
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrowave assisted regioselective halogenation of benzo[b][1,4]oxazin-2-ones via sp2 C–H functionalization†
Sandeep Kumar a,
Princea,
Mohit Guptaa,
Ram Sunil Kumar Laljiab and
Brajendra K. Singh*a
a,
Princea,
Mohit Guptaa,
Ram Sunil Kumar Laljiab and
Brajendra K. Singh*a
aBio-Organic Research Laboratory, Department of Chemistry, University of Delhi, Delhi, 110007, India. E-mail: singhbk@chemistry.du.ac.in
bDepartment of Chemistry, Kirori-mal College, Delhi University, Delhi, 110007, India
First published on 16th January 2023
Abstract
A microwave assisted, palladium-catalyzed regioselective halogenation of 3-phenyl-2H-benzo[b][1,4]oxazin-2-ones has been demonstrated using inexpensive and readily available N-halosuccinimide. The reaction utilizes the nitrogen atom present in the heterocyclic ring as the directing group to afford regioselective halogenated products in good to moderate yields. The established protocol provides wide substrate scope, high functional group tolerance, and high atom and step economy. The reaction proved to be cost-effective and time-saving as it required only a few minutes for completion and is amenable to gram scale. The halogen atoms present in synthesized products provide further scope for post-functionalization. Several post-functionalized products have also been synthesised to demonstrate the high utility of the reaction in the field of drug discovery and late-stage functionalization.
Introduction
Benzoxazin-2-ones are an extremely important class of heterocyclic compounds due to their ubiquity in natural products and pharmaceuticals.1 These compounds display diverse bioactivities such as anti-mycobacterial,2 anti-inflammatory,3 anti-hypertensive,4 anti-fungal,5 D2 receptor antagonist,6 antitumor,7 and inhibitor of platelet aggregation8 (Fig. 1). The core structural framework of benzoxazin-2-ones also proved to be an important building block for the synthesis of many medicines and photoactive materials.9 Owing to their widespread biological activities and applications, the synthesis of heterocyclic compounds carrying a benzoxazin-2-one scaffold is an active research field in pharmaceutical and medicinal chemistry. Organohalogens are widely spread in natural products and have remarkable biological activities.10 Some of these compounds find significant applications in industry and agriculture as well as in everyday life. The halogen functionality is unarguably the most synthetically valuable in organic chemistry due to its convertibility into other functional groups.11 Consequently, organohalogens proved to be extremely valuable building blocks in organic synthesis and used as precursors for many organic transformations such as Grignard reactions,12 nucleophilic substitution reactions13 and organolithium reactions.14 They also provide a versatile platform for many transition metal catalyzed cross-coupling reactions and can further be manipulated into countless valuable functionalities.15 Owing to their remarkable properties and applications, the development of simple protocols for the construction of aryl halides is of extensive interest. The common methods for the preparation of aryl halides include halogenation of electron rich arenes, oxidation of anilines to aromatic diazonium salt followed by halogenation16 and direct ortho-lithiation-halogenation sequence.17 Despite being immensely important industrial methods, these traditional methods for halogenation are associated with numerous shortcomings such as tedious and dangerous reaction procedures, poor regioselectivity, low product yields and toxic, air-sensitive, volatile and corrosive halogenating agents18 that restrict the economy, health and sustainability of common halogenating procedures.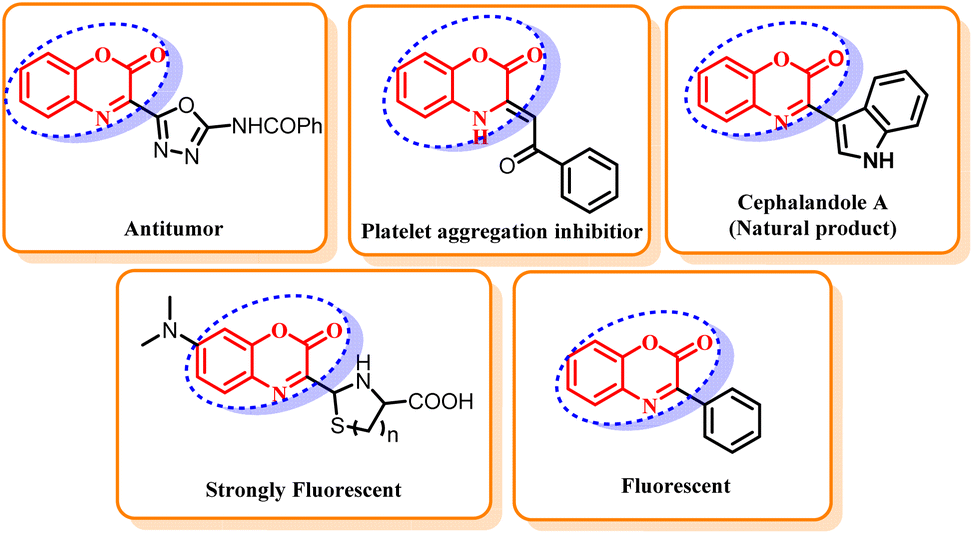 | ||
| Fig. 1 Importance of benzoxazin-2-one scaffold. | ||
During the past few decades, transition metal catalysed C–H activation and C–H functionalization has profoundly revolutionized the synthetic chemistry by providing sustainable tools for the construction of various C–C, C–O, C–N, C–S and C–X bonds.19 These transformations do not require any pre-functionalization of substrates, making the method step and atom economical.20 The coordinating group present in the heterocyclic compounds is commonly utilized to derive ortho-regioselectivity of C–H functionalization reactions.21 As a result, directing group assisted, transition metal catalyzed C–H halogenation proved to be a better alternative to the traditional halogenation reaction as they solve problems of regioselectivity, harsh reaction conditions and often require easy to handle reagents.22 From the perspective of green chemistry, tedious workups and time consuming reaction also limit the sustainability of a protocol. A non-conventional energy source such as microwave is a better alternative to the conventional heating. Microwave irradiation generates heat in the interior of the system and causes effective and uniform heating in a fraction of time. Consequently, it reduces the reaction time considerably and provides cleaner reactions that are easier to work up. Intrigued by the inherent advantages of transition-metal catalyzed transformation, microwave irradiation and our interest in the development of green and sustainable methodologies for the functionalization of biologically active heterocycles,23 we herein report microwave-assisted methodology for the regioselective, palladium catalyzed C–H halogenation of biologically important 3-phenyl-2H-benzo[b][1,4]oxazin-2-ones using N-halosuccinimide (NXS) as an inexpensive and commercially available halogenating agents. Till date, numerous halogenating agents have been developed to overcome the toxicity, regioselectivity and handling difficulty imposed by typical halogenating agents such as elemental chlorine, bromine and iodine.24 For instance, N-chloramines,19 HOCl,19 chlorinated cyclohexadiene,19 N-bromodialkylamines,25 ZrBr4,26 KBr/oxone,27 dioxane dibromide,28 I2/KMnO4,29 I2/H2O2,30 etc., are well known halogenating agents employed for the halogenation of aromatic compounds. Among these halogenating agents, N-halosuccinamides19,27,31 are the reagent of choice owing to their low cost, commercial availability, ease of handling, high compatibility with transition metal catalysts and solubility of succinimide by-products in water.32 To the best of our knowledge, transition-metal catalyzed halogenation of 3-phenyl-2H-benzo[b][1,4]oxazine-2-ones is not reported so far. Herein, we for the first time, are reporting transition-metal catalyzed microwave assisted halogenation of 3-phenyl-2H-benzo[b][1,4]oxazine-2-ones.
Results and discussion
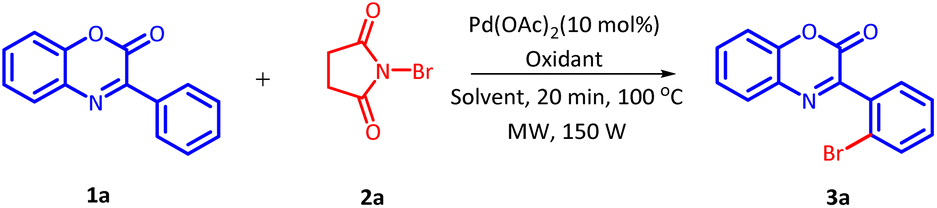
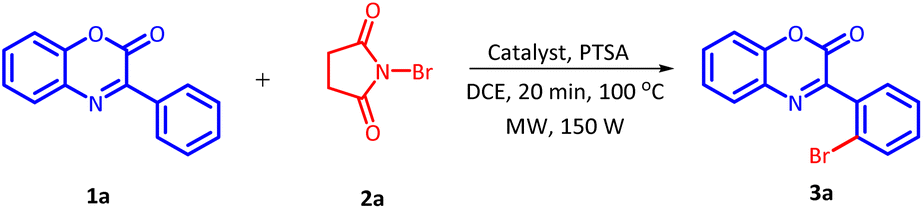
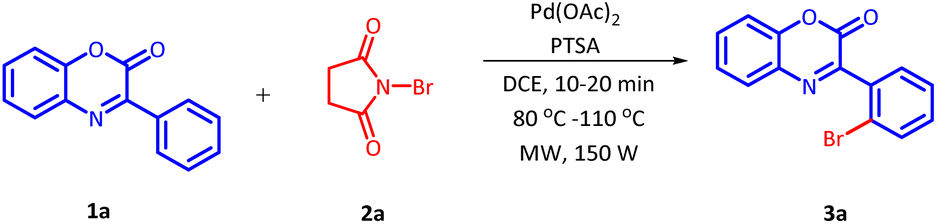
The initial investigation for the regioselective bromination of 3-phenyl-2H-benzo[b][1,4]oxazin-2-one (1a) started by employing commercially available N-bromosuccinimide (NBS) (2a) as the brominating agent, 10 mol% of Pd(OAc)2 as a catalyst and 0.5 equivalents of p-toluenesulphonic acid (PTSA) as an additive. Initially, the reaction mixture was irradiated with microwave at 100 °C for 20 minutes. The reaction proceeded smoothly and delivered the desired, regioselective, mono-brominated product 3a in 62% yield (Table 1, Entry 1). Inspired by these preliminary results, a variety of solvents were screened (Table 1, Entries 2–7). However, none of the solvent provided any improvement in product yield. Solvents such as H2O and DMSO were found completely ineffective for this transformation. Similarly, other solvents also exhibited an adverse influence on the outcome of this reaction. After fixing DCE as a suitable solvent, we next examine the effect of additives on the efficiency of the reaction (Table 1, Entries 8–12). On replacing PTSA with any other additive, an adverse effect on the product yield was recorded.
|
|||
|---|---|---|---|
| S. no. | Solvent (2 mL) | Additive (0.5 equiv.) | Yieldsb (%) |
| a Reagents and conditions: a mixture of 3-phenyl-2H-benzo[b][1,4]oxazin-2-one (1a, 1 equiv.), NBS (2a, 1.2 equiv.), Pd(OAc)2 (10 mol%) and additive (0.5 equiv.) were irradiated with microwave under indicated solvent at indicated temperature for given period of time in sealed reaction vial.b Isolated yields. | |||
| 1 | DCE | PTSA | 62 |
| 2 | Toluene | PTSA | 53 |
| 3 | MeCN | PTSA | 47 |
| 4 | H2O | PTSA | NR |
| 5 | DMF | PTSA | 41 |
| 6 | DMSO | PTSA | NR |
| 7 | AcOH | PTSA | 19 |
| 8 | DCE | Cu(OAc)2 anhyd. | 15 |
| 9 | DCE | Cu(OTf)2 | 39 |
| 10 | DCE | CuO | 43 |
| 11 | DCE | K2S2O8 | 29 |
| 12 | DCE | AgOAc | 42 |
Similarly, when the regioselective bromination of 1a was attempted with different catalysts, no improvement in product yield was perceived (Table 2, Entries 1–10). Except palladium, none of the screened metal could lead to the desired transformation. Moreover, all the screened palladium catalysts delivered the desired product 3a in slightly inferior yield as compared to the palladium acetate.
|
||
|---|---|---|
| S. no. | Catalyst (10 mol%) | Yieldsb (%) |
| a Reagents and conditions: a mixture of 3-phenyl-2H-benzo[b][1,4]oxazin-2-one (1a, 1 equiv.), NBS (2a, 1.2 equiv.), catalyst (10 mol%) and PTSA (0.5 equiv.) were irradiated with microwave under indicated solvent at indicated temperature for given period of time in sealed reaction vial.b Isolated yields. | ||
| 1 | PdCl2 | 25 |
| 2 | Pd(PPh3)4 | 60 |
| 3 | PdCl2(PPh3)2 | 30 |
| 4 | Pd2(dba)3 | 39 |
| 5 | Cu(OAc)2·2H2O | NR |
| 6 | Fe(acac)3 | NR |
| 7 | CoCl2 anhyd. | Trace |
| 8 | NiCl2 | NR |
| 9 | Ru(p-cym)Cl2 dimer | NR |
| 10 | (Cp*RhCl2)2 | NR |
The efficiency of the reaction was slightly affected by the amount of PTSA used in the reaction (Table 3, Entries 1–3). A maximum yield of 69% was recorded on decreasing the amount of PTSA from 0.5 equivalents to 0.25 equivalents (Table 3, Entry 3). However, lower product yield was observed on increasing the amount of PTSA from 0.5 equivalents to 0.7 and 1 equivalent (Table 3, Entries 2–3). To enhance the product yield further, the catalyst loading was inspected next (Table 3, Entries 4–7). Reduced catalytic activity was observed on increasing the catalyst loading from 10 mol% to 15 mol%. Nevertheless, the catalytic activity of palladium acetate increases on decreasing the catalyst loading to 5 mol% and product was obtained in 74% yield (Table 3, Entry 6). Further decreases in catalytic loading showed a deteriorating effect on the reaction efficiency (Table 3, Entry 7). Next, the effect of temperature on this regioselective bromination of 3-phenyl-2H-benzo[b][1,4]oxazin-2-one (1a) was studied (Table 3, Entries 8–10). Multiple by-products were detected at higher temperature leading to inferior product yield (Table 3, Entry 8). Below 100 °C, incomplete conversion of the reaction was recorded with inferior isolated yield even after 30 min of reaction (Table 3, Entries 9 and 10). After setting up the optimum temperature, we subsequently examined the optimum reaction time for this reaction (Table 3, Entries 11–13). A dramatic increase in product yield was observed by shortening the reaction time duration and a maximum of 84% yield of the desired product was achieved in 15 minutes (Table 3, Entry 12). Further decreases in reaction time rendered the reactant unreacted. On prolong heating, no significant change in product yield was observed. In conclusion, the optimum reaction condition for the regioselective bromination of 3-phenyl-2H-benzo[b][1,4]oxazin-2-one (1a) was established as 5 mol% of Pd(OAc)2, 0.25 equivalents of PTSA, 2 mL of DCE, 100 °C temperature under microwave irradiation for 15 minutes. It is worth mentioning here that, when this reaction was carried out under conventional heating the reaction could be completed in 1 hour with 70% yield (Table 3, Entry 14).33
|
|||
|---|---|---|---|
| S. no. | Pd(OAc)2 | PTSA (equiv.) | Yieldsb (%) |
| a Reagents and conditions: a mixture of 3-phenyl-2H-benzo[b][1,4]oxazin-2-one (1a, 1 equiv.), NBS (2a, 1.2 equiv.), Pd(OAc)2 and PTSA as indicated in the parenthesis were irradiated with microwave under indicated solvent at indicated temperature for given period of time in sealed reaction vial.b Isolated yields.c Reaction mixture was irradiated at 110 °C for 15 minutes.d Reaction mixture was irradiated at 90 °C for 30 minutes.e Reaction was irradiated at 80 °C for 30 minutes.f Reaction was irradiated at 100 °C for 25 minutes.g Reaction was irradiated at 100 °C for 15 minutes.h Reaction was irradiated at 100 °C for 10 minutes.i Reaction was carried out at 100 °C under conventional heating for 1 hour. | |||
| 1 | 10 mol% | 0.75 | 60 |
| 2 | 10 mol% | 1.0 | 56 |
| 3 | 10 mol% | 0.25 | 69 |
| 4 | 15 mol% | 0.25 | 46 |
| 5 | 7.5 mol% | 0.25 | 69 |
| 6 | 5 mol% | 0.25 | 74 |
| 7 | 3 mol% | 0.25 | 70 |
| 8 | 5 mol% | 0.25 | 64c |
| 9 | 5 mol% | 0.25 | 70d |
| 10 | 5 mol% | 0.25 | 61e |
| 11 | 5 mol% | 0.25 | 62f |
| 12 | 5 mol% | 0.25 | 84g |
| 13 | 5 mol% | 0.25 | 65h |
| 14 | 5 mol% | 0.25 | 70i |
After having the optimized reaction condition in hand, we subsequently evaluated the substrate scope of this established protocol. A series of differently substituted 3-phenyl-2H-benzo[b][1,4]oxazin-2-ones were allowed to react with NBS under the standard reaction condition (Scheme 1). All the subjected reactants afforded the desired products in good to excellent yields.
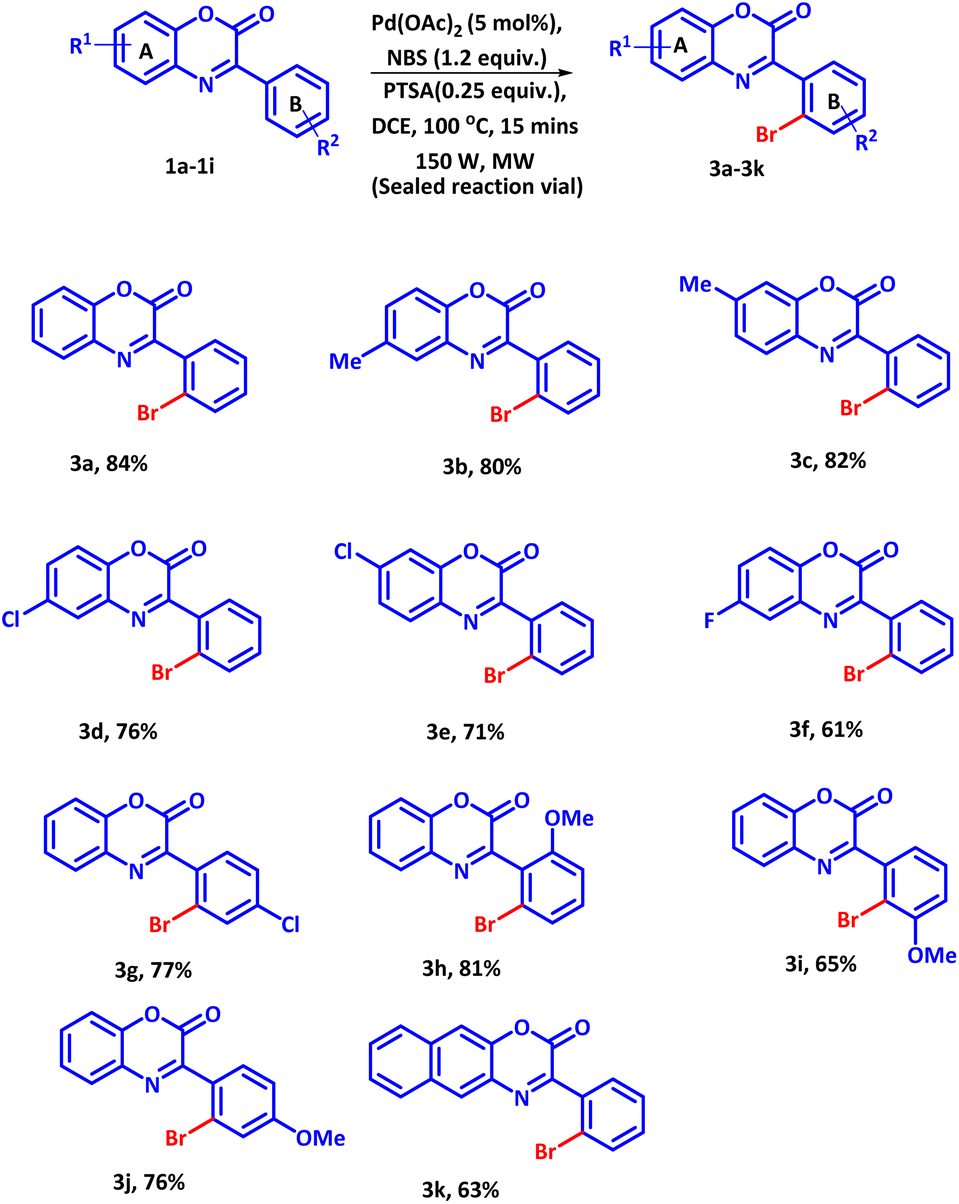 | ||
| Scheme 1 Substrate scope of 3-phenyl-2H-benzo[b][1,4]oxazin-2-ones with NBS. | ||
It was observed that electron donating groups present on ring A delivered better product yields as compared to the electron withdrawing groups (Scheme 1, 3b, 3d and 3f) and (Scheme 1, 3c and 3e). A reverse electronic effect was perceived with substituents present on ring B. Electron-donating groups delivered the product in slightly inferior yield as compared to the electron-withdrawing groups (Scheme 1, 3j and 3g). It is worth mentioning here that with strong electron-withdrawing group such as –NO2 & –CN started material remained unreactive. To investigate the effect of -ortho, -meta & -para substituent on ring B, -ortho, -meta & -para derivatives were subjected to bromination. The -ortho, and -para derivatives delivers better product yields as compared to -meta substituent (Scheme 1, 3h–3j). Moreover, expanding the scope from benzene to naphthalene fused oxazine-2-one system also facilitated the formation of desired product in good yield (Scheme 1, 3k).
The success of the established protocol was further extended to the regioselective iodination of 3-phenyl-2H-benzo[b][1,4]oxazin-2-ones using NIS as iodinating agent (Scheme 2). Owing to the high reactivity of NIS as compared to NBS and NCS, the reaction proceeded to completion in just 5 minutes at slight lower temperature. However, under conventional heating, the reaction could be completed in 30 min with inferior 60% yield. Under this optimized condition, a variety of 3-phenyl-2H-benzo[b][1,4]oxazin-2-ones bearing electron donating and electron-withdrawing groups were reacted with 1.25 equiv. of NIS. All the substrates afforded the regioselective -ortho mono iodo products in moderate to good yields. It was observed that electron-donating groups present on ring A, such as (–CH3) enhanced the product yield whereas, a negative effect on the product yield was observed with electron-withdrawing groups (Scheme 2, 4a–4f). For ring B, a reverse trend was observed. Electron-withdrawing groups enhanced the product yield whereas electron-donating groups exhibited an adverse effect on product yield (Scheme 2, 4g and 4h). Moreover, 3-phenyl-2H-naphtho[2,3-b][1,4]oxazin-2-one also successfully delivered the product in good yield (Scheme 2, 4i), suggesting high compatibility of the established protocol.
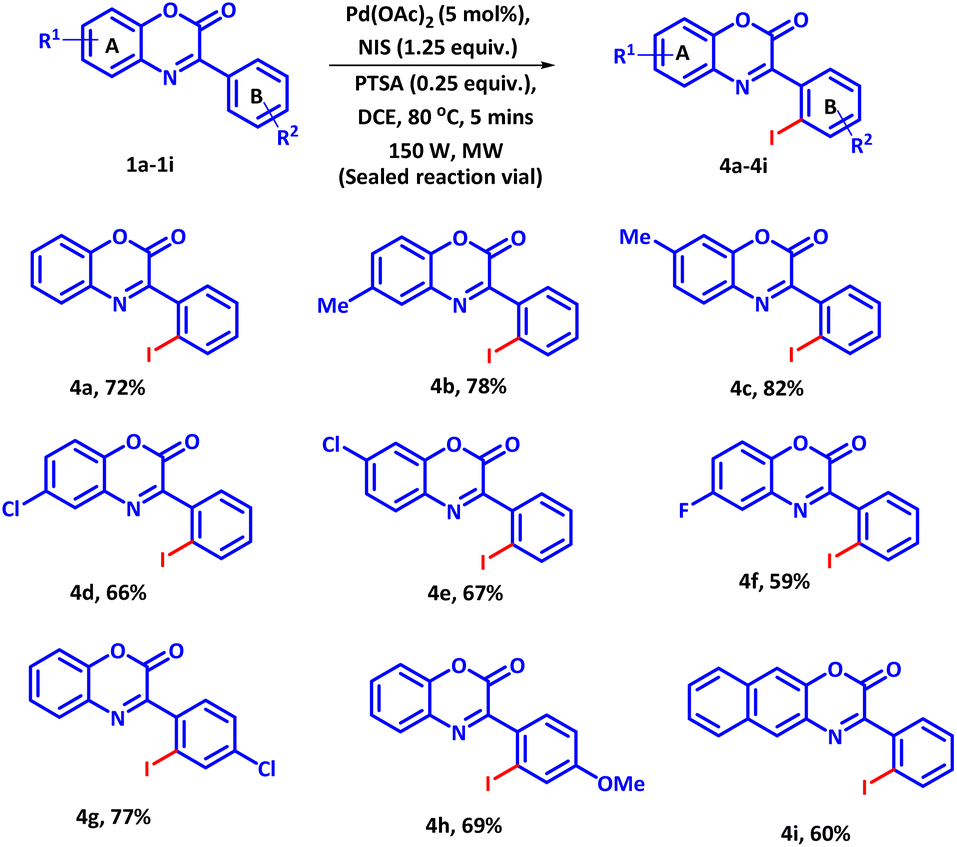 | ||
| Scheme 2 Substrate scope of 3-phenyl-2H-benzo[b][1,4]oxazin-2-ones with NIS. | ||
Next, we evaluated the substrate scope of the established protocol for regioselective chlorination of 3-phenyl-2H-benzo[b][1,4]oxazin-2-ones using NCS as chlorinating agent under microwave condition (Scheme 3). It was observed that reaction proceeded to completion at 120 °C and yielded product 5a with 75% yield in 30 minutes. However, under conventional heating the reaction could be completed in 8 hours with 60% product yield. Moreover, slightly larger amount of PTSA and NCS compared to bromination and iodination were required to afford the best result. The reason for sluggish nature of the reaction is attributed to the low reactivity of NCS and high bond dissociation energy of N–Cl bond. With optimized condition, diversely substituted 3-phenyl-2H-benzo[b][1,4]oxazin-2-ones were subjected to react with NCS. All the reaction proceeded smoothly and afford the products in good to moderate yields. The electron withdrawing groups present on ring A furnished the desired products in lower yields as compared to the electron donating groups (Scheme 3, 5b–5f). Contrary electronic effect was observed with the substituents present on ring B. Electron withdrawing group afforded the product in higher yield as compared to the electron donating group (Scheme 3, 5g and 5h). Moreover, the naphthyl group was also tolerated well under the established reaction condition (Scheme 3, 5i). Contrary electronic effect was observed with the substituents present on ring B. Electron withdrawing group afforded the product in higher yield as compared to the electron donating group (Scheme 3, 5g and 5h). Moreover, the naphthyl group was also tolerated well under the established reaction condition (Scheme 3, 5i).
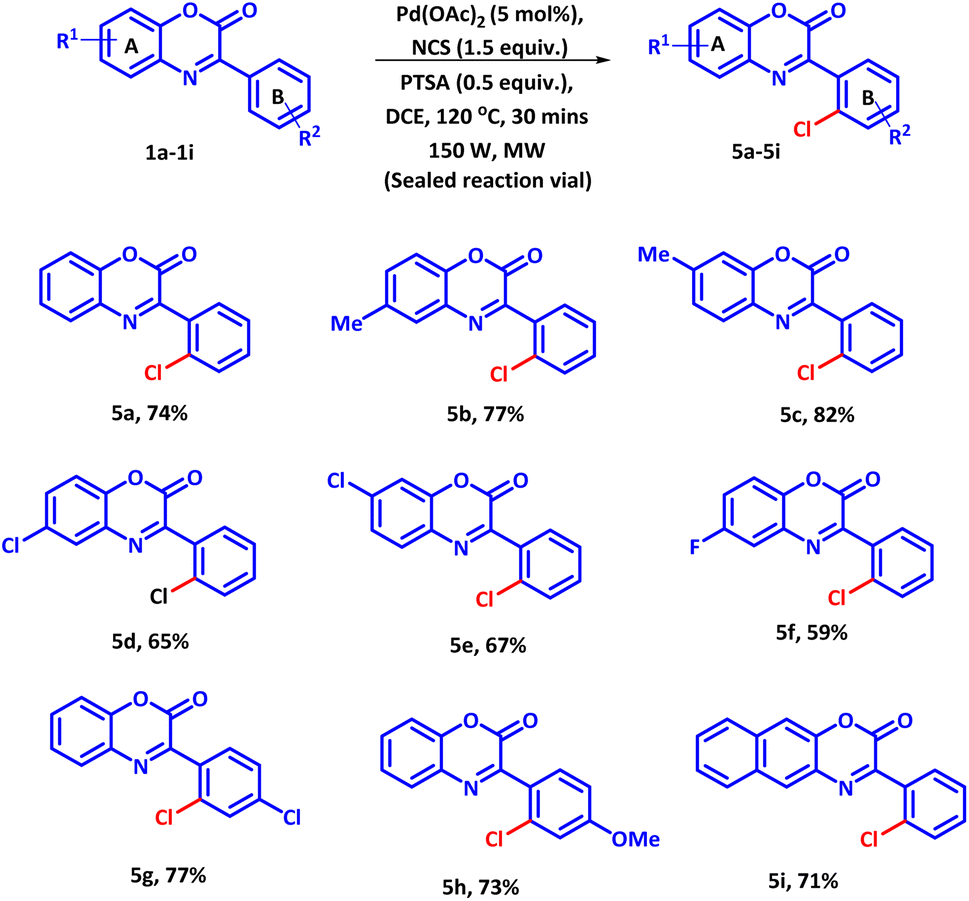 | ||
| Scheme 3 Substrate scope for regioselective chlorination of 3-phenyl-2H-benzo[b][1,4]oxazin-2-ones. | ||
To demonstrate the practicality and efficiency of the established methodology, we conducted scale-up experiment for the regioselective bromination of 3-phenyl-2H-benzo[b][1,4]oxazin-2-one under microwave conditions (Scheme 4). Under the established reaction conditions, 5 mmol of 1a afforded the product 3a in 80% yield, suggesting real time application of the protocol.
 | ||
| Scheme 4 Gram scale synthesis of 3-(2-bromophenyl)-2H-benzo[b][1,4]oxazin-2-one. | ||
To further explore the synthetic application of the established methodology, late stage functionalization was explored. The product 3a was subjected to Suzuki coupling with various aryl boronic acids (Scheme 5). All the boronic acids coupled smoothly and afforded the desired products in excellent yields. Thus, the synthetic applications of the established protocol open avenues for further functionalization or functional group manipulation of biologically active molecules with excellent regioselectivity.
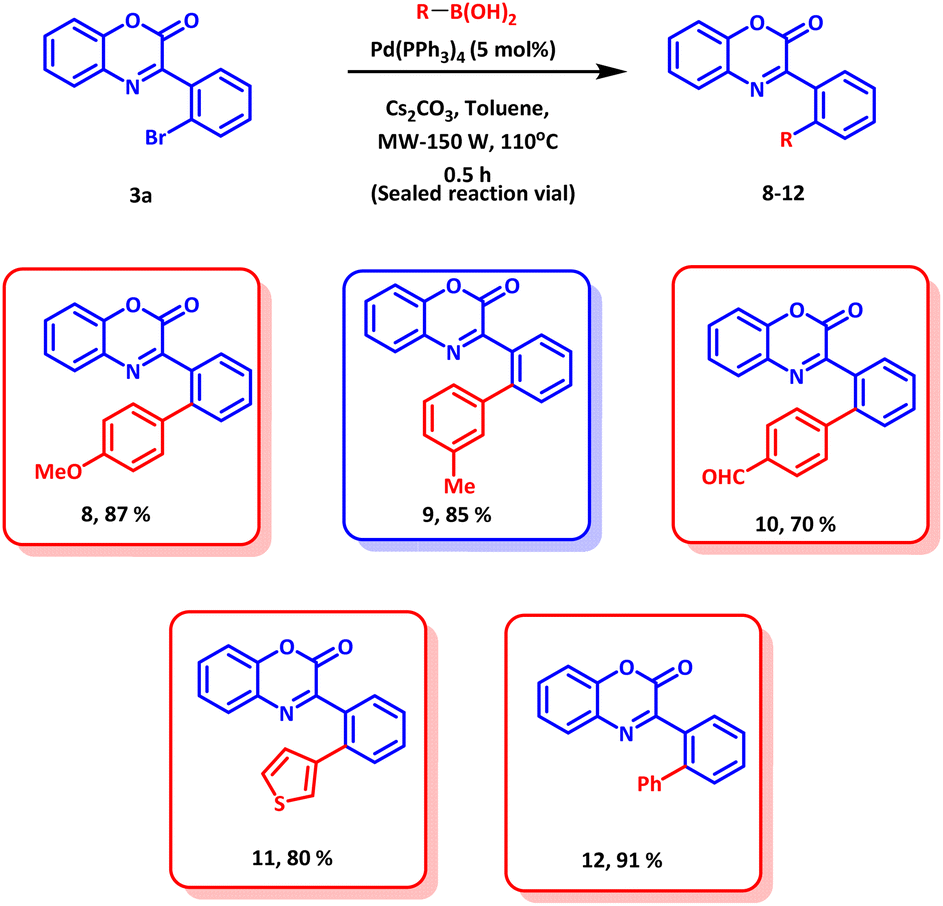 | ||
| Scheme 5 Synthetic applications; late stage functionalizations. | ||
To gain the insight of the reaction mechanism, several control experiments were carried out (Scheme 6). In the first control experiment, the reaction was allowed to run under standard conditions without the addition of bromination agent (NBS). As expectedly, no reaction was observed and the reactant was rendered unreacted (Scheme 6a). Subsequently, the reaction was allowed to run in the absence of the catalyst. A traces amount of unidentified product was observed on thin layer chromatography analysis (Scheme 6b). Evidently, the catalyst is crucial to complete the reaction in a short duration of time. Further, the influence of PTSA on the reaction outcome was examined by conducting the same reaction in the absence of PTSA (Scheme 6c).
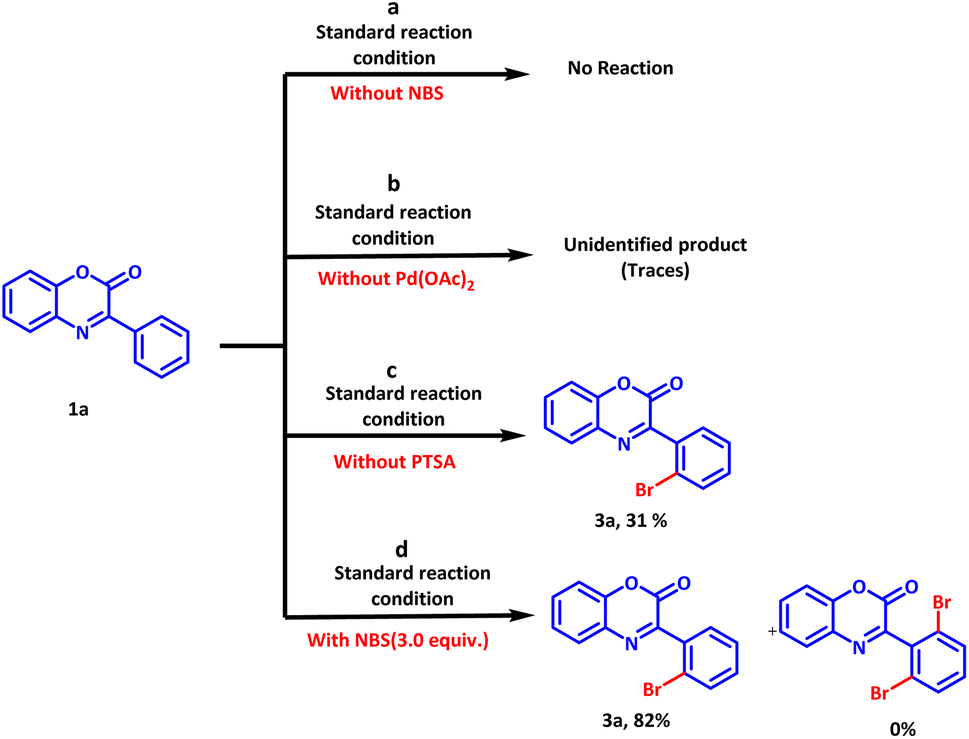 | ||
| Scheme 6 Control experiments to ascertain the reaction mechanism. | ||
A maximum yield of 31% suggested the importance of PTSA in promoting the reaction. However, PTSA alone is unable to promote the reaction. Moreover, formation of dibromo derivative was not observed even with 3.0 equivalents of NBS and only ortho mono bromo derivative was isolated in 82% yield.
Based on our control experiments and available literature reports, a plausible reaction mechanism for the regioselective halogenation is outlined in Scheme 7. Initially, palladium acetate reacts with PTSA to generate Pd(OTs)2 which undergoes C–H activation reaction with the substrate 1a to afford cyclopalladated intermediate 13.34 The intermediate 13 undergoes oxidative addition with halogenating agent NXS (2a–2c) to furnish intermediate 14. The oxidation state of palladium increases from Pd(II) to Pd(IV) in this step. In the next step, reductive elimination occurs to afford the desired product along with the regeneration of Pd(OTs)2 to continue the catalytic cycle.
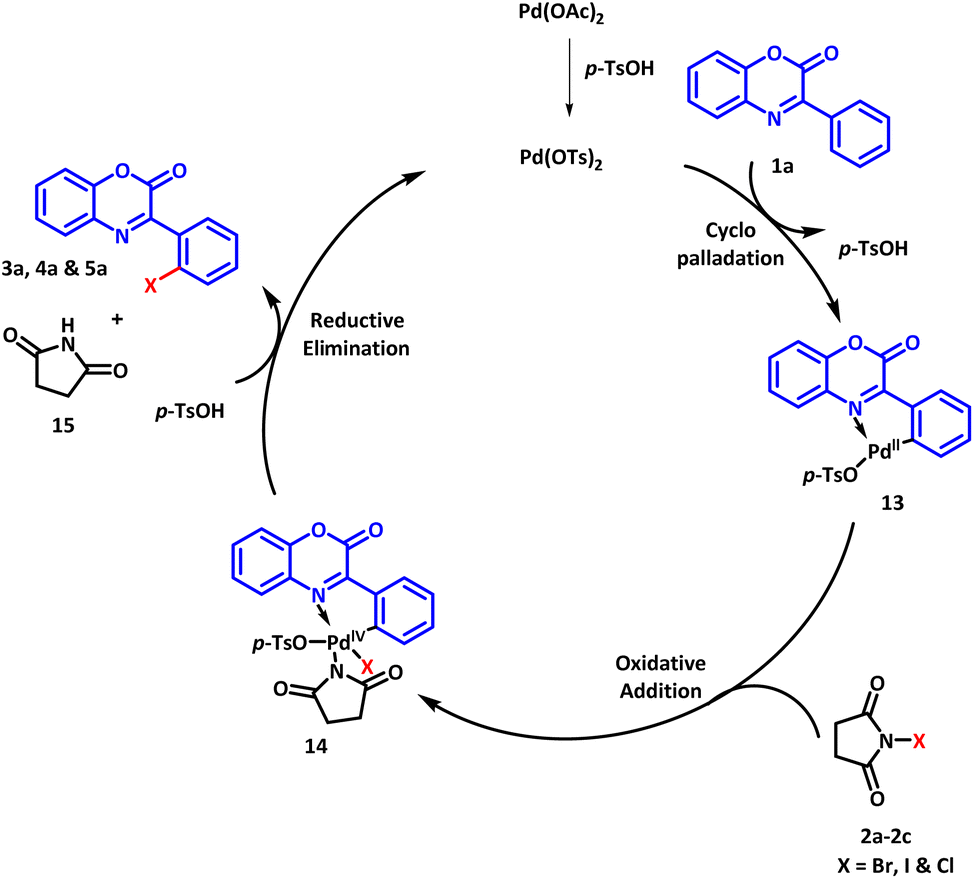 | ||
| Scheme 7 Plausible reaction mechanism. | ||
Conclusion
In summary, we have described microwave assisted, palladium-catalyzed sp2 C–H halogenation of 3-phenyl-2H-benzo[b][1,4]oxazin-2-ones using inexpensive and readily available N-halosuccinimide in high product yields. The coordinating ability of the nitrogen atom present in the heterocyclic ring is utilized to drive excellent regioselectivity at the ortho-position of the benzene ring in the presence of several other potentially reactive positions. Further late stage functionalization of halogenated products using Suzuki reaction demonstrated high synthetic utility in the field of drug discovery and late stage functionalization. The characteristic benefits of the transformation include high functional group tolerance, wide substrate scope, high atom and step economy, cost-effective, time-saving and is amenable to gram scale.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the University of Delhi IoE (Institution of Eminence) grant. We also thank University Science Instrumentation Centre (USIC), University of Delhi for instrumental facilities. Sandeep Kumar acknowledges University Grant Commission for Senior Research Fellowship and Prince thanks CSIR, New Delhi, India for providing Senior Research Fellowship.Notes and references
- (a) K. Krohn, H. A. Kirst and H. Maag, Antibiotics and antiviral compounds: chemical synthesis and modification, VCH, Weinheim, 1993 Search PubMed; (b) J. Teller and E. Schaumann, Hetarenes IV, Six-Membered and Larger 20 Hetero-Rings with Maximum Unsaturation, Houben-Weyl Methods of Organic Chemistry, Georg Thieme, Stuttgart, 1997, vol. E9a, p. 141 Search PubMed; (c) J. Ilaš, P. Š. Anderluh, M. S. Dolenc and D. Kikelj, Tetrahedron, 2005, 31, 7325 CrossRef; (d) B. Achari, S. B. Mandal, P. K. Dutta and C. Chowdhury, Synlett, 2004, 2449 CAS; (e) T. Hasui, N. Matsunaga, T. Ora, N. Ohyabu, N. Nishigaki, Y. Imura, Y. Igata, H. Matsui, T. Motoyaji, T. Tanaka, N. Habuka, S. Sogabe, M. Ono, C. S. Siedem, T. P. Tang, C. Gauthier, L. A. D Meese, S. A. Boyd and S. Fukumoto, J. Med. Chem., 2011, 54, 8616 CrossRef CAS PubMed; (f) H. Matsuoka, N. Ohi, M. Mihara, H. Suzuki, K. Miyamoto, N. Maruyama, K. Tsuji, N. Kato, T. Akimoto, Y. Takeda, K. Yano and T. Kuroki, J. Med. Chem., 1997, 40, 105 CrossRef CAS PubMed; (g) S. M. Bromidge, B. Bertani, M. Borriello, S. Faedo, L. J. Gordon, E. Granci, M. Hill, H. R. Marshall, L. P. Stasi, V. Zucchelli and G. Merlo, Bioorg. Med. Chem. Lett., 2008, 18, 5653 CrossRef CAS PubMed; (h) S. M. Bromidge, B. Bertani, M. Borriello, A. Bozzoli, S. Faedo, M. Gianotti, L. J. Gordon, M. Hill, V. Zucchelli, J. M. Watson and L. Zonzini, Biorg. Med. Chem. Lett., 2009, 19, 2338 CrossRef CAS PubMed; (i) T. Hasui, T. Ohra, N. Ohyabu, K. Asano, H. Matsui, A. Mizukami, N. Habuka, S. Sogabe, S. Endo, C. S. Siedem and T. P. Tang, Bioorg. Med. Chem., 2013, 21, 5983 CrossRef CAS PubMed.
- (a) K. Waisser, M. Peřina, J. Kuneš, V. Klimešová and J. Kaustová, II Farmaco, 2003, 58, 1137 CrossRef CAS PubMed; (b) S. Konda, S. Raparthi, K. Bhaskar, R. K. Munaganti, V. Guguloth, L. Nagarapu and D. M. Akkewar, Bioorg. Med. Chem. Lett., 2015, 25, 1643 CrossRef CAS PubMed.
- (a) V. Mashevskaya, L. V. Anikina, Y. B. Vikharev, V. A. Safin, S. V. Kol'tsova and N. Maslivets, Pharm. Chem. J., 2001, 35, 414 CrossRef; (b) K. Ali, A. Mohammad, K. Abbas, A. Neda, P. Zahra and R. Sara, Indian J. Pharmacol., 2002, 34, 184 Search PubMed.
- F. Touzeau, A. Arrault, G. Guillaumet, E. Scalbert, B. Pfeiffer, M. C. Rettori, P. Renard and J. Y. Mérour, J. Med. Chem., 2003, 46, 1962 CrossRef CAS PubMed.
- K. Waisser, L. Kubicová, V. Buchta, P. Kubanová, K. Bajerová, L. Jirásková, O. Bednařík, O. Bureš and P. Holý, Folia Microbiol., 2002, 47, 488 CrossRef CAS PubMed.
- L. D. Wise, D. J. Wustrow and T. Belliotti, WO Pat.Appl. 9745419, 1997 Search PubMed.
- S. Bondock, S. Adel, H. A. Etman and F. A. Badria, Eur. J. Med. Chem., 2012, 48, 192 CrossRef CAS PubMed.
- P. K. Jaiswal, V. Sharma, S. Kumar, M. Mathur, A. K. Swami, D. K. Yadav and S. Chaudhary, Arch. Pract. Pharm., 2018, 351, 1 Search PubMed.
- (a) Q. A. Chen, M. W. Chen, C. B. Yu, L. Shi, D. S. Wang, Y. Yang and Y. G. Zhou, J. Am. Chem. Soc., 2011, 133, 16432 CrossRef CAS PubMed; (b) M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 6751 CrossRef CAS PubMed; (c) C. Saitz, H. Rodríguez, A. Márquez, A. Cañete, C. Jullian and A. Zanocco, Synth. Commun., 2001, 31, 135 CrossRef CAS; (d) H. Miyabe, Y. Yamaoka and Y. Takemoto, J. Org. Chem., 2006, 71, 2099 CrossRef CAS PubMed; (e) S. Nonell, L. R. Ferreras, A. Cañete, E. Lemp, G. Günther, N. Pizarro and A. L. Zanocco, J. Org. Chem., 2008, 73, 5371 CrossRef CAS PubMed; (f) K. Azuma, S. Suzuki, S. Uchiyama, T. Kajiro, T. Santa and K. Imai, Photochem. Photobiol. Sci., 2003, 2, 443 CrossRef CAS PubMed; (g) M. Hu, J. Fan, H. Li, K. Song, S. Wong, G. Cheng and X. Peng, Org. Biomol. Chem., 2011, 9, 980 RSC.
- (a) G. W. Gribble, Acc. Chem. Res., 1998, 31, 141 CrossRef CAS; (b) N. Winterton, Green Chem., 2000, 2, 173 RSC; (c) G. W. Gribble, J. Chem. Educ., 2004, 81, 1441 CrossRef CAS.
- (a) H. Y. Xiong, D. Cahard, X. Pannecoucke and T. Besset, Eur. J. Org. Chem., 2016, 3625 CrossRef CAS; (b) S. Sathyamoorthi, S. Banerjee, J. D. Bois, N. Z. Burns and R. N. Zare, Chem. Sci., 2018, 9, 100 RSC; (c) R. Das and M. Kapur, Asian J. Org. Chem., 2018, 7, 1524 CrossRef CAS.
- Handbook of Grignard Reagents, ed. G. S. Silverman and P. E. Rakita, Dekker, New York, 1996 Search PubMed.
- (a) Y. L. Janin, Chem. Rev., 2012, 112, 3924 CrossRef CAS PubMed; (b) I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837 CrossRef CAS PubMed.
- N. Sotomayor and E. Lete, Curr. Org. Chem., 2003, 7, 275 CrossRef CAS.
- A. D. Meijere, S. Bräse and M. Oestreich, Metal-Catalyzed Cross-Coupling Reactions and More, Wiley-VCH, New York, 2014 Search PubMed.
- Y. Xiang, P. Y. Caron, B. M. Lillie and R. Vaidyanathan, Org. Process Res. Dev., 2008, 12, 116 CrossRef CAS.
- V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS.
- I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837 CrossRef CAS PubMed.
- (a) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS PubMed; (b) S. Agasti, A. Dey and D. Maiti, Chem. Commun., 2017, 53, 6544 RSC; (c) R. Y. Zhu, M. E. Farmer, Y. Q. Chen and J. Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS PubMed; (d) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138 CrossRef CAS PubMed; (e) P. Gandeepan and C. H. Cheng, Chem.–Asian J., 2015, 10, 824 CrossRef CAS PubMed; (f) J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70 RSC; (g) G. Yan, A. J. Borah and M. Yang, Adv. Synth. Catal., 2014, 356, 2375 CrossRef CAS; (h) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (i) J. W. Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef PubMed; (j) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (k) B. J. Li and Z. J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (l) J. Hu, C. Wang, M. Yu, S. Zhang, N. Chen and H. Du, Eur. J. Org. Chem., 2022, e202101266 CAS.
- (a) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (b) J. W. Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef PubMed.
- (a) R. Das and M. Kapur, Asian J. Org. Chem., 2018, 7, 1524 CrossRef CAS; (b) F. Lied, T. Patra and F. Glorius, Isr. J. Chem., 2017, 57, 945 CrossRef CAS; (c) D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003 CrossRef CAS PubMed; (d) D. Sarkar, A. V. Gulevich, F. S. Melkonyan and V. Gevorgyan, ACS Catal., 2015, 5, 6792 CrossRef CAS.
- (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (b) A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439 CrossRef CAS; (c) X. Chen, X. S. Hao, C. E. Goodhue and J. Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed; (d) A. Sen, Acc. Chem. Res., 1998, 31, 550 CrossRef CAS; (e) B. D. Dangel, J. A. Johnson and D. Sames, J. Am. Chem. Soc., 2001, 123, 8149 CrossRef CAS PubMed; (f) M. Lin, C. Shen, E. A. G. Zayas and A. Sen, J. Am. Chem. Soc., 2001, 123, 1000 CrossRef CAS PubMed.
- (a) M. Gupta, P. Kumar, V. Bahadur, K. Kumar, V. S. Parmar and B. K. Singh, Eur. J. Org. Chem., 2018, 896 CrossRef CAS; (b) P. Kumar, M. Gupta, V. Bahadur, V. S. Parmar and B. K. Singh, Eur. J. Org. Chem., 2018, 1552 CrossRef CAS; (c) M. Gupta, S. Kumar, P. Kumar, A. K. Singh, V. Bahadur and B. K. Singh, ChemistrySelect, 2019, 4, 13992 CrossRef CAS; (d) R. S. K. Lalji, P. Kumar, M. Gupta, V. S. Parmar and B. K. Singh, Adv. Synth. Catal., 2020, 362, 552 CrossRef CAS; (e) P. Kumar, S. Dutta, S. Kumar, V. Bahadur, E. V. V. Eycken, K. S. Vimaleswaran, V. S. Parmar and B. K. Singh, Org. Biomol. Chem., 2020, 18, 7987 RSC; (f) P. Prince, S. Kumar, R. S. K. Lalji, M. Gupta, P. Kumar, R. Kumar and B. K. Singh, Org. Biomol. Chem., 2022, 20, 8944 RSC.
- M. B. Smith, March's Advanced Organic Chemistry Reactions, Mechanisms, and Structure, Wiley, New York, 7th edn, 2013, p. 603 Search PubMed.
- E. Schmitz and I. Pagenkopf, J. Prakt. Chem., 1985, 327, 998 CrossRef CAS.
- T. Stropnik, S. Bombek, M. Kočevar and S. Polanc, Tetrahedron Lett., 2008, 49, 1729 CrossRef CAS.
- N. Narender, P. Srinivasu, M. Ramakrishna Prasad, S. J. Kulkarni and K. V. Raghavan, Synth. Commun., 2002, 32, 2313 CrossRef CAS.
- S. K. Chaudhuri, S. Roy, M. Saha and S. Bhar, Synth. Commun., 2007, 37, 579 CrossRef CAS.
- J. A. Chen, C. S. Lin and L. K. Liu, J. Chin. Chem. Soc., 1996, 43, 95 CrossRef CAS.
- J. Iskra, S. Stavber and M. Zupan, Synthesis, 2004, 1869 CrossRef CAS.
- H. Eguchi, K. Tokumoto and H. Shuyama, Toso Kenkyu Hokoku, 1993, 37, 109 CAS.
- W. Lorpaiboon and P. Bovonsombat, Org. Biomol. Chem., 2021, 19, 7518 RSC.
- (a) C. O. Kappe, B. Pieber and D. Dallinger, Angew. Chem., Int. Ed., 2013, 52, 1088 CrossRef CAS PubMed; (b) A. Sharma, P. Appukkuttan and E. V. Eycken, Chem. Commun., 2012, 48, 1623 RSC; (c) A. D. Ortiz, P. Prieto and A. Hoz, Chem. Rec., 2019, 19, 85 CrossRef PubMed; (d) C. O. Kappe and D. Dallinger, Nat. Rev. Drug Discovery, 2006, 5, 51 CrossRef CAS PubMed; (e) V. P. Mehta and E. V. Eycken, Chem. Rev., 2011, 40, 4925 CAS.
- K. R. Reddy, P. Kannaboina and P. Das, Asian J. Org. Chem., 2017, 6, 534 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07259a |
| This journal is © The Royal Society of Chemistry 2023 |