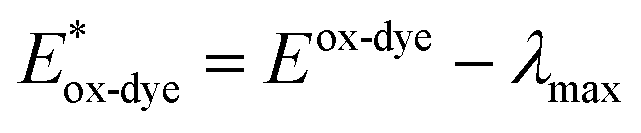DOI:
10.1039/D2RA06767F
(Paper)
RSC Adv., 2023,
13, 1019-1030
Computational study of linear carbon chain based organic dyes for dye sensitized solar cells†
Received
26th October 2022
, Accepted 13th December 2022
First published on 4th January 2023
Abstract
Spectroscopic, electronic and electron injection properties of a new class of linear carbon chain (LCC) based organic dyes have been investigated, by means of density functional theory (DFT) and time-dependent density functional theory (TDDFT), for application in dye-sensitized solar cells (DSSCs). The photophysical properties of LCC-based dyes are tuned by changing the length of the linear carbon chain; UV/VIS absorption is red-shifted with increasing LCC length whereas oscillator strength and electron injection properties are reduced. Excellent nonlinear optical properties are predicted in particular for PY-N4 and PY-S4 dyes in the planar conformation. Results indicate that a LCC-bridge produces better results compared to benzene and thiophene bridges. Simulations of I−-Dye@(TiO2)14 and Dye@(TiO2)14 anatase complexes indicate that designed dyes inject electrons efficiently into the TiO2 surface and can be regenerated by electron transfer from the electrolyte. Superior properties in terms of efficiency are shown by compounds with a pyrrole ring as the donor group and PY-3N is expected to be a promising candidate for applications, however all the investigated dyes could provide a good performance in solar energy conversion. Our study demonstrates that computational design can provide a significant contribution to experimental work; we expect this study will contribute to future developments to identify new and highly efficient sensitizers.
Introduction
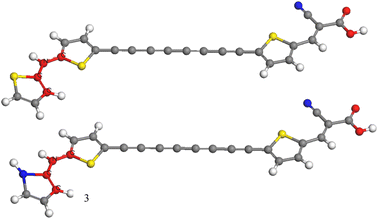
Dye sensitized solar cells, also known as Gratzel cells,1 are considered a mature solar energy conversion technology with promising photovoltaic technologies representing a valid alternative to traditional silicon-based solar cells. DSSCs attract great interest due to their low-cost fabrication along with their peculiar optical and mechanical properties and their high device efficiency.2,3 Photosensitizers represent one of the most essential components of a DSSC device being responsible for the absorption of solar photons and in turn the generation of photocurrent.4 A myriad of photosensitizers have been synthesized and studied in the last decades, more in general they belong to three main classes, ruthenium(II) polypyridyl complexes, zinc(II) porphyrin derivatives and metal-free organic dyes. Among the ruthenium complexes, derivatives containing two isocyanate ligands showed an absorption which is extended up to 800 nm producing very high values of the photocurrent density determined in short circuit conditions (JSC), and efficiency.5–9 Thiocyanate free ruthenium sensitizers have also been prepared, giving rise to a device with considerable photovoltaics properties.10,11 The other important class of metal DSSC sensitizers is based on porphyrin derivatives. The first study on these derivatives showed electron injection to be very fast and comparable to that of ruthenium based DSSCs.12 Further studies revealed that push–pull porphyrins, compounds in which an electron donor group and an electron acceptor group were spaced by the macrocycle, provided several advantages, including the extension of optical absorption, the improving of light harvesting efficiency (LHE) and better electron injection properties.13 This finding paved the way for many push pull porphyrins derivatives to further improve efficiency and stability toward the commercialization.14–23 Both ruthenium and porphyrin dyes have been limited since the difficulty of their synthesis and mainly their undesirable environmental impact, as a consequence metal free organic synthesizers have been extensively studied. This class mainly consist of three regions with different purposes; an electron acceptor moiety (A) which anchor the molecule to the semiconducting electrode providing an efficient electronic coupling between the sensitizer and the substrate, a linker section (π) consisting of a π-conjugated system and an electron donor moiety (D) at the free end, thus they are based on a push–pull structure D–π–A. The three features have been extensively varied in order to improve the photovoltaic properties. Among the donor units triphenylamine have been largely employed,24,25 more in general dyes based on coumarin, indoline, perylene, phenohiazine and carbazole were synthesized to achieve superior properties in terms of stability, cost and efficiency.26–30 Novel dyes based on quinoxaline or quinoxalinoid moieties were prepared by Jiang and coworkers obtaining co-sensitized devices with an efficiency greater than 30% under indoor light conditions.31 Li and coworkers synthesized dyes based on tetraphenylethylene employing different π-bridges, these sensitizers displayed excellent values of efficiency.32 A dye characterized by anthracene donor group, synthesized by Reddy et al., showed an efficiency greater than 20% under low light illumination and revealed a good efficiency when co-sensitized with porphyrin dye.33 Wu and coworkers achieved devices with improved photothermal stability, broaden absorption and higher extinction coefficients by varying the classical D–π–A structure inserting an electron poor moiety into the π-bridge and obtaining the new scheme D–A–π–A.34 Following this approach many dyes have been synthesized using as donor groups benzothiadiazole,35 phenanthrene,36 pyridal[2,1,3]benzotriazole,37 indoline,38 derivatives which displayed excellent stability and good efficiency. Dyes with higher performance were obtained by including a rigid aromatic system in the molecular structure thus increasing the planarity and, in turn, favoring the electron flow between donor and acceptor moieties.39–41 The acceptor-anchoring groups are mainly represented by cyanoacrylic and benzoic acid moieties which are present in the vast majority of DSSC with best performance, see all references reported above. Various π-conjugated bridges have been employed with aim of favoring the electrical communication between donor and acceptor. Good efficiency was obtained using thiophene and dithiophene derivatives in the π-bridge,42 in the latter case an effective reduction of dye aggregation was reported.43 Lin et al. suggested that furan could be a good alternative to thiophene,44 in addiction acene bridge based DSSCs were investigated by using a computational approach, optical and electrochemical results showed that dye with the anthracene moiety was the best candidate for DSSC applications.45 Further theoretical studies suggested that the inclusion of a triple bond linking anthracene with the anchoring group is necessary to make high efficiency sensitizers.46 Li et al. by using a DFT approach explored the role of substituents on tetracene and pentacene in acene-bridged dyes.47 Alkynyl bridge group has been employed and results indicated an improving in the absorption capacity, remarkable values of JSC and more in general a very good photovoltaic performance.48–53 In this work we investigated by means of DFT approach a new class of metal free dyes for applications in DSSCs which is based on a linear carbon chain (LCC) linker between simple donor and acceptor groups. Since the high π-conjugated system LCCs exhibit electrical conduction and very unusual optoelectronic properties,54–62 moreover can be easily synthesized and their long-term stability is obtained.63 We studied UV/VIS and electrochemical properties increasing the length of the bridge, moreover NLO properties were systematically investigated. Designed structures are reported in Fig. 1 in which pyrrole and thiophene derivatives are used as donor moiety, cyanoacrylic acid as acceptor group and the LCCs (C2, C4, C6 C8) as π-bridge. Since thiophene rings have been extensively used as components of the conjugated bridge of DSSC, we inserted them at both ends of the linker. Compounds obtained by replacing LCC with benzene and thiophene rings were studied for comparison, see Fig. 1. Both dye@(TiO2)14 and I−-Dye@(TiO2)14 anatase nanocluster systems were also simulated to show the electronic structure at the interface and the electrolyte contribution to electronic transition and injection. The results obtained in this study can be a useful reference to the synthesis and optimization of a new and more efficient class of metal free dyes for DSSC applications.
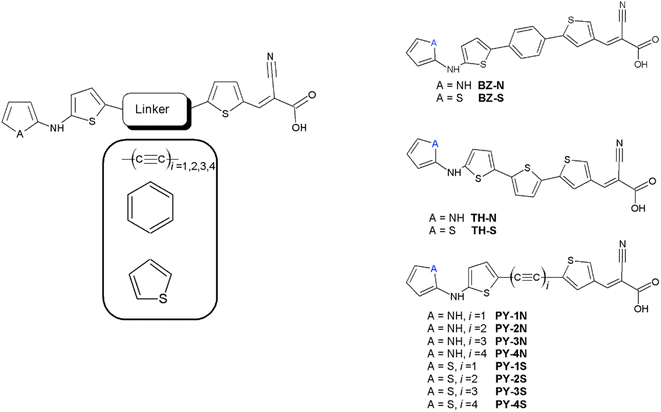 |
| | Fig. 1 Molecular structures of the designed dyes. | |
Computational details
The geometries of all compounds were fully optimized in the framework of the DFT using the hybrid B3LYP functional at quantitative accuracy level of approximation employing the extended basis set 6-311+G(3df,3pd). Vibrational analysis was carried out for all the investigated structures, results provided no imaginary modes, see ESI.† UV/VIS spectra were calculated within the TD-DFT approach, PBE0, B3LYP and CAM-B3LYP functionals were tested on dyes with known absorption spectra, results clearly indicated that, at this level of approximation, both B3LYP and PBE0 underestimated the absorption energy values whereas CAM-B3LYP provided results in good agreement with experimental data. Moreover, it has been reported that long range corrected functionals provide reasonable optical properties for molecules characterized by electronic excitations with large charge transfer (CT) character, likewise the series reported in this study, therefore CAM-B3LYP was chosen for the prediction of UV/VIS spectra.64,65 The solvated model based on density (SMD) was used to model the solvation effect for neutral and cationic dyes, and the lowest 10 energy states were considered in acetonitrile solvent. A comparison with conductor-like polarizable continuum model (CPCM) and IEFPCM methods showed very small differences in λmax values and absorption intensities. Nonlinear optical properties, α and β, were also estimated at quantitative accuracy level of approximation. In particular here we report the isotropically averaged polarizabilities 〈α〉 which is defined as:| |
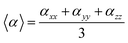 | (1) |
And the magnitude of the first order hyperpolarizability β defined as:
| |
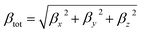 | (2) |
Where βi is given by:
| |
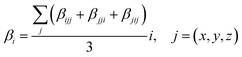 | (3) |
A (TiO2)14 cluster was chosen to model the semiconductor, it was carved out from crystallographic structure of anatase after cleaving the (1 0 1) surface. The ground state geometries of Dye@(TiO2)14, I−-Dye@(TiO2)14 and the absorption spectra were performed by using the computational approach proposed by Xie and coworkers.66 All the calculations were carried out with the Gaussian 16 package67 and Multiwfn 3.8 program68 was used to display the molecular orbitals (MOs) and NLO properties.
Results and discussion
Optimized geometries
To gain insight into the molecular structure all the studied compounds were optimized in the framework of the DFT with the hybrid B3LYP functional and the 6-311+G(3df,3pd) basis set. In general, the degree on conjugation plays a significant role in the performance of the dye, hence planarity was investigated considering the dihedral angle, Φ, between the pyrrole/thiophene moiety of the electron donor group and the thiophene ring inserted as a component of the conjugated bridge, see Fig. 2.
 |
| | Fig. 2 C–N–C–C dihedral angle Φ and partial double bond N–C are highlighted in red (sulphur atoms are colored in yellow). | |
All compounds exhibit a deviation from planarity which can be ascribed to a steric effect between the hydrogen atom in the position 3 of the heterocyclic ring and the sulphur atom of thiophene group inserted in the bridge, see Fig. 1. Φ values were calculated in the range of 16.14–39.70°, lowest and highest values were observed respectively in BZ-N and PY-2S, Table 1. A comparison among the LCC bridged dyes highlights that the steric effect is reduced in the pyrrole derivatives and affects the absorption spectra. It is to note that ground state geometries with no imaginary modes were found for planar structure (Φ = 0) of PY-4S and PY-4N, furthermore the calculated energy were about 1 kcal mol−1 higher than the non-planar structures and the absorption spectra were redshifted of about 15 nm. By contrast imaginary modes were always found in the other planar structures. Further aspects could affect electron transition, here we focus on the distance N–C highlighted in Fig. 2. A shorter distance suggests that electron transfer from donor to acceptor is facilitated, indeed, as listed in Table 1, pyrrole derivatives show the lower values. The analysis of the distance between carbon atoms in LCCs reveals an extended conjugation along the bridge, bond lengths are in the range between 1.22–1.34 Å, see ESI,† and the average value approximates 1.27 Å (the middle value between 1.20 Å of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–, and 1.34 Å of –C
C–, and 1.34 Å of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–) as the LCC become longer, Table 1.
C–) as the LCC become longer, Table 1.
Table 1 Selected bond length (N–C, averaged CC value, Å), dihedral angles (Φ, degree) for optimized dyes calculated at B3LYP/6-311+G(3df,3pd)/SMD level
| Compound |
Φ |
N–C |
CC (Av.) |
| BZ-S |
32.42 |
1.372 |
|
| BZ-N |
16.14 |
1.364 |
|
| PY-4S |
36.24 |
1.361 |
1.272 |
| PY-4N |
25.04 |
1.355 |
1.274 |
| PY-3S |
37.01 |
1.362 |
1.269 |
| PY-3N |
20.95 |
1.355 |
1.269 |
| PY-2S |
39.70 |
1.363 |
1.262 |
| PY-2N |
18.02 |
1.356 |
1.262 |
| PY-1S |
38.48 |
1.364 |
1.218 |
| PY-1N |
20.27 |
1.356 |
1.218 |
| TH-S |
30.81 |
1.368 |
|
| TH-N |
17.77 |
1.361 |
|
GAP
Fig. 3 shows calculated energy levels (from HOMO−4 to LUMO+4) of the investigated dyes. With the exception of BZ-N (−4.85 eV) the HOMO level energies are in all cases below the I−/I3− redox couple (−4.8 eV (ref. 69)), whereas LUMO levels are above the conducting band (CB) of TiO2 (−4.0 eV (ref. 70)) with energy differences much greater than 0.2 eV, approximately the value above which an efficient electron injection is thermodynamically favorable.71 These findings suggest that, apart from BZ-N, the oxidized dyes can be regenerated efficiently by electron transfer from the electrolyte, moreover all dyes are predicted to inject electrons in the CB of TiO2. A favorable photovoltaic conversion of all LCCs series is expected, best results seem to be provided by the compound in the series with the largest gap, PY-1S, characterized by one triple bond and the thiophene ring as donor group. In general, pyrrole ring lowers the HOMO energy thus reducing the energy difference with the redox potential of the electrolyte and affecting negatively the photovoltaic conversion. In addition, dyes with longer LCC bridge show a slight lowering of the LUMO energies, however, as displayed in Fig. 3, such energy values remain sufficiently higher than the CB, hence the injection process should not be affected along the series. Fig. 3 also shows that BZ-S is the dye with largest energy gap, however, it is necessary to investigate further aspects in order to predict which of the designed sensitizers could be the best candidate for photovoltaic applications.
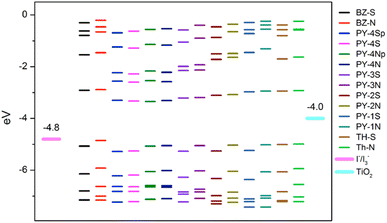 |
| | Fig. 3 Selected MOs energy levels (from HOMO−4 to LUMO+4) for the studied dyes at B3LYP/6-311+G(3df,3dp)/SMD level together with TiO2 CB and I−/I3− redox potential. p = planar structure. | |
UV/VIS absorption, OM, NLO
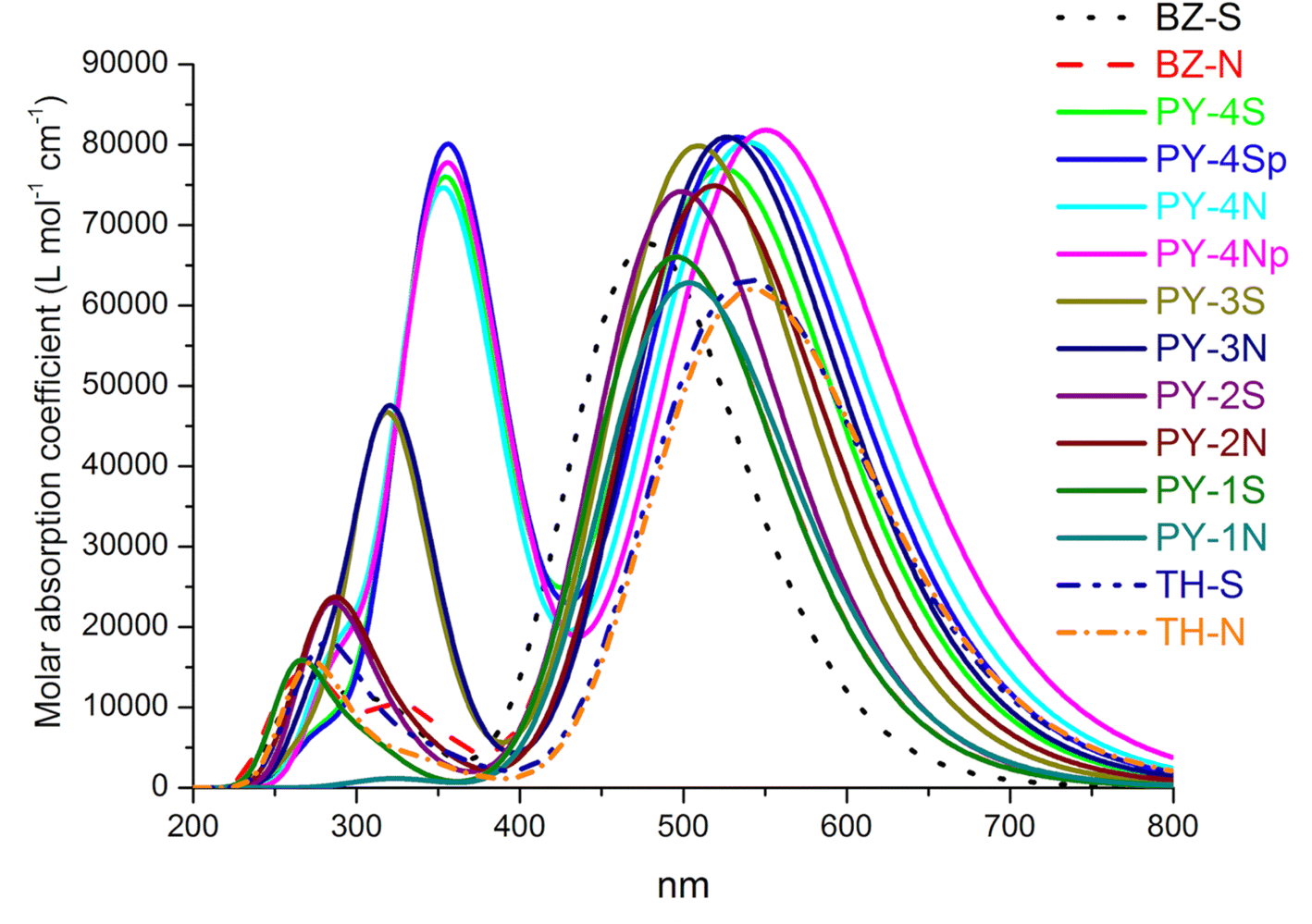
In order to evaluate the performance as sensitizers UV/VIS absorption spectra give more reliable information than energy gap. First of all, we have verified the reliability of the CAM-B3LYP functional for the description of absorption spectra in organic sensitizers. To this purpose we have simulated, after geometry optimization, the absorption spectra of well-known dyes and results were compared to experimental data. In particular we referred to dyes MM6,31 C293 (ref. 40) and D2.52 Given the considerable size of MM6 and C293 the 6-311+G(d,p) basis set was adopted whereas the extended basis set was maintained for D2. The calculated spectra, see ESI Fig. 1,† are in good agreement with experimental values, in particular the main bands of D2, a dye with a structure similar to the LCC studied series, are nicely predicted. As a matter of fact, all the experimental maximum absorption wavelengths are predicted by CAM-B3LYP with a deviation of about 10 nm. The simulated UV/VIS spectra of the investigated dyes are reported in Fig. 4, wavelengths, oscillator strength (f) > 0.2 and the main contributions to transitions are presented in Table 2.
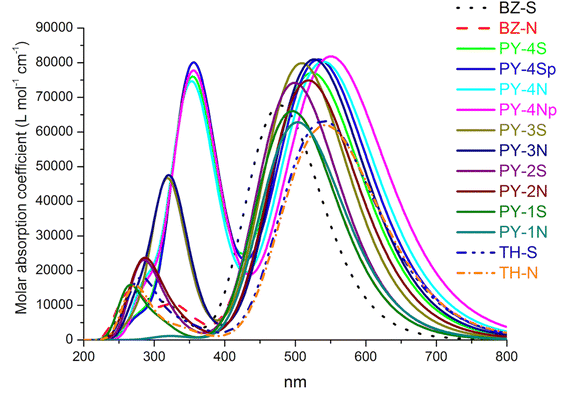 |
| | Fig. 4 Absorption spectra of dyes in acetonitrile at TD-CAM-B3LYP/6-311+G(3df,3pd)/SMD level. p = planar structure. The spectra are Gaussian broaded with 0.3 eV (half width half maximum). | |
Table 2 Absorption wavelengths (nm), oscillator strength and main contributions to the transitions of dyes in acetonitrile at CAM-B3LYP/6-311+G(3df,3pd)/SMD level
| Compound |
λ |
f |
Main contribution to the transition |
| BZ-S |
478.42 |
1.67 |
H → L (76%) |
| |
317.97 |
0.23 |
H → L+1 (40%) |
| BZ-N |
495.89 |
1.64 |
H → L (75%) |
| |
327.35 |
0.23 |
H → L+1 (54%) |
| |
275.02 |
0.25 |
H−2 → L (47%) |
| PY-4S |
534.07 |
0.61 |
H → L+1 (42%) |
| |
519.72 |
1.31 |
H → L (35%) |
| |
358.98 |
1.43 |
H−2 → L+1 (37%) |
| |
332.13 |
0.51 |
H → L+2 (29%) |
| PY-4Sp |
533.19 |
2.00 |
H → L (60%) |
| |
362.85 |
1.44 |
H−2 → L+1 (44%) |
| |
337.58 |
0.68 |
H → L+2 (34%) |
| PY-4N |
546.01 |
0.24 |
H → L+1 (59%) |
| |
538.99 |
1.75 |
H → L (50%) |
| |
366.70 |
0.86 |
H−2 → L+1 (37%) |
| |
343.14 |
1.09 |
H → L+2 (30%) |
| |
287.05 |
0.33 |
H−3 → L (38%) |
| PY-4Np |
550.60 |
2.02 |
H → L (59%) |
| |
371.95 |
0.81 |
H−2 → L+1 (37%) |
| |
346.91 |
1.26 |
H → L+2 (32%) |
| |
289.17 |
0.31 |
H−3 → L (36%) |
| PY-3S |
509.23 |
1.97 |
H → L (70%) |
| |
319.63 |
1.09 |
H−2 → L+1 (18%) |
| PY-3N |
526.71 |
2.00 |
H → L (68%) |
| |
322.03 |
1.12 |
H−3 → L+1 (26%) |
| |
276.57 |
0.20 |
H−2 → L (43%) |
| PY-2S |
498.85 |
1.83 |
H → L (79%) |
| |
279.55 |
0.35 |
H−6 → L (54%) |
| PY-2N |
519.24 |
1.85 |
H → L (76%) |
| |
294.64 |
0.30 |
H−2 → L (25%) |
| |
281.58 |
0.24 |
H−6 → L (68%) |
| PY-1S |
495.87 |
1.60 |
H → L (87%) |
| |
267.25 |
0.27 |
H → L+2 (37%) |
| PY-1N |
503.47 |
1.55 |
H → L (44%) |
| TH-S |
539.93 |
1.56 |
H → L (84%) |
| |
282.44 |
0.20 |
H → L+2 (28%) |
| TH-N |
542.38 |
1.53 |
H → L (86%) |
| |
265.25 |
0.22 |
H → L+3 (28%) |
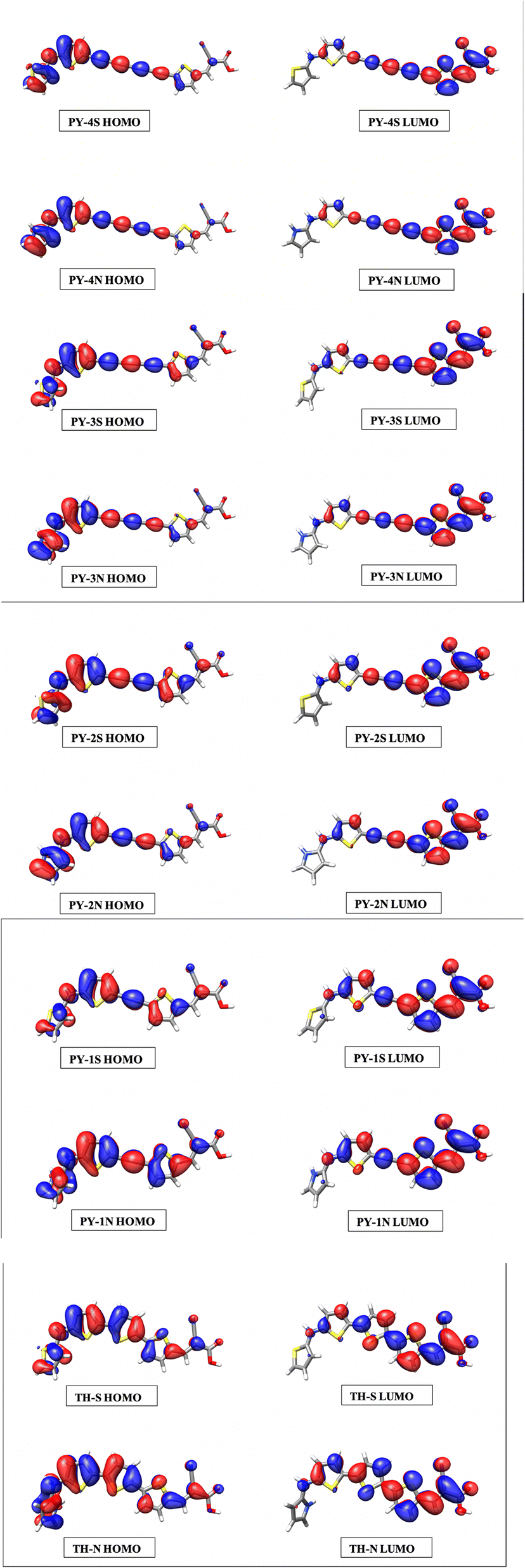
Simulated spectra show a first intense peak between 460–550 nm arising from the intramolecular charge transfer (ICT), characterized by HOMO → LUMO transition, and a second peak located in the range 270–320 nm which can be ascribed to π–π* transitions. With the exception of single triple bond compounds (PY-1S and PY-1N) the introduction of LCC bridge enhances the absorption intensity compared to benzene and thiophene bridges, the highest molar absorption coefficient is found for PY-4N in the planar conformation, more in general expanded π-conjugated systems represented by PY-4S, PY-4N, (4 triple conjugated bonds) PY-3S and PY-3N (3 triple conjugated bonds) show the higher molar absorption coefficient values. The orbital spatial distribution of the frontier molecular orbitals reveals that electron density is clear moving, through the π-linker, from the dye-donor side (HOMO) to the cyanoacrylic acid acceptor end (LUMO) of the dyes, allowing the ICT, see Fig. 5 and ESI.†
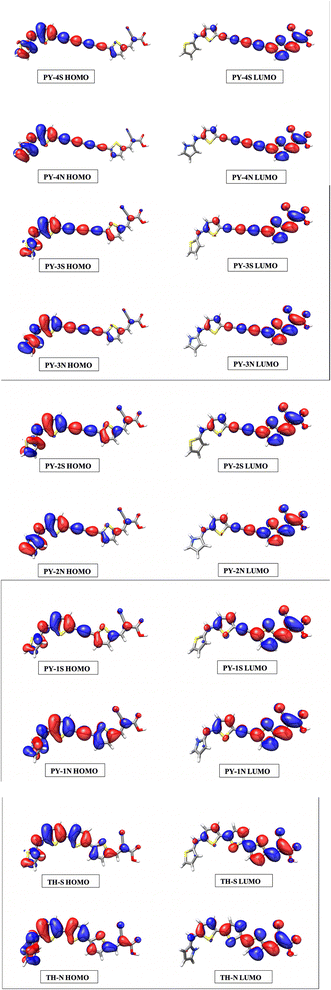 |
| | Fig. 5 Frontiers molecular orbitals of HOMO and LUMO of the dyes in acetonitrile at TD-CAM-B3LYP/6-311+G(3df,3dp)/SMD level. | |
It is to note that overlapped HOMO and LUMO orbitals extend across the π-linker thus indicating, for donor and acceptor moieties, excellent induction and electron-withdrawing properties which facilitate the ICT process. For PY-4S and PY-4N, absorptions included a second intense band at around 350 nm which is related to the extension of the linker conjugation length, in fact blue-shift and a significant decreasing in intensity of this band occur with the shortening of the LCC bridge. PY-1S and PY-1N absorptions are found below 300 nm and are comparable with BZ-S, BZ-N, TH-S and TH-N. All designed dyes are characterized by an extensive electron delocalization and exhibit a large difference between ground and excited state dipole moment, thus possess the requisite properties for non-linear behavior. Calculated values of polarizability (α) and first hyperpolarizability (β) reported in Table 3 confirm this property, interestingly pyrrole give rise to a remarkable enhancement of β values, in particular we expect that PY-4N is superior to other compounds. In the planar conformation PY-4N exhibits the exceptional β value of 290.491 (a. u.).
Table 3 Maximum absorption wavelength (eV), redox potential (eV), ΔGinject, LHE as function of f, polarizability and first hyperpolarizability (a. u.) for the dyes in acetonitrile at TD-CAM-B3LYP/6-311+G(3df,3dp)/SMD level
| Compound |
λmax |
Eox |
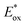
|
LHE |
ΔGinject |
α·10−3 |
β·10−5 |
| BZ-S |
2.59 |
4.96 |
2.37 |
0.978 |
−1.78 |
0.80 |
0.99 |
| BZ-N |
2.50 |
4.82 |
2.32 |
0.977 |
−1.69 |
0.87 |
1.30 |
| PY-4S |
2.32 |
5.19 |
2.80 |
0.951 |
−1.11 |
1.73 |
2.21 |
| PY-4N |
2.27 |
5.05 |
2.85 |
0.982 |
−1.12 |
1.75 |
2.59 |
| PY-3S |
2.44 |
5.18 |
2.75 |
0.989 |
−1.22 |
1.31 |
1.71 |
| PY-3N |
2.35 |
5.01 |
2.66 |
0.990 |
−1.27 |
1.42 |
2.26 |
| PY-2S |
2.49 |
5.15 |
2.67 |
0.985 |
−1.30 |
1.03 |
1.04 |
| PY-2N |
2.39 |
4.97 |
2.58 |
0.986 |
−1.38 |
1.16 |
1.95 |
| PY-1S |
2.50 |
5.09 |
2.59 |
0.977 |
−1.39 |
0.85 |
1.19 |
| PY-1N |
2.46 |
4.92 |
2.50 |
0.972 |
−1.48 |
0.96 |
1.60 |
| TH-S |
2.30 |
4.83 |
2.53 |
0.973 |
−1.41 |
0.97 |
1.49 |
| TH-N |
2.29 |
4.78 |
2.49 |
0.954 |
−1.48 |
1.07 |
1.96 |
Light harvesting efficiency (LHE) and electron injection driving force ΔGinject
To predict the extent of light adsorption and the short circuit photocurrent, LHE was calculated by using the equation:72| | |
LHE(λ) = 1 − 10−ε(λ)Γ
| (4) |
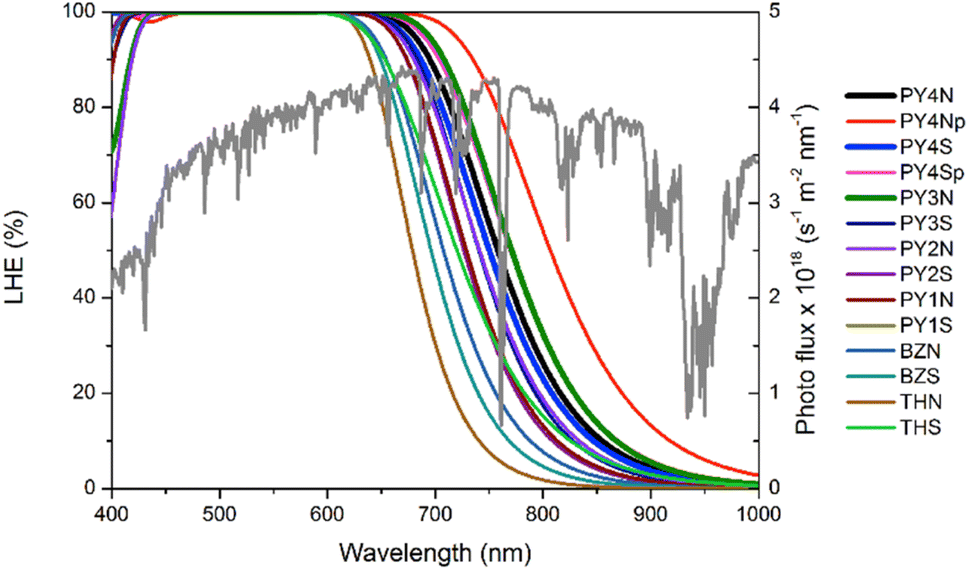
where ε(λ) is the molar absorption coefficient whose value depends on wavelength and Γ is the dye loading amount corresponding to the product of the dye concentration c with TiO2 film thickness b, (Γ = c × b). The LHE curves along with the AM 1.5G solar spectrum are displayed in Fig. 6. Curve coverages of PY-4N in the planar conformation is the broadest of the series, in the other cases PY-3N show the best coverage. More in general pyrrole derivatives exhibit better LHE compared to the analogues with thiophene, along the series the efficiency decreases following the order PY-3 > PY-4 > PY-2 > PY-1.
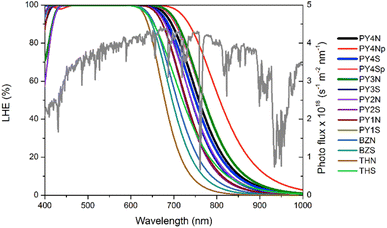 |
| | Fig. 6 LHE curves of all dyes (Γ = 20 nmol cm−2 is taken for PY-4N, PY-4S, PY-3N, PY-3S, Γ = 30 nmol cm−2 is considered for the other dyes). AM 1.5G solar spectrum is reported in green. | |
In Table 3 are listed approximated LHE values calculated as:
| | |
LHE = 1 − 10−A = 1 − 10−f
| (5) |
where
f is the oscillator strength of the dye. These results are in agreement with curves plotted in
Fig. 6,
PY-3S and
PY-3N show the greatest LHE values followed by
PY-4N and
PY-4S, since short circuit photocurrent, which is an important characteristic for the performance of the cell, is proportional to LHE, these dyes are expected to provide the greatest
JSC value and, in turn, the best efficiency in DSSC applications. The short circuit photocurrent can be evaluated as:
| |
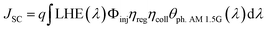 | (6) |
where
q is the elementary charge of electron,
Φinj represents the electron injection efficiency,
ηreg the dye regeneration efficiency,
ηcoll the electron collection efficiency and
θph. AM 1.5G is the photon flux which corresponds to the AM 1.5G solar radiation spectrum.
JSC value is one of the crucial factors determining the overall cell efficiency,
η, which is related to electrical parameters by the following equation:
| |
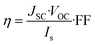 | (7) |
where
Is is the intensity of the incident light,
VOC represents the open circuit voltage and depends on the energy difference between the electrolyte redox potential and the Fermi potential of the semiconductor, FF is the fill factor defined as the ratio between the maximum power (
Vmax·
Jmax) yielded by the device and the maximum theoretical power (
VOC·
JSC).
73,74 Apparently, the differences in the LHE values listed in
Table 3 are very small, as a consequence comparable performance is expected from the whole LCCs series.
PY-4S is the dye with lowest value of the series, but it should be emphasized that in the planar it reveals a light-harvesting-efficiency value equal to 0.99, furthermore in view of the free rotation around the C–N bond, as the slight difference in energy between planar and non-planar conformers suggests, a good efficiency can be hypothesized also for this compound as shown in
Fig. 6.
For high performance of DSSCs an efficient and fast electron injection from the excited state of the dye to the CB of TiO2 surface is required. The ΔGinject is the free energy related to the electron injection process and represents the difference between the excited electron of the sensitizer and the injected electron onto the TiO2 surface. ΔGinject is given by:
| |
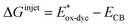 | (8) |
where
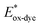
denotes the oxidation potential of the dye in its excited state whereas
ECB represents the reduction potential of the CB of the semiconductor TiO
2 (
ECB = 4.0 eV). In turn,
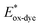
can be computed as:
| |
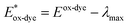 | (9) |
In which Eox-dye is the oxidation potential of the ground state and λmax represents the lowest absorption energy associated with the photo-induced ICT. Table 3 shows that the findings ΔGinject of the investigated dyes are negative, thus spontaneous electron injection from the excited sensitizers to the CB of TiO2 is predicted.
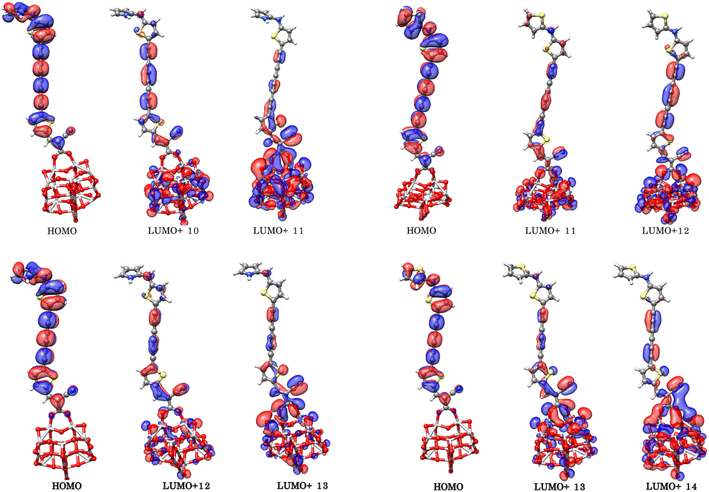
TiO2–dye interaction
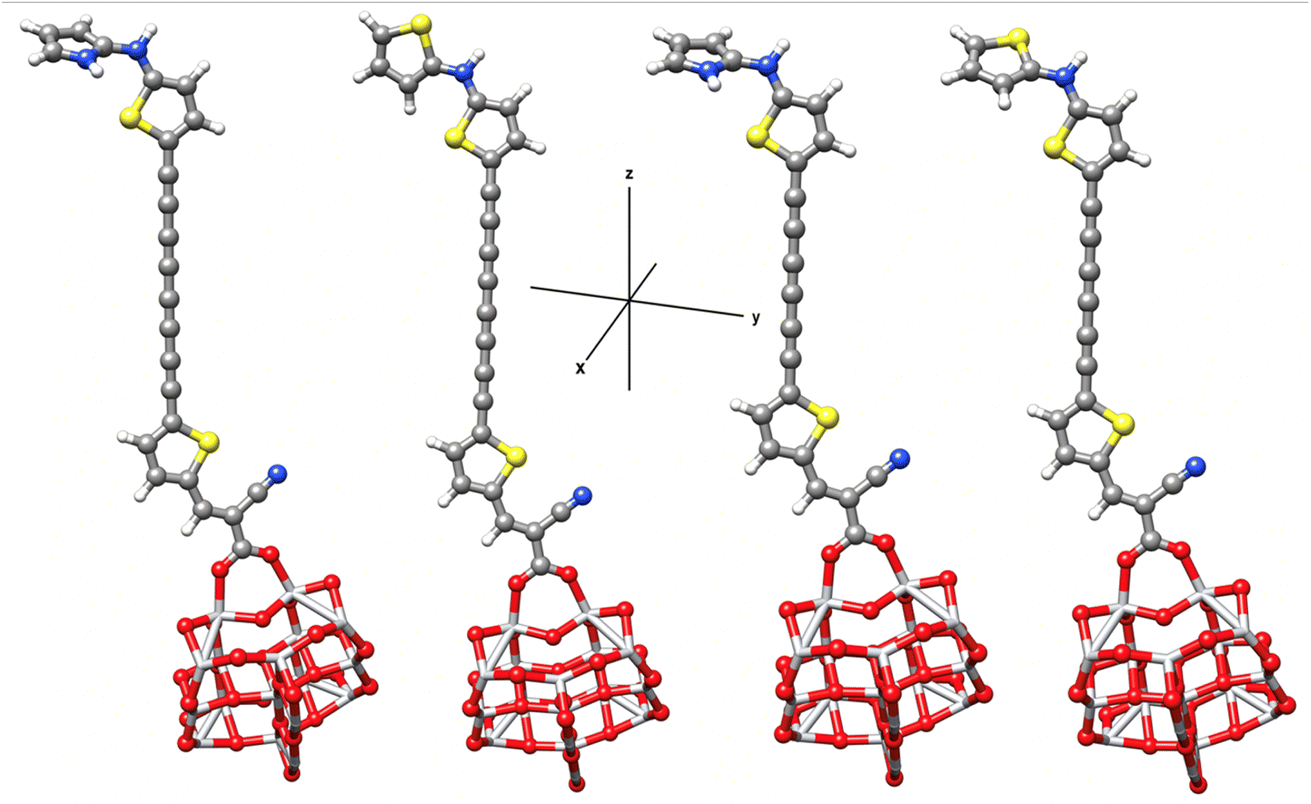
Prediction studies of the dye–semiconductor interface, together with electron transfer from the sensitizers to semiconductor, represent an important step towards the DSSCs optimization. Excitation and injection of electron process is much faster than reduction of oxidized dye,66,75 hence we modeled the oxidized dye–TiO2 adsorption complexes considering the most promising sensitizers PY-4N, PY-4S, PY-3N and PY-3S. Fig. 7 shows the optimized structures of the complexes and in Table 4 are reported the dipole moment, μ, and the interaction energies, Eint, calculated as:
| Eint = Eox-dye@TiO2 − (Eox-dye + ETiO2) |
 |
| | Fig. 7 Optimized geometries of bidentate binding mode of PY-4N, PY-4S, PY-3N and PY-3S on (TiO2)14. | |
Table 4 Eint (kcal mol−1) and dipole moment μ (Debye) for the selected dyes at B3LYP-D3/6-31G(d)/LANL2DZ level. In parenthesis the dipole moment of the corresponding isolated dyes. Maximum absorption wavelengths (eV), oscillator strength and main contributions to the transitions for the selected dyes at TD-M062X/6-31G(d)/LANL2DZ level
| Complexes |
Eint |
μ |
λmax |
f |
Main contribution to the transition |
| PY-4N@(TiO2)14 |
−20.95 |
16.46 (14.60) |
2.38 |
0.96 |
H → L+10 (0.29) |
| |
|
|
|
|
H → L+11 (0.36) |
| PY-4S@(TiO2)14 |
−20.73 |
17.68 (12.39) |
2.41 |
0.45 |
H → L+11 (0.24) |
| |
|
|
|
|
H → L+12 (0.27) |
| PY-3N@(TiO2)14 |
−19.92 |
16.03 (13.95) |
2.35 |
0.93 |
H → L+12 (0.39) |
| |
|
|
|
|
H → L+13 (0.28) |
| PY-3S@(TiO2)14 |
−19.66 |
16.68 (12.27) |
2.49 |
0.87 |
H → L+13 (0.26) |
| |
|
|
|
|
H → L+14 (0.32) |
As shown, the dyes are linked by means of the two carboxylic oxygen atoms bonded with a titanium atom of surface, such bidentate adsorption mode is energetically favored compared to monodentate mode. Calculated Eint values of Dye@(TiO2)14 indicate a very favorable interaction between the dyes and the semiconductor surface, moreover calculated dipole moment along z axis, see Fig. 7 and Table 4, confirms the strong ICT properties and, in turn, the efficient electron injection properties.
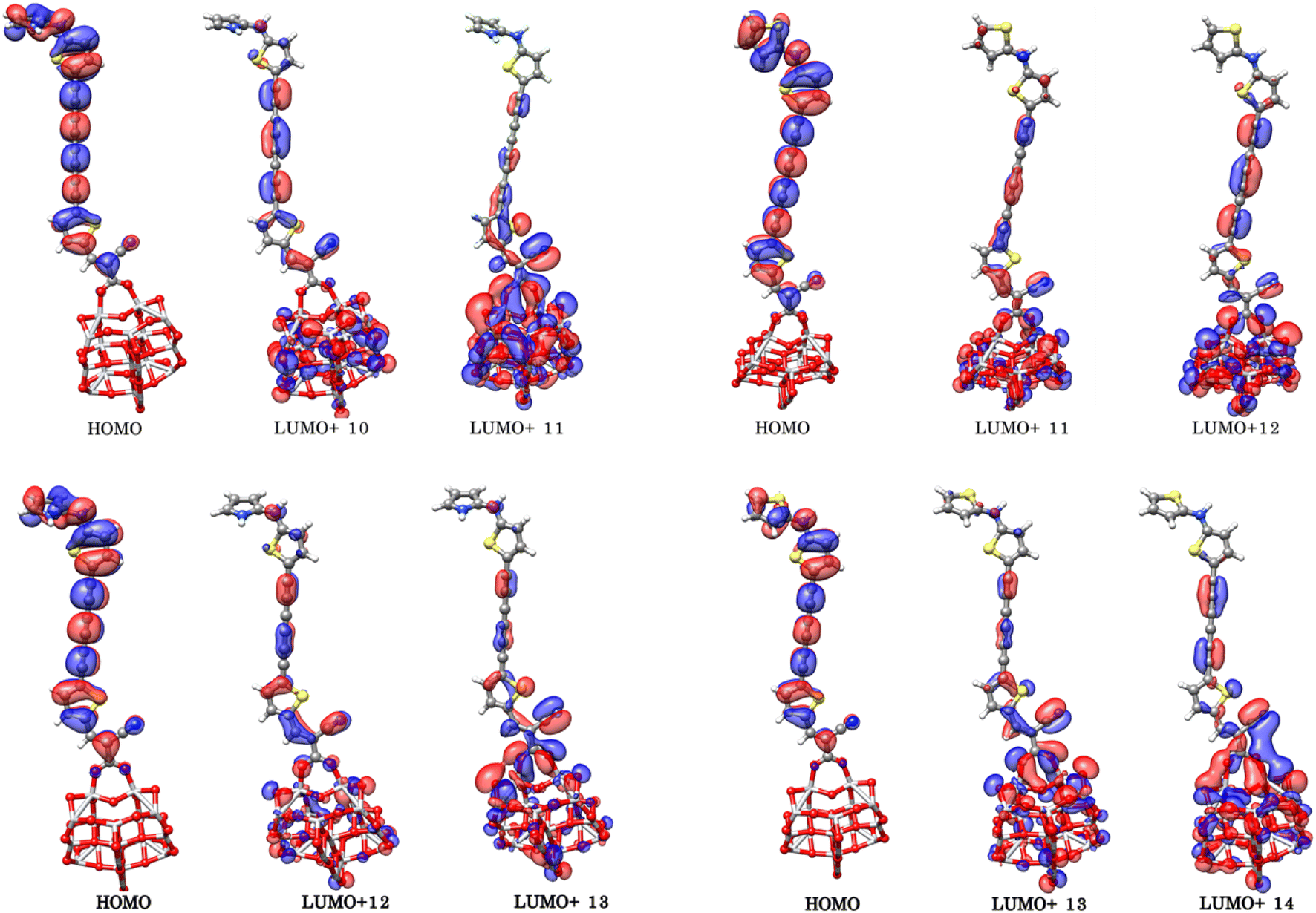
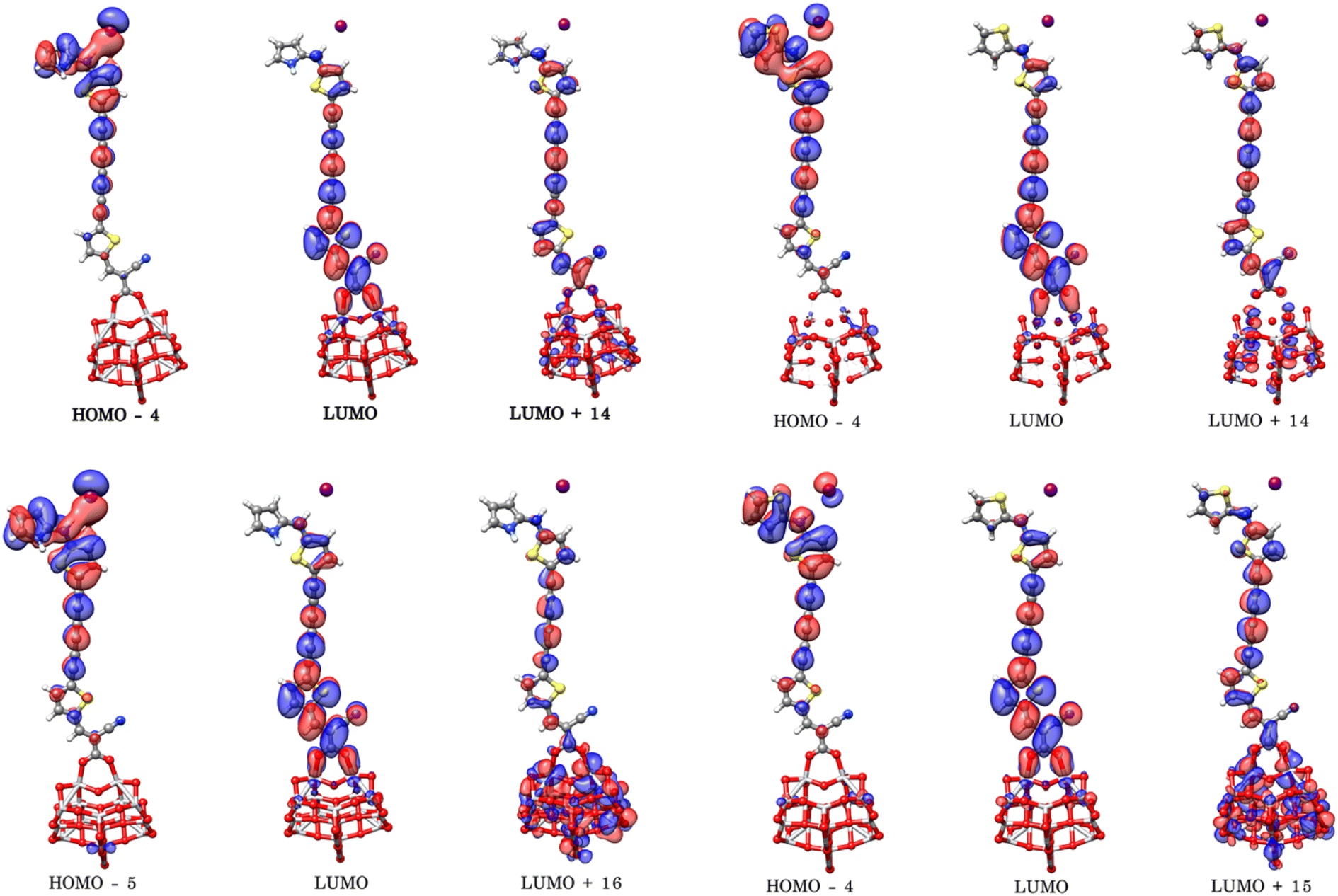
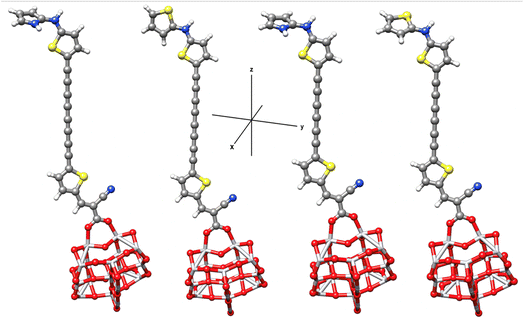
To better understand the electronic coupling between the LUMO on the dye and the CB on TiO2 and electron-transfer features upon excitation, electronic transition energies were calculated at TD-M062X/6-31G(d)/LANL2DZ level. The results are listed in Table 4, electronic transitions isosurfaces for Dyes@(TiO2)14 complexes are shown in Fig. 8. We observe that HOMO levels of Dyes@(TiO2)14 are localized to the sensitizers retaining the HOMO character of the isolated dyes and the values are slightly increased in comparison with free dyes, whereas the LUMO levels, and most of the LUMO+n levels, are centered on the substrate showing the same character as TiO2, therefore have no coupling to the semiconductor CB. Interacting orbitals with considerable contributions from the LUMO of free dyes are found at higher energy level, in particular the patterns of LUMO+10 and LUMO+11 for PY-4N, LUMO+11 and LUMO+12 for PY-4S, LUMO+12 and LUMO+13 for PY-3N and LUMO+12, LUMO+14 for PY-3S clearly indicate that electron distribution is delocalized between the anchoring group and the TiO2 surface and represents electron transfer from dye to the semiconductor by means of the cyanoacrylic group as a result of direct electron transfer.
 |
| | Fig. 8 Electronic transitions of PY-4N@(TiO2)14 (up left), PY-4S@(TiO2)14 (up right), PY-3N@(TiO2)14 (down left) PY-3S@(TiO2)14 (down right) calculated at TD-M062X/6-31G(d)/LANL2DZ level. | |
Electrolyte–dye–TiO2 interaction
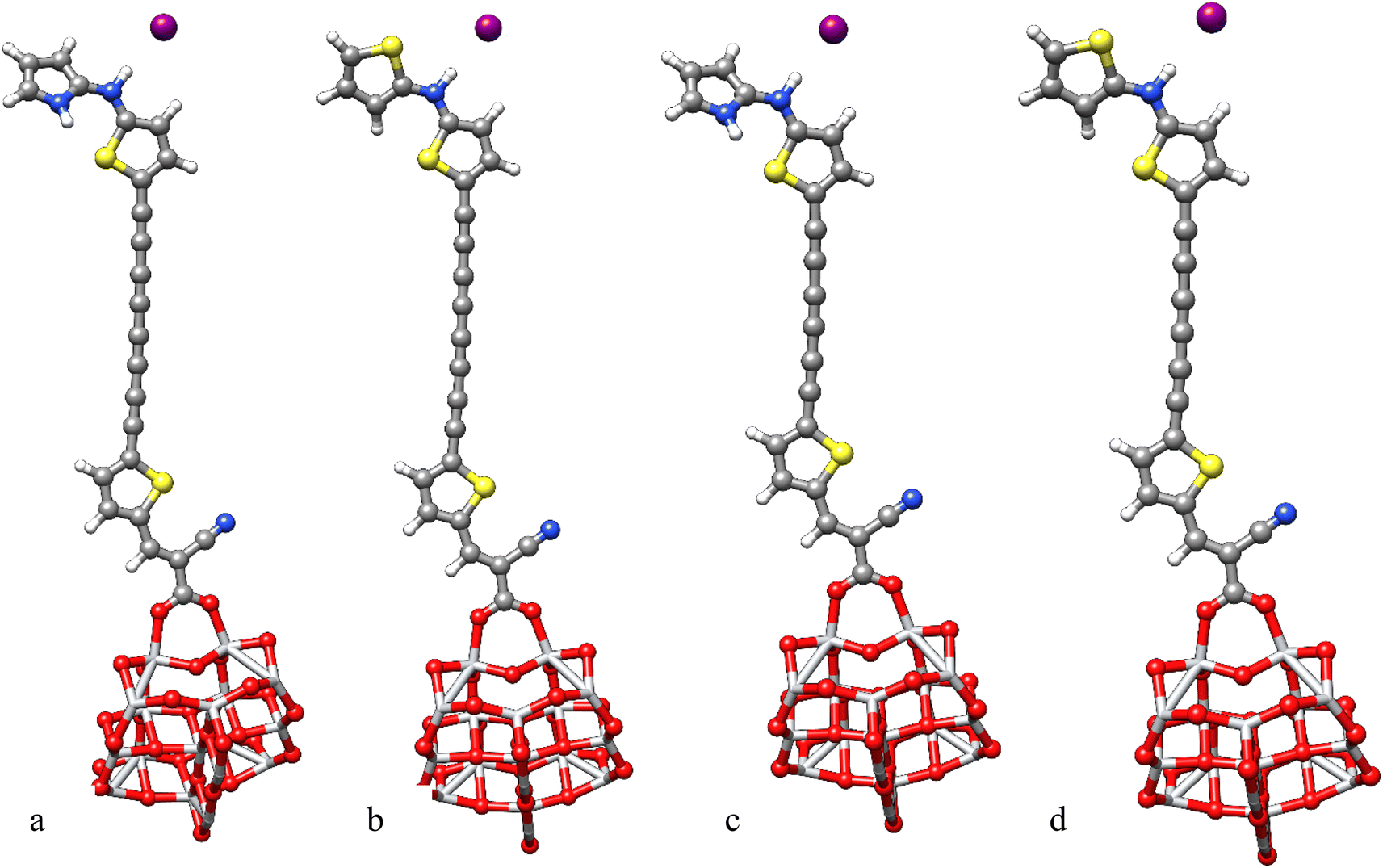
To investigate the change in absorption spectra and molecular orbitals before and after electrolyte interaction, the geometry optimization of electrolyte–Dye–TiO2 systems were performed.66 Electrostatic potential simulations suggest that active electrolyte attacking area is mainly localized around the enaminic H atom hence we sited I− close to this positive area, see Fig. 9. The interaction energies of the complexes I−-Dye@(TiO2)14 reported in Table 5 indicate a favorable interaction between the electrolyte and Dye@(TiO2)14 systems.
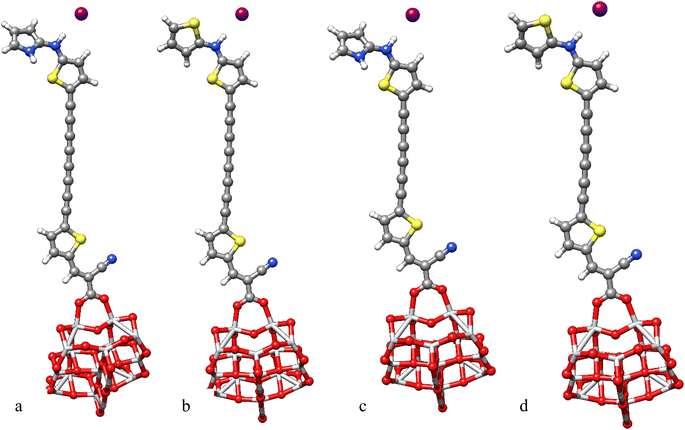 |
| | Fig. 9 Optimized geometries of I−-PY-4N@(TiO2)14 (a), I−-PY-4S@(TiO2)14 (b), I−-PY-3N@(TiO2)14 (c), I−-PY-3S@(TiO2)14 (d) at B3LYP-D3/6-31G(d)/LANL2DZ/SDDALL level. | |
Table 5 Eint (kcal mol−1) for the selected dyes at B3LYP-D3/6-31G(d)/LANL2DZ/SDDALL level. Maximum absorption wavelengths (eV), oscillator strength and main contributions to the transitions for the selected dyes at TD-M062X/6-31G(d)/LANL2DZ/SDDALL level
| Complexes |
Eint |
λmax |
f |
Main contribution to the transition |
| I−-PY-4N@(TiO2)14 |
−11.23 |
2.23 |
0.59 |
H−4 → L (0.33) |
| |
|
|
|
H−4 → L+14 (0.29) |
| I−-PY-4S@(TiO2)14 |
−10.95 |
2.31 |
0.95 |
H−4 → L (0.34) |
| |
|
|
|
H−4 → L+14(0.28) |
| I−-PY-3N@(TiO2)14 |
−9.86 |
2.18 |
0.79 |
H−5 → L (0.35) |
| |
|
|
|
H−5 → L+16 (0.27) |
| I−-PY-3S@(TiO2)14 |
−9.58 |
2.32 |
0.61 |
H−4 → L (0.26) |
| |
|
|
|
H−4 → L+15 (0.23) |
As compare with complexes Dye@(TiO2)14 maximum absorption wavelengths are considerably red-shifted whereas the pattern of molecular orbitals mainly involved in the transition highlight the contribution of the electrolyte to the effective charge transfer in particular for PY-4N and PY-3N, see Table 5, Fig. 10 and ESI† for the UV/VIS absorption spectra.
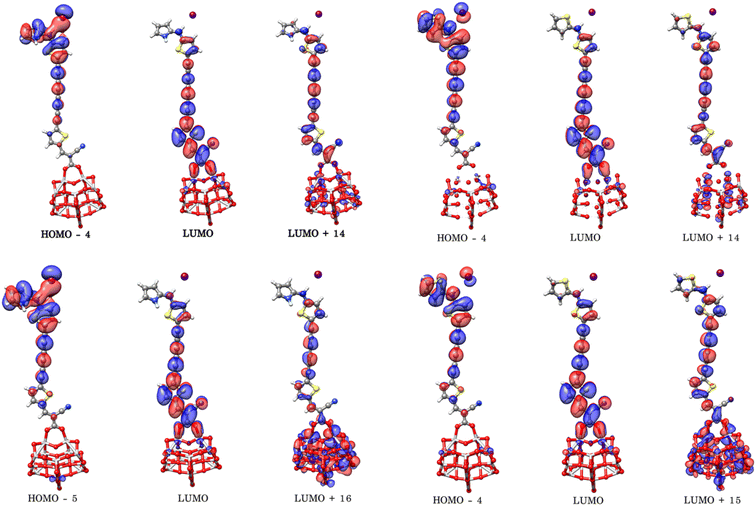 |
| | Fig. 10 Electronic transitions of I−-PY-4N@(TiO2)14 (up left), I−-PY-4S@(TiO2)14 (up right), I−-PY-3N@(TiO2)14 (down left) I−-PY-3S@(TiO2)14 (down right) calculated at TD-M062X/6-31G(d)/LANL2DZ/SDDALL level. | |
Conclusions
In this study we used DFT and TD-DFT calculations to investigate a new class of metal-free LCC based dyes for applications in DSSCs. Our results reveal that insertion of LCC produce a significant effect on absorption spectra and electrochemical properties. With increasing length of LCC the absorption spectra are red-shifted due to the expansion of π-conjugation and the intensity of the two main bands is greatly enhanced in comparison with benzene and thiophene linked dyes. LHE and electron injection properties vary slightly with the length of LCC, PY-3N exhibits good optical properties and the best electrochemical performance, hence is expected to be a promising candidate for DSSC applications. Interestingly planar conformation of PY-4N and PY-4S display an enhanced LHE and superior spectroscopic parameters, moreover ground state geometries with no imaginary modes are obtained only for these two dyes. As expected, excellent NLO properties are predicted for all the member of the LCC series. Energetic and spectroscopic calculations indicate that pyrrole derivatives provide higher performance than analogues with thiophene. Simulations of Dye@(TiO2)14 and I−-Dye@(TiO2)14 anatase nanocluster systems confirmed the efficient electron injection properties and the contribution due to the I− to the effective charge transfer. Our simulations suggest that all of the LCC-bridged sensitizers are able to inject electron into TiO2 and can be regenerated efficiently by electron transfer from the electrolyte. We expect this work will contribute to synthesize a new class of metal-free dyes with application in the DSSC field.
Author contributions
Giuseppe Consiglio: conceptualization, investigation, writing, revision and editing. Adam Gorcyński: investigation, writing, revision and editing. Salvatore Petralia: investigation, writing, revision and editing. Giuseppe Forte: conceptualization, supervision, methodology, writing original draft.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
This research was partially funded by University of Catania (Piano della Ricerca di Ateneo, Linea di Intervento 2, 2018–2020 and PIACERI – GRABIO project).
References
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. L. Parisi, S. Maranghi and R. Basosi, Renew. Sustain. Energy Rev., 2014, 39, 124–138 CrossRef CAS.
- X. Zhou, Y. Zhang, A. L. Porter, Y. Guo and D. Zhu, Scientometrics, 2014, 100, 705–721 CrossRef.
- S. Shalini, R. Balasundaraprabhu, T. Satish Kumar, N. Prabavathy, S. Senthilarasu and S. Prasanna, Int. J. Energy Res., 2016, 40, 1303–1320 CrossRef CAS.
- M. K. Nazeeruddin, P. Péchy and M. Grätzel, Chem. Commun., 1997, 1705, 1705–1706 RSC.
- M. K. Nazeeruddin, P. Péchy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. D. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613–1624 CrossRef CAS PubMed.
- R. Buscaino, C. Baiocchi, C. Barolo, C. Medana, M. Grätzel, M. K. Nazeeruddin and G. Viscardi, Inorg. Chim. Acta, 2008, 361, 798–805 CrossRef CAS.
- W. C. Chen, F. T. Kong, Z. Q. Li, J. H. Pan, X. P. Liu, F. L. Guo, L. Zhou, Y. Huang, T. Yu and S. Y. Dai, ACS Appl. Mater. Interfaces, 2016, 8, 19410–19417 CrossRef CAS PubMed.
- Z. Z. Lu, J. Peng, W. De, C. H. Lin, C. G. Wu, K. C. Ho, Y. C. Lin and K. L. Lu, Eur. J. Inorg. Chem., 2016, 8, 1214–1224 CrossRef.
- K. L. Wu, H. C. Hsu, K. Chen, Y. Chi, M. W. Chung, W. H. Liu and P. T. Chou, Chem. Commun., 2010, 46, 5124–5126 RSC.
- J. F. Huang, J. M. Liu, P. Y. Su, Y. F. Chen, Y. Shen, L. M. Xiao, D. B. Kuang and C. Y. Su, Electrochim. Acta, 2015, 174, 494–501 CrossRef CAS.
- Y. Tachibana, S. A. Haque, I. P. Mercer, J. R. Durrant and D. R. Klug, J. Phys. Chem. B, 2000, 104, 1198–1205 CrossRef CAS.
- Y. C. Chang, C. L. Wang, T. Y. Pan, S. H. Hong, C. M. Lan, H. H. Kuo, C. F. Lo, H. Y. Hsu, C. Y. Lin and E. W. G. Diao, Chem. Commun., 2011, 47, 8910–8912 RSC.
- A. Yella, H. W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. G. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- K. Omata, S. Kuwahara, K. Katayama, S. Qing, T. Toyoda, K. M. Lee and C. G. Wu, Phys. Chem. Chem. Phys., 2015, 15, 10170–10175 RSC.
- G. Consiglio, S. Failla, C. G. Fortuna, L. D'Urso and G. Forte, Comput. Theor. Chem., 2015, 1067, 1–6 CrossRef CAS.
- I. P. Oliveri, G. Forte, G. Consiglio, S. Failla and S. Di Bella, Inorg. Chem., 2017, 56, 14206–14213 CrossRef CAS PubMed.
- S. Aghazada, P. Gao, A. Yella, G. Marotta, T. Moehl, J. Teuscher, J. E. Moser, F. De Angelis, M. Grätzel and M. K. Nazeeruddin, Inorg. Chem., 2016, 55, 6653–6659 CrossRef CAS PubMed.
- N. V. Krishna, J. V. S. Krishna, M. Mrinalini, S. Prasanthkumar and L. Giribabu, ChemSusChem, 2017, 10, 4668–4689 CrossRef CAS PubMed.
- C. C. Chen, J. S. Chen, V. S. Nguyen, T. C. Wei and C. Y. Yeh, Angew. Chem., 2021, 60(9), 4886–4893 CrossRef CAS PubMed.
- J. Lu, S. Liu and M. Wang, Front. Chem., 2018, 6, 541 CrossRef CAS PubMed.
- J. Zou, Y. Tang, G. Baryshnikov, Z. Yang, R. Mao, W. Feng, J. Guan, C. Li and Y. Xie, J. Mater. Chem. A, 2022, 10, 1320–1328 RSC.
- M. Xu, S. Wenger, H. Bala, D. Shi, R. Li, Y. Zhou, S. M. Zakeeruddin, M. Grätzel and P. Wang, J. Phys. Chem. C, 2009, 113, 2966–2973 CrossRef CAS.
- S. Ahmad, E. Guillén, L. Kavan, M. Grätzel and M. K. Nazeeruddin, Energy Environ. Sci., 2013, 6, 3439–3466 RSC.
- C. Zhong, J. Gao, Y. Cui, T. Li and L. Han, J. Power Sources, 2015, 273, 831–838 CrossRef CAS.
- A. Venkateswararao, P. Tyagi, K. R. J. Thomas, P. W. Chen and K. C. Ho, Tetrahedron, 2014, 70, 6318–6327 CrossRef CAS.
- S. S. K. Raavi, P. Docampo, C. Wehrenfennig, M. J. P. Alcocer, G. Sadoughi, L. M. Herz, H. J. Snaith and A. Petrozza, J. Phys. Chem. C, 2014, 118, 16825–16830 CrossRef CAS.
- M. Mao, X. L. Zhang, X. Q. Fang, G. H. Wu, S. Y. Dai, Q. H. Song and X. X. Zhang, J. Power Sources, 2014, 268, 965–976 CrossRef CAS.
- J. Liu, X. Sun, Z. Li, B. Jin, G. Lai, H. Li, C. Wang, Y. Shen and J. Hua, J. Photochem. Photobiol., A, 2014, 294, 54–61 CrossRef CAS.
- M. L. Jiang, J. J. Wen, Z. M. Chen, W. H. Tsai, T. C. Lin, T. J. Chow and Y. J. Chang, ChemSusChem, 2019, 12, 3654–3665 CrossRef CAS PubMed.
- C. T. Li, Y. L. Kuo, C. P. Kumar, P. T. Huang and J. T. Lin, J. Mater. Chem. A, 2019, 7, 23225–23233 RSC.
- K. S. K. Reddy, Y. C. Chen, C. C. Wu, C. W. Hsu, Y. C. Chang, C. M. Chen and C. Y. Yeh, ACS Appl. Mater. Interfaces, 2018, 10, 2391–2399 CrossRef CAS PubMed.
- Y. Wu and W. Zhu, Chem. Soc. Rev., 2013, 42, 2039–2058 RSC.
- S. Chaurasia and J. T. Lin, Chem. Rec., 2016, 16, 1311–1336 CrossRef CAS PubMed.
- R. Y. Huang, W. H. Tsai, J. J. Wen, Y. J. Chang and T. J. Chow, J. Power Sources, 2020, 458, 228063 CrossRef CAS.
- S. Chaurasia, J. S. Ni, W. I. Hung and J. T. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 22046–22057 CrossRef CAS PubMed.
- S. Qu, C. Qin, A. Islam, Y. Wu, W. Zhu, J. Hua, H. Tian and L. Han, Chem. Commun., 2012, 48, 6972–6974 RSC.
- S. Chaurasia, C. J. Liang, Y. S. Yen and J. T. Lin, J. Mater. Chem. C, 2015, 3, 9765–9780 RSC.
- J. Wang, H. Wu, L. Jin, J. Zhang, Y. Yuan and P. Wang, ChemSusChem, 2017, 10, 2962–2967 CrossRef CAS PubMed.
- H. Roohi and N. Mohtamadifar, RSC Adv., 2022, 12, 11557–11573 RSC.
- W. H. Liu, I. C. Wu, C. H. Lai, C.-H. Lai, P. T. Chou, Y. T. Li, C. L. Chen, Y. Y. Hsu and Y. Chi, Chem. Commun., 2008, 41, 5152–5154 RSC.
- Y. Gao, X. Li, Y. Hu, Y. Fan, J. Yuan, N. Robertson, J. Hua and S. R. Marder, J. Mater. Chem. A, 2016, 4, 12865–12877 RSC.
- J. T. Lin, P. C. Chen, Y. S. Yen, Y. C. Hsu, H. H. Chou and M. C. Yeh, Org. Lett., 2009, 11, 97–100 CrossRef CAS PubMed.
- W. Fan, D. Tan and W. Q. Deng, ChemPhysChem, 2012, 13, 2051–2060 CrossRef CAS PubMed.
- R. Tarsang, V. Promarak, T. Sudyoadsuk, S. Namuangruk, N. Kungwan, P. Khongpracha and S. Jungsuttiwong, RSC Adv., 2015, 5, 38130–38140 RSC.
- M. Li, L. Kou, L. Diao, Q. Zhang, Z. Li, Q. Wu, W. Lu and D. Pan, J. Phys. Chem. A, 2015, 119, 3299–3309 CrossRef CAS PubMed.
- B. Nagarajan, S. Kushwaha, R. Elumalai, S. Mandal, K. Ramanujam and D. Raghavachari, J. Mater. Chem. A, 2017, 5, 10289–10300 RSC.
- M. C. Tsai, C. L. Wang, C. W. Chang, C. W. Hsu, Y. H. Hsiao, C. L. Liu, C. C. Wang, S. Y. Lin and C. Y. Lin, J. Mater. Chem. A, 2018, 6, 1995 RSC.
- M. C. Tsai, Y. C. Chiu, M. DeLu, Y. L. Tung, H. C. Tsai, J. R. Chang Chien and C. Y. Lin, ACS Appl. Energy Mater., 2020, 3, 2744 CrossRef CAS.
- M. Al-Eid, S. Lim, K. W. Park, B. Fitzpatrick, C. H. Han, K. Kwak, J. Hong and G. Cooke, Dyes Pigm., 2014, 104, 197–203 CrossRef CAS.
- D. Wang, H. Han, H. Gao, Z. Yang, Y. Xing, H. Cao, W. He, H. Wang, J. Gu and H. Hu, Synth. Met., 2016, 220, 41–47 CrossRef CAS.
- S. S. M. Fernandes, I. Mesquita, L. Andrade, A. Mendes, L. L. G. Justino, H. D. Burrows and M. M. M. Raposo, Org. Electron., 2017, 49, 194–205 CrossRef CAS.
- G. Forte, L. D'Urso, E. Fazio, S. Patanè, F. Neri, O. Puglisi and G. Compagnini, Appl. Surf. Sci., 2013, 272, 76–81 CrossRef CAS.
- E. Fazio, L. D'Urso, G. Consiglio, A. Giuffrida, G. Compagnini, O. Puglisi, S. Patanè, F. Neri and G. Forte, J. Phys. Chem. C, 2014, 118, 28812–28819 CrossRef CAS.
- N. R. Agarwai, A. Lucotti, M. Tommasini, W. A. Chalifoux and R. R. Tykwinski, J. Phys. Chem. C, 2016, 120(20), 11131–11139 CrossRef.
- P. Marabotti, A. Milani, A. Lucotti, L. Brambilla, M. Tommasini, C. Castiglioni, P. Mecik, B. Pigulski, S. Szafert and C. S. Casari, Carbon Trends, 2021, 5, 100115 CrossRef.
- A. D. Slepkov, F. A. Hegmann, S. Eisler, E. Elliott and R. R. Tykwinski, J. Chem. Phys., 2004, 120, 6807–6810 CrossRef CAS PubMed.
- S. Eisler, A. D. Slepkov, E. Elliott, T. Luu, R. McDonald, F. A. Hegmann and R. Tykwinski, J. Am. Chem. Soc., 2005, 127, 2666–2676 CrossRef CAS PubMed.
- I. Ledoux, I. D. W. Samuel, J. Zyss, S. N. Yaliraki, F. J. Schattenmann, R. R. Schrock and R. J. Silbey, Chem. Phys., 1999, 245, 1–16 CrossRef CAS.
- L. K. Zaitriand and S. M. Mekelleche, Mol. Phys., 2020, 118, 1–10 Search PubMed.
- J. Yong Lee, S. B. Suh and K. S. Kim, J. Chem. Phys., 2000, 112, 344–360 CrossRef.
- F. Cataldo, Polyynes: Synthesis, Properties, and Applications, 2005 Search PubMed.
- J. Preat, C. Michaux, D. Jacquemin and E. A. Perpete, J. Phys. Chem. C, 2009, 113, 16821–16833 CrossRef CAS.
- M. Pastore, E. Mosconi, F. De Angelis and M. Gratzel, J. Phys. Chem. C, 2010, 114, 7205–7212 CrossRef CAS.
- M. Xie, F. Q. Bai, J. Wang, C. P. Kong, J. Chen and H. X. Zhang, Comput. Mater. Sci., 2016, 111, 239–246 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox. Gaussian 16 Rev C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. Preat, A. Hagfeldt and E. A. Perpete, Energy Environ. Sci., 2011, 4, 4537–4549 RSC.
- J. B. Asbury, Y. Q. Wang, E. C. Hao, H. N. Ghosh and T. Q. Lian, Res. Chem. Intermed., 2001, 27, 393–406 CrossRef CAS.
- H. W. Ham and Y. S. Kim, Thin Solid Films, 2010, 518, 6558–6563 CrossRef CAS.
- N. Vlachopoulos, P. Liska, J. Augustynski and M. Grätzel, J. Am. Chem. Soc., 1988, 110, 1216–1220 CrossRef CAS.
- L. Mao, S. Dun, H. Ren, J. Jiang, X. Guo, F. Huang, P. Heng, L. Wang, J. Zhang and H. Agren, J. Mater. Chem. C, 2021, 9, 5800–5807 RSC.
- W. Zhang, L. Wang, L. Mao, J. Jiang, H. Ren, P. Heng, H. Agen and J. Zhang, J. Phys. Chem. C, 2020, 124, 3980–3987 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Adam Gorcyński
a,
Adam Gorcyński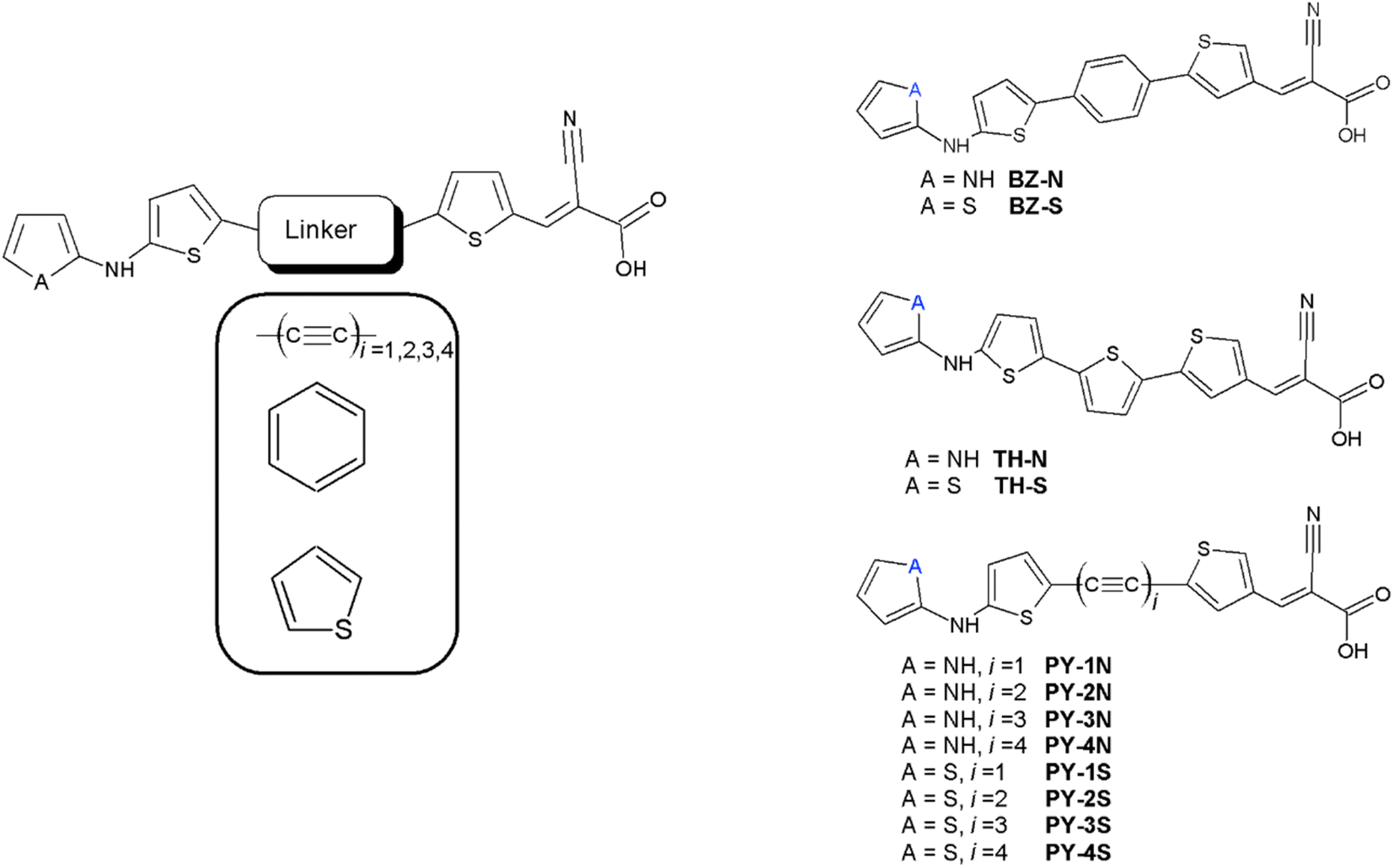
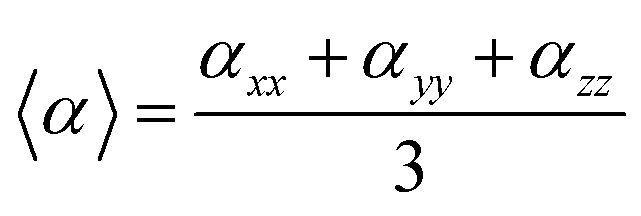
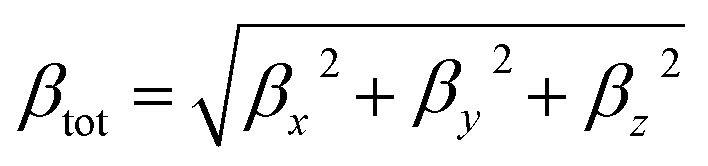
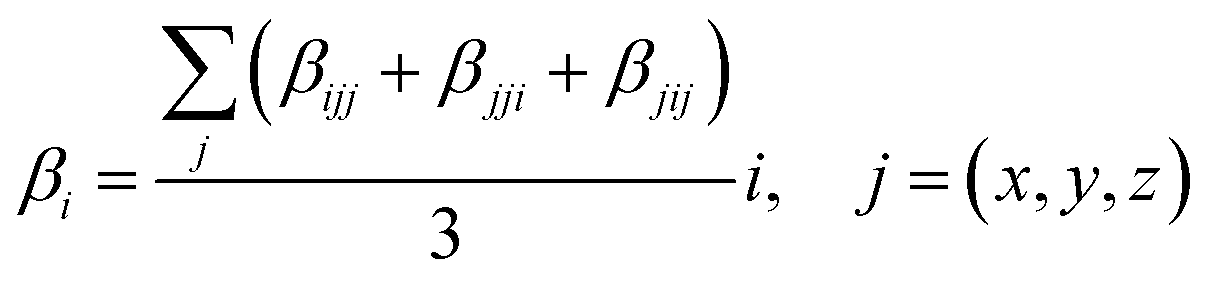

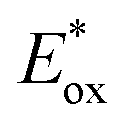
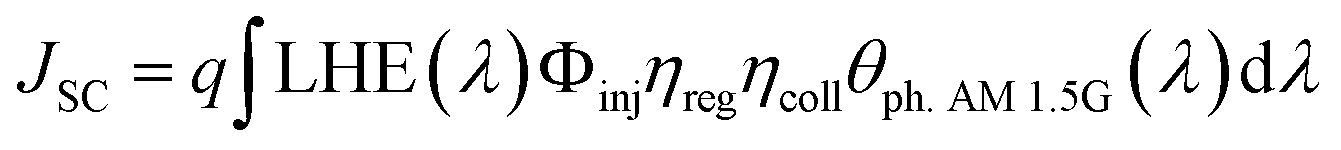
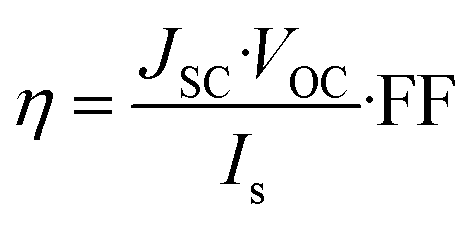
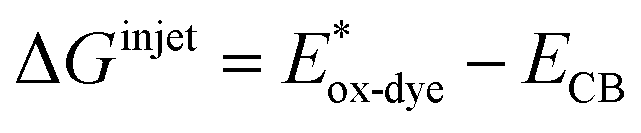
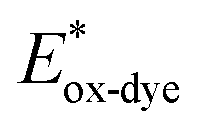 denotes the oxidation potential of the dye in its excited state whereas ECB represents the reduction potential of the CB of the semiconductor TiO2 (ECB = 4.0 eV). In turn,
denotes the oxidation potential of the dye in its excited state whereas ECB represents the reduction potential of the CB of the semiconductor TiO2 (ECB = 4.0 eV). In turn, 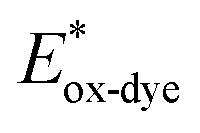 can be computed as:
can be computed as: