
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the photosensitization activities of flavins irradiated by blue LEDs†
Jonathan Ribes ,
Pauline Cossard,
Khaled Al Yaman,
Isabelle Bestel and
Eduard Badarau*
,
Pauline Cossard,
Khaled Al Yaman,
Isabelle Bestel and
Eduard Badarau*
University of Bordeaux, CNRS, CBMN, UMR 5248, Institute of Chemistry and Biology of Membranes & Nano-objects (CBMN), Allée Geoffroy Saint Hilaire, Bât B14, 33600 Pessac, France. E-mail: edouard.badarau@u-bordeaux.fr
First published on 16th January 2023
Abstract
Due to their ability to easily absorb light and to generate highly reactive species, photosensitizers emerged as promising tools in a wide variety of physico-chemical and biological processes. Natural photosensitizers have the benefit of a life-compatible toxicological profile. Porphyrins and flavins are such examples that already proved their efficiency as photo-dynamic therapeutics. The present article describes a reliable, easy-to-implement, readily available and reproducible method that can be used to characterize the photosensitizing activity of flavins. Several key factors were investigated during this study, the optimum parameters were: (i) a blue LED light source (λem = 455 nm) at 6.69 mW; (ii) a pH of 6 mimicking the tumoral environment; (iii) an air-saturated atmosphere reaction medium, (iv) a tetrazolium dye (MTT) was used to monitor the photosensitization efficacy via the generation of the colored MTT-formazan product. This method can be used to rank a series of flavins based on their photosensitizing activities. Such structure–photosensitization activity relationships are essential for the discovery of future potent photosensitizers for photodynamic therapy.
Introduction
Photosensitizers are molecular systems able to efficiently generate an excited state by absorbing light (photons).1 Contrarily to other molecules, the excited photosensitizer has a lifetime (τ) long enough to allow it to react with its neighboring environment, before returning to its original ground-state. The ability of a photosensitizer to easily absorb light is intimately related to its molecular structure: in the case of organic materials, a highly conjugated π-system is essential to promote photoexcitation.2 While the photo-dynamic therapy (PDT) is by far the most well-known and studied application of photosensitizers,3–5 other utilizations are also described in the literature, for example: as catalysts in synthesis6 or pollutants degradations,7 as initiators in crosslinking or degradation of polymers,8 as key elements for the construction of solar energy conversion systems9,10 or as chromophores for optical power limiting systems.7Among the small-molecule photosensitizers, natural photosensitizers have already proved their efficiency, as they are currently used in clinics: porphyrins and flavins are such examples. In its native environment, riboflavin (RF, vitamin B2) and its metabolically active derivatives (flavin mononucleotide – FMN and flavin adenine dinucleotide – FAD) are involved in light-activated physiological processes.11 Activated by the blue light (λ = 445 nm), RF is used in clinics for treating keratoconus12,13 or for sterilizing blood components.14–16
Natural photosensitizers cannot be used in their native state in many applications, and require specific modulations in order to either conjugate them on a material or to modify their physico-chemical properties.17–19 As these structural modifications can impact their photosensitizing properties, it is often necessary to reassess the photosensitizer's activity through a reliable evaluation method of the induced photosensitizing reactions.
The efficiency of flavins as photosensitizers is highly dependent on their ability to promote photosensitizing reactions. In the case of flavins, the most studied photosensitizing reactions are: (a) type I reactions where the activated photosensitizer reacts directly with a substrate and (b) type II reactions, where singlet oxygen (1O2), a highly reactive oxygenated species (ROS), is firstly generated by the activated photosensitizer, followed by the reaction of the 1O2 with a substrate (Fig. 1A).20,21 Depending on the lifetime and the concentrations of the excited species (substrate or oxygen), one or the other pathway will be favored, but in general they co-exist. Compared to the lifetime of singlet oxygen in water (evaluated at 3 μs), in the case of flavins, their lifetime is substantially longer (for example, for triplet RF, 3RF*, τ = 15 μs).22 It should also be mentioned that flavoenzymes can stabilize excited flavins through geometrical and electrostatic interactions, compared to the non-bound flavins.23
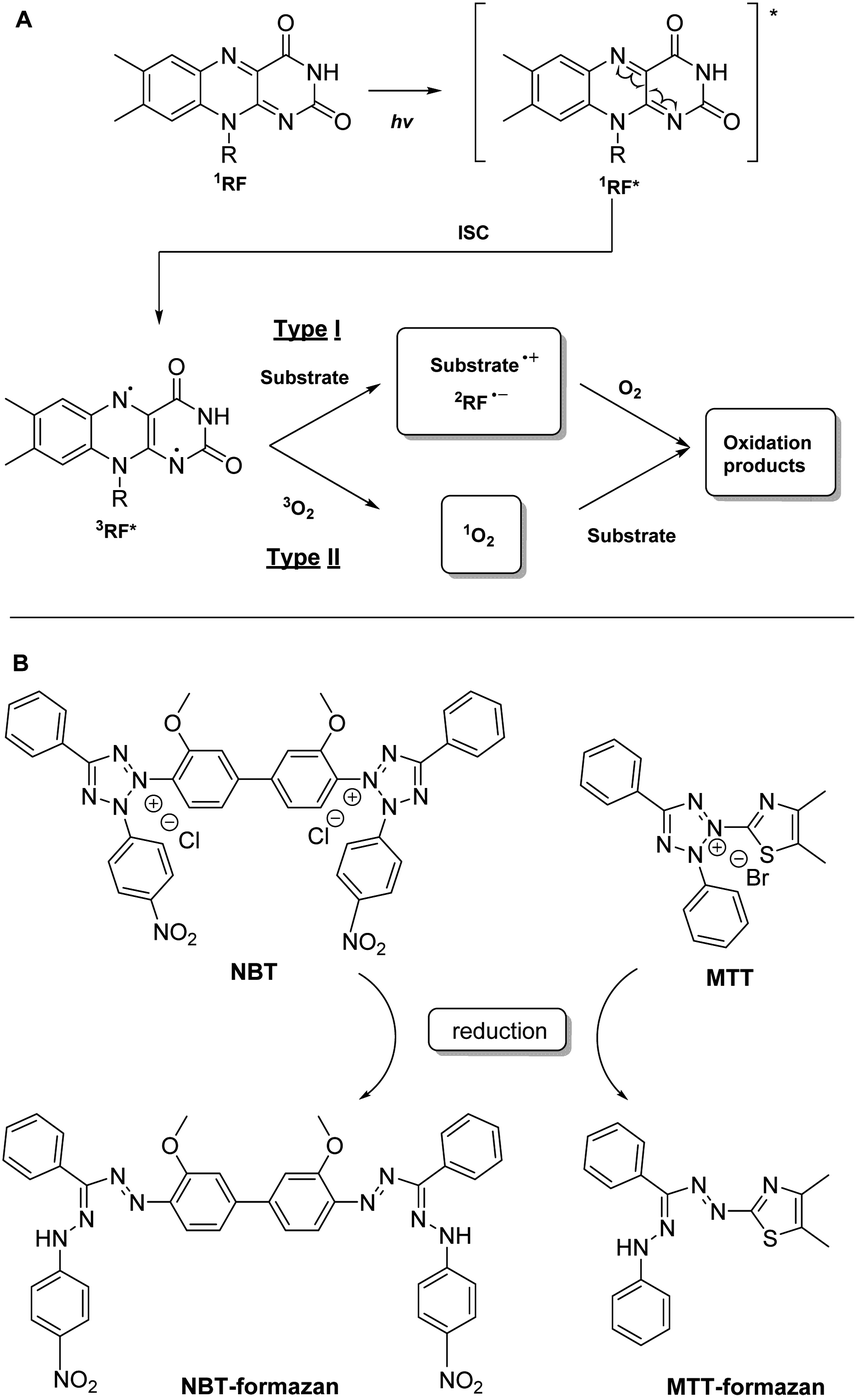 | ||
| Fig. 1 (A) Main photosensitizing reactions pathways described for riboflavin: (a) type I involves direct reaction of the excited photosensitizer with the substrate and (b) type II involves substrate modification by singlet oxygen (1O2) generated by the excited photosensitizer;26,27 (B) chemical structures of NBT and MTT dyes and their reduced forms. | ||
Although complex and focused mainly on a fundamental understanding of the physico-chemical process driving the radical reactions, studies that characterize individually the type I or the type II reactions do exist24–26 and are conducted on simplified model systems. We were interested in the development of a method able to characterize the photosensitizing activity of a sensitizer in a complex biological environment. Consequently, our interest was not in discriminating the type I from the type II reactions, but on the global output, thus summing up both pathways. We intended to develop a reliable and easy-to-implement protocol that can be used to conduct structure–photosensitizing activity relationships (SAR) on flavins. This protocol is essential during medicinal chemistry projects, when a series of flavin analogues are synthesized with the goal of identifying new potent photosensitizers: they should be ranked based on their photosensitizing activities, ideally using a flavin reference during the same study (riboflavin, FMN or FAD, being the most commonly used flavin standards).
An important criterion for our study was the use of in vivo-compatible conditions: an aqueous reaction environment, a pH range and an atmosphere specific to the targeted biological environment, reagents that are non-toxic (or having an acceptable toxicity profile already approved by other cell studies) and a life-compatible source of light.
Several methods are described in the literature for the characterization of the photosensitizing activity. They are focused on either the direct monitoring of the generated ROS28 or indirectly by monitoring the behavior of a probe.28,29 Among them, the colorimetric methods are easy to implement and to adapt to different biological systems. Briefly, a colored indicator will modify its absorbance when reacted with a photosensitizer. Monitoring the hyper/hypsochromic effect of the indicator will thus reveal the photosensitizer's activity.
One such colorimetric test was described for flavins by Liang et al.:30 it involves the use of riboflavin as photosensitizer and nitro-blue-tetrazolium (NBT) as indicator (Fig. 1B). The riboflavin is excited by a blue LED to the excited singlet state, subsequently generating 3RF*, which is reduced to a semiquinone state by methionine. RF semiquinone can reduce NBT, either in ground or in an excited state, to the corresponding formazan (directly via a type I reaction or indirectly via a type II reaction). As the generated formazan is absorbing at 560 nm, its concentration can be directly correlated with the activity of the photosensitizer.
We reproduced this test for riboflavin, but in our hands this protocol was difficult to master. Thus, we started a methodological approach to identify, and eventually optimize, the key parameters governing this reaction.
Results and discussion
Starting from the method previously reported by Liang et al.30 and summarized above, several parameters were investigated during our study: the choice of the indicator, the concentration of the sensitizer, the influence of the irradiation source, the influence of the pH, as well as that of the reaction's atmosphere. These will be detailed in the following sections.1 Design of the blue LED-promoted photosensitization-reaction
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) of the dye is thus a key parameter to take into account when monitoring the dye's concentration using the Beer–Lambert law.
P) of the dye is thus a key parameter to take into account when monitoring the dye's concentration using the Beer–Lambert law.We started our study using an NBT indicator, as previously described in the literature for monitoring riboflavin photosensitizing activity.30 Unfortunately, after reaction with the light-activated riboflavin, the reduced NTB (NBT-formazan) is only partly soluble in water. For example, an important sedimentation of the formed NBT formazan is observed if the reaction mixture is immobilized for 20 min: the absorbance decreased by half compared to the one recorded immediately after the reaction. This was clearly shown by passing the reaction mixture on a polytetrafluoroethylene (PTFE) syringe filter (0.2 μm): the absorbance at 560 nm was completely abolished (∼0.01 μA). Variation of the pH (from 1 to 14) or addition of a co-solvent such as dioxane, tetrahydrofurane (THF), dimethylsulfoxide (DMSO), acetone, ethyl acetate, dimethylformamide (DMF), acetonitrile (ACN), methanol (MeOH), ethanol (EtOH) did not completely dissolve the precipitated formazan, even after a 100-fold dilution of the reaction mixture (Fig. 2A).
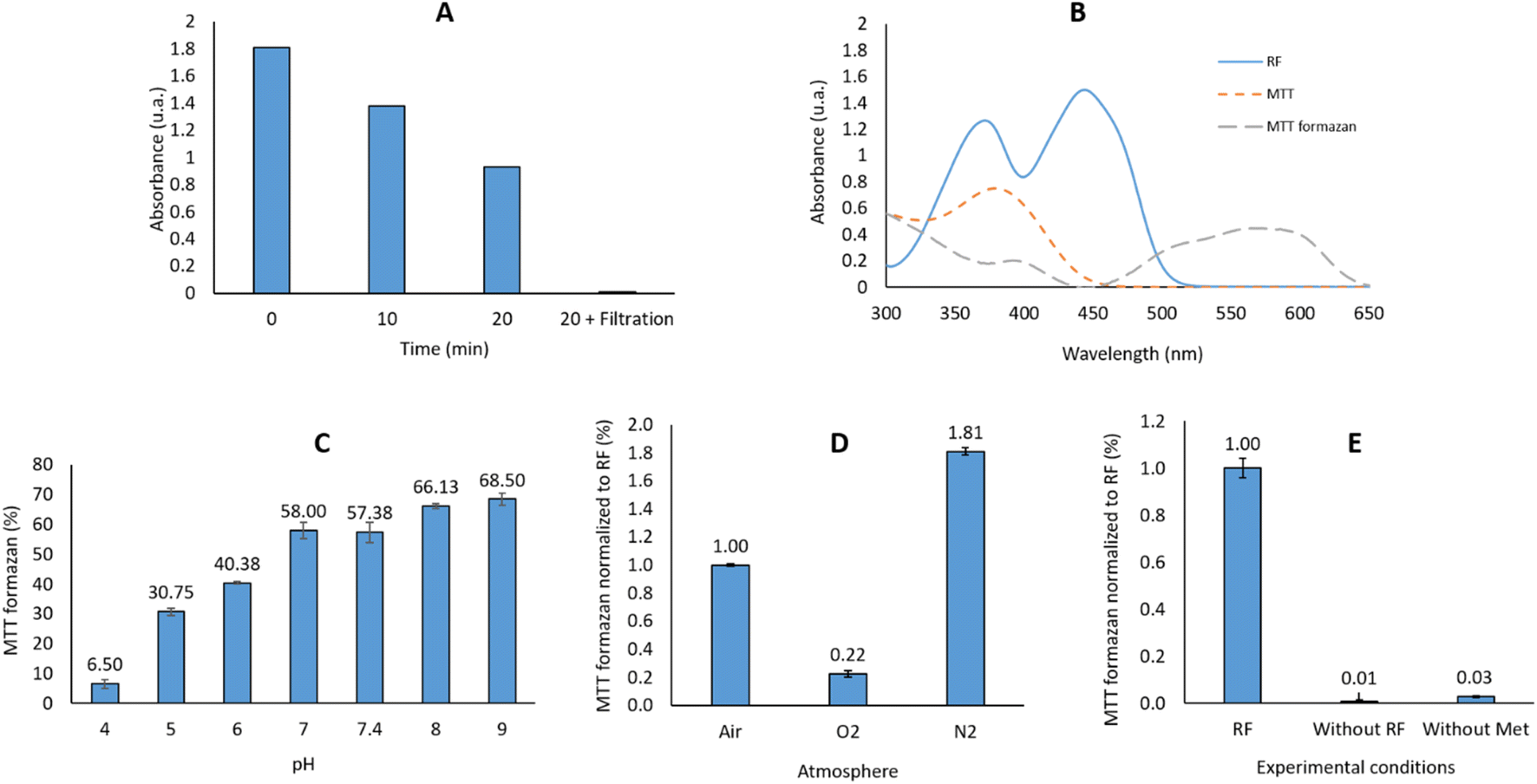 | ||
| Fig. 2 (A) Absorbance at 560 nm after 0, 10 & 20 min of pause, and after filtration on 0.2 μm filters; (B) UV-Vis spectra of RF (12.8 mM in water), MTT (10.9 mM in water) and MTT-formazan (2.19 mM in THF/water 1/1); (C) influence of the pH on the reaction output; (D) influence of the atmosphere on the reaction output; (E) control reactions: without RF or without methionine. | ||
The bis-tetrazolium salts, such as NBT, have another dis-advantage: they can be only partly reduced, resulting in a mixture of mono- and double-reduced formazan products. We therefore selected another tetrazolium salt, this time in the mono-tetrazolium series. Among the commercially available salts used for viability studies (XTT, MTT, MTS, CTC, etc.),31 we selected MTT (Fig. 1B), which has an absorption range different than that of the flavins. Although still precipitating at the end of the reaction, contrarily to NBT-formazan, MTT-formazan was soluble in a wide variety of tested solvents: DMSO, ACN, DMF or THF. We selected THF for dissolving the generated formazan: the reaction mixture was diluted with an equivalent volume of THF and its absorbance at 560 nm before and after filtration on a 0.2 μm syringe filter was identical: A560nm (before) = 0.825; A560nm (after) = 0.814.
Accordingly, MTT was selected as probe to monitor the photosensitizer activity, and THF as co-solvent to dissolve the MTT-formazan generated at the end of the reaction.
A calibration curve was generated using the commercially-available MTT-formazan (ESI – F1†), and it showed that the concentration necessary to induce an absorbance of 0.4 is 1.275 × 10−4 M. As the reaction between the RF and MTT is stoichiometric, the concentration of RF should be the same. This did not cause any issues, as the aqueous solubility of riboflavin is ∼100 mg L−1 (∼2.66 × 10−4 M).32
In conclusion, the selected light source was the Thorlabs monochromatic M455F3 LED (λem = 455 nm), modulated by a cube driver (LEDD1B) to an output of 6.69 mW, equipped with the 0.4 mm optical fiber M28L01. This source allowed to attain an Abs = 0.4 for the resultant MTT-formazan after 3 minutes of irradiation of the reaction mixture.
:1 mixture used in the reaction. The reaction output in our case is thus dependent on the RF activity at different pH. Our results can be correlated with the previously data on 3RF* quenching in water in a pH dependent manner: Huvaere36 reported that at a pH = 4.2 the bimolecular rate constant is k = 4.9 × 107 L mol−1 s−1 while at pH = 7 the rate constant is k = 2.2 × 107 L mol−1 s−1. The initial mechanism described by Huvaere for the interaction of 3RF* and methionine, with the final formation of DMDS (dimethyldisulfide) was questioned later by Bassani et al.37 which proposed another mechanistic pathway for the formation of this odor-repellent product. For a potential usage in the PDT, we decided to use a pH = 6, in order to mimic the slightly acidic tumoral microenvironment. Moreover, this pH allows generation of the 40% reaction output as initially targeted (vide supra), necessary to rank different flavins based on this method. It should be reminded that the addition of THF as co-solvent was mandatory for dissolving the generated MTT formazan.After excitation by light to the singlet state, subsequently generating the triplet state, 3RF* is reduced by the electron donor present in the reaction (methionine) to the radical anion state RF˙−.37–39 This last will trap a proton (H+) and will subsequently generate the semiquinone RFH˙ able to reduce MTT to the corresponding MTT formazan. However, this reaction pattern is modified by the presence of a high concentration of O2:38,40 under an O2-rich medium, the 3RF* quenching by methionine (k = 6.36 × 107 M−1 s−1)41 is highly overpassed by 3RF* quenching by the O2 (k = 0.5–1.7 × 109 M−1 s−1).38 This last reaction will generate 1O2, which is quenched by the presence of a high excess of methionine in the reaction medium (55 eq. vs. 1 eq. MTT/1.1 eq. RF) to a rate previously reported in the range 1.3–1.7 × 107 M−1 s−1.42,43 The final result of such a cascade will be a lower amount of the resultant MTT formazan than in an air-saturated solution.
In an N2-rich medium: a lower concentration of 1O2 will be generated, thus the probability for the 3RF* to be quenched by 1O2 is decreased. The electron transfer from methionine will thus be favored in such an N2-rich compared to an O2-rich medium. No reaction takes place in the absence of methionine (vide infra).
An air-saturated solution will combine both reactions described before: 20% of O2 in air will penalize the reaction output compared to N2-saturated solution (Fig. 2D).
It should also be reminded that the photophysics of a photosensitization reaction is temperature sensitive, depending on several factors like the thermal stability (e.g. deactivation) of the 3PS or 1O2, the temperature-dependent ground-O2 solubility and diffusion in water, etc. For example, Ogilby et al. noted that, contrary to the 1FMN which is less sensitive to temperature variation, the rate constant for quenching 3FMN by ground O2 increases 10 fold upon increasing the temperature from 10 to 43 °C, having as consequence an increased rate of singlet oxygen formation in this temperature range.44 The same increasing photosensitization trend with the temperature increase was described for porphyrin-based photosensitizers (e.g. see work of Kimel et al.).45
As the goal of our study was to develop a straightforward methodology that can be used to compare photosensitization abilities for a series of flavins, this method was optimized at room temperature (20 °C). However, even if riboflavin is a thermostable vitamin,46 different temperatures will impact the reaction output (see ESI – F4† for details), presumably via the mechanisms reminded above.
Based on the previous experiments, in a typical irradiation protocol (see Materials and methods) an aqueous solution of the investigated flavin (1.275 × 10−4 M, 1 eq.) was mixed with methionine (55 eq.) and MTT (1 eq.) and the pH adjusted to 6 by addition of HCl (aq soln 0.1 N). The mixture was subsequently irradiated for 3 minutes by a blue LED (λmax = 455 nm, 6.69 mW) under air atmosphere. The reaction mixture was subsequently diluted by addition of an equivalent amount of THF and heated for 5 min at 50 °C. The absorbance of the solution was measured at λ = 560 nm and reported on a calibration curve prepared from commercially available MTT formazan in THF/H2O 1/1 for concentrations between 0 and 5.34 × 10−5 M (y = 0.0631x − 0.0152, R2 = 0.991).
2 Photo-degradation reactions
It is well known that flavins undergo chemical or metabolic degradation products. If the enzymatic metabolites also generate oxidation products of the isoalloxazine ring, the pH-dependent chemical transformations and the photo-degradation involve mostly ribityl side-chain transformations, the main derivatives being the lumichrome (LC), lumiflavin (LF) and formyllumiflavin (FLF).47–50 In order to assess the possible photo-degradation products generated under the irradiation conditions and their impact on the photosensitizing activity, the same reaction was conducted for an extended period of time using the same LED sources as before. Comparing the reaction mixture with the commercially available LC and LF by thin layer chromatography (TLC) allowed us to qualitatively estimate the output of the reaction. No traces of LC and LF were detected after 3 min irradiation at 6.69 mW with the M455F3 LED. Traces of these derivatives were observed after 3 minutes of irradiation of the riboflavin sample at 32.5 mW; these products became clearly visible after 15 min of reaction. The LC ratio continuously increased over 2 h, until it was the only product of the reaction. The same LC product was obtained only after 3 minutes when the sample was irradiated with the more powerful 1 W LED (Table 2).| Irradiation power (mW) | Irradiation time (min) | LC | LF |
|---|---|---|---|
| a “—” indicates no presence of cpd as observed by TLC; “+” indicates traces of cpds; “++” moderate intensity of TLC spots and “+++” main compounds by TLC. | |||
| 6.69 | 3 | — | — |
| 32.5 | 3 | + | + |
| 32.5 | 15–90 | ++ | + |
| 32.5 | 120 | +++ | — |
| 1000 | 3 | +++ | — |
The photosensitizing activities of the commercially available LC and LF were subsequently evaluated using the previously described protocol. As shown in Fig. 3A, LF is equipotent with the RF, while the LC is devoid of any photosensitizing activity. However, it is important to note that LC does not present the same UV spectra as the flavins: it does not absorb at 450 nm, but at lower wavelengths: 355 nm and 385 nm. As described previously by Weiss,51 LC has the same capacity of inter-system crossing as RF (ΦISC = 0.71 vs. ΦISC = 0.67),52 but it requires irradiation at 347 nm which is highly mutagenic compared to the 455 nm range. In conclusion, the selected 3 min irradiation with a LED power of 6.69 mW was mild enough not to degrade RF and validated the irradiation conditions. Such a methodology can be used to rank flavins and their derivatives based on their photosensitizing activity.
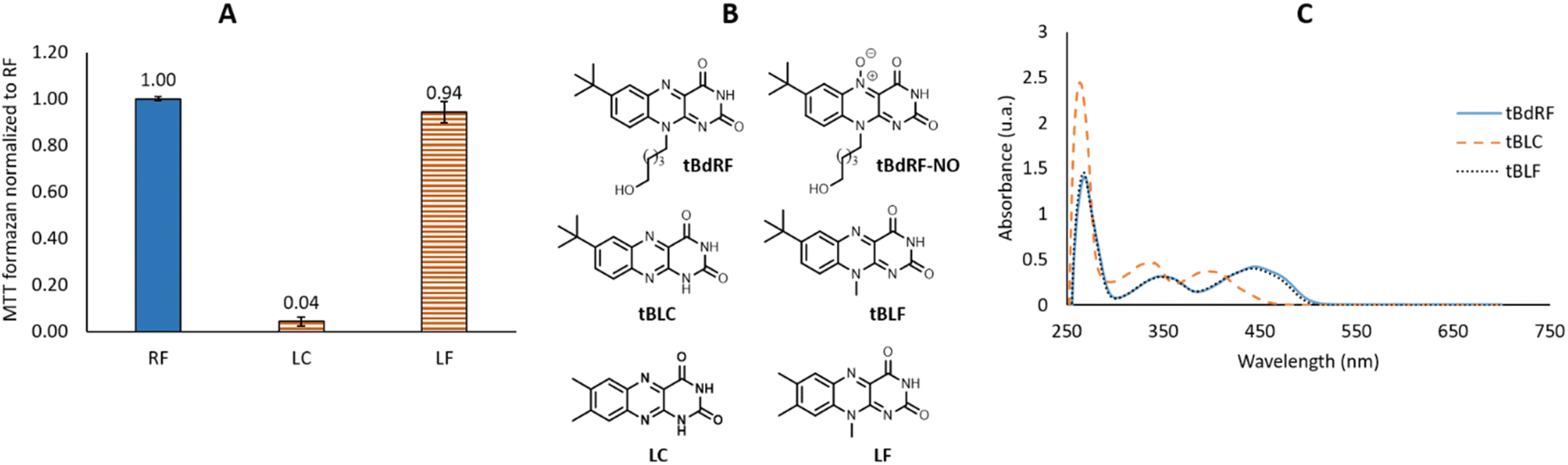 | ||
| Fig. 3 (A) Photosensitizing activities of RF, LC and LF; (B) chemical structures of tBdRF, tBLC, tBLF, LC and LF; (C) UV-Vis spectra of tBdRF, tBLC and tBLF. | ||
We described one such flavin derivative recently: 7-tbutyl-desoxyriboflavin (tBdRF, Fig. 3B). This was conjugated on phospholipid derivatives through a pentyl linker, in order to induce vesicular self-assemblies (e.g. liposomes).19 With this derivative in our library, we were interested in verifying if it would generate the same photo-degradation products as riboflavin. Degradation of riboflavin is described in the literature to be assisted by the C2–OH of the ribityl chain.26 In the case of tBdRF, this hydroxyl moiety is absent from the N-alkyl chain, in consequence, the mechanism of photo-degradation could be different, or could result in other degradation derivatives. In order to investigate this hypothesis, our irradiation protocol with different irradiation sources was employed. However, contrary to LC and LF, the analogues in 7-tbutyl-isoalloxazine series are not commercially available, thus they were synthesized in-house, by analogy with the previously reported methods.53
Briefly, chlorouracil was engaged in a solvent-free reaction with either 4-tbutyl-aniline or 4-(tbutyl)-N-methylaniline. The substitution products were subsequently submitted to an N-oxidation by sodium nitrite in acetic acid. The instable N-oxide intermediate was directly reduced by dithiothreitol (DTT) to the corresponding lumiflavin or lumichrome, depending on the substitution on the N-10 (Scheme 1).
 | ||
| Scheme 1 Synthesis of tBLF and tBLC. | ||
The synthesized compounds in 7-tButyl series (tBLC and tBLF) present the same UV-Vis patterns as LC and LF.
Thus, despite the low hypochromic effect, the tBLF spectrum overlaps the tBdRF (Fig. 3C). Again, as for RF, the tBLC does not absorb at the same wavelength as tBLF (400 nm vs. 440 nm). It was expected that tBLC would not induce any photosensitizing activity using the M455F3 LED (λem = 455 nm).
With both tBLC and tBLF used as controls, we proceeded with the irradiation of tBdRF. Interestingly, no traces of tBLC or tBLF were observed by TLC after 15 min of irradiation using the 1 W LED. Other minor compounds were generated in the reaction mixture, which became predominant only after full consumption of the starting tBdRF: after 8 h of reaction. Only traces of the expected tBLC or tBLF were observed at the end of the reaction. Unfortunately, the 1H, 13C and MS spectra did not allow identification of the main derivatives generated under these irradiation conditions. From a structural point of view, it is important to underline that no aromatic signals were present in the 1H & 13C spectra of the main photo-degradation products. Hydroxyl moieties present in the structure of RF seem thus to mediate a specific photo-degradation mechanism. In the absence of such –OH moieties, the degradation mechanism was different and led to other derivatives than the corresponding tBLF and tBLC. However, in the absence of structural details, no other studies were conducted with these derivatives.
In all cases, independent of the presence of hydroxyl moieties on the N10-alkyl chain, the selected 3 min irradiation with a LED power of 6.69 mW did not conduct to any photo-degradations. The previously identified irradiation conditions were thus validated.
3 Method validation
Other flavin derivatives were evaluated using this protocol in order to assess its utility for characterizing their photosensitizing abilities. For example, we tested the FMN and other derivatives in the tBdRF series belonging to our internal library of flavins. For example, when compared to RF, its biologically active form, FMN, has a 17% lower photosensitizing activity. In order to mimic the micro-tumoral environment, an acidic pH 6 was used during our tests. Under these acidic conditions,54 FMN can generate the photo-inactive LC. This could explain why its activity is lower than RF. Another derivative of the previously reported tBdRF (Fig. 3B) was subsequently investigated: the terminal hydroxyl group was substituted by dimethylamine. This derivative was synthesized after the initial activation of the hydroxyl moiety into a methylsulfonyl (mesylate) leaving group under basic conditions, followed by its substitution by dimethylamine (Scheme 2). Interestingly, using the same irradiation method as before, this derivative has a 51% higher activity than the starting tBdRF (Fig. 4). The amine moiety in its structure seems to play a vital role in mediating the protonation state of the isoalloxazine ring, with an impact on the generation of its active triplet state.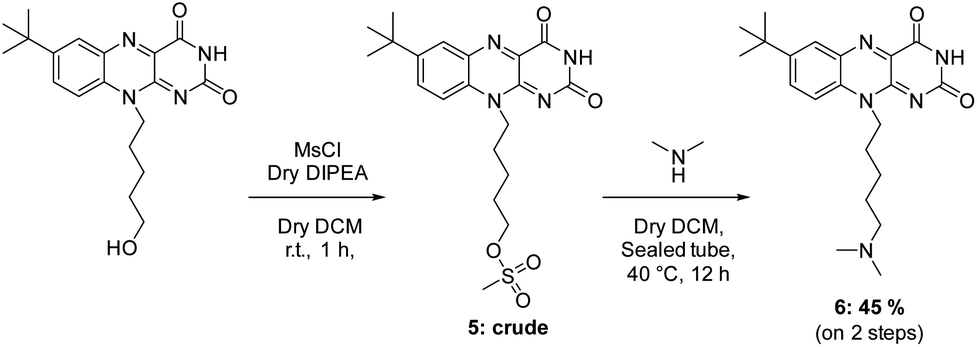 | ||
| Scheme 2 Synthesis of tBdRF-NMe2. | ||
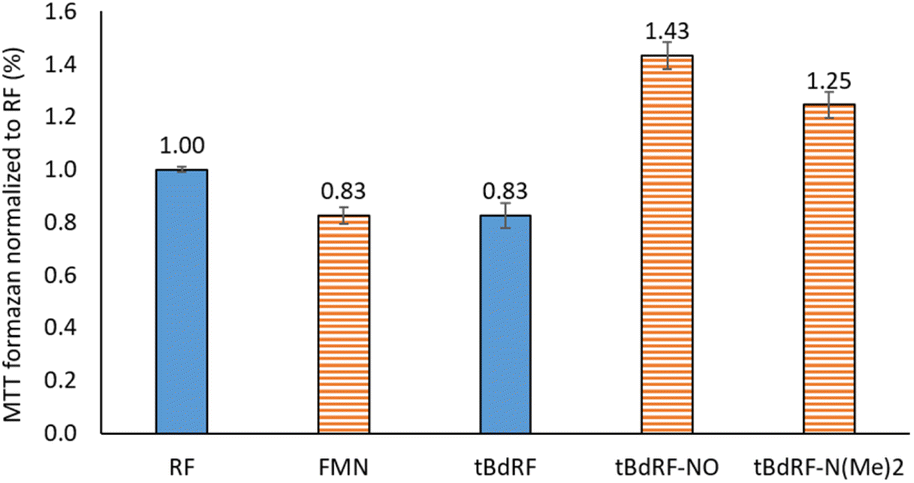 | ||
| Fig. 4 Photosensitizing activities for a panel of flavins. | ||
A final compound was evaluated for its photosensitizing activity: the N-oxide intermediate (Fig. 3B) from our in-house library of flavins. Interestingly, the tBdRF-NO derivative was found to be 72% more active than the starting tBdRF (Fig. 4).
A library of flavin analogues was recently synthesized by our group using a multistep approach. Their photosensitizing activity was evaluated using the methodology reported herein, which allowed identifying lead compounds for potential PDT applications. The best derivative from this series will be conjugated on phospholipids and formulated into liposomes, in order to conduct to a more efficient generation of nanobombs than those we previously described in the literature.19 These results will be disclosed in due time.
Conclusion
The current manuscript presents the design of an easy-to-implement protocol used to investigate the photosensitizing activity of flavins. Several parameters were monitored during this study. They were optimized to deliver a 40% reaction output in order to facilitate a direct comparison of the investigated flavins. The retained parameters were: (i) the light source – a blue LED (λem = 455 nm) at 6.69 mW. A more intense blue LED was observed to induce photo-degradation of the flavins; (ii) the irradiation time – 3 minutes; (iii) the pH – this was adjusted to 6 in order to mimic the tumoral environment; the reaction output was shown to be pH-dependent. For example, compared to a 40% yield at pH = 6, the yields were 7% and 69% at pH = 4 and pH = 9, respectively; (iv) the atmosphere of the reaction medium – the air-saturated medium was found to deliver the optimum reaction rate, intermediate between the O2- (lower yield) and N2-saturated reactions (higher yield); (v) the dye to monitor the sensitization efficacy – a tetrazolium derivative (MTT) was used as the precipitated colored MTT-formazan was soluble several organic solvents (THF was selected for dissolving the precipitated MTT-formazan).The protocol described in the current article is essential for medicinal chemistry projects aiming at identifying potent photosensitizers: the newly synthesized potential photo-sensitizers should be ranked based on their photosensitizing activities. As a proof of concept, this protocol was used to generate preliminary SAR data for a small panel of compounds. Among them, the N-oxide form of a RF analogue was found more photo-active than the parent flavin.
Additionally, a dehydroxylated analogue of riboflavin was found to give photodegradation derivatives other than the expected lumiflavin and lumichrome analogues which were observed for RF. This seems to suggest that, in the absence of an OH group in the C2 position of the ribityl chain, the mechanism of photo-degradation is different than the one observed for RF.
The method described herein could be used to develop structure–photosensitizing activity relationships for libraries of flavin analogues, with the final goal of identifying potent agents for photo-dynamic therapy.
Experimental
1 Materials and methods
All reagents and solvents were purchased from Sigma-Aldrich, Alfa-Aesar, Acros Organics and were used without further purification. The infrared (IR) spectra were recorded a Jasco FT/IR-4600 spectrometer on a single-reflection diamond ATR accessory. 1H-NMR, 13C-NMR were recorded with Bruker Avance 300 spectrometer (at 300 MHz, 75 MHz) in CDCl3-d1, CD3OD-d4, DMSO-d6. Chemical shifts are reported in parts per million (ppm) and the coupling constants are reported in units of Hertz [Hz]. Multiplicities were abbreviated as follows: singlet (s), as: apparent singlet, doublet (d), doubled doublet (dd), triplet (t), quintet (quint), multiplet (m). Low resolution mass spectra (MS), were recorded with a Thermo Q Exactive spectrometer. Melting points (Mp) or degradations temperature (Tdeg) were recorded using a Stuart SMP-30 B-540 apparatus. Flash chromatography was performed on Sigma-Aldrich 52–73 Å (70–230 mesh) silica gel under air pressure. Thin-layer chromatography (TLC) was carried out on Sigma-Aldrich silica gel 60 F254 precoated plates. Visualization was made with ultraviolet light (λ = 254/365 nm) or using an aqueous potassium permanganate staining solution.2 Synthesis
:EA, 99:1 to 9:1) to yield compound 1b as a light brown oil (0.373 g, 81%). C11H17N; M = 163.14 g mol−1; IR: ν 3410, 2955 cm−1; MS (ESI): m/z = 148.2 [M − CH3]+; NMR 1H (CDCl3; 300 MHz): δ (ppm) 1.36 (as, 9H), 2.88 (s, 3H), 3.56 (as, 1H), 6.65 (d, 2H, J = 8.3 Hz), 7.30 (d, 2H, J = 8.6 Hz); NMR 13C (CDCl3; 75 MHz): δ (ppm) 31.0 (CH3), 31.7 (CH3), 33.9 (Cq), 112.3 (CHar), 126.1 (CHar), 140.2 (Cq), 147.1 (Cq).:MeOH, 100:0 to 97:3) to yield compound 2b as a yellow powder (0.103 g, 77%). C15H19N3O2; M = 273.34 g mol−1; Tdeg = 290 °C, IR: ν 3416, 2962, 1728, 1624 cm−1; MS (ESI): m/z = 274.1 [M + H]+, 296.1 [M + Na]+; NMR 1H (DMSO-d6; 300 MHz): δ (ppm) 1.30 (s, 9H), 3.22 (s, 3H), 4.22 (s, 1H), 7.17 (d, 2H, J = 8.3 Hz), 7.45 (d, 2H, J = 8.4 Hz), 10.23 (s, 1H), 10.43 (s, 1H); NMR 13C (DMSO-d6; 75 MHz): δ (ppm) 31.1 (CH3), 34.4 (Cq), 39.6 (CH3), 78.0 (CH), 126.1 (CHar), 126.6 (CHar), 141.5 (Cq), 149.5 (Cq), 151.3 (Cq), 155.1 (Cq), 163.7 (Cq).:EA, 4.6 to EA:MeOH, 95:5) to yield compound tBLF as a yellow powder (0.383 g, 93% after two steps). C15H16N4O2; M = 284.32 g mol−1; Tdeg = 319 °C, IR: ν 3446, 2960, 1712, 1679 cm−1; MS (ESI): m/z = 307.1 [M + Na]+; NMR 1H (DMSO-d6; 300 MHz): δ (ppm) 1.38 (s, 9H), 3.97 (s, 3H), 7.88 (d, 1H, J = 8.8 Hz), 8.05 (m, 2H), 11.39 (s, 1H); NMR 13C (DMSO-d6; 75 MHz): δ (ppm) 30.8 (CH3), 31.9 (CH3), 34.5 (Cq), 116.4 (CHar), 127.2 (CHar), 131.3 (Cq), 133.4 (CHar), 134.6 (Cq), 138.4 (Cq), 149.0 (Cq), 150.7 (Cq), 155.6 (Cq), 159.9 (Cq).:MeOH, 97:3 to 9:1) to yield compound 6 as a dark orange powder (0.429 g, 45% after two steps). C21H29N5O2; M = 383.50 g mol−1; Mp = 175 °C, IR: ν 1659, 1243 cm−1; MS (ESI): m/z = 384.24 [M + H]+; NMR 1H (CD3OD; 300 MHz): δ (ppm) 1.44 (s, 9H), 1.54–1.67 (m, 4H), 1.90 (quint, 2H, J = 7.5 Hz), 2.28 (s, 6H), 2.40 (t, 2H, J = 7.5 Hz), 4.74 (t, 2H, J = 7.7 Hz), 7.93 (d, 1H, J = 9.1 Hz), 8.13 (d, 1H, J = 2.3 Hz, J = 9.0 Hz), 8.17 (s, 1H, J = 2.2 Hz); NMR 13C (CD3OD; 75 MHz): δ (ppm) 25.5 (CH2), 27.8 (CH2), 28.1 (CH2), 31.3 (CH3), 35.7 (Cq), 45.3 (CH3), 46.3 (CH2), 60.3 (CH2), 117.2 (CHar), 129.1 (CHar), 132.2 (Cq), 135.7 (CHar), 137.2 (Cq), 139.0 (Cq), 151.7 (Cq), 151.7 (Cq), 159.1 (Cq), 162.3 (Cq).3 Photosensitizing activity
Stock aqueous solutions of riboflavin (1.275 × 10−4 M, pH 6.0), synthesized isoalloxazine derivatives (1.275 × 10−4 M, pH 6.0), thiazolyl blue tetrazolium bromide (MTT, 9.65 × 10−4 M), and L-methionine (5.66 × 10−2 M) were freshly prepared using ultrapure water before the experiments. Flavins (aq. soln, 2.66 mL, 3.39 × 10−4 mmol) were mixed with 0.34 mL of the second solution (MTT, 3.28 × 10−4 mmol; and L-methionine, 1.52 × 10−2 mmol) in a standard test tube (NAFVSM 621.1225075080.9R000) under air atmosphere.The reaction mixtures were irradiated by direct contact between the reaction vial and a blue LED (λem = 455 nm) at 6.69 mW (M455F3 LED, optical fiber M28L01 0.4 mm and cube driver LEDD1B were purchased from Thorlabs). The output light intensity was measured at a distance of 0.2 cm using the Thorlab PM400 Console and a S120VC probe prior to irradiation experiments. The reaction mixture was irradiated for 3 min. The generated suspension of MTT formazan was dissolved by adding THF (3 mL) and subsequently heated for 5 min at 50 °C. Its absorbance was measured at λ = 560 nm and reported to a calibration curve prepared with commercial MTT formazan in THF/H2O (1/1) for concentrations between 0 and 5.34 × 10−5 M (y = 0.0631x − 0.0152, R2 = 0.9991). The experimental setup is described in ESI – F5.†
Abbreviations
| DCM | Dichloromethane |
| DIPEA | Diisopropylethylamine |
| DTT | Dithiotreitol |
| EA | Ethyl acetate |
| PE | Petroleum ether |
| aq. sat. soln | Saturated aqueous solution |
| MeOH | Methanol |
| TEA | Triethylamine |
| THF | Tetrahydrofurane |
| RF | Riboflavin |
| tBdRF | tert-Butyl desoxyriboflavin |
| Car | Aromatic carbon |
| Cq | Quaternary carbon |
| ESI | Electrospray ionization |
| PTFE | Polytetrafluoroethylene |
| THF | Tetrahydrofurane |
| DMSO | Dimethylsulfoxide |
| DMF | Dimethylformamide |
| ACN | Acetonitrile |
| MeOH | Methanol |
| EtOH | Ethanol |
| NBT | Nitro-blue-tetrazolium |
| XTT | 2,3-Bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| MTS | 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium |
| CTC | 5-Cyano-2,3-ditolyl tetrazolium chloride |
| LED | Light emitting diode |
| PDT | Photo dynamic therapy |
| ROS | Reactive oxygenated species |
| LC | Lumichrome |
| LF | Lumiflavin |
| FLF | Formyllumiflavin |
| TLC | Thin layer chromatography |
| DTT | Dithiothreitol |
| TEMPO | (2,2,6,6-Tetramethyl-piperidin-1-yl)oxyl |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was financially supported by the “Ligue Contre le Cancer” Foundation (Nano-Bombs/2019 to EB). The support of the technical platforms of CBMN for the characterization of the new compounds is also acknowledged.References
- IUPAC – Compendium of Chemical Terminology (Gold Book), https://goldbook.iupac.org Search PubMed.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- T. Hu, Z. Wang, W. Shen, R. Liang, D. Yan and M. Wei, Theranostics, 2021, 11, 3278–3300 CrossRef CAS PubMed.
- Q. Xiao, J. Wu, X. Pang, Y. Jiang, P. Wang, W. A. Leung, L. Gao, S. Jiang and C. Xu, Curr. Med. Chem., 2018, 25, 839–860 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- T. Nyokong and V. Ahsen, Photosensitizers in Medicine, Environment, and Security, Springer Science & Business Media, Berlin, Germany, 2012, ISBN 978-90-481-3870-8 Search PubMed.
- A. S. Dunn, Polym. Int., 1998, 45, 240 CrossRef CAS.
- J.-H. Shon and T. S. Teets, ACS Energy Lett., 2019, 4, 558–566 CrossRef CAS.
- C.-F. Leung and T.-C. Lau, Energy Fuels, 2021, 35, 18888–18899 CrossRef CAS.
- E. Sassetti, M. H. Clausen and L. Laraia, J. Med. Chem., 2021, 64, 5252–5275 CrossRef CAS PubMed.
- D. P. S. O'Brart, J. Optom., 2014, 7, 113–124 CrossRef PubMed.
- F. Raiskup, A. Theuring, L. E. Pillunat and E. Spoerl, J. Cataract Refractive Surg., 2015, 41, 41–46 CrossRef PubMed.
- B. G. Solheim, Transfus. Apheresis Sci., 2008, 39, 75–82 CrossRef PubMed.
- S. Marschner, L. D. Fast, W. M. Baldwin III, S. J. Slichter and R. P. Goodrich, Transfusion, 2010, 50, 2489–2498 CrossRef PubMed.
- L. D. Fast, M. Nevola, J. Tavares, H. L. Reddy, R. P. Goodrich and S. Marschner, Transfusion, 2013, 53, 373–381 CrossRef CAS PubMed.
- F. Marlin, P. Simon, S. Bonneau, P. Alberti, C. Cordier, C. Boix, L. Perrouault, A. Fossey, T. Saison-Behmoaras, M. Fontecave and C. Giovannangeli, ChemBioChem, 2012, 13, 2593–2598 CrossRef CAS PubMed.
- T. Komatsu, M. Moritake, A. Nakagawa and E. Tsuchida, Chem.–Eur. J., 2002, 8, 5469–5480 CrossRef CAS PubMed.
- J. Ribes, N. Beztsinna, R. Bailly, S. Castano, E. Rascol, N. Taib-Maamar, E. Badarau and I. Bestel, Bioconjugate Chem., 2021, 32, 553–562 CrossRef CAS PubMed.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- M. S. Baptista, J. Cadet, P. Di Mascio, A. A. Ghogare, A. Greer, M. R. Hamblin, C. Lorente, S. C. Nunez, M. S. Ribeiro, A. H. Thomas, M. Vignoni and T. M. Yoshimura, Photochem. Photobiol., 2017, 93, 912–919 CrossRef CAS PubMed.
- C. Lu, W. Lin, W. Wang, Z. Han, S. Yao and N. Lin, Phys. Chem. Chem. Phys., 2000, 2, 329–334 RSC.
- Y.-T. Kao, C. Saxena, T.-F. He, L. Guo, L. Wang, A. Sancar and D. Zhong, J. Am. Chem. Soc., 2008, 130, 13132–13139 CrossRef CAS PubMed.
- M. Insińska-Rak and M. Sikorski, Chem.–Eur. J., 2014, 20, 15280–15291 CrossRef PubMed.
- K. Sztandera, M. Gorzkiewicz and B. Klajnert-Maculewicz, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1509 Search PubMed.
- D. Cardoso, S. Libardi and L. Skibsted, Food Funct., 2012, 3, 487–502 RSC.
- C. B. Martin, M.-L. Tsao, C. M. Hadad and M. S. Platz, J. Am. Chem. Soc., 2002, 124, 7226–7234 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- Y. Zhang, M. Dai and Z. Yuan, Anal. Methods, 2018, 10, 4625–4638 RSC.
- J.-Y. Liang, J.-M. P. Yuann, Z.-J. Hsie, S.-T. Huang and C.-C. Chen, J. Photochem. Photobiol., B, 2017, 174, 355–363 CrossRef CAS PubMed.
- J. C. Stockert, R. W. Horobin, L. L. Colombo and A. Blázquez-Castro, Acta Histochem., 2018, 120, 159–167 CrossRef CAS PubMed.
- Pubchem, RF, https://pubchem.ncbi.nlm.nih.gov/compound/493570, accessed 2022-06-01 Search PubMed.
- A. Sengupta, R. V. Khade and P. Hazra, J. Photochem. Photobiol., A, 2011, 221, 105–112 CrossRef CAS.
- P. F. Heelis, Chem. Soc. Rev., 1982, 11, 15–39 RSC.
- J. A. Plumb, R. Milroy and S. B. Kaye, Cancer Res., 1989, 49, 4435–4440 CAS.
- K. Huvaere, M. L. Andersen, M. Storme, J. Van Bocxlaer, L. H. Skibsted and D. De Keukeleire, Photochem. Photobiol. Sci., 2006, 5, 961–969 CrossRef CAS PubMed.
- A. Furet, A. Sicello, B. Guillemat, C. Absalon, E. Langleron and D. M. Bassani, Food Chem., 2022, 372, 131281 CrossRef CAS PubMed.
- H. Görner, J. Photochem. Photobiol., B, 2007, 87, 73–80 CrossRef PubMed.
- P. Mondal, K. Schwinn and M. Huix-Rotllant, J. Photochem. Photobiol., A, 2020, 387, 112164 CrossRef CAS.
- L. Valle, F. E. M. Vieyra and C. D. Borsarelli, J. Phys. Org. Chem., 2016, 29, 629–635 CrossRef CAS.
- D. R. Cardoso, D. W. Franco, K. Olsen, M. L. Andersen and L. H. Skibsted, J. Agric. Food Chem., 2004, 52, 6602–6606 CrossRef CAS PubMed.
- A. Michaeli and J. Feitelson, Photochem. Photobiol., 1994, 59, 284–289 CrossRef CAS PubMed.
- I. B. C. Matheson and J. Lee, Photochem. Photobiol., 1979, 29, 879–881 CrossRef CAS.
- M. Westberg, M. Bregnhøj, M. Etzerodt and P. R. Ogilby, J. Phys. Chem. B, 2017, 121, 2561–2574 CrossRef CAS PubMed.
- V. Gottfried and S. Kimel, J. Photochem. Photobiol., B, 1991, 8, 419–430 CrossRef CAS PubMed.
- E. Choe, R. Huang and D. B. Min, J. Food Sci., 2005, 70, R28–R36 CrossRef CAS.
- W. Holzer, J. Shirdel, P. Zirak, A. Penzkofer, P. Hegemann, R. Deutzmann and E. Hochmuth, Chem. Phys., 2005, 308, 69–78 CrossRef CAS.
- G. E. Treadwell, W. L. Cairns and D. E. Metzler, J. Chromatogr. A, 1968, 35, 376–388 CrossRef CAS PubMed.
- W. M. Moore, J. T. Spence, F. A. Raymond and S. D. Colson, J. Am. Chem. Soc., 1963, 85, 3367–3372 CrossRef CAS.
- P. Hemmerich, C. Veeger and H. C. S. Wood, Angew. Chem., Int. Ed., 1965, 4, 671–687 CrossRef CAS PubMed.
- M. S. Grodowski, B. Veyret and K. Weiss, Photochem. Photobiol., 1977, 26, 341–352 CrossRef CAS.
- W. Moore, J. McDaniels and J. Hen, Photochem. Photobiol., 1977, 25, 505–512 CrossRef CAS PubMed.
- S.-Y. Ju and F. Papadimitrakopoulos, J. Am. Chem. Soc., 2008, 130, 655–664 CrossRef CAS PubMed.
- S. L. J. Tan, J. M. Kan and R. D. Webster, J. Phys. Chem. B, 2013, 117, 13755–13766 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Calibration curve for MTT-formazan used to calculate the photosensitizing activity of flavins, the pH dependence of the absorbance for the MTT formazan, the control reactions in the absence of methionine and of riboflavin, as well as the 1H- and 13C-NMR spectra of the newly synthesized compounds. See DOI: https://doi.org/10.1039/d2ra07379j |
| This journal is © The Royal Society of Chemistry 2023 |