surface, resulting in the rapid catalytic reaction. As a common Pt-based electrocatalyst, commercial Pt/C refers to commercial carbon supported Pt nanoparticles (NPs). To date, commercial Pt/C catalyst has been regarded as one of the most effective catalysts in PEMFCs, while its limited activity and poor stability are very difficult to meet the extensive applications.9 The main reason is that Pt is low abundance in nature and high price, and it is also very difficult to tune its morphology and structure, which cannot meet high-efficiency electrocatalysts in PEMFCs. Moreover, both active Pt NPs and carbon carriers are easily prone to be oxidized and dissolved under the electrochemical tests, leading to an irreversible structure transition compared to the initial structure. In contrast, the current densities of Pt-based alloys are tens or even hundreds of times than that of commercial Pt/C based on the previous reports.10–13 As an effective electrocatalyst, Pt-based alloys include Pt-based binary alloys, Pt-based multi-element alloys, and Pt-based high-entropy alloys. With the development of the technology, the following two strategies have been used to prepare a series of Pt-based electrocatalysts: (1) alloying Pt with a second element, including Ru, Pd, Co, Ni, Fe and Cu, could reduce the usage of Pt and enhance its catalytic performance.14–18 That is because that the formation of binary or multi-element alloys can prevent the gradual deterioration of Pt and improve its CO tolerance. The enhanced CO tolerance usually involves in bifunctional mechanism and d-band theory. The former one emphasizes that the adsorbed CO species (COad) could react with oxygen-containing species at oxophilic neighbour sites, and the latter one is an electronic effect, which can downshift the d-band center of Pt, thus weakening the interaction between Pt and COad. (2) The improvement can also be made by fabricating Pt-based composites with carbon-based materials or metal oxides.19–21 In the composites, the synergy between Pt and carbon-based materials or metal oxides is believed to weaken the binding strength of CO. The synergy first involves absorbed hydroxide (OHad) species, which are obtained by promoting the dissociation of H2O are essential for the complete oxidation of CO intermediate. Next, another effective method is to employ the charge-transfer mechanism in the composites. The appropriate supports are good for anchoring Pt NPs, and making them uniformly dispersed. Such Pt-based composites can transfer electron from Pt atoms to supports, thus receiving strong binding of single Pt and positively charged Pt atoms, which leads to producing more vacant d orbitals in single Pt atoms. In addition, to generate more unsaturated active sites and effectively improve the Pt utilization rate, many approaches have been developed to synthesize high surface-area structures, such as porous structure, nanowires, nanosheets and nanoframes. Nevertheless, it is still critical challenges to develop novel anti-toxic Pt-based electrocatalysts, including developing macroscopic preparation techniques, regulating their size, morphology and structure, and investigating their structure–activity relationships.
CO poisoning has been considered a notorious catalyst-deactivating process to limit the commercialization of PEMFCs.22–24 In order to design a novel anti-toxic Pt-based electrocatalyst, the electrochemistry behavior (e.g., adsorption and oxidation) of CO on the Pt surface must be understood. In an acid electrolyte, the adsorption and oxidation of CO usually occur on the (100) and (110) facets of Pt. For CO adsorption, CO competes with fuels or other intermediates for the active sites, and CO adsorption heat is much higher than that of fuels or other intermediates, indicating CO is more easily adsorbed on the Pt active sites. In addition, there are different modes of CO adsorption on the Pt electrode, including linear or top bond, bridge or double bond, as well as hollow or triple bond.25–28 In contrast, the linear bond type dominates the mode of CO adsorption on the Pt electrode, and the adsorption isotherm is a Tempkin isotherm, as follows:


In this review, we will concisely summarize the latest developments of Pt-based electrocatalysts and its anti-CO poisoning mechanism in the following strategies, including (1) modifying the electronic structure of Pt by alloying Pt with transition metals to weaken the strong adsorption of CO and alleviate its CO poisoning, resulting in the improved activity and stability. (2) Synthesizing Pt-based composites to improve the conductivity and resistance against corrosion and oxidation. Importantly, utilizing the synergistic effect between Pt and carriers provides extra catalytic performance. In addition, we will discuss the advantages and challenges of Pt-based electrocatalysts for future development in PEMFCs. We hope that this review will call for more efforts to explore the design of Pt-based electrocatalysts to inhibit CO poisoning and propose new strategies for usage in PEMFCs (Fig. 1).
 | ||
| Fig. 1 Design strategies of Pt-based electrocatalysts and its outstanding strategies for CO tolerance. | ||
2. Alloying Pt to modify its electronic structure
Based on the previous reports, the generated CO intermediate during the anodizing process can easily occupy the Pt active sites, thereby leading to decreased catalytic performance.31,32 Generally, the electrochemistry behaviors of CO include the adsorption and oxidation on the Pt surface. The linear bond type dominates the mode of CO adsorption, and the adsorption mode and coverage of CO determine the rate of CO oxidation. Therefore, increasing attention has focused on the adsorption and electrooxidation of CO for unravelling the anti-CO poisoning mechanism itself and accelerating the fabrication of anti-poisoning Pt-based electrocatalysts. For example, in situ electrochemical shell isolated nanoparticle-enhanced Raman spectroscopy is employed to explore the adsorption and electrooxidation of CO on the Pt(hkl) single crystal electrodes, and the decreasing sequence of CO electrooxidation activity is Pt(111) > Pt(100) > Pt(110) in acidic medium (Fig. 2a and b).33 At the same time, both isotope substitution experiments and DFT calculations clarify that the formation of OHad and COOHad species could boost the CO electrooxidation process (Fig. 2c). As a result, fully understanding the adsorption process and electrooxidation mechanism of CO on the Pt electrode has long been the focus of solving the poisoning mechanisms of Pt-based electrocatalysts. | ||
| Fig. 2 (a) Schematic showing the in situ SHINERS study of the CO electrooxidation process on a Pt (100) surface. Large gold spheres indicate the nanoparticles; (b) CVs of CO electrooxidation on Pt(111) (black curve), Pt(100) (blue curve) and Pt(110) (red curve) single-crystal electrode surfaces in 0.1 M HClO4 solution saturated with CO vs. RHE. Sweep rate was 50 mV s−1; (c) the proposed mechanism of CO electrooxidation on Pt(100) surface. Reproduced with permission from ref. 34. Copyright 2020, Wiley-VCH. | ||
Alloying Pt with the incorporation of a second metal (e.g., Pd, Ru, Co, Ni, Bi, Pb) can not only increase the catalytic performance but also reduce the usage of Pt.34,35 The improved performance arises from the improvement of CO tolerance on the Pt electrode, which usually involves d-band theory and bifunctional mechanism.12,36 Based on d-band theory, both alloy-induced compressive strain and ligand-induced increases in the orbital overlap between the surface and ligand metal have the effect of widening the d-band of Pt, thereby causing the d-band center to shift down in energy to maintain its filling. The downshift in energy causes the occupancy of the anti-bonding orbitals of the adsorbate to increase, thus leading to their weaker adsorption for CO. Therefore, modifying Pt with a second metal could effectively adjust its electronic structure based on electron or ligand effects, including electron donation or reverse donation, thus reducing the strength of Pt–CO bond. More importantly, the adsorption energy of CO is related to the degree of CO coverage, and the oxidation rate of CO is determined by the electronic modification. At lower potentials, the d-band theory mechanism plays a dominant role because of the weak CO adsorption. The bifunctional mechanism occurs in the formation of oxygen-containing species in the oxygenophilic site of the second site at higher overpotentials, whose potential is less than that of Pt, which favors the oxidative removal of CO.37–40 Aside from the compositions, geometry and size effects of the catalyst also play an important role in improving the catalytic performance and weakening the Pt–CO bond. By adjusting the size and structure of Pt-based alloys, it can not only promote the exposure of Pt atoms, but also induce the formation of unsaturated coordination sites, thus greatly causing surface strain and space electric domain sites.
2.1 Pt-based binary alloys
In recent years, considerable interest has focused on Pt-based binary alloys owing to their great progress in both basic research and practical applications. The advantages of alloying Pt with a second metal can not only significantly reduce the usage of Pt but also effectively modify its electronic structure and change the coordination environment, thus contributing to the superior catalytic efficiency, selectivity and stability. Based on the guideline, different metal elements (e.g., noble metal and transition metal elements) and geometric structures (e.g., nanoplates, nanowires and porous structure) have been investigated to improve the CO tolerance and electrocatalytic performance of Pt. Both metal elements and geometric structures could predominate the catalytic effect under different conditions. For example, the surface component of Pt-based binary alloys with the same nanostructure is the key factor to determining the catalytic performance. In addition, the nanostructure of Pt-based binary alloys with the same surface component will predominate the catalytic effect.Alloying Pt with noble metal elements, such as Pd, Au, Ag, Ir, and Ru, can effectively adjust the electronic structure and geometric structure of Pt. Among the noble metal elements, many efforts have focused on the synthesis of PtRu bimetallic alloys due to the famous bifunctional mechanism and electronic effect, which can effectively weaken the CO adsorption and enhance the CO oxidation rate.41–43 For instance, Wang et al. have prepared porous flower-like Pt72Ru28 nanoalloys assembled with sub-4.0 nm NPs by a facile aqueous method.44 Thanks to the optimized molar ratio of Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ru and unique nanostructures, the specific activity and mass activity of Pt72Ru28 nanoalloys can reach 10.98 mA cm−2 and 1.70 A mgPt−1 for methanol oxidation reaction (MOR), respectively. The enhanced oxidizing ability of Pt72Ru28 nanoalloys toward CO, HCOH, and HCOOH intermediates significantly makes them exhibit long-term durability for MOR. Another good example involves the fabrication of PtRu bimetallic NPs with adjustable alloying degree via a co-reduction method.45 Based on in situ attenuated total reflection infrared (ATR-IR) spectroscopy, PtRu/PC–H can easily cleave C–H bond and accelerate the oxidative removal of COad at a lower potential, indicating the enhanced CO tolerance. Therefore, porous PtRu bimetallic nanoalloys have long-term solvent durability after immersion in water for 16 months. In addition, Cai et al. have prepared PtRu/C by using a dimethylamine borane (DMAB)-based aqueous phase process.46 The as-prepared PtRu/C exhibits superior MOR performance and output power density due to the enhanced CO resistance and CO2 generation. Importantly, in situ ATR-IR spectroscopy was used to corroborate the improved MOR performance and output power density at a molecular level. Based on the above, PtRu bimetallic alloys have been considered as one of the most promising catalysts in PEMFCs. In the bifunctional mechanism, Pt can provide the active sites, and the neighbouring Ru can activate water and produce reactive oxides for the oxidative removal of COad adjacent to the Pt active sites. Interestingly, the well-defined facets enclosed PtRu nanocrystals exhibited the different CO-poisoning tolerance. Dong et al. have synthesized ultrathin PtRu nanocrystals with tuneable facets by a one-step wet chemical approach.47 They found that the (111)-terminated PtRu nanocrystals showed much higher mass activity toward MOR, which was 2.28 times higher than (100)-terminated PtRu nanocrystals, indicating the superior CO tolerance of PtRu (111) facets relative to (100) facets.
:Ru and unique nanostructures, the specific activity and mass activity of Pt72Ru28 nanoalloys can reach 10.98 mA cm−2 and 1.70 A mgPt−1 for methanol oxidation reaction (MOR), respectively. The enhanced oxidizing ability of Pt72Ru28 nanoalloys toward CO, HCOH, and HCOOH intermediates significantly makes them exhibit long-term durability for MOR. Another good example involves the fabrication of PtRu bimetallic NPs with adjustable alloying degree via a co-reduction method.45 Based on in situ attenuated total reflection infrared (ATR-IR) spectroscopy, PtRu/PC–H can easily cleave C–H bond and accelerate the oxidative removal of COad at a lower potential, indicating the enhanced CO tolerance. Therefore, porous PtRu bimetallic nanoalloys have long-term solvent durability after immersion in water for 16 months. In addition, Cai et al. have prepared PtRu/C by using a dimethylamine borane (DMAB)-based aqueous phase process.46 The as-prepared PtRu/C exhibits superior MOR performance and output power density due to the enhanced CO resistance and CO2 generation. Importantly, in situ ATR-IR spectroscopy was used to corroborate the improved MOR performance and output power density at a molecular level. Based on the above, PtRu bimetallic alloys have been considered as one of the most promising catalysts in PEMFCs. In the bifunctional mechanism, Pt can provide the active sites, and the neighbouring Ru can activate water and produce reactive oxides for the oxidative removal of COad adjacent to the Pt active sites. Interestingly, the well-defined facets enclosed PtRu nanocrystals exhibited the different CO-poisoning tolerance. Dong et al. have synthesized ultrathin PtRu nanocrystals with tuneable facets by a one-step wet chemical approach.47 They found that the (111)-terminated PtRu nanocrystals showed much higher mass activity toward MOR, which was 2.28 times higher than (100)-terminated PtRu nanocrystals, indicating the superior CO tolerance of PtRu (111) facets relative to (100) facets.
In addition to the corporation of noble metal elements, many transition metal elements, such as Cu, Co, Ni, Fe, and Zn, have been employed to alloy Pt for improving the CO tolerance of Pt-based binary alloys.17,48,49 For example, ultrastable self-supported PtCu nanowires with abundant Cu-vacancies were prepared by combining a co-reduction method and an electrochemical dealloying strategy (Fig. 3a).50 A series of CO poisoning measurements and DFT results proved that the optimized PtCu (111) with Cu-vacancy defects could effectively weaken the adsorption of CO and simultaneously enhance the adsorption of OHad, resulting in outstanding MOR performance (Fig. 3b–g). In addition, Yang et al. have successfully prepared Pt–Co nanoframes by etching Co from solid rhombic dodecahedra.51 The MOR mass activity of Pt–Co nanoframes can reach 4.28 A mgPt−1 in alkaline media, which is 4 times higher than that of commercial Pt/C. The enhanced MOR performance can be attributed to the robust surface and optimized binding strength of CO. Similarly, PtNi colloidal nanocrystal clusters (CNCs) assembled by many PtNi nanocrystals were synthesized by a simple one-pot solvothermal method.52 The corporation of Ni could adjust the catalytic structure and downshift the d-band center, which contributes to weakening the CO adsorption and improving MOR performance.
 | ||
| Fig. 3 (a) Schematic illustration of the surface structure evolution of d-Pt2Cu1 NWs with the electrochemical dealloying process; (b–d) electronic structure analyses of Pt(111), Pt2Cu1(111), and Pt2Cu1(111) with Cu-vacancy surfaces. Models for the adsorption of CO (left sides of (b–d)). Charge density differences of CO (right sides of (b–d)). The yellow and blue shadows represent charge accumulation and depletion in the space, respectively. Calculated projected density of states (PDOSs) of coupling CO (below sides of (b–d)) on different surfaces. (e) CO stripping performed at a scan rate of 20 mV s−1 in a N2-saturated 0.1 M HClO4 solution. (f) MOR CVs normalized to the Pt mass recorded in a N2-saturated 0.1 M HClO4 + 0.5 M CH3OH solution at 50 mV s−1; the inset CV curves show the MOR histograms of the onset potential. (g) Chronoamperometric (CA) measurements at 0.7 V vs. RHE for 3600 s. Reproduced with permission from ref. 50. Copyright 2021, American Chemical Society. | ||
Bi as the representative main group element has been employed to alloy Pt for improving the CO tolerance of Pt. The incorporation of Bi can regulate the electronic structure of Pt and easily generate metal oxides on the surface of Pt-based alloys, which can promote the bifunctional mechanism. Based on the previous reports, considerable studies mainly focused on the addition of Bi to alloy Pt, which could enable PtBi alloys with high tolerance to CO poisoning. Based on this guideline, PtBi–Pt core–shell nanoplates with controlled composition and structure have been synthesized by using a simple solution-phase reaction.53 The incorporation of Bi species not only accelerates the formation of ordered intermetallics but also downshifts the d-band center, thereby resulting the enhanced MOR activity and stability. Moreover, the core–shell structure contributes to the strain effect due to lattice mismatch and contraction between Pt and Bi, further promoting the MOR performance. Similarly, Guo et al. have developed a new class of 2D nanoplates by a facile one-pot approach, which is composed of an intermetallic PtBi core and ultrathin Pt shell.54 Thanks to the strain effect, the as-prepared catalysts also exhibit enhanced MOR performance in both acid and alkaline electrolytes. In addition, significant studies have proved that Bi can irreversibly adsorb on the Pt electrode. For example, Shin et al. have obtained Bi-modified Pt supported on carbon black by mixing Bi2O3, 40 wt% commercial Pt/C and 0.5 M H2SO4.55 Bi is irreversibly adsorbed on Pt/C, and the amount of Bi can be tuned by the concentration of Bi3+ solution. Therefore, the current density of Bi-modified Pt/C for formic acid oxidation reaction (FAOR) is 13 times higher than that of Pt/C. The enhanced FAOR activity can be attributed to the fact that the adsorbed Bi works as a third body, making Pt ensembles and thus significantly decreasing CO intermediate’ generation. Another interesting finding is that porous Pt nanoframes decorated with Bi(OH)3 have been fabricated by a two-step method. On the one hand, incorporating Bi species could effectively tune the geometric structure and downshift the Pt d-band center. On the other hand, the decoration of Bi(OH)3 can favor the oxidative removal of CO by using the formation of OHad species.56 In general, the decoration of Bi species could greatly enhance the electrocatalytic performance of Pt by geometrically separating adjacent Pt surface sites and adsorbing Bi adatoms on the Pt surface.
In order to further enhance the utilization of Pt atoms, accelerate the electron and mass transfer, and expose more active sites, many efforts have been paid attention to the design and fabrication of various Pt-based nanostructures, such as one-dimensional nanowires, two-dimensional nanoplates, and three-dimensional polyhedra and nanoframes.23,57–59 For example, Yang and co-authors have developed bimetallic Pt95Co5 nanowires by a one-pot surfactant-free method.60 The formation of one-dimensional nanowires could not only reduce the cost but also contribute to the superior electrochemical properties due to the modification of Co, which can improve the tolerance to carbonaceous species, such as COad, CHOad, and COOHad intermediates. Moreover, Tang et al. employed a facile one-pot hydrothermal method to prepare porous Pt3Cu nanobowls that are assembled by ultrafine NPs (≈2.9 nm).61 Interestingly, N,N′ -methylenebisacrylamide works as a crucial structure-directing agent, which plays an important role in the deposition and growth of well-defined bowl-shaped nanostructures. Therefore, the self-supported Pt3Cu nanobowls show significant MOR activity and anti-poisoning capability. A series of characterizations and measurements further confirm that highly open geometric configuration, the number of the energetic low-coordinated sites and compressive lattice strain contribute to the high utilization efficiency of Pt and induce the compressive lattice strain, thus tuning the Pt d-band center. In addition, TEM image of Pt3Cu nanobowls after the stability test shows the perfect maintenance of their initial bowl-like nanostructure, illustrating the bowl-like nanostructure could not only reduce the Pt usage but also contribute to the superior CO tolerance.
In addition to the above-mentioned strategies, there is an important approach to improve the Pt utilization and its anti-poisoning properties, which involves the design of point defects (atomic vacancies) and surface defects (grain boundaries). That is because the generated defects could regulate the surface electronic structure of Pt and contribute greatly to improving their adsorption capacity for CO intermediate.62,63 For example, porous PtAg nanoflowers have been successfully prepared by combining liquid reduction and chemical etching method. Wang et al. first synthesized different element proportions of PtAg alloys by adjusting the feeding ratios of Pt and Ag precursors, resulting in uneven element distribution.64 Next, a chemical etching method was used to corrode silver by the addition of nitric acid, finally leading to the formation of porous PtAg nanoflowers. More importantly, the molar ratio of Pt:Ag can greatly affect the size and morphology of porous PtAg nanoflowers. Further chemical etching method determines the forming process of porous crystal (Fig. 4a). Therefore, porous PtAg nanoflowers manifest a mass activity of 1136 mA mgPt−1, which is 2.6 times higher than that of commercial Pt/C. Both CO-stripping voltammograms and forward/reverse peak current density ratio exhibit high anti-CO poisoning capability due to the incorporation of Ag, which could alter the Pt electronic structure and promote the oxidative removal of CO species (Fig. 4b). Therefore, it is a very effective strategy to adjust the adsorption/desorption properties of CO intermediate by fabricating porous nanostructures with abundant surface defects. Another impressive sample focused on the fabrication of bimetallic PtBi nanorings with inhomogeneous tensile strain and abundant low coordination atoms.65 PtBi nanoplates were first prepared by a facile solvothermal method, and the porous nanoplates were generated by a self-etching effect with Br− and O2 pairs as etching agents. The mass activity and specific activity of PtBi nanorings for MOR can reach 6.42 A mg−1 and 11.93 mA cm−2, respectively. Both CO stripping measurements and DFT results have demonstrated that the improved MOR performance of PtBi NRs could be ascribed to the tensile strain and ligand effect, which arise from the well-defined crystal structure and modification of Pt electronic structure owning to the doping of Bi (Fig. 4c–g). In addition, the morphology of the PtBi NRs were well maintained after the CA test, indicating their excellent structural stability.
 | ||
| Fig. 4 (a) The schematic diagram of the synthesis of E-PtAg NFs; (b) CO-stripping voltammograms in 0.1 M HClO4 solution at a scan rate of 50 mV s−1 for E-Pt1Ag2 NFs/C, E-Pt1Ag3 NFs/C, and commercial Pt/C, respectively; reproduced with permission from ref. 64. Copyright 2022, Wiley-VCH. (c) Schematic illustration of the formation mechanism of PtBi NRs; (d–f) HAADF-STEM images of time-dependent products obtained at various reaction time; (g) free energy diagram for MOR on Pt, PtBi, and PtBi ε = 4% surfaces. Reproduced with permission from ref. 65. Copyright 2022, Wiley-VCH. | ||
Except for bimetallic Pt-based alloys, the single-atom alloy (SAA) strategy could significantly reduce the amount of Pt loading and promote the catalytic performance by effectively decreasing the binding strength of CO.22,66 Therefore, one of the biggest advantages of SAA catalysts is that each Pt atom is highly exposed on the surface of electrocatalysts, which can improve the Pt utilization rate and make them highly active catalysts for electrochemical reactions. To promote the fast conversion of CO to CO2 at low temperature in fuel cells, Flytzani-Stephanopoulos and co-works have systematically investigated the interaction between CO and different Pt ensembles on Pt–Cu SAA by using a variety of surface science and catalytic techniques.24 They found that CO is weakly adsorbed on single Pt atoms, while both the larger Pt ensembles and monometallic Pt exhibit stronger adsorption capacity for CO. As a result, more CO-free Pt sites are available on the SAA electrode compared to larger Pt ensembles and monometallic Pt, thereby contributing to obtaining higher hydrogenation and hydrogen electro-oxidation activity. In addition, single Pt atoms dispersed on Ru NPs have the advantage of providing OHad species based on the bifunctional mechanism, which can easily remove the CO intermediate from the active sites. For example, Tilley et al. successfully synthesized single Pt atoms on the surface of Ru by the slow reduction of a Pt(II) precursor with oleylamine. More importantly, they employ a high temperature treatment to drive the rearrangement of Pt islands on the branched Ru, resulting in the formation of discrete Pt atoms on the Ru electrode.67 Therefore, the forming OHad species adjacent to the active sites of Ru can easily remove the CO intermediate when CO molecules adsorb on the Pt atoms, thus leading to improved MOR activity and stability. DFT calculations further confirm that the structural properties of single Pt atoms could strongly promote the adsorption of CH3OH and reduce the adsorption strength of CO intermediate, thereby limiting their CO poisoning and improving the MOR performance.
In summary, alloying Pt with a second element (Pd, Zn, Co, Cu, Au etc.) has considered as one of the most effective ways to tune its geometric structure, electronic structure and interactions with reaction intermediates.68 Importantly, the second element and special nanostructures can induce synergistic effects to abundant active sites, high mass utilization, and fast mass transfer and weaken the affinity of Pt to the poisonous species. Despite the success, the second element is easily oxidized and dissolved, thus leading to unstable nanostructures, which could reduce the CO tolerance of Pt-based binary alloys. As a result, optimizing the amount and species of the second element is especially important for the anti-CO poisoning.
2.2 Pt-based multi-element alloys
Based on the previous reports, the increased activity and stability of Pt-based electrocatalysts by alloying Pt with transition metals could be attributed to downshifting the Pt d-band center, thus optimizing the adsorption of CO intermediate. Therefore, the catalytic activity and stability of Pt-based electrocatalysts can be further enhanced by the synergistic interaction between different active components, which have many advantages for the design of Pt-based multi-element alloys.69–72 First, the incorporation of less-expensive metals could greatly reduce the usage of Pt. Second, the multi-element properties can effectively provide multiple sites to adsorb different intermediates, respectively. Third, the electronic structures of Pt active sites can be finely adjusted by the surrounding atoms. For example, Xie and Lin et al. have successfully prepared trimetallic PtNiCo branched nanocages by etching Ni-rich cores of PtNiCo nanomultipods (Fig. 5a).73 The mass activity of trimetallic PtNiCo nanocages is 2.8 A mgPt−1 in acidic medium, which is much higher than that of commercial Pt/C. According to in situ FTIR spectra, trimetallic PtNiCo nanocages show the weaker interaction of Pt-COad binding strength. As a result, the alloying effects of Co and Ni could optimize the Pt d-band center and improve the CO tolerance of Pt, leading to the enhanced MOR performance on the trimetallic PtNiCo nanocages. (Fig. 5b and c). MOR durability tests were conducted by CA tests for 3600 s. PtNiCo nanocages possessed the highest mass activity compared to PtNi/C or Pt/C even after the durability testing, demonstrating the enhanced CO tolerance is beneficial to improve the catalytic stability of PtNiCo nanocages. | ||
| Fig. 5 (a) SEM image of trimetallic PtNiCo nanomultipods with a core–shell structure; (b) comparison of specific activity and mass activity at 0.9 V (vs. RHE); (c) the oxygen reduction reaction (ORR) specific activity for catalysts associated with the shift in d-band center vs. Pt/C; reproduced with permission from ref. 73. Copyright 2021, Royal Society of Chemistry. (d) The synthetic schematic diagram of Au@Pt–Pd H-Ss; (e) SEM image of the Au@Pt–Pd H-Ss; (f) schematic diagram of the growth mechanism of Au@Pt–Pd. Reproduced with permission from ref. 81. Copyright 2021, Wiley-VCH. | ||
The rational design of ternary Pt-based electrocatalysts is of vital significance for the practical applications of fuel cells. Tian and his co-authors have developed a series of two-dimensional ternary PtBiM nanoplates (M = Co, Ni, Cu, Zn, Sn) via a facile solvothermal method.74 Both MOR activity and durability exhibit significant improvement with the incorporation of metal compositions. Notably, Pt3Bi3Zn nanoplates exhibited a much slower decay than that of the other catalysts after one thousand cycles, which further indicated a significant improvement in the stability. The prominent performance can be attributed to the optimized electronic structure of Pt and generated OHad species, which will result in weak adsorption between Pt and CO and the oxidative removal of CO. Up to now, single-atom tailoring metal alloys have been extensively used to maximize the Pt active center because of its unsaturated coordinative environment and unique electronic structure, which benefit the oxidative removal of CO. For instance, Sun and his co-authors have developed PtNi NPs with the modification of Ru single atoms by combining chemical etching and a selective atomic layer deposition (ALD) technique.75 Impressively, the mass activity of Ru-ca-PtNi hybrid catalysts can reach 2.01 A mg−1 for MOR, and only 16% activity loss of its initial activity can be observed during the tests, indicating a significant durability. DFT calculation and FTIR analysis prove that the confined Ru atoms in cavities could efficiently promote the removal of CO by tuning the Pt d-band center. On the basis of ternary alloys, the addition of special functional metal has been believed to make the ternary alloys have the corresponding functions. Based on the guideline, Xie et al. have fabricated 1.5 nm-thin quarter metallic PtCoNiRh nanowires with high atomic-exposure by using an oleylamine system.76 PtCoNi nanowires were used as a control, and Rh as a highly anticorrosive metal was used to construct Pt–Rh sites on the surface of quatermetallic PtCoNiRh nanowires. Compared to PtCoNi nanowires, the mass activity and specific activity of PtCoNiRh nanowires show the significant improvements in MOR. Moreover, the onset potential of MOR is distinctly negative, and the prominent CO tolerance also can be observed on the PtCoNiRh nanowires. Both in situ FTIR study and DFT calculation have proved that the incorporation of Rh could change the CO binding from linear mode to bridge mode, and Pt–Rh sites belong to the thermodynamical sites for adsorption and dehydrogenation of CH3OH. It is generally accepted that the enhancement of catalytic activity stems from the lattice strain caused by the lattice mismatch between Pt and other metals, which can weaken the interaction between Pt atoms and the adsorbed species, and thus release more active sites on the surface. A variety of elements doped in Pt lattice can cause lattice strain due to their different lattice and directly affect the local coordination environment mainly by regulating the non-uniform elements, thus greatly changing the adsorption properties of nanostructures.
Remarkably, many studies have focused on the design of the structure and morphology of Pt-based multi-element alloys based on the incorporation of three or even more elements, especially core–shell structures, ordered alloys, graded interfacial nanolayers, and open-structure nanoframes.77–79 Among the unique catalytic structure, fabricating shape-controlled core–shell structures with functional Pt surfaces could provide an effective routine to improve the catalytic properties of Pt-based alloys, which will involve the synergistic effect of multi-component alloys. However, how to develop a facile approach is the critical factor for the fabrication of multi-component core–shell structures, which usually include the shape control of metallic core and thickness control of Pt-based shell. Based on the above, Fang et al. have prepared octahedral CuNi@Pt–Cu core–shell nanocrystals.80 In the core–shell structure, the formation of Pt-based shell should precisely control the experimental parameters, including Pt precursor/capping ligand, temperature and heating rate. The resultant CuNi@Pt–Cu catalysts exhibit superior MOR activity compared to commercial Pt/C. That is because that the onset potential of CO electrooxidation peaks on the CuNi@Pt–Cu electrode is more negative than that on the commercial Pt/C, indicating a weaker CO adsorption affinity. Similarly, Jiang et al. have successfully fabricated anisotropic Au@Pt–Pd hemispherical nanostructures, in which Au NPs are the core, and Pt–Pd NPs are the shell (Fig. 5d and e).81 In the synthetic process, BO2− with ultra-low concentration will result in their non-uniform adsorption on Au seed surface. Therefore, both the non-uniform adsorption of BO2− and rapid growth of Pt–Pd atoms promote the formation of anisotropic hemispherical nanostructures, as shown in (Fig. 5f). Compared to commercial Pt/C, Au@Pt–Pd hemispherical nanostructures manifest superior electrocatalytic activity and durability for alcohols oxidational and FAOR. The great improvement could be attributed to the existence of surface defects and unsaturated coordination environment, which can weaken the adsorption of CO, thus improving their catalytic performance. It is well known that small organic molecules oxidation reaction at the anode belongs to a surface structure-sensitive reaction, and thus the surface crystal structure of Pt-based electrocatalysts can determine their activity and selectivity. For instance, Pt (110) facets can effectively promote the cleavage of C–C bond in acidic solution.82 In addition to the surface crystal structure and compositions, strain engineering can tune the Pt electronic structure and adsorption energy of CO intermediate. Based on the theoretical guidance, Cui et al. have constructed a class of core–shell hexagonal nanoplates (PtPd@PtIr1 HNPs) enclosed by Pt (110) facets with a 7.2% tensile strain. PtPd@PtIr1 HNPs show a top-ranked activity and a Faraday efficiency of 57.93% for C1 pathway for ethanol oxidation reaction (EOR).83 DFT results confirm that the strong synergy between Ir1 sites and tensile strain contributes to breaking the C–C bond of CH2CO* and facilitating oxidative removal of CO. As a result, it can be seen that shape-controlled multi-element nanocrystals with low-coordination active sites could effectively tune their catalytic properties.
2.3 Pt-based high-entropy alloys
It is well known that alloying Pt strategy with relatively small amounts of secondary elements has long been used to confer desirable properties to Pt-based alloys. A novel alloying strategy has been developed to combine multiple elements in high concentrations to generate high-entropy alloys (HEAs). In general, HEAs contain five or more metal elements with similar atomic ratios, and each component has a different atomic size, thereby easily leading to lattice distortion. Compared to conventional alloys, major effects of HEAs include high entropy, lattice distortion, slow diffusion, synergic effect, and high organizational stability.84–86 Therefore, HEAs with unique structural properties and a significant high-entropy effect will break through the bottleneck of electrochemical catalytic materials in fuel cells.In the past few years, HEAs as catalytic materials have arisen extensive interest owing to their unique characteristics. Moreover, the catalytic properties of HEAs can be tuned by adjusting their compositions and phase structure. Specially, for the uniform and ultrasmall (∼3.4 nm) HEAs, their dissimilar element interaction could exhibit novel catalytic properties. For example, Wang and co-authors have prepared the uniform and ultrasmall Pt18Ni26Fe15Co14Cu27 HEAs by a simple low-temperature oil phase strategy.87 The transition metal elements (Ni, Fe, Co, and Cu) are selected because they are in high abundance and can easily form solid solutions with Pt. Therefore, Pt18Ni26Fe15Co14Cu27 HEAs show the mass activity of 15.04 A mgPt−1 for MOR, and the If/Ib (If represents the forward current density, and Ib represents the reverse current density) ratio of Pt18Ni26Fe15Co14Cu27 HEAs (3.26) is much higher than that of commercial Pt/C (2.31), indicating the superior CO tolerance of Pt18Ni26Fe15Co14Cu27 HEAs. DFT results have proved that the active sites on the HEA surfaces exhibit very varied adsorption preferences for CO intermediate. Therefore, a larger energy barrier of 0.81 eV and activation energy (0.94 eV) are obtained for the formation of CO, resulting in suppressing the CO poisoning. Compared to conventional alloys with typically disordered nanocrystals, the intermetallics possess highly ordered structures, which can effectively clarify the structure–property relationship in practical applications. Therefore, how to combine the advantages of intermetallics and HEAs have become promising candidate as multifunctional electrocatalysts. Based on the design, Quan group has successfully synthesized PtRhBiSnSb HEAs with hexagonal close-packed structure by a facile wet-chemistry method, in which the formation of PtBi-type (PtRh)(BiSnSb) arises from the substitution of Sn/Sb atoms at the Bi position and Rh atoms at the Pt position (Fig. 6a).88 The as-obtained PtBi-type HEAs exhibit the advantages of HEAs and intermetallics, thereby demonstrating the isolated and combined multimetallic elements, stronger interatomic interaction, enlarged Pt–Pt distance, and better etching resistance. As a result, PtRhBiSnSb HEAs show not only ultra-high mass activities in MOR, EOR and GOR (Fig. 6c) but also long-term stability. The oxidation peaks of COad at 0.7 V vs. RHE are significantly suppressed on the PtRhBiSnSb and PtBiSnSb electrodes compared to the Pt/C, which can be attributed to the isolated Pt sites on their surface without contiguous Pt active sites and enlarged Pt–Pt distance. DFT calculation has further proved that COad species have been significantly inhibited at 0.7 V owning to the optimal electronic structure of Pt and enlarged Pt–Pt distance, resulting in stronger selectivity and durability for the CO2 reaction pathways (Fig. 6b). In addition, exposure and modification of oxophilic Bi/Sn/Sb species also indirectly weaken the CO poisoning on PtBi-type HEAs (Fig. 6d–g). Impressively, the hexagonal shape and multimetallic compositions of PtRhBiSnSb HEAs can be basically maintained, showing their excellent structural and compositional stability. As we known, Pt-group metals (PGMs) contain six neighbouring elements in the periodic table, and each PGM could efficiently accelerate catalytic reactions. Therefore, alloying Pt with PGMs will promote the formation of Pt-based HEAs with an excellent structure for complex or multistep reactions, which involve many reactants or intermediates. For example, Kitagawa and his co-authors have developed Pt-based HEAs with all six PGMs (denoted as PGM-HEAs) by using facile chemical synthesis.89 They found that PGM-HEAs show the record-high activity and stability for EOR. The enhanced EOR performance can be attributed to alloying six elements together, which might provide an optimal surface for the adsorption/desorption of reactants or CO intermediate.
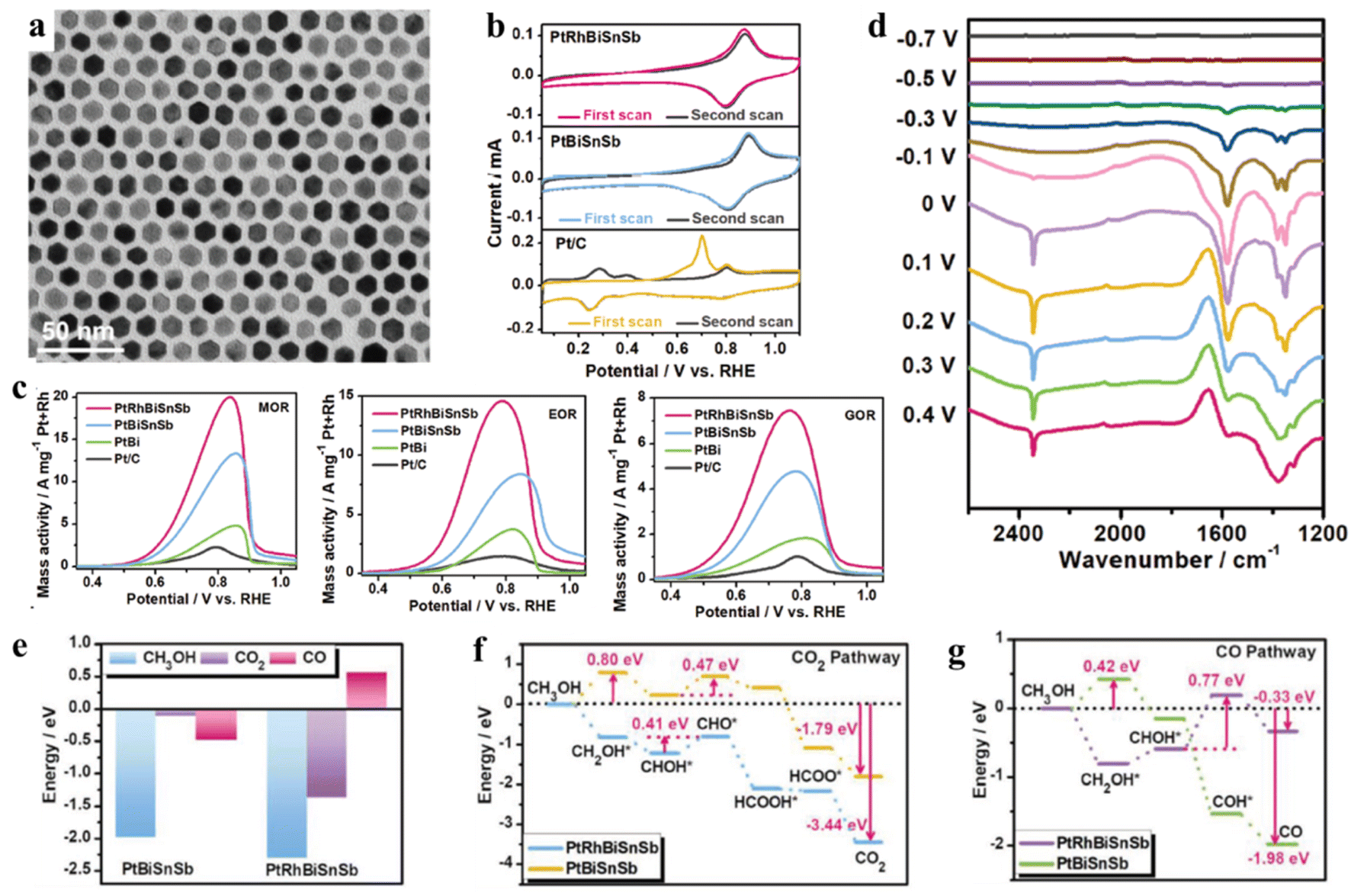 | ||
| Fig. 6 (a) TEM image; (b) CO-stripping curves of PtRhBiSnSb HEI nanoplates, PtBiSnSb nanoplates, and Pt/C catalysts recorded in Ar-saturated 1.0 M KOH at a scan rate of 50 mV s−1; (c) MOR, EOR and GOR positive-going polarization curves of different catalysts recorded at a scan rate of 50 mV s−1; (d) the in situ FTIR spectra of PtRhBiSnSb HEI nanoplates recorded in Ar-saturated 1.0 M KOH + 1.0 M CH3OH solution using a Hg/HgO as the reference electrode; (e) the adsorption energy comparison of CH3OH, CO2, and CO on PtRhBiSnSb HEI nanoplates and PtBiSnSb nanoplates. (f and g) The reaction energy comparison of CO2 pathway (g) and CO pathway (h) for MOR process on PtRhBiSnSb HEI nanoplates and PtBiSnSb nanoplates. Reproduced with permission from ref. 88. Copyright 2022, Wiley-VCH. | ||
The above results indicate that Pt-based HEAs formed by incorporating different metal elements can effectively improve their catalytic performance and anti-CO poisoning pathway. However, the design of Pt-based HEAs don't focus on the precise control of elemental component and species at the atomic level, including the modification of different ligands, multiatomic sites and nanoclusters with atomic precision and single atom deposition. Some innovative synthesis strategies are still needed to further develop various advanced Pt-based HEAs. In addition, it is greatly need to investigate the synergy in Pt and other added elements due to the lack information at the atomic level. More importantly, it is crucial process to optimize the design of current reactors and systems for practical applications. Except for the specific shortcomings of the current synthetic Pt-based HEAs method, it is still urgent to investigate their catalytic mechanism in the reaction process.
3. Pt-based composites
According to the above discussion, it is feasible to alloy Pt with metal elements to tune the adsorption/desorption of CO on the Pt-based electrocatalysts. In addition to the alloying strategy, fabricating Pt with carbon-based materials or metal oxides have been considered as one of the most effective way to improve catalytic performance and promote the wide applications of Pt-based electrocatalysts in the field of catalysis.28,52,90 In the composites, the synergy between Pt and carriers is believed to mainly contribute to the enhanced performance due to the optimal electronic structure of Pt and the formation of OHad species adjacent the Pt active sites, which can be determined by both selecting the optimized carriers and constructing suitable interface structure.91 The former one could weaken the binding strength of CO owning to the formation of electron-rich structure on the Pt electrode, and latter one is essential for the complete oxidation of CO intermediate.92,93 In addition, appropriate carriers can favor the better dispersion of Pt on their surface, which can promote electron transfer between Pt atoms and carriers, thus receiving strong binding of single Pt and positively charged Pt atoms. Therefore, the fabrication of Pt-based composites provides an important opportunity to understand the catalytic process and mechanism and explores the relationship between structure–activity-performance and CO intermediate.94–963.1 Carbon-supported Pt-based electrocatalysts
To improve the utilization and catalytic performance of Pt, carbon-based materials with high surface area have been considered as the promising support. That is because carbon-based materials with a large specific surface area could benefit the dispersion of Pt NPs and lower their cost.97,98 Many carbon-based materials, such as graphene, carbon nanotubes (CNTs), carbon black (CB), and carbon clothes etc., have received increasing attention because of their intrinsic properties, such as a large surface area, porous structure and good electric conductivity. As a very valuable catalyst support, graphene materials with special electronic structure can result in uneven distribution of surface charge, which can promote the formation of a large number of active sites. In addition, their conjugate structure and cavity structure make them possess excellent transport performance and semiconductor properties. Therefore, the accurate design and manufacture of novel graphene-supported Pt catalysts with precise atomic compositions and efficient morphology control have extensive practical prospects.For example, Li et al. synthesized single-nickel-atom-alloyed platinum hexagonal nanocrystals (NiPtSAA/GDY) by a simple electrochemical deposition strategy, which induces the porous GDY hexagonal ring by the domain-limiting effect (Fig. 7a).99 Both the blue and red lines represent the lattice spacing of Pt hexagonal nanocrystals (0.230 nm, HPtNs/GDY, Fig. 7b). The green circle in Fig. 7c represents the Ni atoms, and different lines represent the lattice spacing of NiPtSAA/GDY (0.212 nm). Notably, the GDY can easily tune the size and morphology of Pt NPs, and alloying a single Ni atom with Pt NPs will promote their lattice distortion, thus leading to reducing the reaction kinetic barrier. Therefore, the low valence of Pt in NiPtSAA/GDY shows a high resistance to CO poisoning and enhanced MOR performance. Similarly, Sun et al. have proposed a facile treatment strategy for controlling PtRu/PC-H nanocrystals with high alloying temperature.100 First, a simple hydrogen co-reduction strategy was performed at 300 °C, and then homogeneous PtRu NPs on porous graphitic carbon surfaces were obtained by high temperature-treatment at 700 °C. In situ FTIR spectroscopy shows the appearance of a CO2 signal (2345 cm−1) at −150 mV on PtRu-PC-H, which is much lower than that of PtRu/PC-L and Pt/PC, indicating the decreased CO oxidation potential. As an effective carrier, another important carbon-based material is carbon nanotubes, which possess a one-dimensional hollow tubular structure, and their properties mainly depend on the curled way of the graphene sheet, which will determine the metal properties or semiconductor properties. At the same time, as an excellent catalytic carrier, carbon nanotubes also have a great specific surface, stable chemical properties, excellent mechanical properties, and unique pore structure. Based on the above, Hou et al. have developed a facile strategy to synthesize a free-standing 3D interconnected embossed graphene (e-RGO)-single wall carbon nanotubes (SWCNTs) paper supported Pt NPs (Pt/e-RGO-SWCNT). SWCNTs were assembled to insert through the film to promote the penetration of electrolyte into the inner structure of three-dimensional porous electrode.101 Benefiting from the structural properties, Pt/e-RGO-SWCNT exhibits a current density of 191.7 mA mg−1, demonstrating excellent MOR performance. The generation of OHad species promotes the oxidative removal of CO on the Pt/e-RGO-SWCNT, thus contributing to the improved MOR performance (Fig. 7d–f). Moreover, a bottom-up approach has been developed to prepare ultrafine Pt NPs dispersed on three-dimensional low-defect carbon nanotube/nitrogen-doped graphene hybrid aerogel (Pt/LDCNT-NG).102 The main synthetic process contains two steps: (1) the self-assembly of three-dimensional LDCNT-NG aerogels and dispersion of LDCNT on GO surface via π–π supramolecular interaction; (2) controllable growth of Pt NPs on the LDCNT-NG backbones. Therefore, Pt/LDCNT-NG manifests high mass activity and reliable long-term stability. They found that N dopants can accelerate the formation of a large number of OHad species, thereby removing CO-like poisoning intermediates. In general, increasing attention has been paid to the interaction between Pt and carbon nanotubes, which can determine the forming process, structure, and dispersion of Pt NPs. It is well-known that carbon nanotubes as a carrier can alter the catalytic system's Galvani potential and lower the Fermi level, thus benefiting the electron transfer at the electrode–electrolyte interface. As a result, to obtain highly catalytic catalysts, the effects and interaction mechanism need to be clearly understood.
 | ||
| Fig. 7 (a and b) HAADF-STEM images of HPtNP/GDY; (c) HAADF-STEM images of NiPtSAA/GDY; reproduced with permission from ref. 99. Copyright 2020, Elsevier. (d) SEM images of the cross-section of Pt/e-RGO-SWCNT. (e) The histograms of ECSAs of as-prepared samples. (f) CVs of Pt/e-RGO-SWCNT catalyst toward MOR in 0.5 M H2SO4 + 1.0 M CH3OH at 50 mV s−1 with different deformed states: as-prepared, folded, and rolled. Note that the rolled film was immobilized on insulating plastic substrates. Reproduced with permission from ref. 94. Copyright 2019, Elsevier. (g) DFT-computed permeation energy barriers for H2 and CO gases vs. Kav of the different types of defective graphene. (h) Correlation between particle diameter and EMSA of PtRu@C/C catalysts including the available volume fraction (Kav = available volume/total volume) values estimated from DFT calculations. (i) CO tolerance factor of the corresponding catalysts calculated from the CA currents in H2/CO gas mixture (100 ppm CO) and pure H2 gas atmospheres, respectively. Reproduced with permission from ref. 106. Copyright 2022, Elsevier. | ||
In recent years, many efforts have focused on the fabrication of carbon-based materials by converting metal–organic frameworks (MOFs) into N-doped structures owing to their superior structural properties, including a large specific surface area, high porosity as well as adjustable pore structures.103 Based on the guideline, Pt-based electrocatalysts on N-doped CNT coated by CoP NPs with high specific surface areas (Pt–CoP-NCZ/CNT) have been developed by in situ growth of CoZn-ZIF on CNT materials. Pt–CoP-NCZ/CNT catalysts possess superior MOR activity and long-term stability in both acid medium and alkaline medium.104 The excellent MOR performance can be mainly attributed to the N-doped structure and the introduction of Zn2+. The former benefits the assembly of Pt NPs, and the latter can suppress the Co agglomeration, thereby reducing CO poisoning. Similarly, Hu et al. have developed an in situ reduction-fusion method to synthesize different Pt–Co nanostructures embedded on porous N-doped carbon carriers (PtCo@NCs).105 A pyrolysis process of ZnCo-MOF precursor was precisely controlled, in which the metallic Co source serves as the conductive carbon carrier. The PtCo@NCs manifests a mass activity of 2.30 A mgPt−1 for MOR, demonstrating an excellent MOR performance. The CO tolerance of PtCo@NCs has been significantly enhanced owing to the synergistic effect in the Pt–Co sites, which can accelerate the rate-determining step of methanol dehydrogenation and oxidative removal of CO. Encapsulating Pt NPs with carbon-based materials have been considered as one of the most promising strategies for inhibiting CO poisoning. Meanwhile, the CA test results showed that PtCo@NCs with precisely controlled nanostructures possessed the optimized durability compared to other PtCo catalysts and commercial Pt/C. Moreover, Jung et al. have fabricated PtRu NPs by using a molecular sieve effect, in which PtRu NPs were encapsulated by carbon shells with controlled pore thickness and size.106 The carbon shell plays an important role in the hybrid system. On the one hand, carbon shell could efficiently promote the hydrogen oxidation reaction (HOR) by selectively permeating H2 gas, thus decreasing CO poisoning effect (Fig. 7g). On the other hand, carbon shell also can effectively prevent the Ru dissolution, and the ligand effect by alloying Pt NPs with Ru was still operative. Therefore, carbon shell-encapsulated PtRu NPs show the improved activity and stability (Fig. 7h and i).
Although the carbon carriers or carbon layers have achieved great progress in anti-CO poisoning, carbon-based materials are easily oxidized into CO or CO2 in the electrochemical reactions. Then, the oxidized carbon carriers could cause the aggregation of Pt electrocatalysts. More seriously, CO produced from carbon-based materials will further accelerate the poisoning of Pt. To solve the above issues, increasing attention has been paid to investigating the mixture of carbon and semiconductor carriers. For example, Yang and his co-authors successfully synthesized the atomic carbon layers on the surface of SiC NPs by a facile dry-etching approach, and Pt NPs were then anchored on their surface.22 A porous carbon layer with an enlarged surface area can be generated by selectively removing silica atoms from the surface of SiC NPs. In addition, the etching time can determine the thickness of atomic carbon layers. Significantly, Pt/SiC catalysts show high MOR activity, long-term stability and enhanced anti-CO poisoning ability.107 DFT calculation and XAFS measurements were used to clarify the catalytic mechanism. They found that the reduction of CO adsorption energy and enhanced amount of OHad species on the atomic carbon layers promote the high MOR performance. As a result, this novel catalyst can achieve the integration of maximum MOR performance with minimized CO positioning.
Besides the success of fabricating carbon-based carriers, there are still challenges and urgent needs for developing advanced carbon-based materials: (1) Pt-based nanocrystals were generally physically mixed with carbon-based materials or deposited on the carriers, which leads to the random distribution between Pt NPs and carriers. (2) Carbon-based materials are prone to the structural changes during reaction process, such as corrosion and agglomeration, which is not beneficial for the catalytic stability. Future works need to focus on the structural stability of carbon-based materials. (3) The big electrolyte–electrode area can increase the side reactions.
3.2 Metal oxides or hydroxides supported Pt-based electrocatalysts
Based on the previous reports, combining Pt NPs with metal oxides has been widely used in practical applications because it can promote high activity and selectivity for significant electrochemical reactions. Therefore, metal oxides or hydroxides as the semiconductor carriers have received more and more attention due to their low cost and strong bonding force with Pt. More importantly, the synergy between Pt and metal oxides can weaken the binding strength of CO due to the formation of OHad and the modified electronic structure of Pt. However, as a semiconductor, the conductivity of metal oxides is very low, and they also cannot be operated in acidic solution. To solve the issues, a facile strategy has been developed to fabricate two-dimensional Pt/SnO2/rGO nanocomposites by a facile photo-assisted reduction method.108 In the work, the photo-excited electrons from SnO2 NPs can promote the reduction of ultrafine Pt NPs with an average size of 1–2 nm on the surface of SnO2. Therefore, Pt/SnO2/rGO nanocomposites manifest higher MOR activity and better long-term stability for MOR compared to Pt/rGO catalysts. With the incorporation of SnO2, the OHad species can be easily generated adjacent to the Pt active sites, thus benefiting the oxidative removal of CO-like species and then release the Pt active sites for further MOR. Similarly, Pt3Sn NPs enriched with Pt3Sn/SnO2 interfaces have been synthesized by using a surface restructuring engineering under H2 and air atmospheres.109 The polyol method was first used to deposit Pt2Sn NPs on nitrogen-doped graphene (NG). Next, SnO2/Pt3Sn interfaces were fabricated through thermal treatment method in a H2 atmosphere, followed by annealing under air atmosphere. The incorporation of Sn could not only tune the Pt electronic structure but also increase the atomic distance of Pt–Pt, thereby lowering the CO affinity on the Pt active sites. Importantly, the ultra-small SnO2 can promote the formation of OHad species at low potentials. Thanks to the unique structure, Pt3Sn@u-SnO2/NG catalysts exhibit superior EOR and MOR activity and excellent stability. Besides, other transition metal oxides, such TiO2, CeO2, and Co3O4, were used as the optimal alternatives due to their strong metal-support interactions (SMSI).110–113 Among these metal oxides, TiO2 carriers could fix Pt NPs on its surface, and tune the d-band center of Pt due to its wide band gap. More importantly, hydrophilic TiO2 carriers are conducive to the formation of OHad in both acid and alkaline media. Based on the above, Yan et al. have fabricated novel porous UV-Pt@TiO2/graphene hybrids by a simple hydrothermal synthesis.114 They found that TiO2 nanorods with the (001) and (110) facets could strengthen their trapping ability for Pt NPs, thus improving their adsorption for methanol molecules and weakening the CO poisoning compared to TiO2 NPs. At the same time, the strong metal-support interaction facilitates the adjustment of surface properties and electronic structure of Pt, thereby demonstrating superior catalytic activity and long-term durability for MOR.Recently, considerable works have proved that sub-nanometre clusters possess better catalytic performance than nanometre-sized crystals. Therefore, combining metal oxides and Pt sub-nanometre clusters has been considered as the promising strategy for the improvement of CO tolerance on the Pt electrode. For example, Zhang et al. have fabricated single Pt atoms that uniformly disperses on a FeOx carriers with a large surface area.110 DFT results confirm that the electron transfer from Pt atoms to FeOx surface promotes the formation of vacant d orbitals. The generated vacant d orbitals not only benefit the strong binding and stability of single Pt atom, but also provide positively charged Pt atoms, leading to reducing the CO adsorption energy and the activation barrier of CO oxidation. As a result, Pt1/FeOx catalyst shows excellent catalytic activity. To investigate the catalytic activity in monatomic catalysts that really comes from the separated sub-atoms, Parkinson et al. have employed atomically resolved scanning probe microscopy, surface-sensitive spectroscopy, and DFT calculation to investigate CO oxidation on a Pt/Fe3O4 electrode.115 They have proved that individual CO molecules can adsorb on a sub-nanocluster, and the oxidation of CO can occur from the metastable (PtCO)2. Generally, ultrathin structure (<2 nm) can benefit the exposure of Pt active sites and high utilization of Pt. Moreover, Mo as an oxophilic metal could effectively alleviate the CO poisoning due to the formation of OHad at lower potential. To combine these two advantages, Guo et al. have reported a series of sub-monolayer YOx/MoOx-surface co-decorated Pt nanowires (YOx/MoOx-Pt NWs) by developing a new strategy (Fig. 8a).116 The forming mechanism of YOx/MoOx-Pt NWs has been investigated systematically by collecting the intermediates during the preparation process. The addition of CTAB plays an important role in the formation of Pt NWs, and carbonyl group decomposed from the Mo(CO)6 and its concentration determine the formation of ultrathin NWs and their uniformity, respectively (Fig. 8b–d). In addition, FTIR spectroscopy confirms that YOx/MoOx improves the anti-CO poisoning ability of YOx/MoOx-Pt NWs. Interestingly, the advanced systems can inhibit the CO poisoning of YOx/MoOx-Pt NWs by a new decoupling process, in which the free energy of COad and COOHad can be decoupled on the YOx/MoOx-Pt NWs due to the high oxygen affinity of Mo and Y (Fig. 8e). The CO onset oxidation potential of 5% MoOx-Pt and 22% YOx/MoOx-Pt shifts negatively compared to commercial Pt/C, indicating the enhanced CO tolerance due to the high affinity of Y and Mo to oxygen. Importantly, YOx/MoOx-Pt NWs can be well preserved after the stability tests, while commercial Pt/C exhibited severe aggregation under the same condition. As a result, it can be seen that both the enhanced CO tolerance and stable nanostructures benefit the excellent catalytic performance.
 | ||
| Fig. 8 (a) Atomic-resolution HAADF-STEM image (inset is the corresponding SAED pattern) of 22% YOx/MoOx-Pt NWs; (b) Pt mass-normalized MOR; (c) linear sweep voltammogram (LSV) curves, (d) the If/Ib ratios derived from the MOR and mass activities after different scan cycles. (e) CO stripping curves of 22% YOx/MoOx-Pt, 5% MoOx-Pt, and commercial Pt/C. Reproduced with permission from ref. 116. Copyright 2021, Wiley-VCH. | ||
The oxophilic metals, such as Bi, Pb, Ru, and Rh, could effectively alleviate the CO poisoning due to the formation of OHad at lower potential.117–120 In particular, some of them (e.g. Bi) can interact with Pt to form intermetallic compounds with long-range ordered nature and stable compositions. For example, hexagonal PtBi intermetallic nanoplates with the modification of trace Pb atoms (PtBi@6.7%Pb) via a facile wet-chemistry method (Fig. 9).120 DFT results confirm that Pb oxyhydroxide on the surface can not only promote the electron transfer but also inhibit the CO poisoning. Therefore, the mass activity of PtBi@6.7%Pb can reach 51.07 A mgPt−1 with excellent durability and high CO tolerance in the alkaline electrolyte. At the same time, the p–d coupling of Bi and Pb sites can improve the MOR process with low reaction barriers. In addition, it has been reported that the incorporation of Bi(OH)3 species can adjust the geometric structure and downshift the d-band center of Pt. Especially, the surface decoration of Bi(OH)3 species can accelerate the decomposition of H2O to generate OHad, resulting the removal of CO and other intermediates.63,121–123 On this basis, porous Pt nanoframes decorated with Bi(OH)3 species have been prepared by using a two-step method.124 The porous structure can improve the utilization efficiency of Pt, and Bi(OH)3 species can enhance the anti-CO poisoning. CO stripping experiments and in situ ATR-IR confirm the key role of Bi(OH)3 species. Therefore, The mass activity of Pt–Bi(OH)3 catalyst is 13.5 times higher than that of Pt/C, and the long-term durability of EOR on the Pt–Bi(OH)3 is much stable in both alkaline and acidic condition. In recent years, many efforts have focused on the decoration of Pt surface with oxides or other compounds, thus forming inverse heterogeneous nanostructures with an inverse interface structure. Therefore, a bismuth oxyhydroxide (BiOx (OH)y)-Pt inverse interface has been developed via an electrochemical reconstruction strategy.125 The as-prepared (BiOx (OH)y)-Pt inverse interface could induce the electron deficiency adjacent Pt active sites, resulting in decreasing the CO adsorption and strengthening OHad adsorption, leading to high MOR activity and good stability. In general, this work emphasizes the importance of inverse interface structure and its electronic effect on Pt.
 | ||
| Fig. 9 (a) The schematic illustration showing the atomic arrangement of Pt/Bi/Pb atoms in the PtBi@6.7%Pb nanoplates; (b) the positive-going MOR polarization curves of different catalysts recorded at a scan rate of 50 mV s−1; (c) long-term chronoamperometry of PtBi@6.7%Pb nanoplates and Pt/C recorded at −0.3 V vs. SCE at 60 °C; (d) CO-stripping curves of PtBi@6.7% Pb nanoplates and Pt/C catalysts; (e) the in situ FTIR spectra of PtBi@6.7% Pb nanoplates recorded in Ar-saturated 1 M KOH + 1 M CH3OH solution. Reproduced with permission from ref. 120. Copyright 2022, Wiley-VCH. | ||
Although metal hydroxides show great advantages for CO tolerance on the Pt electrode, poor electronic conductivity is their inevitable defect, which can affect the electron transfer and limit the catalytic activity. In addition, the interaction between metal hydroxides and OHad species is neither too strong nor too weak, which can benefit the removal of CO intermediate. Based on the above, amorphous Ni(OH)2 and graphene have been mixed to support Pt NPs.126 As a result, the Pt-based ternary hybrids show superior activity and durability for MOR. The incorporation of amorphous Ni(OH)2 species significantly accelerates the dissociative adsorption of H2O and then promotes the oxidative removal of CO via the Langmuir–Hinshelwood reaction pathway. Similarly, three-dimensional porous Pt/Ni(OH)2/NG catalysts have been prepared by a facile route.127 In the composites, the N-doped graphene structure can promote the electron transfer and electrolyte diffusion, and Pt NPs can expose more active sites. Importantly, Ni(OH)2 species provide a large number of reactive oxygen species. Therefore, the mass activity of Pt/Ni(OH)2/NG can reach 2.99 A mgPt−1, and their catalytic durability and CO tolerance were also significantly improved due to the abundant OHad species adjacent to the Pt active sites.
Based on the above, the synergy between Pt and metal oxides or hydroxides can weaken the binding strength of CO due to the formation of OHad and the modified electronic structure of Pt. However, the conductivity of metal oxides or hydroxides as the semiconductor carriers is much lower compared to carbon-based carriers. The usage of carbon-based materials could effectively improve the conductivity of metal oxides or hydroxides. Moreover, the structural stability of composites is its inherent defect. For example, Pt-based composites were generally physically mixed with carriers or deposited on the carriers, which leads to the random distribution between Pt NPs and carriers. Modifying metal oxides or hydroxides on the Pt surface could promote the formation of strong coupling interface structure, and no significant reduction of conductivity can be observed due to the trace metal oxides or hydroxides.
4. Summary and outlooks
In summary, fabricating Pt-based electrocatalysts with high CO tolerance is essential for the widespread commercialization of PEMFCs. Two effective strategies have been developed to improve the CO tolerance of Pt-based electrocatalysts, including alloying Pt with a second element and fabricating Pt-based composites with geometry and interface engineering. Alloying strategy not only could reduce the usage of Pt but also tune the Pt electronic structure based on the electron or ligand effects, including electron donation or reverse donation, thus reducing the strength of Pt–CO bond. Importantly, alloying strategy can effectively tune the geometric structure of Pt, which make Pt-based electrocatalysts exhibit a much stable nanostructure. Despite the success, the second element is easily oxidized and dissolved under long-term electrochemical reactions, thus leading to unstable nanostructures, which could reduce the CO tolerance of Pt-based alloys. In the Pt-based composites, the synergistic effect between Pt and carriers is believed to mainly optimize the Pt electronic structure and promote the formation of OHad species adjacent the Pt active sites. The former one could weaken the binding strength of CO, and the latter one is essential for the complete oxidation of CO intermediate. As a result, Pt-based electrocatalysts synthesized by two strategies have excellent electrochemical performance, as shown in Table 1.| Strategies | Catalysts | Electrolytes | Mass activity (A mg−1) | Ref. |
|---|---|---|---|---|
| Pt-based binary alloys | Pt72Ru28 | 0.1 M HClO4 + 0.5 M CH3OH | 1.70 | 44 |
| PtRu/C(DMAB) | 0.5 M H2SO4 + 1 M CH3OH | 0.53 | 46 | |
| d-Pt2Cu1 | 0.1 M HClO4 + 0.5 M CH3OH | 2.23 | 50 | |
| Pt–Co nanoframes | 1 M KOH + 1 M CH3OH | 4.28 | 51 | |
| PtNi CNCs | 0.5 M H2SO4 + 0.5 M CH3OH | 0.69 | 52 | |
| PtBi nanoplates/C | 0.1 M HClO4 + 0.1 M CH3OH | 1.10 | 53 | |
| Pt95Co5 | 0.5 M H2SO4 + 1 M CH3OH | 0.49 | 60 | |
| Pt3Cu NBs | 0.5 M H2SO4 + 1 M CH3OH | 0.53 | 61 | |
| PtAg nanoflowers | 0.1 M HClO4 + 0.5 M CH3OH | 1.13 | 64 | |
| PtBi nanorings | 1 M KOH + 1 M CH3OH | 6.42 | 65 | |
| Pt-based multi-element alloys | h-PtNiCo/C | 0.1 M HClO4 + 0.5 M CH3OH | 2.82 | 73 |
| Pt3Bi3Zn NPLs | 0.1 M HClO4 + 0.5 M CH3OH | 3.29 | 74 | |
| Ru-ca-PtNi | 0.1 M HClO4 + 0.5 M CH3OH | 2.01 | 75 | |
| PtCoNiRh NWs | 0.1 M HClO4 + 0.5 M CH3OH | 1.36 | 76 | |
| Cu–Ni@Pt–Cu | 0.1 M HClO4 + 1 M CH3OH | 0.99 | 80 | |
| Au@Pt–Pd | 1 M KOH + 1 M CH3OH | 4.38 | 81 | |
| Pt-based high-entropy alloys | Pt18Ni26Fe15Co14Cu27 | 1 M KOH + 1 M CH3OH | 15.04 | 87 |
| PtRhBiSnSb | 1 M KOH + 1 M CH3OH | 19.53 | 88 | |
| Carbon-supported Pt-based electrocatalysts | NiPtSAA/GDY | 1 M KOH + 1 M CH3OH | 4.40 | 99 |
| PtRu/PC-H | 0.1 M HClO4 + 0.5 M CH3OH | 1.67 | 100 | |
| CoP-NCZ/CNT | 1 M KOH + 1 M CH3OH | 3.62 | 104 | |
| PtCo@NCs | 0.1 M HClO4 + 1 M CH3OH | 2.30 | 105 | |
| Pt/SiC | 0.5 M H2SO4 + 0.5 M CH3OH | 0.65 | 22 | |
| Metal oxides or hydroxides supported Pt-based electrocatalysts | Pt/SnO2/rGO | 0.5 M H2SO4 + 0.5 M CH3OH | 0.63 | 108 |
| UV-Pt@TiO2/graphene | 0.5 M H2SO4 + 1 M CH3OH | 1.94 | 114 | |
| YOx/MoOx-Pt NWs | 0.1 M HClO4 + 0.5 M CH3OH | 2.1 | 116 | |
| PtBi@6.7%Pb | 1 M KOH + 1 M CH3OH | 13.93 | 120 | |
| Pt/Ni(OH)2/NG | 1 M KOH + 1 M CH3OH | 2.99 | 127 |
Despite the great progress has been achieved in Pt-based alloys and composites as highly effective catalysts against CO poisoning, there are still great challenges that need to be resolved: (1) it is difficult to choose the type and number of metallic elements and precisely control their proportions. (2) Strict environmental conditions, such as acid solution, alkali solution and high temperature, can easily cause the agglomeration and elemental dissolution of Pt-based alloys and composites. (3) The limitations of computational software and models make it difficult to theoretically calculate the top-level design of Pt-based electrocatalysts. (4) The complexity of surface compositions, phase state, and catalytic reactions makes it difficult to monitor the real-time evolution behavior on the Pt electrode.
Based on the above discussion, controllable synthesis of Pt-based electrocatalysts and its investigation of CO tolerance can be mainly focused on five aspects, as follows: (1) the optimal catalytic system can be constructed by means of theoretical calculation, including the selection of type, quantity and proportion of metallic elements. (2) Focusing on the design and synthesis of stable structures, including intermetallic compounds, high-entropy alloys, and strong coupling interface structures etc. (3) Combining in situ characterizations, the real-time structural evolution process can be observed, thereby guaranteeing the detection of the intrinsic active sites and catalytic mechanism. (4) It is also important to design and develop simpler synthesis processes to reduce costs and improve the utilization of components in the catalyst. (5) Large-scale synthesis paths need to be developed for the next-generation fuel cells. Based on the study of Pt-based electrocatalysts, we believe that many effective approaches can be developed to design Pt-based electrocatalysts with high CO tolerance, which can be widely used in PEMFCs in the future.
Conflicts of interest
There are no conflicts to declare.References
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- C. Z. Li, Q. Yuan, B. Ni, T. He, S. M. Zhang, Y. Long, L. Gu and X. Wang, Nat. Commun., 2018, 9, 3702 CrossRef.
- S. Wang and S. P. Jiang, Natl. Sci. Rev., 2017, 4, 163–166 CrossRef CAS.
- H. Yang, A. C. Zhang, Y. F. Bai, M. Y. Chu, H. Li, Y. Liu, P. Zhu, X. L. Chen, C. W. Deng and X. L. Yuan, Inorg. Chem., 2022, 61, 14419–14427 CrossRef CAS PubMed.
- M. A. Abdelkareem, K. Elsaid, T. Wilberforce, M. Kamil, E. T. Sayed and A. Olabi, Nat. Energy, 2021, 6, 451 CrossRef.
- J. N. Zhou, C. H. Zeng, H. Ou, Q. Y. Yang, Q. Y. Xie, A. Zeb, X. M. Lin, Z. Ali and L. Hu, J. Mater. Chem. C, 2021, 9, 11030–11058 RSC.
- S. N. Upadhyay and S. Pakhira, J. Mater. Chem. C, 2021, 9, 11331–11342 RSC.
- F. Xiao, Y. C. Wang, Z.-P. Wu, G. Chen, F. Yang, S. Zhu, K. Siddharth, Z. Kong, A. Lu, J. C. Li, C. J. Zhong, Z. Y. Zhou and M. Shao, Adv. Mater., 2021, 33, 2006292 CrossRef CAS PubMed.
- J. Tollefson, Nature, 2008, 451, 877 CrossRef CAS PubMed.
- J. Choi, J. Cho, C. W. Roh, B. S. Kim, M. S. Choi, H. Jeong, H. C. Ham and H. Lee, Appl. Catal., B, 2019, 247, 142–149 CrossRef CAS.
- L. C. Lin, C. H. Kuo, Y. H. Hsu, L. C. Hsu, H. Y. Chen, J. L. Chen and Y. T. Pan, Appl. Catal., B, 2022, 317, 121767 CrossRef CAS.
- M. Gong, D. Xiao, Z. Deng, R. Zhang, W. Xia, T. Zhao, X. Liu, T. Shen, Y. Hu, Y. Lu, X. Zhao, H. Xin and D. Wang, Appl. Catal., B, 2020, 282, 119617 CrossRef.
- J. Zhai, J. Gao, J. Zhang, D. Liu, S. Gao, Y. Yan, K. Zhang, K. Cai, F. Yu, M. Lin and J. Li, Chem. Eng. J., 2022, 442, 136172 CrossRef CAS.
- Y. Lu, S. Du and R. Steinberger-Wilckens, Appl. Catal., B, 2016, 187, 108–114 CrossRef CAS.
- Y. X. Xiao, J. Ying, G. Tian, Y. Tao, H. Wei, S. Y. Fan, Z. H. Sun, W. J. Zou, J. Hu, G. G. Chang, W. Li, X. Y. Yang and C. Janiak, Appl. Catal., B, 2019, 258, 118080 CrossRef.
- Y. Y. Zhou, C. H. Liu, J. Liu, X. L. Cai, Y. Lu, H. Zhang, X. H. Sun and S. D. Wang, Nano-Micro Lett., 2016, 8, 371–380 CrossRef PubMed.
- T. W. Song, M. X. Chen, P. Yin, L. Tong, M. Zuo, S. Q. Chu, P. Chen and H. W. Liang, Small, 2022, 18, 2202916 CrossRef CAS PubMed.
- J. Lee, C. Yeon, J. Oh, G. Han, J. Do Yoo, H. J. Yun, C. W. Lee, K. T. Lee and J. Bae, Appl. Catal., B, 2022, 316, 121645 CrossRef CAS.
- D. Sebastián, I. Suelves, E. Pastor, R. Moliner and M. J. Lázaro, Appl. Catal., B, 2012, 132–133, 13–21 Search PubMed.
- X. Yang, J. Zheng, M. Zhen, X. Meng, F. Jiang, T. Wang, C. Shu, L. Jiang and C. Wang, Appl. Catal., B, 2012, 121–122, 57–64 CrossRef CAS.
- Y. M. Xi, Y. Zhang, X. T. Cai, Z. X. Fan, K. F. Wang, W. R. Dong, Y. Shen, S. X. Zhong, L. Yang and S. Bai, Appl. Catal., B, 2022, 305, 121069 CrossRef CAS.
- G. Bai, C. Liu, Z. Gao, B. Lu, X. Tong, X. Guo and N. Yang, Small, 2019, 15, 202951 Search PubMed.
- W. Chen, J. Cao, W. Fu, J. Zhang, G. Qian, J. Yang, D. Chen, X. Zhou, W. Yuan and X. Duan, Angew. Chem., Int. Ed., 2022, 61, e202200190 CAS.
- J. Liu, F. R. Lucci, M. Yang, S. Lee, M. D. Marcinkowski, A. J. Therrien, C. T. Williams, E. C. H. Sykes and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2016, 138, 6396–6399 CrossRef CAS PubMed.
- P. Ferrari, L. M. Molina, V. E. Kaydashev, J. A. Alonso, P. Lievens and E. Janssens, Angew. Chem., Int. Ed., 2016, 55, 11059–11063 CrossRef CAS PubMed.
- J. Zhang, S. Deo, M. J. Janik and J. W. Medlin, J. Am. Chem. Soc., 2020, 142, 5184–5193 CrossRef CAS PubMed.
- P. Trens, R. Durand, B. Coq, C. Coutanceau, S. Rousseau and C. Lamy, Appl. Catal., B, 2009, 92, 280–284 CrossRef CAS.
- V. F. Valdés-López, T. Mason, P. R. Shearing and D. J. L. Brett, Prog. Energy Combust. Sci., 2020, 79, 100842 CrossRef.
- M. J. Hazlett, M. Moses-Debusk, J. E. Parks, L. F. Allard and W. S. Epling, Appl. Catal., B, 2016, 202, 404–417 CrossRef.
- J. Yeston, Science, 2016, 9, 2155–5435 Search PubMed.
- H. Chen, J. Liu, X. Wu, C. Ye, J. Zhang, J. L. Luo and X.-Z. Fu, Small, 2022, 18, 202204100 Search PubMed.
- A. M. Baena-Moncada, A. Bazan-Aguilar, E. Pastor and G. Á. Planes, J. Power Sources, 2019, 437, 226915 CrossRef CAS.
- M. Su, J. C. Dong, J. B. Le, Y. Zhao, W. M. Yang, Z. L. Yang, G. Attard, G. K. Liu, J. Cheng, Y. M. Wei, Z. Q. Tian and J. F. Li, Angew. Chem., Int. Ed., 2020, 59, 23554–23558 CrossRef CAS PubMed.
- L. Huang, S. Zaman, X. Tian, Z. Wang, W. Fang and B. Y. Xia, Acc. Chem. Res., 2021, 54, 311–322 CrossRef CAS PubMed.
- W. Liang, Y. Wang, L. Zhao, W. Guo, D. Li, W. Qin, H. Wu, Y. Sun and L. Jiang, Adv. Mater., 2021, 33, e2100713 CrossRef PubMed.
- Z. Wang, H. Wang, Z. Zhang, G. Yang, T. He, Y. Yin and M. Jin, ACS Nano, 2017, 11, 163–170 CrossRef CAS PubMed.
- S. Q. Liu, H. R. Wen, G. Ying, Y. W. Zhu, X. Z. Fu, R. Sun and C. P. Wong, Nano Energy, 2018, 44, 7–14 CrossRef CAS.
- A. L. Wang, L. Zhu, Q. Yun, S. Han, L. Zeng, W. Cao, X. Meng, J. Xia and Q. Lu, Small, 2020, 16, e2003782 CrossRef PubMed.
- C. Fang, T. Bi, Q. Ding, Z. Cui, N. Yu, X. Xu and B. Geng, ACS Appl. Mater. Interfaces, 2019, 11, 20117–20124 CrossRef CAS PubMed.
- A. L. Wang, H. Xu, J. X. Feng, L. X. Ding, Y. X. Tong and G. R. Li, J. Am. Chem. Soc., 2013, 135, 10703–10709 CrossRef CAS PubMed.
- A. Rodríguez-Gómez, E. Lepre, L. Sánchez-Silva, N. López-Salas and A. Raquelde la Osa, J. Energy Chem., 2022, 66, 168–180 CrossRef.
- J. S. Guo, G. Q. Sun, S. G. Sun, S. Y. Yan, W. Q. Yang, J. Qi, Y. S. Yan and Q. Xin, J. Power Sources, 2007, 168, 299–306 CrossRef CAS.
- S. L. Lu, K. Eid, D. H. Ge, J. Guo, L. Wang, H. J. Wang and H. W. Gu, Nanoscale, 2017, 9, 1033–1039 RSC.
- W. Y. Zhao, B. Ni, Q. Yuan, P. L. He, Y. Gong, L. Gu and X. Wang, Adv. Energy Mater., 2017, 7, 1601593 CrossRef.
- J. M. Zhang, X. M. Qu, Y. Han, L. F. Shen, S. H. Yin, G. Li, Y. X. Jiang and S. G. Sun, Appl. Catal., B, 2020, 263, 118345 CrossRef CAS.
- Q. J. Wang, Y. W. Zhou, Z. Jin, C. G. Chen, H. Li and W. B. Cai, Catalysts, 2021, 11, 925 CrossRef CAS.
- L. Huang, X. P. Zhang, Q. Q. Wang, Y. J. Han, Y. X. Fang and S. J. Dong, J. Am. Chem. Soc., 2018, 140, 1142–1147 CrossRef CAS PubMed.
- T. Fu, J. Huang, S. Lai, S. Zhang, J. Fang and J. Zhao, J. Power Sources, 2017, 365, 17–25 CrossRef CAS.
- Y. S. Kang, K. H. Choi, D. Ahn, M. J. Lee, J. Baik, D. Y. Chung, M. J. Kim, S. Y. Lee, M. Kim, H. Shin, K. S. Lee and Y.-E. Sung, J. Power Sources, 2016, 303, 234–242 CrossRef CAS.
- K. L. Wang, D. Y. Huang, Y. C. Guan, F. Liu, J. He and Y. Ding, ACS Catal., 2021, 11, 14428–14438 CrossRef CAS.
- S. P. Chen, M. F. Li, M. Y. Gao, J. B. Jin, M. A. van Spronsen, M. B Salmeron and P. D. Yang, Nano Lett., 2020, 3, 1974–1979 CrossRef PubMed.
- P. P. Yang, X. L. Yuan, H. C. Hu, Y. L. Liu, H. W. Zheng, D. Yang, L. Chen, M. H. Cao, Y. Xu, Y. L. Min, Y. G. Li and Q. Zhang, Adv. Funct. Mater., 2018, 28, 1704774 CrossRef.
- X. L. Yuan, X. J. Jiang, M. H. Cao, L. Chen, K. Q. Nie, Y. Zhang, Y. Xu, X. H. Sun, Y. G. Li and Q. Zhang, Nano Res., 2019, 12, 429–436 CrossRef CAS.
- Y. N. Qin, M. C. Luo, Y. J. Sun, C. J. Li, B. L. Huang, Y. Yang, Y. J. Li, L. Wang and S. J. Guo, ACS Catal., 2018, 8, 5581–5590 CrossRef CAS.
- M. Choi, C. Ahn, H. Lee, J. Kim, S. Oh, W. Hwang, S. Yang, J. Kim, O. Kim, I. Choi, Y. Sung, Y. Cho, C. Rhee and W. Shin, Appl. Catal., B, 2019, 253, 187–195 CrossRef CAS.
- M. Y. Chu, J. L. Huang, J. Gong, Y. Qu, G. L. Chen, H. Yang, X. C. Wang, Q. X. Zhong, C. W. Deng, M. H. Cao, J. X. Chen, X. L. Yuan and Q. Zhang, Nano Res., 2022, 15, 3920–3926 CrossRef CAS.
- Y. C. Yan, H. Shan, G. Li, F. Xiao, Y. Y. Jiang, Y. Y. Yan, C. H. Jin, H. Zhang, J. B. Wu and D. R. Yang, Nano Lett., 2016, 16, 7999–8004 CrossRef CAS PubMed.
- A. Mahmood, D. Q. He, S. H. Talib, Y. He, Z. Q. Song, Z. B. Li, D. X. Han and L. Niu, Adv. Funct. Mater., 2022, 32, 2205223 CrossRef CAS.
- E. B. Zhu, X. C. Yan, S. Y. Wang, M. J. Xu, C. Wang, H. T. Liu, J. Huang, W. Xue, J. Cai, H. Heinz, Y. J. Li and Y. Huang, Nano Lett., 2019, 19, 3730–3736 CrossRef CAS PubMed.
- Q. Q. Lu, L. T. Sun, X. Zhao, J. S. Huang, C. Han and X. R. Yang, Nano Res., 2018, 11, 2562–2572 CrossRef CAS.
- Z. B. Zhang, J. Li, S. Y. Liu, X. Zhou, L. Xu, X. L. Tian, J. Yang and Y. W. Tang, Small, 2022, 18, 2202782 CrossRef CAS PubMed.
- D. Kim, M. Chung, S. Kim, K. Yun, W. Cha, R. Harder and H. Kim, Nano Lett., 2019, 19, 5044–5052 CrossRef CAS PubMed.
- Y. J. Chen, J. J. Pei, Z. Chen, A. Li, S. F. Ji, H. P. Rong, Q. Xu, T. Wang, A. J. Zhang, H. L. Tang, J. F. Zhu, X. D. Han, Z. B. Zhuang, G. Zhou and D. S. Wang, Nano Lett., 2022, 22, 7563–7571 CrossRef CAS PubMed.
- M. Qiao, H. Wu, F. Y. Meng, Z. B. Zhuang and J. X. Wang, Small, 2022, 18, 2106643 CrossRef CAS PubMed.
- S. M. Han, Y. Ma, Q. B. Yun, A. L. Wang, Q. S. Zhu, H. Zhang, C. H. He, J. Xia, X. M. Meng, L. Gao, W. B. Cao and Q. P. Lu, Adv. Funct. Mater., 2022, 2208760 CrossRef CAS.
- L. Zhang, H. S. Liu, S. H. Liu, M. N. Banis, Z. X. Song, J. J. Li, L. J. Yang, M. Markiewicz, Y. Zhao, R. Y. Li, M. Zheng, S. Y. Ye, Z. J. Zhao, G. A. Botton and X. L. Sun, ACS Catal., 2019, 9, 9350–9358 CrossRef CAS.
- A. R. Poerwoprajitno, L. Gloag, J. Watt, S. Cheong, X. Tan, H. Lei, H. A. Tahini, A. Henson, B. Subhash, N. M. Bedford, B. K. Miller, P. B. O'Mara, T. M. Benedetti, D. L. Huber, W. Zhang, S. C. Smith, J. J. Gooding, W. Schuhmann and R. D. Tilley, Nat. Catal., 2022, 5, 231–237 CrossRef CAS.
- X. F. Ren, Q. Y. Lv, L. F. Liu, B. H. Liu, Y. Wang, A. M. Liu and G. Wu, Sustainable Energy Fuels, 2020, 4, 15–30 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- X.-X. Li, P.-Y. Zhu, Q. Li, Y.-X. Xu, Y. Zhao and H. Pang, Rare Met., 2020, 39, 680–687 CrossRef CAS.
- J. Liang, N. Li, Z. Zhao, L. Ma, X. Wang, S. Li, X. Liu, T. Wang, Y. Du, G. Lu, J. Han, Y. Huang, D. Su and Q. Li, Angew. Chem., Int. Ed., 2019, 58, 2–9 CrossRef.
- R. M. Aran-Ais, F. Dionigi, T. Merzdorf, M. Gocyla, M. Heggen, R. E. Dunin-Borkowski, M. Gliech, J. Solla-Gullon, E. Herrero, J. M. Feliu and P. Strasser, Nano Lett., 2015, 15, 7473–7480 CrossRef CAS PubMed.
- H. R. Ma, Z. P. Zheng, H. S. Zhao, C. Shen, H. M. Chen, H. Q. Li, Z. M. Cao, Q. Kuang, H. X. Lin and Z. X. Xie, J. Mater. Chem. A, 2021, 9, 23444 RSC.
- H. Tian, D. X. Wu, J. Li, J. M. Luo, C. M. Jia, Z. X. Liu, W. Huang, Q. Chen, C. M. Shim, P. L. Deng, Y. J. Shen and X. L. Tian, J. Energy Chem., 2022, 70, 230–235 CrossRef CAS.
- F. P. Kong, X. Z. Liu, Y. J. Song, Z. Y. Qian, J. J. Li, L. Zhang, G. P. Yin, J. J. Wang, D. Su and X. L. Sun, Angew. Chem., Int. Ed., 2022, 61, e202207524 CAS.
- W. Wang, X. W. Chen, X. Zhang, J. Y. Ye, F. Xue, C. Zhen, X. Y. Liao, H. Q. Li, P. T. Li, M. C. Liu, Q. Kuang, Z. X. Xie and S. F. Xie, Nano Energy, 2020, 71, 104623 CrossRef CAS.
- S. Xie, S.-I. Choi, N. Lu, L. T. Roling, J. A. Herron, L. Zhang, J. Park, J. Wang, M. J. Kim, Z. Xie, M. Mavrikakis and Y. Xia, Nano Lett., 2014, 14, 3570–3576 CrossRef CAS PubMed.
- J. Park, L. Zhang, S.-I. Choi, L. T. Roling, N. Lu, J. A. Herron, S. Xie, J. Wang, M. J. Kim, M. Mavrikakis and Y. Xia, ACS Nano, 2015, 9, 2635–2647 CrossRef CAS PubMed.
- Y. Xia, K. D. Gilroy, H. C. Peng and X. Xia, Angew. Chem., Int. Ed., 2017, 56, 60–95 CrossRef CAS PubMed.
- C. Li, X. B. Chen, L. H. Zhang, S. H. Yan, A. Sharma, B. Zhao, A. Kumbhar, G. W. Zhou and J. Y. Fang, Angew. Chem., Int. Ed., 2021, 60, 7675–7680 CrossRef CAS PubMed.
- W. K. Liang, Y. W. Wang, L. Zhao, W. Guo, D. Li, W. Qin, H. H. Wu, Y. H. Sun and L. Jiang, Adv. Mater., 2021, 33, 2100713 CrossRef CAS PubMed.
- H. Peng, J. Ren, Y. Wang, Y. Xiong, Q. Wang, Q. Li, X. Zhao, L. Zhan, L. Zheng and Y. Tang, Nano Energy, 2021, 88, 106307 CrossRef CAS.
- G. L. Zhang, D. J. Cao, S. Y. Guo, Y. Fang, Q. Wang, S. Cheng, W. S. Zuo, Z. Z. Yang and P. Cui, Small, 2022, 18, 2202587 CrossRef CAS PubMed.
- P. Wang, Y. Q. Bu, J. B. Liu, Q. Q. Li, H. T. Wang and W. Yang, Mater. Today, 2020, 37, 64–73 CrossRef CAS.
- Z. W. Chen, L. X. Chen, Z. Gariepy, X. Yao and C. V. Singh, Trends Chem., 2022, 4, 577–579 CrossRef CAS.
- M. Bondesgaard, N. L. N. Broge, A. Mamakhel, M. Bremholm and B. B. Iversen, Adv. Funct. Mater., 2019, 29, 1905933 CrossRef CAS.
- H. D. Li, Y. Han, H. Zhao, W. J. Qi, D. Zhang, Y. D. Yu, W. W. Cai, S. X. Li, J. P. Lai, B. L. Huang and L. Wang, Nat. Commun., 2020, 11, 5437 CrossRef CAS PubMed.
- W. Chen, S. P. Luo, M. Z. Sun, X. Y. Wu, Y. S. Zhou, Y. J. Liao, M. Tang, X. K. Fan, B. L. Huang and Z. W. Quan, Adv. Mater., 2022, 34, 2206276 CrossRef CAS PubMed.
- D. S. Wu, K. H. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 13833–13838 CrossRef CAS PubMed.
- Y. J. Wang, W. Y. Long, L. L. Wang, R. S. Yuan, A. Ignaszak, B. Z. Fang and D. P. Wilkinson, Energy Environ. Sci., 2018, 11, 258–275 RSC.
- B. W. Zhang, Y. X. Jiang, J. Ren, X. M. Qu, G. L. Xu and S. G. Sun, Electrochim. Acta, 2015, 162, 254–262 CrossRef CAS.
- J. Zheng, G. Li, J. M. Zhang, N. Y. Cheng, L. F. Ji, J. Yang, J. L. Zhang, B. W. Zhang, Y. X. Jiang and S. G. Sun, ACS Catal., 2021, 11, 9317–9332 CrossRef.
- J. Zheng, G. Li, J. M. Zhang, N. Y. Cheng, L. F. Ji, J. Yang, J. L. Zhang, B. W. Zhang, Y. X. Jiang and S. G. Sun, Sci. China: Chem., 2023, 66, 279–288 CrossRef CAS.
- K. Kodama, T. Nagai, A. Kuwaki, R. Jinnouchi and Y. Morimoto, Nat. Nanotechnol., 2021, 16, 140–147 CrossRef CAS PubMed.
- C.-Y. Ahn, J. E. Park, S. Kim, O.-H. Kim, W. Hwang, M. Her, S. Y. Kang, S. B. Park, O. J. Kwon, H. S. Park, Y. Cho and Y.-E. Sung, Chem. Rev., 2021, 121, 15075–15140 CrossRef CAS PubMed.
- X. Guo, M. G. Lia, L. Y. Qiu, F. Y. Tian, L. He, S. Geng, Y. Q. Qiu, Y. Song, W. W. Yang and Y. S. Yu, Chem. Eng. J., 2023, 453, 1397996 Search PubMed.
- Q. C. Tran, V.-D. Dao, H. Y. Kim, K.-D. Jun and H.-S. Choi, Appl. Catal., B, 2021, 204, 365–373 CrossRef.
- G. Han, M. G. Li, W. Y. Zhang, L. He, F. Y. Tian, Y. Q. Liu, Y. S. Yu, W. W. Yang and S. J. Guo, Adv. Mater., 2022, 34, 2202943 CrossRef CAS PubMed.
- L. Hui, Y. R. Xue, C. Y. Xing, Y. X. Liu, Y. C. Du, Y. Fang, H. D. Yu, C. Zhang, F. He and Y. L. Li, Nano Energy, 2022, 95, 106984 CrossRef CAS.
- J. M. Zhang, X. M. Qu, Y. Han, L. F. Shen, S. H. Yin, G. Li, Y. X. Jiang and S. G. Sun, Appl. Catal., B, 2020, 263, 118345 CrossRef CAS.
- Q. Zhang, F. Yue, L. J. Xu, C. X. Yao, R. D. Priestley and S. F. Hou, Appl. Catal., B, 2019, 257, 117886 CrossRef CAS.
- M. M. Yan, Q. G. Jiang, T. Zhang, J. Y. Wang, L. Yang, Z. Y. Lu, H. Y. He, Y. S. Fu, X. Wang and H. J. Huang, J. Mater. Chem. A, 2018, 6, 18165–18172 RSC.
- J. Fonseca, T. H. Gong, L. Jiao and H. L. Jiang, J. Mater. Chem. A, 2021, 9, 10562–10611 RSC.
- W. Zhan, L. Ma, M. Y. Gan, J. J. Ding, S. C. Han, D. Y. Wei, J. Shen and C. L. Zhou, Int. J. Hydrogen Energy, 2020, 45, 15630–15641 CrossRef.
- G. F. Hu, L. Shang, T. Sheng, Y. G. Chen and L. Y. Wang, Adv. Funct. Mater., 2020, 30, 2002281 CrossRef CAS.
- J. Min, S. Kim, A. A. Jeffery, H. Shin, Y. S. Kang, Y. Kim, J. Jang, S. Lee, S. Park, G. Park, S. J. Yoo, S. Yim and N. Jung, Mater. Today Energy, 2022, 29, 101124 CrossRef CAS.
- G. L. Bai, C. Liu, Z. Gao, B. Y. Lu, X. L. Tong, X. Y. Guo and N. J. Yang, Small, 2019, 15, 1902951 CrossRef PubMed.
- S. L. Wu, J. Liu, D. W. Liang, H. M. Sun, Y. X. Ye, Z. F. Tian and C. H. Liang, Nano Energy, 2016, 26, 699–707 CrossRef CAS.
- Z. C. Wang, L. Wang, W. B. Zhu, T. Zeng, W. Wu, Z. Lei, Y. Y. Tan, H. F. Lv and N. C. Cheng, Nanoscale Adv., 2021, 3, 5062–5067 RSC.
- B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- M. Y. Gao, F. Y. Tian, Z. Guo, X. Zhang, Z. J. Li, J. Zhou, X. Zhou, Y. S. Yu and W. W. Yang, Chem. Eng. J., 2022, 446, 137127 CrossRef.
- B. D. Chandler, Nat. Chem., 2017, 9, 108–109 CrossRef CAS PubMed.
- Z. X. Luo, G. Q. Zhao, H. G. Pan and W. P. Sun, Adv. Energy Mater., 2022, 12, 2201395 CrossRef CAS.
- K. F. Zhang, J. Qiu, J. Wu, Y. Q. Deng, Y. H. Wu and L. F. Yan, J. Mater. Chem. A, 2022, 10, 4254 RSC.
- M. Meier, J. Hulva, Z. Jakub, F. Kraushofer, M. Bobić, R. Bliem, M. Setvin, M. Schmid, U. Diebold, C. Franchini and G. S. Parkinson, Sci. Adv., 2022, 8, eabn4580 CrossRef CAS PubMed.
- M. G. Li, Z. L. Zhao, W. Y. Zhang, M. C. Luo, L. Tao, Y. J. Sun, Z. H. Xia, Y. G. Chao, K. Yin, Q. H. Zhang, L. Gu, W. W. Yang, Y. S. Yu, G. Lu and S. J. Guo, Adv. Mater., 2021, 33, 2103762 CrossRef CAS PubMed.
- Y. X. Liang, Y. J. Sun, X. Y. Wang, E. G. Fu, J. Zhang, J. L. Du, X. D. Wen and S. J. Guo, Nanoscale, 2018, 10, 11357–11364 RSC.
- S. P. Chen, M. F. Li, X. Zhang, Z. J. Li, J. Zhou, X. Zhou, Y. S. Yu and W. W. Yang, Nano Res., 2022, 15, 1809–1816 CrossRef.
- Y. J. Sun, Y. X. Liang, M. C. Luo, F. Lv, Y. N. Qin, L. Wang, C. Xu, E. G. Fu and S. J. Guo, Small, 2017, 14, 1702259 CrossRef PubMed.
- W. Chen, S. P. Luo, M. Z. Sun, M. Tang, X. K. Fan, Y. Cheng, X. Y. Wu, Y. J. Liao, B. L. Huang and Z. W. Quan, Small, 2022, 18, 2107803 CrossRef CAS PubMed.
- B. Lan, Q. L. Wang, Z. X. Ma, Y. J. Wu, X. L. Jiang, W. S. Jia, C. X. Zhou and Y. Y. Yang, Appl. Catal., B, 2022, 300, 120728 CrossRef CAS.
- M. G. Li, Z. G. Xia, M. C. Luo, L. He, L. Tao, W. W. Yang, Y. S. Yu and S. J. Guo, Small Sci., 2021, 13, 2100061 CrossRef.
- Y. L. Yuan, Q. Y. Wang, Y. Qiao, X. L. Chen, Z. L. Yang, W. C. Lai, T. W. Chen, G. H. Zhang, H. G. Duan, M. Liu and H. W. Huang, Adv. Energy Mater., 2022, 12, 2200970 CrossRef CAS.
- X. L. Yuan, B. Jiang, M. H. Cao, C. Y. Zhang, X. Z. Liu, Q. H. Zhang, F. L. Lyu, L. Gu and Q. Zhang, Nano Res., 2020, 13, 265–272 CrossRef CAS.
- X. C. Wang, M. Xie, F. L. Lyu, Y. M. Yiu, Z. Q. Wang, J. T. Chen, L. Y. Chang, Y. J. Xia, Q. X. Zhong, M. Y. Chu, H. Yang, T. Cheng, T. K. Sham and Q. Zhang, Nano Lett., 2020, 20, 7751–7759 CrossRef CAS PubMed.
- G. Yuan, L. Wang, X. W. Zhang and Q. F. Wang, J. Colloid Interface Sci., 2019, 536, 189–195 CrossRef CAS PubMed.
- K. F. Zhang, H. F. Wang, J. Qiu, J. Wu, H. J. Wang, J. W. Shao, Y. Q. Deng and L. F. Yan, Chem. Eng. J., 2021, 421, 127786 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |