
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo nanoparticles supported on mixed magnesium–lanthanum oxides: effect of calcium and barium addition on ammonia synthesis catalyst performance†
Hubert Ronduda a,
Magdalena Zyberta,
Wojciech Patkowskia,
Dariusz Moszyńskib,
Aleksander Albrechtb,
Kamil Sobczakc,
Artur Małolepszyd and
Wioletta Raróg-Pilecka*a
a,
Magdalena Zyberta,
Wojciech Patkowskia,
Dariusz Moszyńskib,
Aleksander Albrechtb,
Kamil Sobczakc,
Artur Małolepszyd and
Wioletta Raróg-Pilecka*a
aWarsaw University of Technology, Faculty of Chemistry, Noakowskiego 3, Warsaw, 00-664, Poland. E-mail: wioletta.pilecka@pw.edu.pl; Tel: +48 22 234 57 66
bWest Pomeranian University of Technology in Szczecin, Faculty of Chemical Technology and Engineering, 42 Piastów Ave, Szczecin, 71-065, Poland
cUniversity of Warsaw Biological and Chemical Research Centre, Żwirki i Wigury 101, Warsaw, 02-089, Poland
dWarsaw University of Technology, Faculty of Chemical and Process Engineering, Waryńskiego 1, Warsaw, 00-645, Poland
First published on 6th February 2023
Abstract
The synthesis of ammonia in the Haber–Bosch process produces millions of tons of ammonia annually needed for producing fertilisers required to feed the growing population. Although this process has been optimised extensively, it still accounts for about 2% of global energy consumption. It is, therefore, desirable to develop an efficient ammonia synthesis catalyst. Over the last decades, many attempts have been made to improve the ammonia synthesis catalyst efficiency under mild conditions. Here, we studied the effect of adding Ca and Ba to the cobalt ammonia synthesis catalyst. The combination of the different experimental results allows concluding that Ca served as an inactive additive, whereas Ba served as an electronic promoter. The Ca addition did not change the textural, structural, and chemisorptive properties of the Ca-doped Co catalyst. On the other hand, the Ba addition had a major effect on the nature of active Co sites. It contributed to the formation of new active sites for hydrogen and nitrogen adsorption and dissociation. Barium addition also contributed to the generation of new basic sites, particularly the strong ones. These unique characteristics were ascribed to the formation of Co(core)–BaO(shell) structures. It is likely that the donation of electrons from BaO to N2 via Co markedly promoted ammonia synthesis. This catalyst exhibited ammonia synthesis activity 4 times higher than that of the undoped Co catalyst and 2 times higher than that of the industrial Fe catalysts under identical conditions.
1. Introduction
The promotional effect is one of the crucial topics for researchers working on the development of heterogeneous catalysts since they have started to be used in industrial processes.1 Scientific studies on promotion effects began in about 19202 and tremendous advances have been achieved over the century. Although classifying promoters into those exhibiting a structural or modifying mode of action is known and generally accepted, generalising the effects of the selected promoter is usually impossible. Promoter effects tend to be specific to a particular catalyst formulation, which usually is complex for industrial catalysts. Excellent examples of the diverse influence of promoters on the properties of catalysts can be found among the ammonia synthesis reaction catalysts based on iron, ruthenium or cobalt.The fused iron catalyst is the first and most commonly used catalyst in industrial ammonia production.3–13 The essential component of this catalyst is Fe3O4 melted with selected non-reducible and catalytically inactive additives such as Al2O3, K2O, CaO, MgO, and SiO2 present in small quantities of a few weight percentages.3 Each plays a different role, from improving the catalytic activity of iron through increasing catalyst thermal stability or resistance toward impurities in the gas to prolonging the catalyst life.6–13 The activating effect on iron is exerted by promoters (activators)—both acidic and basic oxides. However, the most substantial influence is the combination of oxides, e.g., Al2O3 and K2O, which indicates the complex relationship between the structural and electronic effects induced by promoters.1 Aluminium oxide is a typical example of a structural promoter affecting the textural parameters (i.e., increasing the specific surface area of the catalyst and stabilising the catalyst surface during operation). The mechanism of its operation has not been clearly explained so far. The most common model assumes that the alumina is located on the iron surface. During reduction, the surface of Fe crystallites is covered with a thin layer of aluminium oxide (1–2 monolayers), which effectively separates the iron crystallites, thereby preventing their sintering.12 Two mechanisms were proposed in the case of potassium. According to Ertl et al.,14 a local increase in N2 adsorption energy is observed as a consequence of the transfer of some of the charge from potassium to the substrate (iron), which gives rise to the Feδ––Kδ+ dipole. The molecule adsorbed near such a dipole binds more strongly with the substrate, weakening the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond. Consequently, an acceleration of the dissociative nitrogen chemisorption, i.e., the rate-determining step of the ammonia synthesis reaction, is observed. According to Somorjai et al.,15 potassium facilitates the desorption of ammonia from the catalyst surface and unblocks active sites, increasing the NH3 synthesis reaction rate. However, in some cases, it is challenging to assign a specific function to the promoter as it is for calcium oxide, where the exact mode of action and function have not been identified so far. On the one hand, it can be considered an electronic promoter increasing the catalyst activity because the triply promoted (Al2O3, K2O and CaO) iron catalyst is more active than the system without calcium oxide.16 However, this effect may be because CaO favours uniform distribution of aluminium in the magnetite, lowering the melting point of the system and thus indirectly affecting the catalyst's texture. Hence, CaO shows some features of a structural promoter as well. Despite this discrepancy, studies show an indisputable positive effect of CaO on the iron catalyst properties—it improves the iron catalyst's mechanical strength and thermal stability and also extends its operation time.16
N bond. Consequently, an acceleration of the dissociative nitrogen chemisorption, i.e., the rate-determining step of the ammonia synthesis reaction, is observed. According to Somorjai et al.,15 potassium facilitates the desorption of ammonia from the catalyst surface and unblocks active sites, increasing the NH3 synthesis reaction rate. However, in some cases, it is challenging to assign a specific function to the promoter as it is for calcium oxide, where the exact mode of action and function have not been identified so far. On the one hand, it can be considered an electronic promoter increasing the catalyst activity because the triply promoted (Al2O3, K2O and CaO) iron catalyst is more active than the system without calcium oxide.16 However, this effect may be because CaO favours uniform distribution of aluminium in the magnetite, lowering the melting point of the system and thus indirectly affecting the catalyst's texture. Hence, CaO shows some features of a structural promoter as well. Despite this discrepancy, studies show an indisputable positive effect of CaO on the iron catalyst properties—it improves the iron catalyst's mechanical strength and thermal stability and also extends its operation time.16
In the case of promoted Ru catalysts, the structural factor may be important. For example, Kitano et al.17 showed that the activity of Ru-loaded Ca(NH2)2 in ammonia synthesis was significantly enhanced by the introduction of Ba into the Ca(NH2)2 matrix. The unique structural change, i.e., the Ru–Ba core–shell structure and the mesoporous structure of the support self-organized during the H2 pretreatment, was a key factor for the high catalytic performance. Nevertheless, for the Ru systems, the structural influence of promoters is not as significant as that for the Fe catalysts. The electronic promotion effect was found to play a crucial role in the activity of Ru-based catalysts. Hence, the promoters able to donate electrons can remarkably increase the activity of Ru-based catalysts. Among them, alkali and alkaline earth metals are the most commonly reported in the literature.18–28 However, it is essential that the effect of a promoter is also related to the support. For the carbon-supported ruthenium catalysts, among the alkali earth metals, the heavier elements were more effective (Ba > Sr > Ca > Mg). For the alkali metal elements, Rb was more effective than Cs (Rb > Cs > K).29,30 In general, the effect of promoters on the NH3 synthesis reaction rate increases with the decreasing electronegativity of the promoter; especially, barium has proved to be an effective promoter for ruthenium, improving its catalytic activity and Ru metal dispersion. It also prevents the sintering of Ru nanoparticles (NPs), inhibits the methanation reaction of activated carbon and enhances the thermal stability of the catalyst,25–27 whereas potassium only enhances the activity of the catalyst.25 Interestingly, Rossetti et al.27 found a simultaneous action of three promoters (K + Ba + Cs), maximising the activity and thermal resistance to methanation and metal sintering of the Ru/C catalyst. Szmigiel et al.31 revealed that caesium and barium are effective promoters for the MgO-supported ruthenium catalysts. In this study, the influence of the support type (MgO or C) was insignificant for the Ba-doped system—the kinetic characteristics of the Ba–Ru/C and Ba–Ru/MgO were similar. However, this factor was of great importance in the case of caesium promotion, where the TOF for the Cs–Ru/C system was much higher than for the Cs–Ru/MgO system. In the case of Ru/Al2O3 catalysts, the influence of alkali metals was more substantial than that of alkaline earth metals. Alkali metal enhanced not only the TOF of the catalyst but also the dispersion of Ru. The promoter effect increased with decreasing electronegativity as follows: Cs > Rb > K.32
A cobalt catalyst for ammonia synthesis is also an example of how a catalyst cannot perform effectively without appropriately selected promoters. The low activity of pure cobalt has increased so far by adding rare earth elements (lanthanum and cerium) and barium.33–44 Studies revealed that lanthanum is a typical structural promoter, developing the active phase surface and preventing its sintering during catalyst operation.33 The addition of cerium had a similar effect34–36 combined with stabilising the hcp Co phase (the most active phase of Co in the NH3 synthesis reaction). Cerium oxide can also be used as a support for cobalt due to its ability to provide a higher metal dispersion. Barium is mainly regarded as a modifying (electronic) promoter, but the structural effects of Ba addition were also observed.35,36 Moreover, for the carbon-supported cobalt catalysts,40 a strong inhibiting effect of Ba on the support methanation was revealed. As in the case of the triply-promoted ruthenium catalyst, also for the doubly promoted cobalt catalyst, the synergistic interaction of two promoters (Ba + Ce) was observed36 and associated with in situ formation of a third promoter, barium cerate (BaCeO3). Its strong basic properties, a tendency to stabilise the Co hcp cobalt phase and its ability to diversify the structure of adsorption sites on the cobalt surface, accelerated the ammonia synthesis. According to the latest research,44 the promoting effect of barium is related to the reduction of the spin polarisation of the neighbouring Co atoms defining the active site for N2 dissociation.
In this paper, we present the continuation of our comprehensive studies concerning cobalt catalysts supported on mixed MgO–La2O3 oxides.45–50 This study addresses the effect of Ca and Ba addition on the physicochemical properties and catalytic performance of the Co ammonia synthesis catalysts. This work aims to determine the location, state, and effect of dopants on the catalytic property of the Co/MgO–La2O3 systems. Calcium and barium were selected taking into account their chemical nature (Ca and Ba are basic elements from the alkaline earth metal group) and their functions in other ammonia synthesis catalysts, i.e., strong electron donating properties of barium and the stabilising effect of calcium oxide, previously known and described above. Optimising the catalyst composition in terms of the type and content of dopants is crucial from a technological point of view for developing efficient and cost-effective catalysts. The activity of the Co catalysts was tested in ammonia synthesis at 370–470 °C under pressures of 6.3 and 9.0 MPa. The detailed characterisation studies using N2 physisorption, X-ray powder diffraction (XRPD), scanning transmission electron microscopy (STEM) coupled with energy dispersive X-ray (EDX), X-ray photoelectron spectroscopy (XPS), and temperature-programmed desorption (TPD) were conducted in order to get a deeper insight into the effect of adding Ca and Ba to the cobalt ammonia synthesis catalyst.
2. Experimental section
2.1. Catalyst preparation
A mixed magnesium–lanthanum oxide support from our previous work45 was used here to prepare a series of cobalt catalysts. The MgO–La2O3 support was prepared by co-precipitation from a MgNO3·6H2O and La(NO3)3·6H2O solution using K2CO3 as a precipitation agent.45 The detailed synthesis and characterisation of this support have been reported in ref. 45.The Co/MgO–La2O3 catalyst was prepared by wet impregnation. A 40 wt% Co loading was chosen based on our previous work.50 Typically, a suitable amount of Co(NO3)2·6H2O (Acros Organics, 98+%) was dissolved in distilled water under stirring. Then, the oxide support was added to the above precursor solution. The mixture was stirred for 15 minutes to obtain a homogeneous mixture and then kept at room temperature overnight. Water was removed using a rotary evaporator under reduced pressure, and the obtained solid was dried in air overnight at 120 °C. The solid was calcined in air at 500 °C for 18 h with a ramp of 5 °C min−1. The sample was labelled as Co. Ca- or Ba-doped Co/MgO–La2O3 catalysts were prepared by the wet impregnation method. The Ca and Ba content was fixed at 0.5, 1, 3, 5, 7, and 9 wt%. Typically, a suitable amount of Ca(NO3)2·6H2O or Ba(NO3)2·6H2O (Chempur, 99+%) was dissolved in distilled water under stirring. Then, the powder Co sample was added to the above precursor solution. The mixture was stirred for 15 minutes to obtain a homogeneous mixture and then kept at room temperature overnight. Water was removed using a rotary evaporator under reduced pressure, and finally, the obtained solid was dried overnight at 120 °C. The samples were labelled as xCa–Co and xBa–Co, where x denotes the theoretical Ca and Ba content in weight percentage. The Ca and Ba doubly doped catalyst was prepared using the same procedure as that used to prepare the singly doped catalyst. The Ca and Ba content was fixed at 5 wt%. The sample was labelled as (5Ca + 5Ba)–Co. The precursor samples were activated at 600 °C in hydrogen before the catalytic reaction and characterisation (see the Section below for details). This activation temperature was chosen based on the TPR/TG-MS results (Fig. S1†).
Unless otherwise specified (e.g., the as-prepared catalysts and catalyst precursors), the Co catalysts mentioned in this paper refer to the reduced catalysts (freshly reduced catalysts). The full list of the prepared samples with their chemical composition is reported in Table S1.†
2.2. Catalyst characterisation
Temperature-programmed reduction was carried out on a NETZSCH STA449C thermal analyser equipped with a quadrupole mass spectrometer (NETZSCH QMS Aëolos 403C) detector. The precursor sample (50 mg) was heated up to 600 °C with a ramp of 3 °C min−1 under H2 + Ar (H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ar = 1:1) flow of 100 cm3 min−1 and kept in the isothermal conditions for about 3 h. The signals at m/z = 16 (CH4), 18 (H2O), 30 (NO), and 44 (CO2) were registered.
:Ar = 1:1) flow of 100 cm3 min−1 and kept in the isothermal conditions for about 3 h. The signals at m/z = 16 (CH4), 18 (H2O), 30 (NO), and 44 (CO2) were registered.
Nitrogen physisorption was performed on a Micromeritics ASAP2020 instrument at −196 °C. Prior to the analysis, the precursor sample (500 mg) was reduced in situ at 600 °C for 10 h under H2 flow (40 cm3 min−1) and purged at 620 °C for 2 h under He flow (40 cm3 min−1). Then, the sample was degassed at 200 °C under high vacuum for 2 h. The surface area was calculated using the Brunauer–Emmett–Teller (BET) method in the relative partial pressure range (P/P0) from 0.05 to 0.3.
X-ray powder diffraction (XRPD) experiments were carried out in the Anton Paar XRK 900 reaction chamber mounted to a Philips X'pert PRO MPD diffractometer working in Bragg–Brentano geometry and equipped with a CoKα radiation source (λKα1 = 1.789010 Å and λKα2 = 1.792900 Å). XRPD measurements were performed to identify the phase composition of the catalyst precursors and catalysts. The precursor sample was reduced in situ at 600 °C for 18 h under H2 flow of 100 cm3 min−1 to obtain the reduced catalyst. Diffractograms were recorded in the 2θ range of 20–140°. Data were collected under N2 flow at 25 °C. PANalytical High Score Plus software with the ICDD PDF-4+ 2021 database was used for data analysis. The semi-automatic Rietveld refinement procedure implemented in the software was utilised to determine the concentration of crystalline phases and estimate average crystallite size values for them. Both size and strain were considered to contribute to the broadening of reflections.
TEM investigations were performed on a Talos F200X (FEI) microscope operated at 200 kV. The measurements were performed in TEM and scanning TEM (STEM) modes using a high-angle annular dark-field (HAADF) detector and energy-dispersive X-ray spectroscopy on a Brucker BD4 spectrometer. The precursor sample was reduced ex situ at 600 °C for 18 h under H2 flow (40 cm3 min−1), then crushed and powdered. The powdered catalyst was dispersed in ethanol under ambient conditions, and several drops of the resulting dispersion were dropped on a carbon-coated copper-mesh TEM grid and dried out overnight at room temperature. STEM-EDX images were used to determine the Co nanoparticle size distributions. The particle size histograms were obtained by assuming the shape of Co nanoparticles as spherical in the calculation. The obtained histograms were fitted to log-normal distributions.
X-ray photoelectron spectroscopy (XPS) analyses were carried out for the reduced catalysts. The measurements were conducted using Al Kα (hν = 1486.6 eV) radiation in a Prevac system equipped with a Scienta SES 2002 electron energy analyser operating at constant transmission energy (Ep = 50 eV). The pressure in the analysis chamber was kept under 10−9 mbar. The reduction of precursors was conducted in a High-Pressure Cell (HPC) of an ultra-high vacuum (UHV) system. A small tablet of the precursor sample, about 10 mm in diameter, was placed on a sample holder and introduced into the HPC. H2 (99.999 vol%) was passed through the sample with a flow rate of 20 cm3 min−1 to obtain the reduced catalyst. The sample was in this experiment only heated to 550 °C because of the instrument limitation. The process was carried out for 5 h. After reduction, the HPC was evacuated, and the sample was transferred under UHV to the analysis chamber of the electron spectrometer.
H2 temperature-programmed desorption was performed on a Micromeritics AutoChem II 2920 instrument. Prior to the analysis, the precursor sample (500 mg) was reduced in situ at 600 °C for 18 h under H2 flow (40 cm3 min−1) and purged at 620 °C for 2 h under Ar flow (40 cm3 min−1). After cooling to 150 °C, H2 was introduced at a flow rate of 40 cm3 min−1 for 15 min, then cooled to 0 °C and kept at that temperature for 15 min. Next, the sample was purged with Ar at 0 °C until the baseline was stable. H2-TPD was carried out in a flow of Ar (40 cm3 min−1) with a ramp of 5 °C min−1 up to 800 °C, and the TCD signal was recorded continuously. The error in the amount of H2 desorbed is ± 3%.
N2 temperature-programmed desorption was carried out on a Micromeritics AutoChem II 2920 instrument. Prior to the analysis, the precursor sample (500 mg) was reduced in situ at 600 °C for 18 h under H2 flow (40 cm3 min−1) and purged at 620 °C for 2 h under He flow (40 cm3 min−1). After cooling to 200 °C, N2 was introduced at a flow rate of 40 cm3 min−1 for 14 h and cooled to 0 °C. Next, the sample was purged with He at 0 °C until the baseline was stable. N2-TPD was carried out in a flow of He (40 cm3 min−1) with a ramp of 5 °C min−1 up to 800 °C, and the TCD signal was recorded continuously.
CO2 temperature-programmed desorption was performed on a Micromeritics AutoChem II 2920 instrument. Prior to the analysis, the precursor sample (500 mg) was reduced in situ at 600 °C for 18 h under H2 flow (40 cm3 min−1) and purged at 620 °C for 2 h under He flow (40 cm3 min−1). After cooling to 40 °C, CO2 was introduced at a flow of 40 cm3 min−1 for 2 h. Next, the sample was purged with He flow at 40 °C until the baseline was stable. CO2-TPD was carried out in a flow of He (40 cm3 min−1) with a ramp of 5 °C min−1 up to 700 °C, and the TCD signal was recorded continuously. The error in the amount of CO2 desorbed is ± 3%.
2.3. Catalytic activity evaluation
Ammonia synthesis activity was evaluated in a tubular flow reactor.51 Prior to the catalytic test, the sample (500 mg) was reduced in situ at the following temperatures for the time indicated in the brackets: 470 °C (72 h); 520 °C (24 h); 550 °C (48 h); 600 °C (72 h) in a mixture of 75% H2 and 25% N2 (70 dm3 h−1) at atmospheric pressure. After the catalyst activation, the system was pressurised to 6.3 or 9.0 MPa in the reaction mixture (H2 + N2 or H2 + N2 + NH3) and heated to different target temperatures (370, 400, 430, and 470 °C). The resulting NH3 gas was analysed interferometrically. The ammonia synthesis rate was calculated from the mass balance for a plug-flow differential reactor, as defined by the equation described in ref. 46. The error of the determined reaction rate is below 1%.46 A thermal stability test was performed by repeating the catalytic test at 370 °C and 9.0 MPa through overheating at 600 °C for three days in the reaction mixture (H2 + N2) at atmospheric pressure.3. Results and discussion
3.1. Catalytic behaviour in ammonia synthesis
First, the effect of the amount of Ca and Ba on the performance of Co catalysts for ammonia synthesis was systematically examined (Fig. 1). The catalysts doped with calcium displayed similar activity to the undoped Co catalyst across the examined Ca content range. Compared with the Ca-doped catalysts, the Ba-doped catalysts showed much greater ammonia synthesis activities across the examined Ba content range. The ammonia synthesis rate gradually increased from 2.0 to 9.6 gNH3 gcat−1 h−1 when the Ba content increased from 0 to 5 wt%. A further increase in the Ba content from 5 to 9 wt% caused a decrease in the catalyst activity. | ||
| Fig. 1 Effect of the amount of Ca and Ba on the performance of Co catalysts for ammonia synthesis at 470 °C and 6.3 MPa. | ||
As discussed earlier, the role of Ca and Ba in ammonia synthesis is an ongoing debate. Therefore, in this work, the Co, 5Ca–Co and 5Ba–Co catalysts were selected to study the effect of Ca and Ba addition on the activity and stability of the Co ammonia synthesis catalyst. The doubly doped catalyst was designed to study if Ca and Ba can synergistically improve the catalyst activity for ammonia synthesis.
Next, the ammonia synthesis activity of the Co, 5Ca–Co, 5Ba–Co, and (5Ca + 5Ba)–Co catalysts was examined under different reaction conditions (Fig. 2, S2, and S3†). Fig. 2a and S2† show the temperature dependence of the ammonia synthesis rate of the Co catalysts. At 9.0 MPa (Fig. 2a), the 5Ba–Co catalyst showed higher activity than the Co, 5Ca–Co, and (5Ca + 5Ba)–Co catalysts across the examined temperature range (370–470 °C). At 400 °C and 9.0 MPa, 5Ba–Co showed an ammonia synthesis rate of 4.24 gNH3 gcat−1 h−1, which was about 1.5 times higher than that of (5Ca + 5Ba)–Co and 4 times than that of Co and 5Ca–Co (Table 1). Table 1 also shows that 5Ba–Co exhibited a much larger TOF value than Co and 5Ca–Co. The catalytic activity of the Co catalysts remained stable within three days of the test (Fig. S3†).
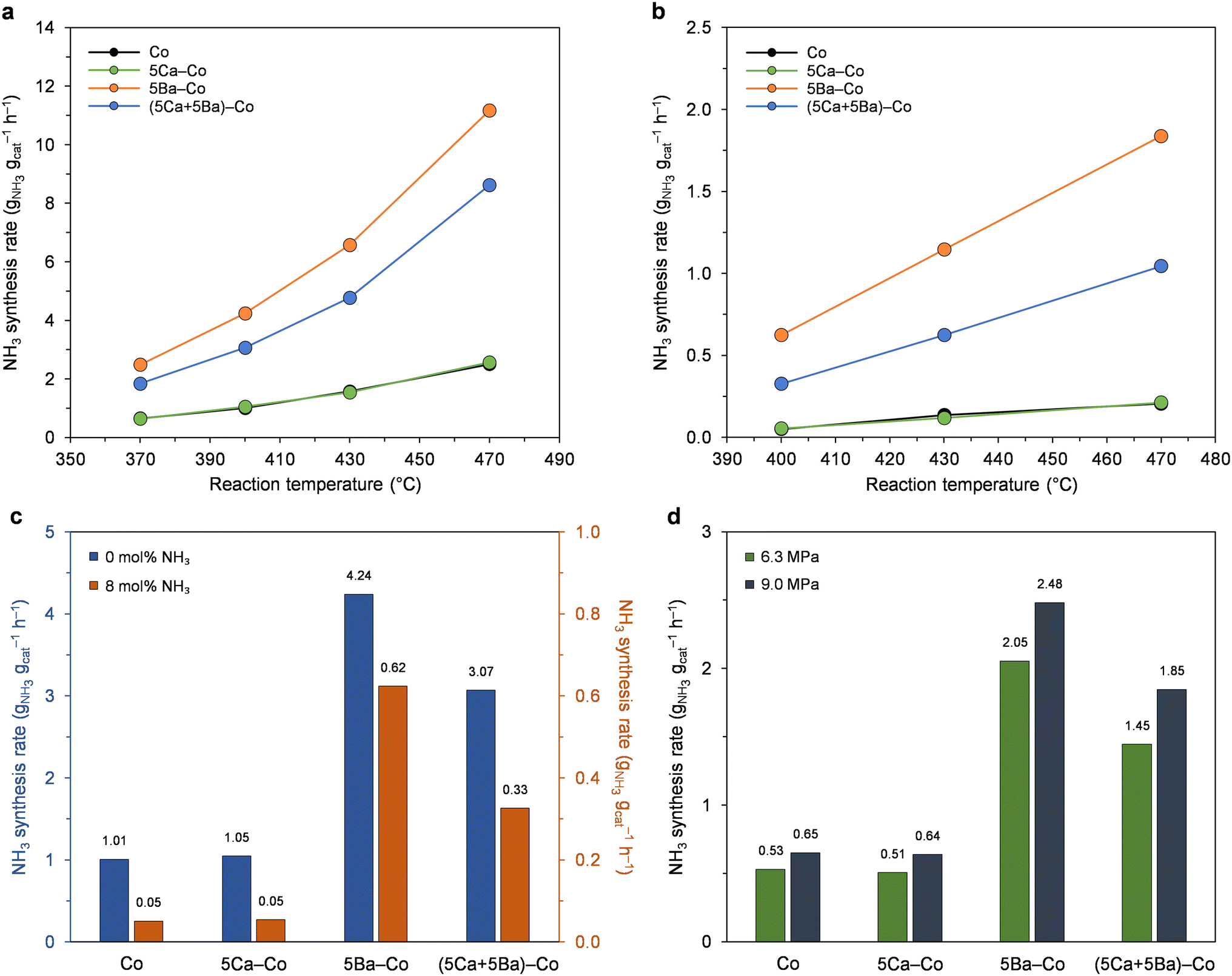 | ||
| Fig. 2 Catalytic performance of Co catalysts in ammonia synthesis. (a) Temperature dependence of the ammonia synthesis rate at 9.0 MPa. The rates were measured in the reaction mixture without ammonia. (b) Temperature dependence of the ammonia synthesis rate at 9.0 MPa. The rates were measured in the reaction mixture with 8 mol% ammonia. (c) Ammonia synthesis rate at 400 °C and 9.0 MPa without (blue bars) and with (orange bars) ammonia in the reaction mixture. (d) Ammonia synthesis rate at 370 °C and 6.3 MPa (green bars) and 9.0 MPa (grey bars). The rates were measured in the reaction mixture without ammonia. | ||
| Catalyst | NH3 synthesis ratea (gNH3 gcat−1 h−1) | TOFb (s−1) | Eappc (kJ mol−1) |
|---|---|---|---|
| a NH3 synthesis rate at 400 °C and 9.0 MPa.b TOF was calculated from the total number of hydrogen adsorption sites (Table 2) and the NH3 synthesis rate shown in Table 1. The error range was ± 4%.c Apparent activation energy calculated from Arrhenius plots (Fig. S4) of the NH3 reaction rate in the temperature range of 370–470 °C under 9.0 MPa (Fig. 2a). The error range was ± 3%. | |||
| Co | 1.01 | 0.050 | 54.0 |
| 5Ca–Co | 1.05 | 0.051 | 55.0 |
| 5Ba–Co | 4.24 | 0.260 | 59.6 |
| (5Ca + 5Ba)–Co | 3.07 | 0.181 | 61.0 |
Fig. S4† presents the Arrhenius plots for ammonia synthesis performed over the Co catalysts. The calculated apparent activation energies (Eapp) were similar and in the range of 54 to 61 kJ mol−1 (Table 1). This implies that the changes in the activity of the Co catalysts are not a result of differences in the apparent activation energies but rather result from the higher intrinsic activity of the active sites.52
The reaction rates significantly decreased when ammonia was introduced to the reaction mixture (Fig. 2b). By comparing the NH3 synthesis rates measured in the reaction mixture without and with the addition of ammonia (Fig. 2c), a significant decrease of 20-fold in the ammonia formation rate for Co and 5Ca–Co was observed. On the contrary, the 5Ba–Co catalyst showed the lowest decrease of 7-fold in ammonia formation rate, indicating that the inhibition of the ammonia formation rate by the product (NH3) is less pronounced in the presence of the barium. Thus, it seems that Ba addition facilitates product (NH3) desorption from the Co surface, increasing the reaction rate, even near equilibrium. Similar conclusions were drawn for a previously reported Ba-doped Co catalyst.48 The TPSR measurements revealed that the presence of the Ba promoter leads to a decrease in the adsorption strength of the NH3 species on the Co, thus increasing the number of active Co sites accessible to the reactants during the reaction. Moreover, the results of the kinetic study revealed that the negative reaction order with respect to NH3 (−1.18) obtained for the undoped Co catalyst was reduced to (−0.92) with the addition of barium promoter.48 These findings suggest that the presence of barium promoter facilitates the desorption of NH3 from the Co surface.
Next, the effect of reaction pressure on the NH3 synthesis rate of the Co catalysts was examined (Fig. 2d). The NH3 synthesis rate increased with the increase of reaction pressure from 6.3 to 9.0 MPa (Fig. 2a and S2†). At 370 °C (Fig. 2d), the ammonia synthesis rate was increased, on average, by 25%, indicating that these catalysts did not suffer from hydrogen inhibition and the N2 activation was promoted with increasing hydrogen partial pressure.53,54
To examine the effects that Ca and Ba have on the ammonia synthesis activity over the Co catalysts, the detailed characterisation of undoped and doped Co catalysts was performed.
3.2. Effect of Ca and Ba addition
First, the chemical and physical properties of Co, 5Ca–Co, 5Ba–Co, and (5Ca + 5Ba)–Co were compared (Table 2). The surface areas of Co and 5Ca–Co were similar, whereas 5Ba–Co and (5Ca + 5Ba)–Co had about 3 and 2 times lower surface area than Co, respectively. Accordingly, the Co particles of 5Ba–Co and (5Ca + 5Ba)–Co were about 2 times larger than those of Co and 5Ca–Co.| Catalyst | Surface areaa (m2 gcat−1) | H2 chemisorptionb (μmol gcat−1) | Particle sizec (nm) | Crystallite sized (nm) | Density of basic sitese (μmol m−2) |
|---|---|---|---|---|---|
| a Estimated from the BET method.b Estimated by using H2-TPD.c Average Co particle size estimated from the STEM-EDX images (Fig. 5–8), and the corresponding particle size histograms are presented in Fig. S7.d Average Co crystallite size estimated from the FWHM value obtained from full pattern refinement utilising the Rietveld method ('size and strain' in High Score Plus software).e Estimated by using CO2-TPD. | |||||
| Co | 23.4 | 164 | 25 | 15 | 7.8 |
| 5Ca–Co | 22.3 | 169 | 26 | 17 | 9.0 |
| 5Ba–Co | 8.4 | 133 | 52 | 25 | 13.1 |
| (5Ca + 5Ba)–Co | 11.5 | 138 | 43 | 24 | 11.7 |
The structure of the Co catalysts was investigated using XRPD. The XRPD patterns of the as-prepared Co catalysts showed the presence of MgCo2O4 (cubic, PDF 04-020-2004), MgO (cubic, PDF 01-074-1225) and small amounts of LaCoO3 (rhombohedral, PDF 04-007-8070) and/or La2(CO3)O2 (orthorhombic, PDF 04-012-3839). Additionally, all the doped catalyst precursors showed a high content of Mg(OH)2 (hexagonal, 04-011-5938). Besides that, for the 5Ba–Co and (5Ca + 5Ba)–Co catalysts, the Ba(NO3)2 phase (cubic, PDF 00-024-0053) was observed (Fig. S5†).
After the reduction in hydrogen at 600 °C, the MgO phase (cubic, PDF 01-074-1225) was the main constituent of the Co catalysts (Fig. 3). In all the catalysts, the concentration of MgO reached about 50 wt%. The catalysts also showed the presence of Co metal (cubic, PDF 01-077-7452) and La2O3 (hexagonal, PDF 04-007-0681). For the Ca-doped catalysts, that is, 5Ca–Co and (5Ca + 5Ba)–Co, the CaO phase (cubic, PDF 01-080-7710) was observed. The composition of catalysts with barium, 5Ba–Co and (5Ca + 5Ba)–Co, was complex. Barium in the form of BaLa2O4 (orthorhombic, PDF 00-042-1500) was observed in a small amount in both these samples. Traces of BaO2 (tetragonal, PDF 04-001-7134) could also occur. Supposedly the second Co metal phase (hexagonal, PDF 04-015-9337) was present in the doubly doped catalyst. Some low-intensity reflections were left unassigned, indicating the possibility of other crystalline phases. The concentrations of the identified crystallographic phases are presented in Table 3.
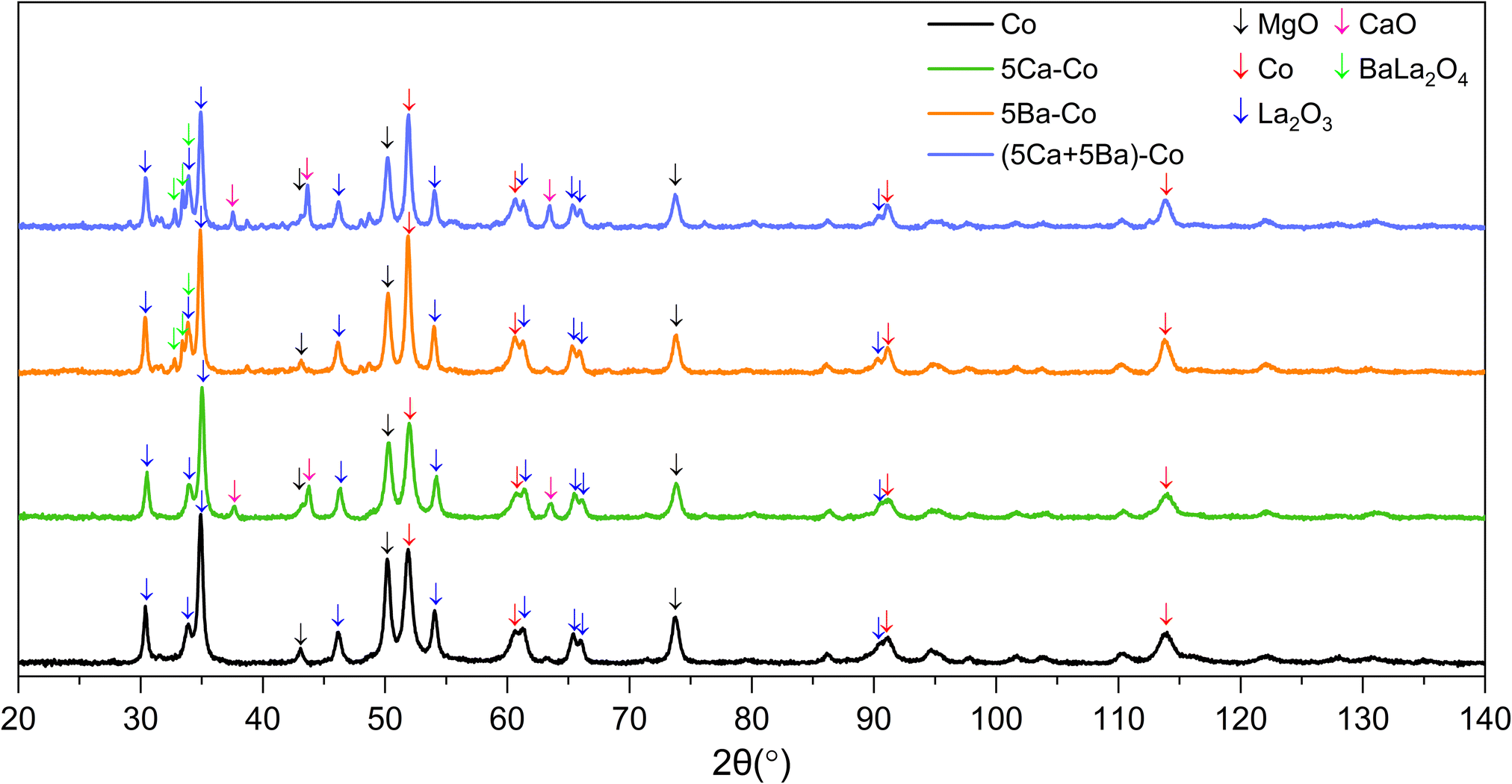 | ||
| Fig. 3 Diffractograms of the Co catalysts collected under N2 flow at 25 °C. | ||
| Catalyst | MgO | Co | La2O3 | CaO | BaLa2O4 | Traces (uncertain) |
|---|---|---|---|---|---|---|
| Co | 50 | 33 | 17 | — | — | — |
| 5Ca–Co | 49 | 29 | 16 | 6 | — | — |
| 5Ba–Co | 46 | 32 | 18 | — | 3 | BaO2 (tetragonal) |
| (5Ca + 5Ba)–Co | 43 | 26 | 15 | 9 | 4 | Co (hexagonal), BaO2 (tetragonal) |
The measured average Co crystallite size was the smallest for the Co and 5Ca–Co catalysts and the largest for the 5Ba–Co and (5Ca + 5Ba)–Co catalysts, in agreement with the STEM results (Table 2). The absolute values were, however, lower for XRPD measurements since particles observed by microscopy might have been composed of multiple crystallites.
Next, the surface structure was examined by STEM/TEM observations and elemental distribution mapping (Fig. 4–8 and Fig. S6 and S7†). The TEM observations revealed that the morphologies of the Co and 5Ca–Co catalysts differed from those of 5Ba–Co and (5Ca + 5Ba)–Co. Fig. 4 shows that the Co and 5Ca–Co catalysts contained rather regular particles of 30–50 nm with a spherical shape, while the 5Ba–Co and (5Ca + 5Ba)–Co catalysts possessed irregularly shaped particles with various diameters of 50–150 nm. EDX studies (Fig. 5–8) were performed in STEM mode to determine the distribution of Ca and Ba elements. For the 5Ca–Co catalyst, small Ca-containing precipitates were observed (Fig. 6), while for the 5Ba–Co catalyst, the encapsulation of the Co nanoparticles with Ba compounds was clearly observed (Fig. 7). For the (5Ca + 5Ba)–Co catalyst, Ca compounds were presented in the form of nanoparticles with a fairly large diameter of 50 nm and more, while the Co nanoparticles were encapsulated with Ba compounds (Fig. 8).
 | ||
| Fig. 4 TEM images of the Co catalysts. (a) Co. (b) 5Ca–Co. (c) 5Ba–Co. (d) (5Ca + 5Ba)–Co. | ||
 | ||
| Fig. 5 STEM-HAADF image (a) of the Co catalyst and corresponding EDX distribution maps of (b) O, (c) Mg, (d) La, and (e) Co. | ||
 | ||
| Fig. 6 STEM-HAADF image (a) of the 5Ca–Co catalyst and corresponding EDX distribution maps of (b) O, (c) Mg, (d) La, (e) Co, (f) Ca, and (g) Ca and Co. | ||
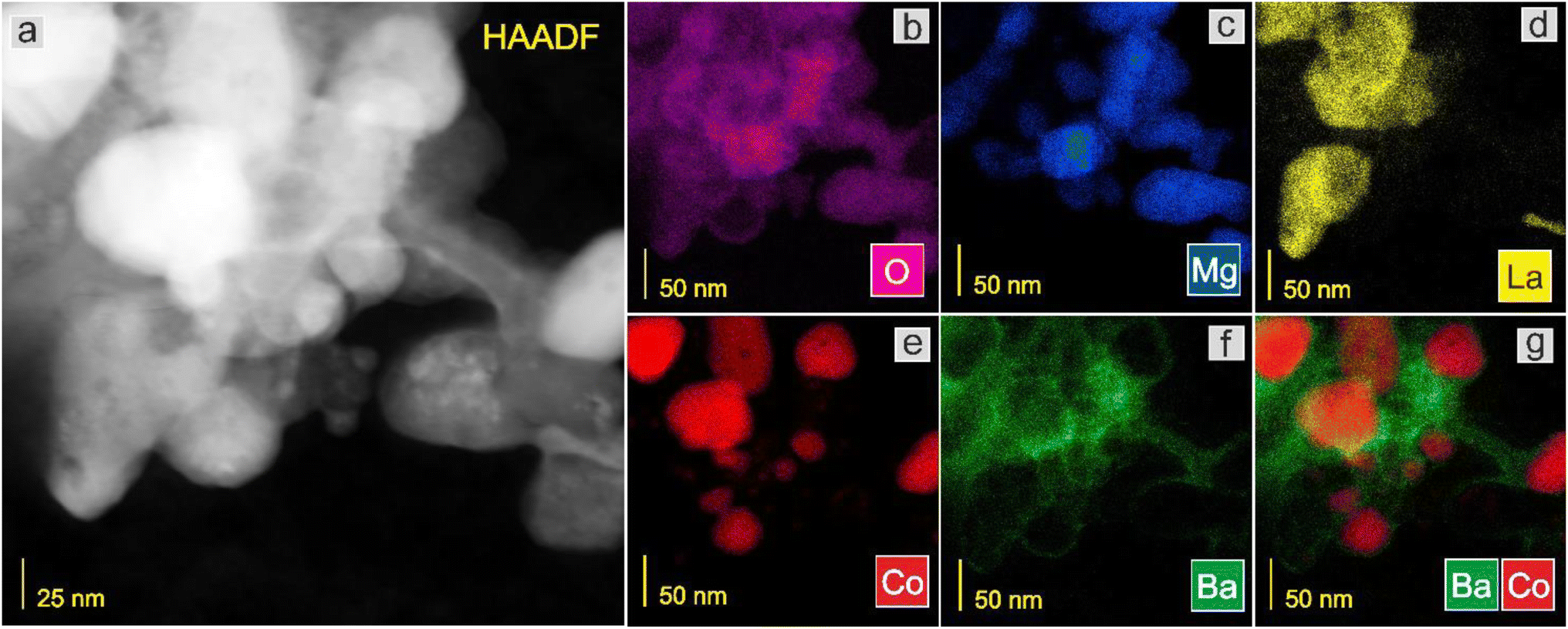 | ||
| Fig. 7 STEM-HAADF image (a) of the 5Ba–Co catalyst and corresponding EDX distribution maps of (b) O, (c) Mg, (d) La, (e) Co, (f) Ba, and (g) Ba and Co. | ||
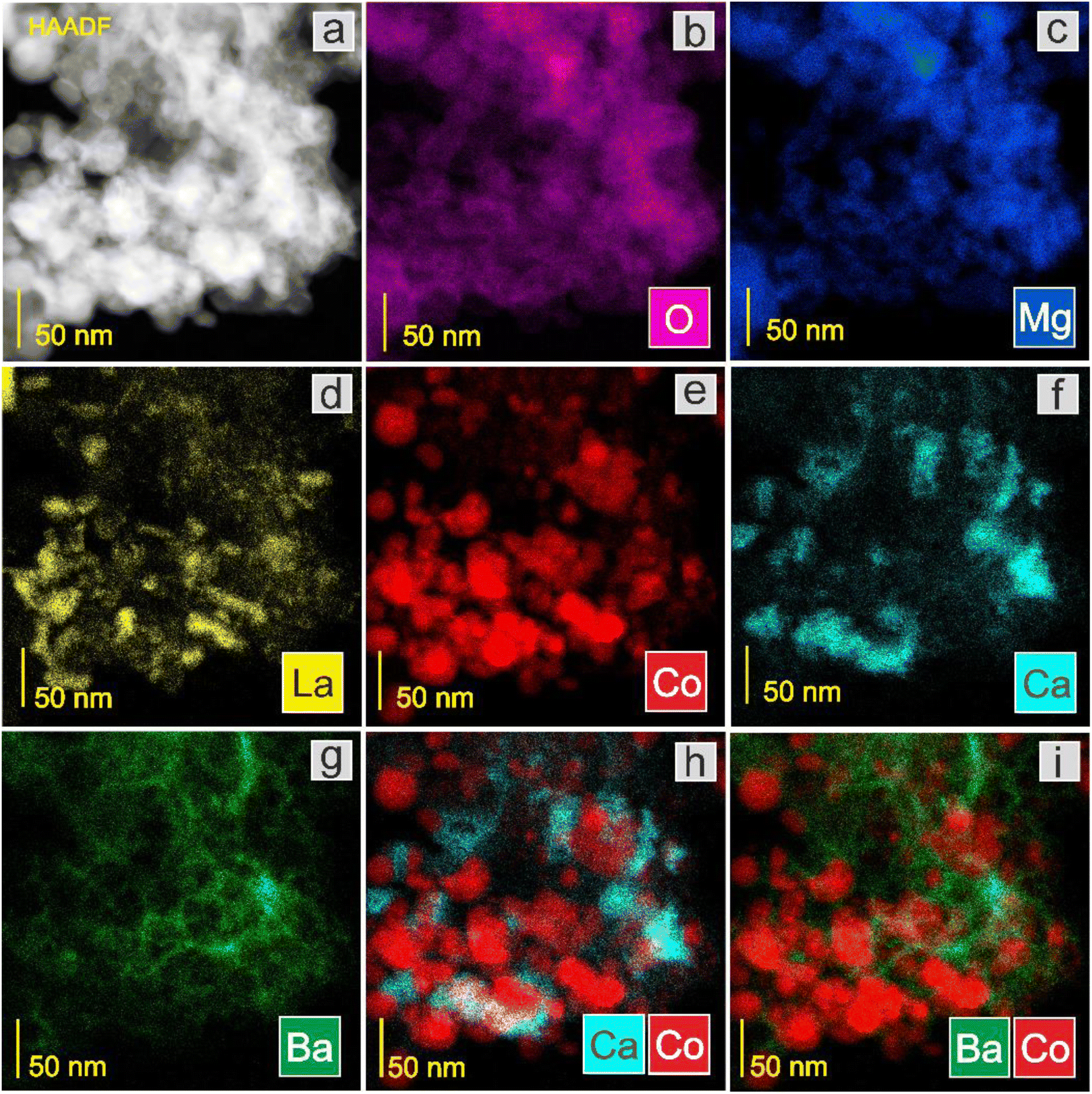 | ||
| Fig. 8 STEM-HAADF image (a) of the (5Ca + 5Ba)–Co catalyst and corresponding EDX distribution maps of (b) O, (c) Mg, (d) La, (e) Co, (f) Ca, (g) Ba, (h) Ca and Co, and (i) Ba and Co. | ||
The surface composition of the catalyst was determined using X-ray photoelectron spectroscopy. Initially, the surface was examined for the catalyst precursors. Next, the catalyst precursors were exposed to hydrogen for 5 h at 550 °C. Oxygen, magnesium, lanthanum and cobalt were found on the surface of all samples. The presence of barium and/or calcium was demonstrated for the catalysts doped with these elements. The K atoms were observed on the catalyst surfaces after the reduction process. This was due to the use of potassium carbonate as a precipitating agent in the support preparation process.45 Quantitative surface composition analysis was performed based on the intensity of the corresponding XPS lines. A simplified model of the surface structure, assuming a homogeneous distribution of all elements in a near-surface region, was used to calculate the surface concentration of the main identified elements. The derived atomic concentrations were converted to weight concentrations and are shown in Table 4.
| Sample | O | Mg | La | Co | Ba | Ca | |
|---|---|---|---|---|---|---|---|
| Co | Precursor | 34 | 15 | 28 | 23 | — | — |
| Catalyst | 21 | 19 | 21 | 32 | — | — | |
| 5Ca–Co | Precursor | 55 | 13 | 11 | 12 | — | 9 |
| Catalyst | 22 | 14 | 21 | 29 | — | 3 | |
| 5Ba–Co | Precursor | 48 | 18 | 18 | 9 | 7 | — |
| Catalyst | 25 | 19 | 9 | 7 | 32 | — | |
| (5Ca + 5Ba)–Co | Precursor | 58 | 10 | 10 | 7 | 5 | 10 |
| Catalyst | 22 | 9 | 8 | 4 | 46 | 2 |
The activation process resulted in a significant decrease in the oxygen content on the surface of the catalysts, mainly due to the reduction/decomposition of oxides, nitrates, and carbonates present in the precursors (Fig. S5†). The weight concentration of magnesium in the near-surface region varied depending on the dopant type. However, it did not generally change after the reduction process for each sample. It might indicate that the reduction process of catalyst precursors occurs so that the concentration of magnesium atoms (generally bound in magnesium oxide MgO) is constant in the surface region. Due to the nature of surface analysis by electron spectroscopy methods, this result also implies that the spatial distribution of magnesium ions in the near-surface region of the materials did not change.
A rather contrasting picture is drawn based on the analysis of the surface concentration of lanthanum. For the undoped catalyst, the lanthanum concentration reached 28 wt% in the precursor and slightly decreased to 21 wt% after the reduction process. For the 5Ca–Co catalyst, the concentration of lanthanum in the region analysed by XPS was only 11 wt% for the precursor but increased almost twice after the reduction process. This result indicates either the diffusion of lanthanum atoms into the near-surface layer or the unveiling of this element initially screened by atoms of another located on top of them. Analysis of the calcium concentration indicates that a significant portion of this element was removed from the volume analysed by XPS. With these two pieces of information, it can be suggested that the calcium atoms initially present on the support surface were partially removed, exposing the lanthanum compounds from deeper layers. Possibly, Ca ions diffused into the bulk of the support. Analysis of the lanthanum concentration on the surface of 5Ba–Co showed opposite changes observed for 5Ca–Co. The initial concentration of lanthanum atoms was 18 wt%, eventually decreasing to 9 wt% after the reduction of the precursor. At the same time, for 5Ba–Co, a very high (from 7 to 32 wt%) increase in barium concentration was observed in the volume analysed by XPS. The observed changes can be explained by the intense diffusion of barium atoms to the surface, indicating that these atoms were largely deposited above the lanthanum atoms, screening La 3d electrons observed in the XPS spectrum. The change in lanthanum concentration was negligible (decrease from 10 to 8 wt%) for (5Ca + 5Ba)–Co. The calcium concentration in the near-surface layer of (5Ca + 5Ba)–Co decreased from 10 to only 2 wt%, while barium concentration increased from only 5 to 46 wt%. Presumably, the diffusion processes described above for the 5Ca–Co and 5Ba–Co catalysts occurred simultaneously in the catalyst doubly doped with barium and calcium.
Another issue is the behaviour of cobalt during the precursor reduction. Fig. 9 shows the binding energy region containing spectra of the Co 3p and Mg 2p photoelectron lines. The intensity of these spectra has been normalised to the intensity observed at the maximum of the XPS Mg 2p line. The maxima of the Co 3p lines observed for all the catalysts were located at a binding energy of about 59 eV. This value is characteristic of metallic cobalt.55 The Co 3p line intensity in relation to the Mg 2p line varied depending on the dopant type. The highest relative intensity was observed for the undoped catalyst. The weight percentage of Co in the near-surface region given by XPS (32 wt%; Table 4) is quite similar to the bulk composition given by XRF (44 wt%; Table S1, entry 1†). This suggests the homogeneous distribution of Co on the support surface and excludes surface segregation. For the 5Ca–Co catalyst, the metallic cobalt accounted for 29 wt%. The surface composition of the Co and 5Ca–Co catalysts seems to be very similar, which is even observed in their almost identical physicochemical properties. On the contrary, the Ba-doped catalysts showed a low cobalt concentration. In the 5Ba–Co catalyst, cobalt constituted 7 wt%, whereas, in the (5Ca + 5Ba)–Co catalyst, cobalt accounted for only 4 wt% of the near-surface region. These values and a significant reduction in the ratio of the intensity of the Co 3p lines to that of the Mg 2p lines (Fig. 9) lead to the conclusion that barium atoms covered the surface of the cobalt nanoparticles during the precursor reduction process.
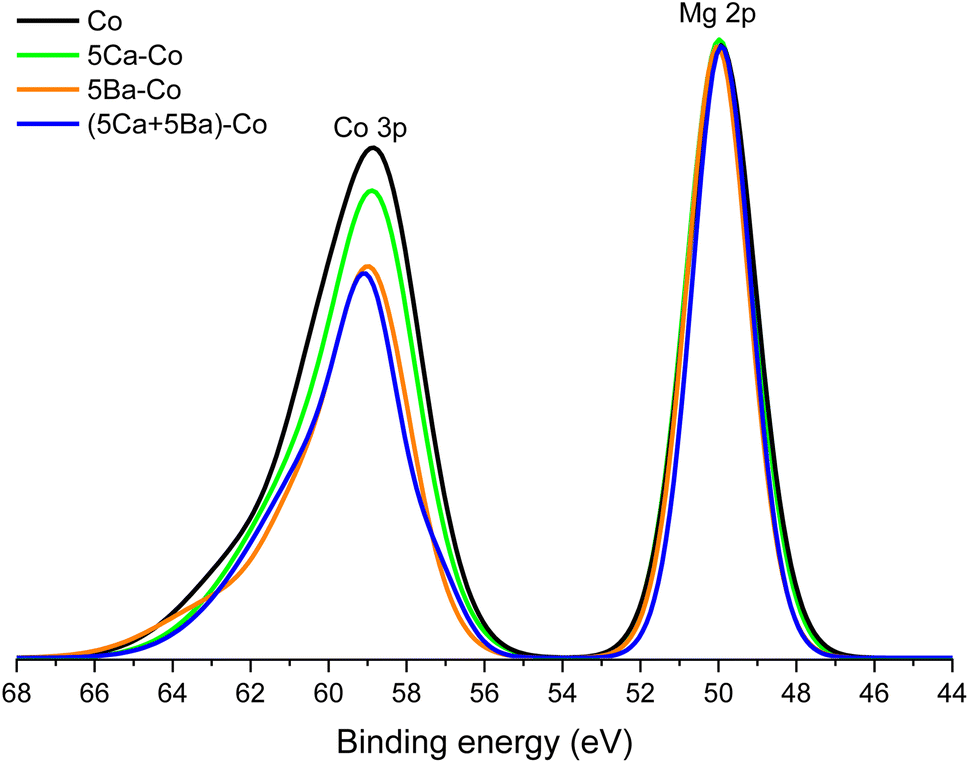 | ||
| Fig. 9 Envelope of XPS Co 3p and Mg 2p transitions acquired for the Co catalysts. | ||
Next, the formation mechanism of the Co(core)–BaO(shell) structures observed for 5Ba–Co and (5Ca + 5Ba)–Co was addressed. The obtained results can be interpreted in terms of a surface enrichment effect.41 Barium in the catalyst precursor mainly existed in the form of nitrate (Fig. S5†). During the reduction step, together with the Ba(NO3)2 decomposition, Ba(OH)2 could form and decompose to BaO.43 Because the Ba compounds generally have lower melting points than their Ca counterparts, they tend to segregate to the surface, partially covering the surface of Co NPs (Table 4 and Fig. 7 and 8) and decomposing to form BaO.43 The formation of such core–shell structures partially covering the Co surface was considered to markedly accelerate the cleavage of the triple NN bond.43 Calcium in the catalyst precursor mainly existed in the form of nitrate, which melted and decomposed to CaO. Ca(OH)2 could also form and decompose to CaO (Fig. 3). As mentioned earlier, compared with Ba compounds, the Ca compounds have higher melting points, thus exhibiting a decreased tendency to segregate to the surface. Therefore, calcium in the form of CaO formed agglomerates (Fig. 3, 6, and 8). Calcium could also be incorporated into the MgO oxide structure because of the similar ionic radii of Ca2+ (100 pm) and Mg2+ (72 pm).56,57 The schematic representations of the proposed surface structures for the Co catalysts are presented in Scheme 1.
 | ||
| Scheme 1 Schematic representations of the proposed surface structures for the Co catalysts. (a) Co. (b) 5Ca–Co. (c) 5Ba–Co. (d) (5Ca + 5Ba)–Co. | ||
Next, the adsorption properties of the Co catalysts were examined by TPD, and the hydrogen TPD and nitrogen TPD profiles are shown in Fig. 10. There were three types of hydrogen desorption peaks in the TPD profiles (Fig. 10a). The low-temperature (at about 100 °C) and medium-temperature (at about 520 °C) peaks were due to the desorption of hydrogen that was weakly and intermediately bound to the surface Co sites, respectively. The presence of the desorption peak above 700 °C could be associated with a strong chemisorption state of hydrogen on the catalysts.45 Two distinct hydrogen desorption peaks were observed for the undoped Co catalyst at 109 and 765 °C. With the addition of Ca, the peak positions almost remained unchanged, indicating that Ca had a minor effect on the nature of active Co sites. The hydrogen TPD profiles changed in terms of both the peak position and intensity in the presence of barium, manifesting the electronic interactions between the Ba and Co. Three desorption peaks were observed for 5Ba–Co at 101, 520, and 771 °C. This means that the Ba addition had a major effect on the nature of active Co sites. Notably, the electronic interactions between Ba and Co played an important role, providing new active Co sites for hydrogen adsorption and dissociation. It also modulated, in this case, decreased the adsorption strength of Co towards hydrogen, as reflected by shifting of the peaks to lower temperatures. For the (5Ca + 5Ba)–Co catalyst, the hydrogen TPD profile was similar to that observed for 5Ba–Co, indicating that barium played a dominant role in modulating the affinity of Co towards hydrogen. The observed decrease in the number of active Co surface sites for hydrogen adsorption and dissociation in 5Ba–Co and (5Ca + 5Ba)–Co (Table 2) suggests that Ba covered (wetted) the Co surface. These results agree with our previous results and literature data.41–43
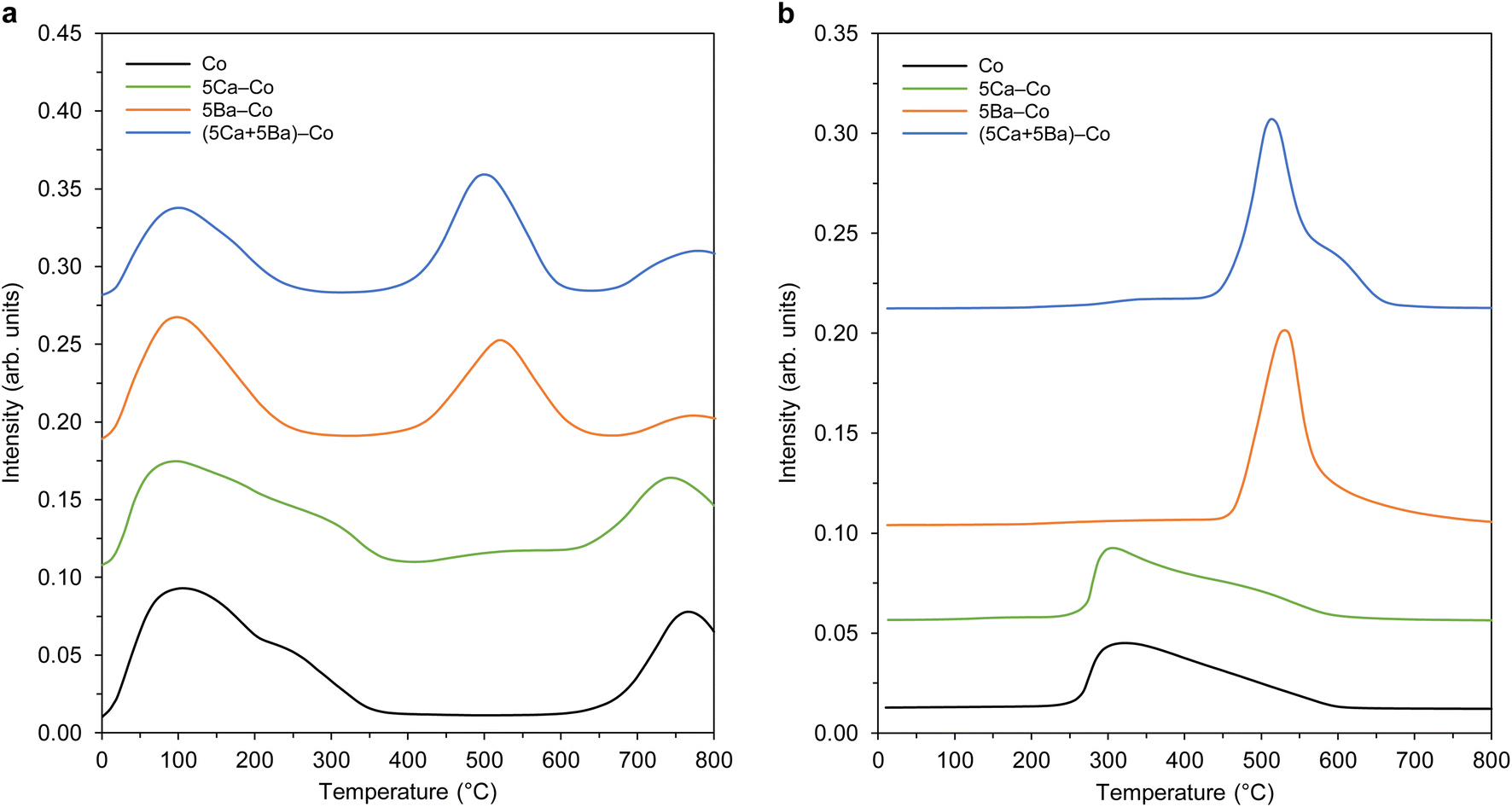 | ||
| Fig. 10 TPD profiles of the Co catalysts. (a) Hydrogen TPD profiles. (b) Nitrogen TPD profiles. | ||
For the nitrogen TPD profiles (Fig. 10b), one broad desorption peak was observed for the undoped Co catalyst in the temperature range of 230–600 °C. Similar to the hydrogen adsorption behaviour, the Ca addition did not affect the nature of active Co sites. On the contrary, the Ba addition caused significant changes in the nitrogen adsorption properties. A distinct desorption peak at 500 °C was observed for 5Ba–Co. This means that the presence of barium affected both the nature and number of the active sites for nitrogen adsorption and dissociation. Both the number of active Co sites and their adsorption strength towards nitrogen increased. For (5Ca + 5Ba)–Co, as it was for hydrogen TPD studies, the nitrogen TPD profile is similar to that observed for 5Ba–Co. This means that barium also played a dominant role in modulating the affinity of Co towards nitrogen. Similar results were obtained for a Co/Ce/Ba catalyst.36
Taken together, the results from the TPD studies (Fig. 10) show that the calcium had a minor effect on the nature of the active sites. On the contrary, the electronic interactions between the Ba promoter and active Co phase modulated the adsorption strengths of hydrogen and nitrogen on the Co surfaces. The modulating role of barium on the electronic nature of Co has also been investigated by Sato et al.43 The spectroscopic and DFT investigations led to the conclusion that N2 adsorption on the Co surface weakens the nitrogen–nitrogen triple bond to the strength of a double bond due to the electron donation from Ba2+ of BaO via adjacent Co atoms. These characteristics resulted in a remarkable increase in the ammonia synthesis activity.43
Next, the carbon dioxide TPD profiles of the Co catalysts were measured to evaluate the surface basicity,58 another important determinant of ammonia-synthesis ability. The carbon dioxide TPD profiles are shown in Fig. 11. In general, three types of basic sites were identified: (1) weak basic sites, with a maximum at about 100 °C, related to the hydroxyl groups on the catalyst surface; (2) medium basic sites, with a maximum in the range of 150–300 °C, assigned to the metal–oxygen pairs; and (3) strong basic sites, with maxima above 400 °C, ascribed to the low-coordinated surface O2− species.59 It is worth noting that only medium and strong basic sites exhibit electron-donating ability, which can donate electrons to Co and promote the dissociative adsorption of nitrogen.60,61 Hence, the presence of these basic sites might be another factor contributing to the catalyst's activity toward ammonia synthesis. With the addition of Ca, the new strong basic sites were generated, as confirmed by the appearance of the desorption peak at 570 °C (Fig. 11). The addition of Ba had a major effect on the basic properties of 5Ba–Co. That is, the number of weak basic sites was increased, accompanied by the generation of new strong basic sites, which was reflected by the appearance of desorption peaks at 414, 489, and 610 °C (Fig. 11). In the case of (5Ca + 5Ba)–Co, the basic property was similar to that of 5Ba–Co, with the difference that the number of strong basic sites was increased in 5Ba–Co. The total amount of CO2 desorbed per surface area unit was used to assess the basicity of the Co catalysts. The basicity of the Co catalysts increased in the order Co (7.8 μmol m−2) < 5Ca–Co (9.0 μmol m−2) < (5Ca + 5Ba)–Co (11.7 μmol m−2) < 5Ba–Co (13.1 μmol m−2) (Table 2).
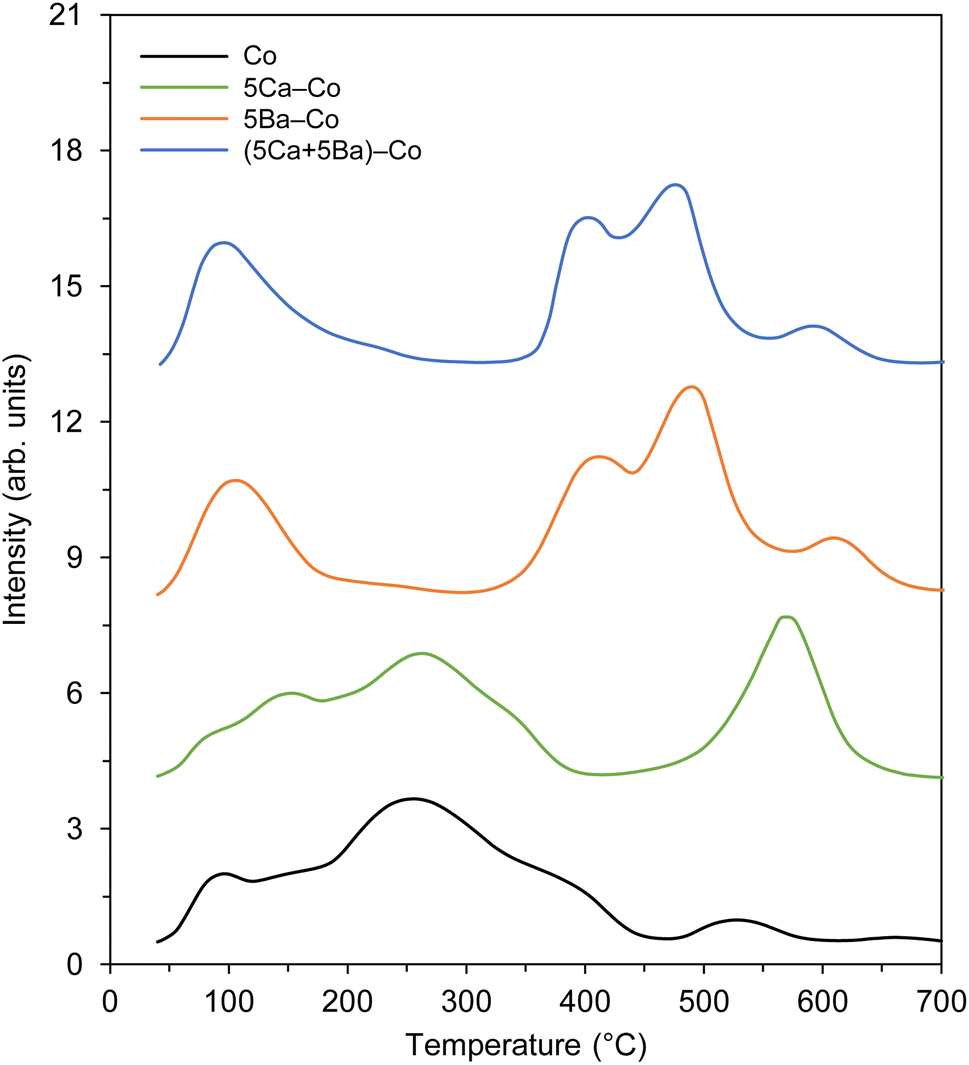 | ||
| Fig. 11 Carbon dioxide TPD profiles of the Co catalysts. | ||
The combination of the different experimental results allows a consistent interpretation, with the conclusions as follows:
(1) The Ca addition did not change the textural, structural and chemisorptive properties of the Ca-doped Co catalyst. Consequently, the activity of the Ca-doped catalyst remained unchanged compared to the undoped Co catalyst (Table 1). This indicates that the role of calcium in the cobalt ammonia synthesis catalyst was ambiguous. However, it appears that Ca was an inactive additive that only had a minor impact on the cobalt catalyst properties. These results contrast with the previous report by Inoue et al.,62 who revealed that the Co catalyst activity could be directly boosted by electron donation from a 12CaO·7Al2O3 electride material. The high catalytic activity was attributed to the enhancement of N2 dissociation by electron injection from 12CaO·7Al2O3 to the nanosized Co particles. This highlights the critical importance of the proper combination of the supports and dopants for their resultant promotion effects.
(2) Ba served as an electronic promoter for Co. It modified the electronic structure of Co, thereby affecting the adsorption properties of the reactants (hydrogen and nitrogen). The Ba addition also contributed to the generation of new basic sites, particularly the strong ones. These all resulted in the formation of a small number of highly active Co sites for ammonia synthesis (average TOF of 0.260 s−1; Table 1). The electronic promotion effect of barium was the reason for the substantial increase in the ammonia synthesis activity observed for the Co catalyst promoted with barium.
(3) The use of both Ca and Ba as the dopants resulted in poorer ammonia synthesis performance compared to the Ba-doped Co catalyst (average TOF of 0.181 s−1; Table 1). This suggests that the presence of Ca was the cause of the poorer performance of the doubly doped Co catalyst. The most likely explanation is the excessive encapsulation of the active Co sites by BaO overlayers (Table 4). The co-presence of barium and calcium salts might favour increased wettability of the cobalt surface by barium compounds during the catalyst reduction, which resulted in the excessive blocking of the active catalytic sites (Table 4).
3.3. Practical considerations
For practical application, superior catalyst activity and stability are important. 5Ba–Co showed good performance in all these areas (Fig. 2, S2, and S3†). Table 5 compares the ammonia synthesis performance of the 5Ba–Co catalyst and the industrial iron catalysts. 5Ba–Co showed higher activity than the conventional magnetite-based catalysts under identical conditions. At 400 °C and 9.0 MPa in the reaction mixture without ammonia, the NH3 synthesis rate of 5Ba–Co was 4.24 gNH3 gcat−1 h−1, which was about 1.5 times that of the Fe catalysts. The difference in activity between these catalysts was even more pronounced when ammonia was introduced to the reaction mixture. The activity of 5Ba–Co was about 2 times that of the Fe catalysts.| Catalyst | NH3 synthesis ratea (gNH3 gcat−1 h−1) | Reference | |
|---|---|---|---|
| 0 mol% NH3 | 8 mol% NH3 | ||
| a NH3 synthesis rate at 400 °C and 9.0 MPa. The reaction rates were measured in the reaction mixture without and with the addition of 8 mol% ammonia. | |||
| 5Ba–Co | 4.24 | 0.63 | This work |
| Industrial Fe catalyst (PS-3-INS) | 2.56 | 0.28 | This work |
| Industrial Fe catalyst (KM1) | 3.09 | 0.33 | 39 |
Although the catalyst cost is often a topic of discussion, in practice, it is a small percentage of the overall cost of ammonia production.63 A calculation described in ref. 63 indicated that for the industrial Fe catalysts, the catalyst cost comprises less than 0.6% of the total cost of ammonia production. For the Ru/C catalyst, the calculations by Yoshida et al.64 showed that catalyst cost comprises less than 9% of the total cost of ammonia production. From this, it can be concluded that utilising the Co catalysts for ammonia synthesis will not be a problem in terms of cost.
The possibility of recycling used catalysts is also an important aspect of the practical application. For the used iron ammonia synthesis catalysts, the recycling is based on reoxidation of the metallic iron to iron oxide, followed by remelting and annealing.63 However, in fact, most of the waste iron catalysts are not recycled because the recycling method is problematic and the cost is high compared with the manufacturing process using cheap natural magnetite.63 Co catalysts have not been commercialised for ammonia synthesis yet. Thus the recycling method for the waste cobalt catalysts from the petrochemical industry was studied. The traditional recycling method is based on the pyrometallurgy and hydrometallurgy processes.65 One such method, developed by Gulf Chemical and Metallurgical Corporation, was designed to recover Mo, V, Ni, and Co from the waste catalysts. The hydrometallurgy process can also recover Co from waste Co–Mn catalysts. Using that method, more than 95 wt% of Co in the catalyst is dissolved, and most of it is precipitated as a high-grade cobalt sulphide.
4. Conclusions
In summary, the effect of calcium and barium addition on the activity of Co catalysts for ammonia synthesis has been investigated. For the Ca-doped Co catalysts, the activity remained unchanged compared to the undoped Co catalyst, regardless of the Ca content. The Ba addition resulted in a significant increase in the ammonia synthesis rate compared to the undoped catalyst. The ammonia synthesis rate increased with increasing Ba content and reached a maximum at 5 wt% Ba. A further increase in the Ba content caused a decrease in the activity. The combination of the different experimental results allows concluding that Ca served as an inactive additive, whereas Ba served as an electronic promoter. The Ca addition did not change the textural, structural and chemisorptive properties of the Ca-doped Co catalyst. On the other hand, the Ba addition had a major effect on the nature of active Co sites. It contributed to the formation of new active sites for hydrogen and nitrogen adsorption and dissociation. Barium addition also contributed to the generation of new basic sites, particularly the strong ones. These unique characteristics were ascribed to the formation of Co(core)–BaO(shell) structures. It was likely that the donation of electrons from the BaO to N2 via Co markedly promoted ammonia synthesis. The performance of the Ba-doped Co ammonia synthesis catalyst was increased by 4 times compared to the undoped catalyst and 2 times compared to the industrial iron catalysts. Taking all this together, it can be concluded that these findings open a new pathway for the preparation of efficient cobalt catalysts for ammonia synthesis under environmentally benign conditions, which is an important step toward realising a sustainable energy economy.Author contributions
H. R. conceived the idea for this study and designed the experiments. H. R. and M. Z. prepared the samples. H. R. and M. Z. performed the gas sorption experiments. H. R., M. Z., W. P., and W. R.-P. performed the catalytic measurements. D. M. and A. A. carried out the XRPD measurements. D. M. performed the XPS measurements. K. S. conducted the STEM and TEM characterisation. A. M. performed the XRF measurements. M. Z., D. M., and W. R.-P. provided suggestions and feedback on the study. H. R. prepared the manuscript with input from M. Z., D. M., and K. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by POB Technologie Materiałowe of Warsaw University of Technology within the Excellence Initiative: Research University (IDUB) programme.References
- G. J. Hutchings, Promotion in Heterogeneous Catalysis: a Topic Requiring a New Approach?, Catal. Lett., 2001, 75, 1–12, DOI:10.1023/A:1016784122682
.
- S. J. Thomson, Introductory lecture. Promotion in heterogeneous catalysis: retrospect and prospect, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 1893–1914, 10.1039/F19878301893
- J. Humphreys, R. Lan and S. Tao, Development and Recent Progress on Ammonia Synthesis Catalysts for Haber–Bosch Process, Adv. Energy Sustainability Res., 2021, 2, 2000043, DOI:10.1002/aesr.202000043
- H. Liu, Ammonia synthesis catalyst 100 years: practice, enlightenment and challenge, Chin. J. Catal., 2014, 35, 1619–1640, DOI:10.1016/S1872-2067(14)60118-2
- V. S. Marakatti and E. M. Gaigneaux, Recent Advances in Heterogeneous Catalysis for Ammonia Synthesis, ChemCatChem, 2020, 12, 5838–5857, DOI:10.1002/cctc.202001141
- A. Nielsen and H. Bohlbro, Investigation on surface properties of reduced iron catalysts for the synthesis of ammonia and correlation with crystal sizes and high pressure conversion activities, J. Am. Chem. Soc., 1952, 74(4), 963–966, DOI:10.1021/ja01124a027
- H. D. Vandervell and K. C. Waugh, On the role of promoters in promoted iron catalysts used in the industrial synthesis of ammonia, Chem. Phys. Lett., 1990, 171, 462–468, DOI:10.1016/0009-2614(90)85247-A
- R. Krabetz and C. Peters, The function of the promotors in the technical ammonia catalyst, Angew. Chem., Int. Ed., 1965, 4, 341–347, DOI:10.1002/anie.196503411
- H. Topsøe, J. A. Dumesic and M. Boudart, Alumina as a textural promoter of iron synthetic ammonia catalysts, J. Catal., 1973, 28, 477–488, DOI:10.1016/0021-9517(73)90141-3
- M. E. Dry, J. A. K. du Plessis and G. M. Leuteritz, The influence of structural promoters on the surface properties of reduced magnetite catalysts, J. Catal., 1966, 6, 194–199, DOI:10.1016/0021-9517(66)90049-2
- D. R. Strongin, S. R. Bare and G. A. Somorjai, The effects of aluminum oxide in restructuring iron single crystal surfaces for ammonia synthesis, J. Catal., 1987, 103, 289–301, DOI:10.1016/0021-9517(87)90121-7
- Z. Kowalczyk, S. Jodzis, J. Środa, R. Diduszko and E. Kowalczyk, Influence of aluminium and potassium on activity and texture of fused iron catalysts for ammonia synthesis, Appl. Catal., A, 1992, 87, 1–14, DOI:10.1016/0926-860X(92)80169-D
- K. Altenburg, H. Bosch, J. G. Van Ommen and P. J. Gellings, The role of potassium as a promoter in iron catalysts for ammonia synthesis, J. Catal., 1980, 66, 326–334, DOI:10.1016/0021-9517(80)90037-8
- G. Ertl, S. B. Lee and M. Weiss, Kinetics of nitrogen adsorption on Fe(111), Surf. Sci., 1982, 114, 515–526, DOI:10.1016/0039-6028(82)90702-6
- D. R. Strongin and G. A. Somorjai, The effects of potassium on ammonia synthesis over iron single-crystal surfaces, J. Catal., 1988, 109, 51–60, DOI:10.1016/0021-9517(88)90184-4
- A. Nielsen, An investigation on promoted iron catalysts for the synthesis of ammonia, J. Gjellerups Forlag, Copenhagen, 1956 Search PubMed
- M. Kitano, Y. Inoue, M. Sasase, K. Kishida, Y. Kobayashi, K. Nishiyama, T. Tada, S. Kawamura, T. Yokoyama, M. Hara and H. Hosono, Self-organized Ruthenium–Barium Core–Shell Nanoparticles on a Mesoporous Calcium Amide Matrix for Efficient Low-Temperature Ammonia Synthesis, Angew. Chem., Int. Ed., 2018, 57, 2648–2652, DOI:10.1002/anie.201712398
- K. Aika and A. Ozaki, Activation of nitrogen by alkali metal-promoted transition metal: III. On the adsorption of nitrogen over the alkali metal-promoted ruthenium catalyst, J. Catal., 1974, 35, 61–65, DOI:10.1016/0021-9517(74)90183-3
- S. Murata and K. Aika, Preparation and characterization of chlorine-free ruthenium catalysts and the promoter effect in ammonia synthesis.: 1. An alumina-supported ruthenium catalyst, J. Catal., 1992, 136, 110–117, DOI:10.1016/0021-9517(92)90110-4
- K. Aika, T. Takano and S. Murata, Preparation and characterization of chlorine-free ruthenium catalysts and the promoter effect in ammonia synthesis: 3. A magnesia-supported ruthenium catalyst, J. Catal., 1992, 136, 126–140, DOI:10.1016/0021-9517(92)90112-U
- K. Aika, Role of alkali promoter in ammonia synthesis over ruthenium catalysts—Effect on reaction mechanism, Catal. Today, 2017, 286, 14–20, DOI:10.1016/j.cattod.2016.08.012
- J. Huang, M. Yuan, X. Li, Y. Wang, M. Li, J. Li and Z. You, Cs-Promoted ruthenium catalyst supported on Ba5Ta4O15 with abundant oxygen vacancies for ammonia synthesis, Appl. Catal., A, 2021, 615, 118058, DOI:10.1016/j.apcata.2021.118058
- M. Guraya, S. Sprenger, W. Raróg-Pilecka, D. Szmigiel, Z. Kowalczyk and M. Muhler, The effect of promoters on the electronic structure of ruthenium catalysts supported on carbon, Appl. Surf. Sci., 2004, 238, 77–81, DOI:10.1016/j.apsusc.2004.05.214
- W. Raróg, Z. Kowalczyk, J. Sentek, D. Skłądanowski and J. Zieliński, Effect of K, Cs and Ba on the kinetics of NH3 synthesis over carbon-based ruthenium catalysts, Catal. Lett., 2000, 68, 163–168, DOI:10.1023/A:1019024629261
- Z. Kowalczyk, S. Jodzis, W. Raróg, J. Zieliński and J. Pielaszek, Effect of potassium and barium on the stability of a carbon-supported ruthenium catalyst for the synthesis of ammonia, Appl. Catal., A, 1998, 173, 153–160, DOI:10.1016/S0926-860X(98)00175-6
- L. Forni, D. Molinari, I. Rossetti and N. Pernicone, Carbon-supported promoted Ru catalyst for ammonia synthesis, Appl. Catal., A, 1999, 185, 269–275, DOI:10.1016/S0926-860X(99)00144-1
- I. Rossetti, N. Pernicone and L. Forni, Promoters effect in Ru/C ammonia synthesis catalyst, Appl. Catal., A, 2001, 208, 271–278, DOI:10.1016/S0926-860X(00)00711-0
- I. Rossetti, F. Mangiarini and L. Forni, Promoters state and catalyst activation during ammonia synthesis over Ru/C, Appl. Catal., A, 2007, 323, 219–225, DOI:10.1016/j.apcata.2007.02.022
- K. Aika, T. Kawahara, S. Murata and T. Onishi, Promoter Effect of Alkali Metal Oxides and Alkali Earth Metal Oxides on Active Carbon-Supported Ruthenium Catalyst for Ammonia Synthesis, Bull. Chem. Soc. Jpn., 1990, 63, 1221–1225, DOI:10.1246/bcsj.63.1221
- S. Ghosh, S. S. Acharyya, T. Kaneko, K. Higashi, Y. Yoshida, T. Sasaki and Y. Iwasawa, Confined Single Alkali Metal Ion Platform in a Zeolite Pore for Concerted Benzene C–H Activation to Phenol Catalysis, ACS Catal., 2018, 8, 11979–11986, DOI:10.1021/acscatal.8b03002
- D. Szmigiel, H. Bielawa, M. Kurtz, O. Hinrichsen, M. Muhler, W. Raróg, S. Jodzis, Z. Kowalczyk, L. Znak and J. Zieliński, The Kinetics of Ammonia Synthesis over Ruthenium-Based Catalysts: The Role of Barium and Cesium, J. Catal., 2002, 205, 205–212, DOI:10.1006/jcat.2001.3431
- K. Aika, K. Shimazaki, Y. Hattori, A. Ohya, S. Ohshima, K. Shirota and A. Ozaki, Support and promoter effect of ruthenium catalyst: I. Characterization of alkali-promoted ruthenium/alumina catalysts for ammonia synthesis, J. Catal., 1985, 92, 296–304, DOI:10.1016/0021-9517(85)90264-7
- M. Zybert, A. Tarka, B. Mierzwa, L. Kępiński and W. Raróg-Pilecka, Promotion effect of lanthanum on the Co/La/Ba ammonia synthesis catalysts—the influence of lanthanum content, Appl. Catal., A, 2016, 515, 16–24, DOI:10.1016/j.apcata.2016.01.036
- M. Karolewska, E. Truszkiewicz, B. Mierzwa, L. Kępiński and W. Raróg-Pilecka, Ammonia synthesis over cobalt catalysts doped with cerium and barium. Effect of the ceria loading, Appl. Catal., A, 2012, 445–446, 28–286, DOI:10.1016/j.apcata.2012.08.028
- W. Raróg-Pilecka, M. Karolewska, E. Truszkiewicz, E. Iwanek and B. Mierzwa, Cobalt Catalyst Doped with Cerium and Barium Obtained by Co-Precipitation Method for Ammonia Synthesis Process, Catal. Lett., 2011, 141, 678–684, DOI:10.1007/s10562-011-0564-8
- A. Tarka, W. Patkowski, M. Zybert, H. Ronduda, P. Wieciński, P. Adamski, A. Sarnecki, D. Moszyński and W. Raróg-Pilecka, Synergistic Interaction of Cerium and Barium-New Insight into the Promotion Effect in Cobalt Systems for Ammonia Synthesis, Catalysts, 2020, 10, 658, DOI:10.3390/catal10060658
- A. Tarka, M. Zybert, H. Ronduda, W. Patkowski, B. Mierzwa, L. Kępiński and W. Raróg-Pilecka, On Optimal Barium Promoter Content in a Cobalt Catalyst for Ammonia Synthesis, Catalysts, 2022, 12, 199, DOI:10.3390/catal12020199
- S. Hagen, R. Barfod, R. Fehrmann, C. J. H. Jacobsen, H. T. Teunissen, K. Ståhl and I. Chorkendorff, New efficient catalyst for ammonia synthesis: barium-promoted cobalt on carbon, Chem. Commun., 2002, 11, 1206–1207, 10.1039/B202781J
- W. Raróg-Pilecka, E. Miśkiewicz, L. Kępiński, Z. Kaszkur, K. Kielar and Z. Kowalczyk, Ammonia synthesis over barium-promoted cobalt catalysts supported on graphitised carbon, J. Catal., 2007, 249, 24–33, DOI:10.1016/j.jcat.2007.03.023
- A. Tarka, M. Zybert, E. Truszkiewicz, B. Mierzwa, L. Kępiński, D. Moszyński and W. Raróg-Pilecka, Effect of a Barium Promoter on the Stability and Activity of Carbon-Supported Cobalt Catalysts for Ammonia Synthesis, ChemCatChem, 2015, 7, 2836–2839, DOI:10.1002/cctc.201500309
- M. Zybert, M. Wyszyńska, A. Tarka, W. Patkowski, H. Ronduda, B. Mierzwa, L. Kępiński, A. Sarnecki, D. Moszyński and W. Raróg-Pilecka, Surface enrichment phenomenon in the Ba-doped cobalt catalyst for ammonia synthesis, Vacuum, 2019, 168, 108831, DOI:10.1016/j.vacuum.2019.108831
- B. Lin, Y. Liu, L. Heng, J. Ni, J. Lin and L. Jiang, Effect of barium and potassium promoter on Co/CeO2 catalysts in ammonia synthesis, J. Rare Earths, 2018, 36, 703–707, DOI:10.1016/j.jre.2018.01.017
- K. Sato, S.-I. Miyahara, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura, K. Inazu, H. Mohri, T. Iwasa, T. Taketsugu and K. Nagaoka, Barium Oxide Encapsulating Cobalt Nanoparticles Supported on Magnesium Oxide: Active Non-Noble Metal Catalysts for Ammonia Synthesis under Mild Reaction Conditions, ACS Catal., 2021, 11, 13050–13061, DOI:10.1021/acscatal.1c02887
- A. Cao, V. J. Bukas, V. Shadravan, Z. Wang, H. Li, J. Kibsgaard, I. Chorkendorff and J. K. Nørskov, A spin promotion effect in catalytic ammonia synthesis, Nat. Commun., 2022, 13, 2382, DOI:10.1038/s41467-022-30034-y
- H. Ronduda, M. Zybert, W. Patkowski, A. Tarka, P. Jodłowski, L. Kępiński, A. Sarnecki, D. Moszyński and W. Raróg-Pilecka, Tuning the catalytic performance of Co/Mg-La system for ammonia synthesis via the active phase precursor introduction method, Appl. Catal., A, 2020, 598, 117553, DOI:10.1016/j.apcata.2020.117553
- H. Ronduda, M. Zybert, W. Patkowski, A. Tarka, A. Ostrowski and W. Raróg-Pilecka, Kinetic studies of ammonia synthesis over a barium-promoted cobalt catalyst supported on magnesium–lanthanum mixed oxide, J. Taiwan Inst. Chem. Eng., 2020, 114, 241–248, DOI:10.1016/j.jtice.2020.09.020
- H. Ronduda, M. Zybert, W. Patkowski, A. Ostrowski, P. Jodłowski, D. Szymański, L. Kępiński and W. Raróg-Pilecka, Boosting the Catalytic Performance of Co/Mg/La Catalyst for Ammonia Synthesis by Selecting a Pre-Treatment Method, Catalysts, 2021, 11, 941, DOI:10.3390/catal11080941
- H. Ronduda, M. Zybert, W. Patkowski, A. Ostrowski, P. Jodłowski, D. Szymański, L. Kępiński and W. Raróg-Pilecka, A high performance barium-promoted cobalt catalyst supported on magnesium–lanthanum mixed oxide for ammonia synthesis, RSC Adv., 2021, 11, 14218–14228, 10.1039/D1RA01584B
- H. Ronduda, M. Zybert, W. Patkowski, A. Ostrowski, P. Jodłowski, D. Szymański, L. Kępiński and W. Raróg-Pilecka, Development of cobalt catalyst supported on MgO–Ln2O3 (Ln = La, Nd, Eu) mixed oxide systems for ammonia synthesis, Int. J. Hydrogen Energy, 2022, 47, 6666–6678, DOI:10.1016/j.ijhydene.2021.12.022
- H. Ronduda, M. Zybert, W. Patkowski, K. Sobczak, D. Moszyński, A. Albrecht, A. Sarnecki and W. Raróg-Pilecka, On the effect of metal loading on the performance of Co catalysts supported on mixed MgO–La2O3 oxides for ammonia synthesis, RSC Adv., 2022, 12, 33876–33888, 10.1039/D2RA06053A
- Z. Kowalczyk, Effect of potassium on the high pressure kinetics of ammonia synthesis over fused iron catalyst, Catal. Lett., 1996, 37, 173–179, DOI:10.1007/BF00807750
- S. Hagen, R. Barfod, R. Fehrmann, C. J. H. Jacobsen, H. T. Teunissen and I. Chorkendorff, Ammonia synthesis with barium-promoted iron–cobalt alloys supported carbon, J. Catal., 2003, 214, 327–335, DOI:10.1016/S0021-9517(02)00182-3
- B. Lin, L. Heng, B. Fang, H. Yin, J. Ni, X. Wang, J. Lin and L. Jiang, Ammonia Synthesis Activity-Supported Ruthenium Catalyst Enhanced by Alumina Phase Transformation, ACS Catal., 2019, 9, 1635–1644, DOI:10.1021/acscatal.8b03554
- B. Fang, F. Liu, C. Zhang, C. Li, J. Ni, X. Wang, J. Lin, B. Lin and L. Jiang, Sacrificial Sucrose Strategy Achieved Enhancement of Ammonia Synthesis Activity over a Ceria-Supported Ru Catalyst, ACS Sustainable Chem. Eng., 2021, 9, 8962–8969, DOI:10.1021/acssuschemeng.1c01275
- A. Sarnecki, P. Adamski, A. Albrecht, A. Komorowska, M. Nadziejko and D. Moszyński, XPS study of cobalt-ceria catalysts for ammonia synthesis—The reduction process, Vacuum, 2018, 155, 434–438, DOI:10.1016/j.vacuum.2018.06.034
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767, DOI:10.1107/S0567739476001551
- D. Müller, C. Knoll, T. Ruh, W. Artner, J. M. Welch, H. Peterlik, E. Eitenberger, G. Friedbacher, M. Harasek, P. Blaha, K. Hradil, A. Werner and P. Weinberger, Calcium Doping Facilitates Water Dissociation in Magnesium Oxide, Adv. Sustainable Syst., 2018, 2, 1700096, DOI:10.1002/adsu.201700096
- X. Wang, L. Lv, Q. Zhang, Y. Zhang, J. Wang and M. Shen, The different NOx trap performance on ceria and barium/ceria containing LNT catalysts below 200 °C, Catal. Sci. Technol., 2013, 3, 200–207, 10.1039/C2CY20547E
- M. Gu, S. Dai, R. Qiu, M. E. Ford, C. Cao, I. E. Wachs and M. Zhu, Structure–Activity Relationships of Copper- and Potassium-Modified Iron Oxide Catalysts during Reverse Water–Gas Shift Reaction, ACS Catal., 2021, 11, 12609–12619, DOI:10.1021/acscatal.1c03792
- Z. Wang, B. Liu and J. Lin, Highly effective perovskite-type BaZrO3 supported Ru catalyst for ammonia synthesis, Appl. Catal., A, 2013, 458, 130–136, DOI:10.1016/j.apcata.2013.03.037
- H. Ronduda, M. Zybert, A. Dziewulska, W. Patkowski, K. Sobczak, A. Ostrowski and W. Raróg-Pilecka, Ammonia synthesis using Co catalysts supported on MgO–Nd2O3 mixed oxide systems: Effect of support composition, Surf. Interfaces, 2023, 36, 102530, DOI:10.1016/j.surfin.2022.102530
- Y. Inoue, M. Kitano, M. Tokunari, T. Taniguchi, K. Ooya, H. Abe, Y. Niwa, M. Sasase, M. Hara and H. Hosono, Direct Activation of Cobalt Catalyst by 12CaO·7Al2O3 Electride for Ammonia Synthesis, ACS Catal., 2019, 9, 1670–1679, DOI:10.1021/acscatal.8b03650
- H. Liu, Ammonia Synthesis Catalysts: Innovation and Practice, World Scientific, 2013 Search PubMed
- M. Yoshida, T. Ogawa, Y. Imamura and K. M. Ishihara, Economies of scale in ammonia synthesis loops embedded with iron- and ruthenium-based catalysts, Int. J. Hydrogen Energy, 2021, 46, 28840–28854, DOI:10.1016/j.ijhydene.2020.12.081
- C. J. Ferron, Recycling of cobalt from alloy scrap, spent batteries or catalysts and metallurgical residues—an Overview, in Ni-Co 2013, eds. T. Battle, M. Moats, V. Cocalia, H. Oosterhof, S. Alam, A. Allanore, R. Jones, N. Stubina, C. Anderson and S. Wang, Springer, Cham, 2013. DOI:10.1002/9781118658826.ch3
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00133d |
| This journal is © The Royal Society of Chemistry 2023 |