
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling the reactivity of phthalonitriles for the efficient synthesis of chiral phthalocyanines with self-assembly abilities†
Irene
Paramio
 a,
Tomás
Torres
abc and
Gema
de la Torre
*ab
a,
Tomás
Torres
abc and
Gema
de la Torre
*ab
aDepartment of Organic Chemistry, Universidad Autónoma de Madrid, Campus de Cantoblanco, C/Francisco Tomás y Valiente 7, 28049 Madrid, Spain. E-mail: gema.delatorre@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, Campus de Cantoblanco, 28049-Madrid, Spain
cInstituto Madrileño de Estudios Avanzados (IMDEA)-Nanociencia, Campus de Cantoblanco, 28049-Madrid, Spain
First published on 9th November 2023
Abstract
Binaphthol-bridged phthalonitriles are common precursors for the preparation of chiral phthalocyanines (Pcs). However, self-condensation of these phthalonitriles usually leads to the target Pcs in extremely low yields. In this work, we report a smart approach to enhance the reactivity towards self-condensation of binaphthol-bridged phthalonitriles, which relies on the addition of another highly reactive phthalonitrile that initiates the macrocyclization process, but it is eventually released. After optimization of the reaction conditions, ethynyl-containing binaphthyloxy-bridged Zn(II)Pc (AA)2-2 has been obtained in remarkable yields. This Pc synthon has been further endowed with hydrophilic cationic tails, giving the strongly amphiphilic Zn(II)Pc 1, which has shown the ability to form nanostructures in aqueous solutions.
Introduction
Phthalocyanines1,2 are fascinating organic compounds with high thermal and optical stability, strong absorption in the visible region, remarkable photophysical properties, semiconducting behaviour and organization abilities, which enable them to be used as targets of choice for a variety of biomedical applications,3 as photocatalysts,4 or as active molecules for a variety of optoelectronic devices.5Self-condensation of phthalonitriles is by far the most widely used method for the synthesis of Pcs,6–10 which in many cases is initiated by a nucleophilic attack of a lithium alkoxide (or another strong nucleophile) on the electrophilic carbon of the nitrile, which generates nucleophilic nitrogen that proceeds with the self-condensation process. Not less important is the template method, which utilizes a transition metal salt to coordinate to the nitrogen of the nitrile functions, causing the necessary polarization that induces self-condensation. The major drawback of this method is that it usually requires high temperature, which shifts the different equilibria towards the formation of a highly conjugated macrocycle, but does not allow the development of chemoselective conditions that lead to the formation of Pc rings containing different chemical functionalities at the constituent isoindoles in a controlled fashion. Some approaches have been addressed to control the cross-reactivity between different phthalonitrile units, using steric constrictions,11–15 by preorganization of phthalonitrile units,16–20 by sequential assembly,21–26 or even using other types of precursors with complementary reactivity.27–31 However, controlling the reactivity of the phthalonitriles on the basis of the different electronic characteristics of the substituents that can modify the involved electrophilicity/nucleophilicity is underdeveloped.32 One of the outstanding strategies to prepare Pcs with certain chemo- and regiocontrol is the use of bisphthalonitriles linked by constrained binding groups.16–20 In particular, the introduction of optically active binaphthyl units as bridges deserves special mention since they lead to optically active Pcs thanks to the intrinsic atropisomerism of the binaphthol subunit.1,17–20,33–35 Binaphthyl-containing Pcs were formerly described by Kobayashi and co-workers,33 and, since then, several Pcs and related compounds endowed with optically pure binaphthyl cores have been prepared, most of them presenting an unsymmetric functionalization of the Pc core in an AABB pattern (AA coding for the binaphthol-linked diisoindole part) (Fig. 1).17–20 Surprisingly, the self-condensation of AA-bisphthalonitriles leading to symmetrically substituted (AA)2-Pc derivatives with two bridging binaphthol units (Fig. 1) is rather unfavoured and these types of compounds have been prepared in extremely low yields.17,33,34
 | ||
| Fig. 1 B3LYP/6-31G(d,p)-optimized structures of non-functionalized AABB and (AA)2 symmetry Zn(II)Pcs. | ||
Recently we have reported efficient conditions for the preparation of binaphthol-bridged AABB Zn(II)Pcs in high yields.18 Using this methodology, we have been able to synthesize a variety of derivatives with highly directional amphiphilicity, holding both non-polar aromatic heads and hydrophilic tails located either at the binaphthol or at the periphery of the Pc core.19,20 These molecules have demonstrated abilities to form chiral nanoparticles in aqueous medium triggered by hydrophobic forces. In particular, the molecules functionalized with cationic residues form homogeneously sized aggregates that perform as nanostructured photosensitizers for the inactivation of bacteria.19,20 On the basis of these results, we have undertaken the preparation of other archetypal chiral amphiphilic Pcs with self-organizing abilities, consisting of a symmetric bis(binaphthol)-(AA)2 Zn(II)Pc central core endowed with hydrophilic cationic tails at the binaphthol units (1, Fig. 2), which could lead to nanostructures differing in shape or size with respect to those formed by unsymmetric AABB Pcs. Towards this aim, we have envisioned the previous preparation of Zn(II)Pc synthons ((AA)2-1 and (AA)2-2 in Scheme 1), holding either bromine or protected acetylene moieties, respectively, which could be starting materials for further Sonogashira reactions to introduce the targeted triply cationic addenda. However, the synthesis of (AA)2-1 and (AA)2-2 is challenging due to the above-mentioned low reactivity towards self-condensation previously found for these types of binaphthol-linked bisphthalonitriles.
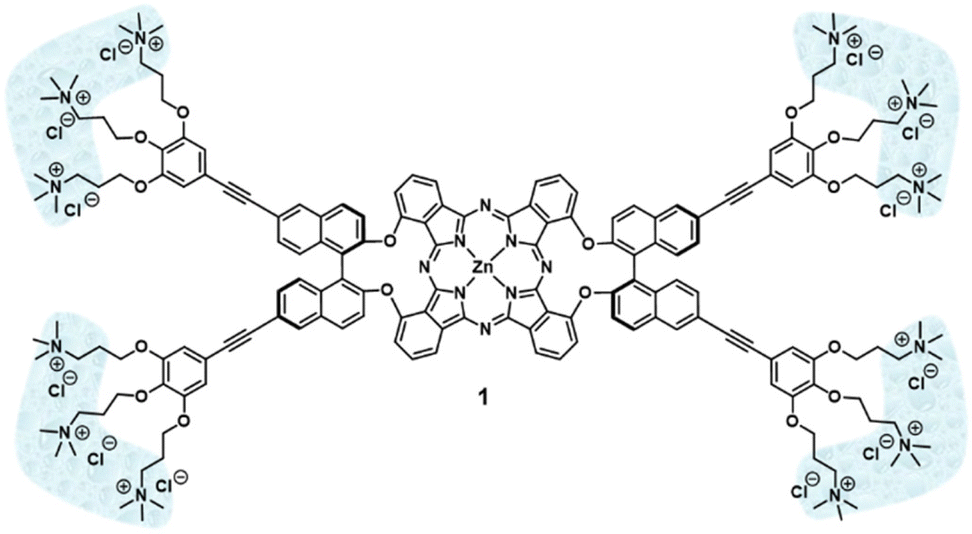 | ||
| Fig. 2 Structure of symmetric Zn(II)Pc 1. | ||
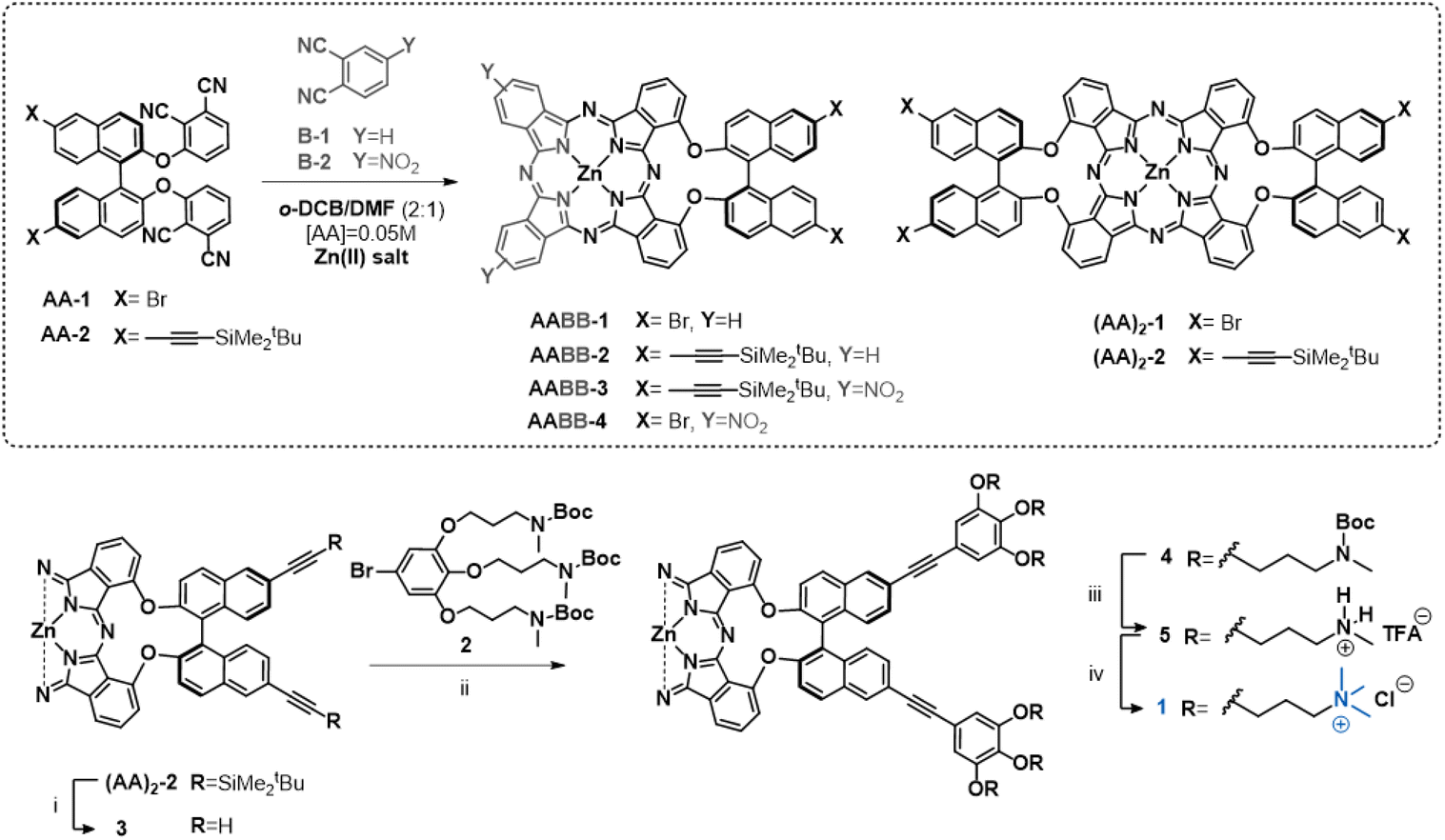 | ||
Scheme 1 Cross-cyclotetramerization reaction between AA and B (top). Synthetic route towards product 1 (bottom): (i) TBAF in THF, 0 °C to rt for 2 h; (ii) Pd(PPh3)4 in DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et3N (2:1), 80 °C; (iii) TFA in DCM, rt for 5h, (iv) MeI and 1,2,2,6,6-pentamethylpiperidine (PMP) in DMF at rt for 16 h, then Dowex resin in Milli-Q water for 2 h. :Et3N (2:1), 80 °C; (iii) TFA in DCM, rt for 5h, (iv) MeI and 1,2,2,6,6-pentamethylpiperidine (PMP) in DMF at rt for 16 h, then Dowex resin in Milli-Q water for 2 h. | ||
Herein we report an elegant approach to enhance the reactivity of the required precursors AA-1 and AA-2 (Scheme 1), which is based on experimental evidence compiled in previous works.18 We had observed that cross-condensation between these types of AA bisphthalonitriles and another phthalonitrile (B) containing electron-withdrawing groups yielded the targeted unsymmetric AABB Pcs accompanied by a considerable amount of the symmetric (AA)2 Zn(II)Pc. Based on this premise, we have explored the use of commercially available 4-nitrophthalonitrile as an “activating” agent for the nitrile moieties of bisphthalonitriles AA-1 and AA-2 at an initial stage of the macrocyclization process to facilitate their further self-condensation. This strategy has allowed us to obtain the Pc synthons (AA)2-1 and (AA)2-2 in good yields and subsequently converted them into the targeted Pc 1, which has proved its ability to form nanostructures in aqueous solutions.
Results and discussion
For the synthesis of the symmetric Zn(II)Pcs (AA)2-1 and (AA)2-2, we first, indeed, tested the self-condensation of AA-118 and AA-218 under the conditions previously applied for the preparation of unsymmetric binaphthyloxy AABB Zn(II)Pcs,18–20 that is, heating them in o-DCB/DMF (2:1) as a solvent in the presence of Zn(OAc)2. The products (AA)2-1 and (AA)2-2 could be isolated in low yields, 2% and 7%, respectively (Table 1, entries 1 and 2). Changing either the solvent to dimethylaminoethanol (DMAE), or the activation conditions (namely MW instead of temperature), or even adding a base such as 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) did not produce a significant increase in the yield (Tables S1 and S2, ESI†). However, in our previous work18 we had observed that (AA)2-1 was obtained as a secondary product in 14% yield when AA-1 was cross-reacted with a phthalonitrile functionalized with two electron-withdrawing ester moieties, in a 1:2 ratio, under the conditions mentioned above. From this result it could be inferred that the electron-deficient phthalonitrile someway activates the self-condensation of AA-1. Therefore, we carried out mixed condensation between one equivalent of bisphthalonitrile AA-1 or AA-2 and two equivalents of the commercially available, electron-deficient 4-nitrophthalonitrile B-2, under the same conditions. The reaction gave (AA)2-1 and (AA)2-2 in 14% and 21% yield, respectively (Table 1, entries 3 and 4). Note that the self-condensation of both AA-1 and AA-2 was favoured to the point that the major isolated product was the symmetric (AA)2 ZnPc derivative.
:1), at 150 °C for 16 h
| Entry | AA (eq.) | B (eq.) | Zn(II) salt (eq.) | (AA)2 (%) | AABB (%) |
|---|---|---|---|---|---|
| a AABB Zn(II)Pc is not isolated from the reaction mixture, but its formation was confirmed by MS analysis. | |||||
| 1 | AA-1 (1.0) | — | Zn(OAc)2 (0.75) | 2 | —a |
| 2 | AA-2 (1.0) | — | Zn(OAc)2 (0.75) | 7 | — |
| 3 | AA-1 (1.0) | B-2 (2.0) | Zn(OAc)2 (1.5) | 14 | —a |
| 4 | AA-2 (1.0) | B-2 (2.0) | Zn(OAc)2 (1.5) | 21 | 8 |
| 5 | AA-1 (1.0) | B-1 (2.0) | Zn(OAc)2 (1.5) | 1 | 21 |
| 6 | AA-2 (1.0) | B-1 (2.0) | Zn(OAc)2 (1.5) | 5 | 30 |
| 7 | AA-2 (1.0) | B-2 (2.0) | ZnCl2 (1.5) | — | 5 |
| 8 | AA-2 (1.0) | B-2 (2.0) | Zn(OMe)2 (1.5) | 4 | — |
| 9 | AA-2 (1.0) | B-2 (0.5) | Zn(OAc)2 (0.5) | 16 | — |
| 10 | AA-2 (1.0) | B-2 (0.5) | Zn(OAc)2 (1.5) | 4 | — |
To assess if the presence of the electron-withdrawing group is necessary to confer this “activation” ability to the phthalonitrile, we carried out mixed condensation of either AA-1 or AA-2 with non-functionalized phthalonitrile (B-1). In this case, the formation of AABB-Zn(II)Pcs resulting from the cross-condensation is strongly favoured. For AA-1, the symmetric ZnPc (AA)2-1 was isolated in only 1% yield, while AABB-1 was obtained in 21% yield (entry 5). Similarly, bisphthalonitrile AA-2 led to a mixture of (AA)2-2 and the corresponding unsymmetric Pc AABB-2 in 5% and 30% yields, respectively (entry 6). As reactions with AA-2 always proceeded with better conversions (probably due to its better solubility), our subsequent efforts were focused on the reactions with this phthalonitrile.
The above results indicate that, under these conditions, electron-deficient phthalonitriles favourably give rise to cross-reactions with binaphthyloxy phthalonitriles but show less reactivity towards self-condensation when the two precursors are present in the solution. To get more insights into the mechanistic pathway and rationalize the outcomes of the reaction, we turned to the cross-reaction between AA-2 and B-2, but changed the Zn(II) salt to ZnCl2 (entry 7) in order to assess if the type of metallic salt has a role in the resulting ratio of Zn(II)Pcs. Under these conditions, (AA)2-2 was not formed, pointing to a plausible participation of the acetate ligands linked to the zinc cation. Considering that alkoxides are commonly used as catalysts for the macrocyclization reaction, we utilized Zn(OMe)2 (entry 8), but (AA)2-2 was isolated in only 4% yield. Taken together, these results point to a plausible mechanism (Scheme 2) in which both acetate anions and electron-deficient 4-nitrophthalonitrile are necessary to activate the self-condensation of AA-2. Apparently, the favoured attack of Zn(OAc)2 on the more reactive B-2 could result in the formation of an isoindolenine derivative (Act-B-2, step 1 in Scheme 2) that would attack on the bisphthalonitrile derivative AA-2 (or AA-1, step 2) to generate an activated trimeric species (Act-AA) with a strongly nucleophilic nitrogen. This intermediate could (i) undergo addition to another AA bisphthalonitrile (step 3A), generating an open species (Act-(AA)2 in Scheme 2) that would undergo cyclotetramerization with the release of the Act-B-2 isoindolenine (step 4) or (ii) self-condensate to give (AA)2-2 (or (AA)2-1, step 3B) with the extrusion of two units of Act-B-2 as leaving groups. Noteworthily, this release is plausible since the negative charge is stabilized due to the resonance effect of the nitro group. Note that B-1 is plausibly less prone to react with Zn(OAc)2 than B-2, and also the corresponding negatively charged isoindolenine form would be less stable, which could explain that, in this case, the exclusive reaction of Zn(OAc)2 with B-1 is not the main pathway, and several activated and non-activated B-1 derivatives are present in the solution, allowing for the formation of Zn(II)Pcs with adjacent BB units (Table 1, entries 5 and 6).
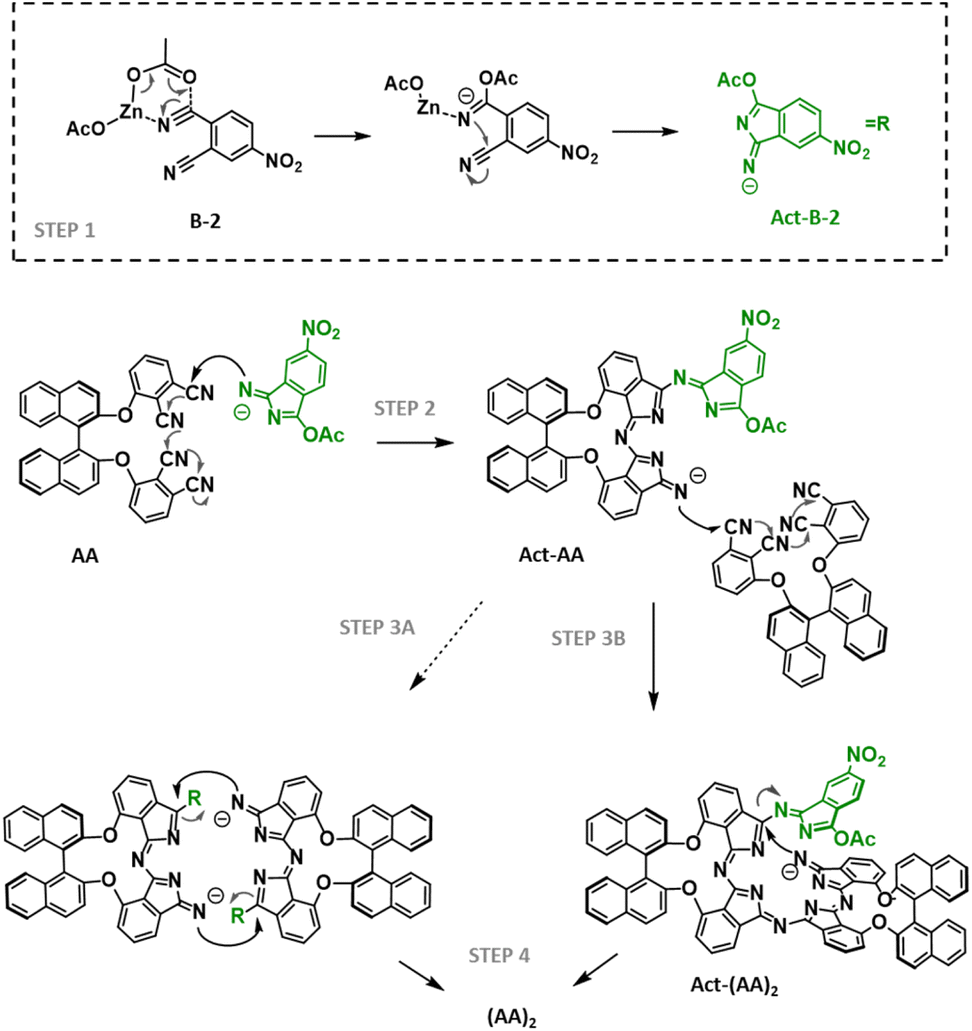 | ||
| Scheme 2 Postulated cyclotetramerization mechanism of B-2 and generic AA. The Zn(II) complex is omitted from the structures for simplification. | ||
To confirm this hypothesis, we carried out an additional reaction between AA-2 and a sub-stoichiometric amount of B-2 and Zn(OAc)2 (0.5 eq. each). Under these conditions, the reaction afforded the target (AA)2-2 in a remarkable 16% yield (entry 9), while the reaction between AA-2, 0.5 eq. of B-2 and an excess of Zn(OAc)2 (1.5 eq.) provided (AA)2-2 in only 4% yield (entry 10). These observations support the suggested pathway: a stoichiometric ratio of Zn(OAc)2 and B-2 results in the major conversion into Act-B-2, which preferably undergoes addition to AA-2 rather than reacting with itself.‡ Unfortunately, the reaction cannot be carried out using lower amounts of B-2, since the minimum amount of Zn(OAc)2 to have the necessary stoichiometric ratio of Zn2+ for the formation of the Zn(II)Pcs must be 0.5 eq. with respect to AA-2.§
Once the synthesis of (AA)2-2 was optimized, we proceeded with the preparation of the target molecule 1 (Fig. 2). For this purpose, the Boc-protected, triamine ligand 2 was synthesized following reported procedures.20 On the other hand, the symmetric Pc (AA)2-2 was deprotected using TBAF and, due to the low stability of the resulting product (3), it was subsequently reacted with 2 under optimized Sonogashira conditions (Scheme 1).20 Zn(II)Pc 4 was isolated in 27% yield from a reaction mixture containing other Zn(II)Pcs with a different degree of functionalization of the terminal ethynyl groups. The lack of solubility of 4, even in coordinating solvents such as DMSO, complicated the characterization through 1H NMR, but the structure was confirmed by MS. Then, 4 was subjected to a deprotection step of the tert-butyloxycarbonyl using TFA, which gave the corresponding trifluoroacetate salt of the multicationic Zn(II)Pc 5 in 55% yield. Quaternization of the terminal amines with MeI in the presence of the voluminous base 1,2,2,6,6-pentamethylpiperidine (PMP) and subsequent counterion exchange (iodine was substituted with chlorine atoms) afforded the final Zn(II)Pc 1 in 98% yield. Both products 5 and 1 showed solubility in DMSO-d6 and could be characterized by 1H NMR and MS. The 1H NMR of 1 was performed in DMSO-d6/D2O to obtain a better resolved spectrum (ESI†).
The next step was the spectroscopic characterization of 1. First, ground-state absorption experiments were performed in DMSO as a coordinating solvent that hampers the aggregation of Zn(II)Pcs through coordination to the metal. The typical Q band for monomeric species and the B-band transition were observed at 691 nm and 323 nm respectively (ESI†). For the verification of the Lambert–Beer law, an analysis of linear regression between the intensity of the Q-band and the concentration of 1 showed R2 values close to 1. Upon excitation at 650 nm, the fluorescence spectrum shows a weak emission with 0.08 ± 0.01 quantum yield.
Due to its pronounced amphiphilicity, 1 was expected to aggregate in aqueous solution. To study the aggregation process, we monitored the changes in UV-vis and fluorescence spectra upon increasing the percentage of Milli-Q water (i.e., in which aggregation is expected) in a solution of 1 in DMSO (where the Pc is in a monomeric state). First, upon increasing the ratio of water from 0% to 20%, an increase in the intensity of the main absorption and emission bands was observed, probably due to a change in the solvation sphere of the molecule. Then, from 20% to 80% water content, a progressive reduction in the intensity of the Q-band takes place, together with a low decrease in the intensity of the fluorescence. Eventually, at 100% water content (where 1 was perfectly soluble), an abrupt reduction and broadening of the Q-band was observed, together with the emergence of a new band at 640 nm which is typical of H-type aggregates. This behaviour is concordant with the almost complete quenching of the emission in fluorescence experiments over pure aqueous solutions.
The presence of the two (R)-binaphthol units in 1 makes the molecule chiral in nature, which allowed us to follow the aggregation process by CD spectroscopy (Fig. 3c and d). In DMSO solution, CD signals with a negative sign and low intensity appear in the Soret and Q-band regions. Upon addition of water, new bisignated CD curves arise with increased intensity, associated with the formation of self-assembled species.
 | ||
| Fig. 3 (a) Absorption and (b) fluorescence spectral changes upon increasing the water ratio in solutions of 1 (5 × 10−6 M) in DMSO:water mixtures (λexcitation = 650 nm). CD spectra of 1 (c) in DMSO and water at 20 °C and (d) on heating the solution from 20 °C to 80 °C at 1 °C min−1 ([1] = 15 × 10−5 M) for temperature stability studies in pure water. (e) and (f) TEM images of 1 prepared using a water solution in ([1] = 15 × 10−5 M). | ||
To obtain information about the morphology of the aggregates in aqueous media, Milli-Q aqueous solutions were studied by transmission electronic microscopy (TEM). 1 was found to form small particles in the 11–25 nm range (Fig. 3e and f). The formation of aggregates in aqueous solution can be explained considering the non-directional hydrophobic forces to have a stronger contribution than the ionic repulsion between ammonium moieties. However, the growth of aggregates is probably restricted by the steric hindrance originating from the binaphthol units, which are almost perpendicular to the Pc plane (Fig. 4), inhibiting the π–π stacking of the hydrophobic cores. Therefore, at the initial stages of the supramolecular polymerization, hydrophobic forces and π–π stacking boost the formation of small nuclei, but when more Pc units are included in the supramolecular entity, steric and ionic repulsions may cause the aggregate to stop growing, resulting in the observed small nanoparticles.
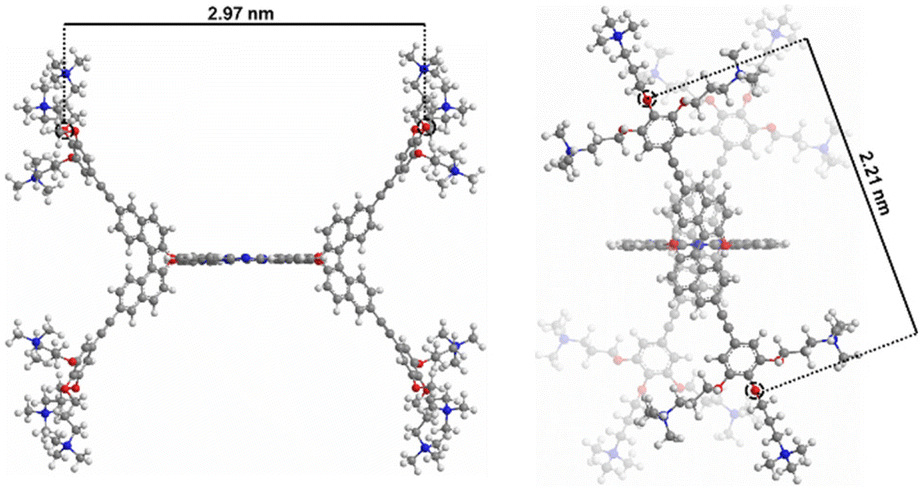 | ||
| Fig. 4 B3LYP/6-31G(d,p)-optimized 3D structure of 1. Distances in nanometres between signalled oxygen atoms (in red) are shown: front view (left) and side view (right). | ||
Conclusions
In this work, we describe the chemically controlled and efficient synthesis of bis(binaphthol)-(AA)2 Zn(II)Pcs. We have demonstrated that the reactivity of phthalonitriles can be regulated as a function of the electronic character provided by the substituents in the ring and through a judicious selection of conditions, which can boost the yield of the desired compounds. Specifically, we have used 4-nitrophthalonitrile as a “catalytic species” that activates the difficult self-condensation of binaphthyloxy-linked bisphthalonitriles. A careful selection of experiments has allowed us to postulate a mechanistic pathway that would explain the observed results. Under the optimized conditions, we have achieved yields up to 21% in the formation of the elusive bis(binaphthyloxy)-bridged Pc (AA)2-2. This Pc has been used as a starting material for the preparation of a new type of amphiphilic Zn(II)Pc, namely, the multicationic Zn(II)Pc 1, which has shown an interesting self-assembly behaviour in aqueous media, giving rise to homogeneous nanoparticles. These interesting nanostructures are the targets for biomedical applications, considering their size and the photosensitizing abilities of the Pc components, and therefore, studies in this regard are envisioned.Author contributions
I. P. performed the experimental work and contributed to the elaboration of the manuscript. T. T. supervised the work and elaborated the manuscript. G. d. l. T. proposed and supervised the project and elaborated the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish MCIN/AEI/10.13039/501100011033 (PID2020-116490GB-I00, PID2020-115801RB-C21), the Comunidad de Madrid and the Spanish State through the Recovery, Transformation and Resilience Plan [“Materiales Disruptivos Bidimensionales (2D)” (MAD2D-CM) (UAM1)-MRR Materiales Avanzados], and the European Union through the Next Generation EU funds is acknowledged. IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant SEV2016-0686).References
- H. Lu and N. Kobayashi, Optically Active Porphyrin and Phthalocyanine Systems, Chem. Rev., 2016, 116, 6184–6261 CrossRef CAS.
- D. Gounden, N. Nombona and W. E. van Zyl, Recent Advances in Phthalocyanines for Chemical Sensor, Non-Linear Optics and Energy Storage Applications, Coord. Chem. Rev., 2020, 420, 213359 CrossRef CAS.
- (a) V. Almeida-Marrero, E. van de Winckel, E. Anaya-Plaza, T. Torres and A. de la Escosura, Porphyrinoid Biohybrid Materials as an Emerging Toolbox for Biomedical Light Management, Chem. Soc. Rev., 2018, 47, 7369–7400 RSC; (b) P. C. Lo, M. S. Rodríguez-Morgade, R. K. Pandey, D. K. P. Ng, T. Torres and F. Dumoulin, The Unique Features and Promises of Phthalocyanines as Advanced Photosensitisers for Photodynamic Therapy of Cancer, Chem. Soc. Rev., 2020, 49, 1041–1056 RSC; (c) I. Paramio, T. Torres and G. de la Torre, Self-Assembled Porphyrinoids: One-Component Nanostructured Photomedicines, ChemMedChem, 2021, 16, 2441–2451 CrossRef CAS PubMed; (d) A. Galstyan, Turning Photons into Drugs: Phthalocyanine–Based Photosensitizers as Efficient Photoantimicrobials, Chem. – Eur. J., 2021, 27, 1903–1920 CrossRef CAS PubMed; (e) B. Zheng, Q. He, X. Li, J. Yoon and J. Huang, Phthalocyanines as Contrast Agents for Photothermal Therapy, Coord. Chem. Rev., 2021, 426, 213548 CrossRef CAS.
- M. Moreno-Simoni, T. Torres and G. de la Torre, Subphthalocyanine Capsules: Molecular Reactors for Photoredox Transformations of Fullerenes, Chem. Sci., 2022, 13, 9249–9255 RSC.
- (a) G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, An Exciting Twenty-Year Journey Exploring Porphyrinoid-Based Photo- and Electroactive Systems, Coord. Chem. Rev., 2021, 428, 213605 CrossRef CAS; (b) M. Urbani, G. de la Torre, M. K. Nazeeruddin and T. Torres, Phthalocyanines and Porphyrinoid Analogues as Hole- and Electron-Transporting Materials for Perovskite Solar Cells, Chem. Soc. Rev., 2019, 48, 2738–2766 RSC.
- K. M. Kadish, K. M. Smith and R. Guilard, The Porphyrin Handbook: Phthalocyanines: Synthesis; The Porphyrin Handbook, Academic Press, 2003 Search PubMed.
- L. Breloy, O. Yavuz, I. Yilmaz, Y. Yagci and D.-L. Versace, Design, Synthesis and Use of Phthalocyanines as a New Class of Visible-Light Photoinitiators for Free-Radical and Cationic Polymerizations, Polym. Chem., 2021, 12, 4291–4316 RSC.
- G. Zanotti, P. Imperatori, A. M. Paoletti and G. Pennesi, Sustainable Approaches to the Synthesis of Metallophthalocyanines in Solution, Molecules, 2021, 26, 1760 CrossRef CAS.
- F. Cong, H. Jiang, X. Du, W. Yang and S. Zhang, Facile, Mild-Temperature Synthesis of Metal-Free Phthalocyanines, Synthesis, 2021, 2656–2664 CrossRef CAS.
- D. Langerreiter, M. A. Kostiainen, S. Kaabel and E. Anaya-Plaza, A Greener Route to Blue: Solid-State Synthesis of Phthalocyanines, Angew. Chem., Int. Ed., 2022, 61, 202209033 CrossRef.
- R. P. Kingsborough and T. M. Swager, A Highly Conductive Macrocycle-Linked Metallophthalocyanine Polymer, Angew. Chem., Int. Ed., 2000, 39, 2897–2900 CrossRef CAS.
- N. Kobayashi, H. Miwa and V. N. Nemykin, Adjacent versus Opposite Type Di-Aromatic Ring-Fused Phthalocyanine Derivatives: Synthesis, Spectroscopy, Electrochemistry, and Molecular Orbital Calculations, J. Am. Chem. Soc., 2002, 124, 8007–8020 CrossRef CAS PubMed.
- N. Kobayashi, T. Ashida, T. Osa and H. Konami, Phthalocyanines of a Novel Structure: Dinaphthotetraazaporphyrins with D2h Symmetry, Inorg. Chem., 1994, 33, 1735–1740 CrossRef CAS.
- K. Sakamoto, E. Ohno-Okumura, T. Kato, M. Watanabe and M. J. Cook, Investigation of Zinc Bis(1,4-Didecylbenzo)-Bis(2,3-Pyrido) Porphyrazine as an Efficient Photosensitizer by Cyclic Voltammetry, Dyes Pigm., 2008, 78, 213–218 CrossRef CAS.
- E. Fazio, J. Jaramillo-García, G. de la Torre and T. Torres, Efficient Synthesis of ABAB Functionalized Phthalocyanines, Org. Lett., 2014, 16, 4706–4709 CrossRef CAS.
- M. Kimura, H. Ueki, K. Ohta, H. Shirai and N. Kobayashi, Self-Organization of Low-Symmetry Adjacent-Type Metallophthalocyanines Having Branched Alkyl Chains, Langmuir, 2006, 22, 5051–5056 CrossRef CAS PubMed.
- N. Kobayashi, Optically Active ‘Adjacent’ Type Non-Centrosymmetrically Substituted Phthalocyanines, Chem. Commun., 1998, 487–488 RSC.
- M. Revuelta-Maza, T. Torres and G. de la Torre, Synthesis and Aggregation Studies of Functional Binaphthyl-Bridged Chiral Phthalocyanines, Org. Lett., 2019, 21, 8183–8186 CrossRef CAS PubMed.
- M. A. Revuelta-Maza, E. de las Heras, M. Agut, S. Nonell, T. Torres and G. de la Torre, Self-Assembled Binaphthyl-Bridged Amphiphilic AABB Phthalocyanines: Nanostructures for Efficient Antimicrobial Photodynamic Therapy, Chem. – Eur. J., 2021, 27, 4955–4963 CrossRef CAS.
- I. Paramio, A. Salazar, M. Jordà-Redondo, S. Nonell, T. Torres and G. de la Torre, Nanostructured AABB Zn(II) Phthalocyanines as Photodynamic Agents for Bacterial Inactivation, Adv. Ther., 2023, 2300116 CrossRef.
- K. J. M. Nolan, M. Hu and C. C. Leznoff, “Adjacent” Substituted Phthalocyanines, Synlett, 1997, 593–594 CrossRef.
- T. Fukuda and N. Kobayashi, Efficient Synthesis of a Donor-Acceptor Phthalocyanine Having Adjacently-Fused Pyrazine Rings, Chem. Lett., 2002, 31, 866–867 CrossRef.
- A. Díaz-Moscoso, G. J. Tizzard, S. J. Coles and A. N. Cammidge, Synthesis of meso–Substituted Tetrabenzotriazaporphyrins: Easy Access to Hybrid Macrocycles, Angew. Chem., Int. Ed., 2013, 52, 10784–10787 CrossRef.
- D. S. Andrianov, V. B. Rybakov and A. V. Cheprakov, Between Porphyrins and Phthalocyanines: 10, 20-diaryl-5, 15-tetrabenzodiazaporphyrins, Chem. Commun., 2014, 50, 7953–7955 RSC.
- A. Díaz-Moscoso, D. L. Hughes, M. Bochmann, G. J. Tizzard, S. J. Coles and A. N. Cammidge, Synthesis of Meso-Substituted Subphthalocyanine–Subporphyrin Hybrids: Boron Subtribenzodiazaporphyrins, Angew. Chem., Int. Ed., 2015, 54, 7510–7514 CrossRef.
- F. Alkorbi, A. Díaz-Moscoso, J. Gretton, I. Chambrier, G. J. Tizzard, S. J. Coles, D. L. Hughes and A. N. Cammidge, Complementary Syntheses Giving Access to a Full Suite of Differentially Substituted Phthalocyanine-Porphyrin Hybrids, Angew. Chem., Int. Ed., 2021, 60, 7632–7636 CrossRef CAS PubMed.
- C. C. Leznoff, S. Greenberg, B. Khouw and A. B. P. Lever, The Syntheses of Mono- and Disubstituted Phthalocyanines Using a Dithioimide, Can. J. Chem., 1987, 65, 1705–1713 CrossRef CAS.
- J. G. Young and W. Onyebuagu, Synthesis and Characterization of Di-Disubstituted Phthalocyanines, J. Org. Chem., 1990, 55, 2155–2159 CrossRef CAS.
- P. Stihler, B. Hauschel and M. Hanack, Synthesis of a Bisdienophilic Phthalocyanine and of Precursors for Repetitive Diels-Alder Reactions Based on Hemiporphyrazines and Phthalocyanines, Chem. Ber., 1997, 130, 801–806 CrossRef CAS.
- W. J. Youngblood, Synthesis of a New Trans-A2B2 Phthalocyanine Motif as a Building Block for Rodlike Phthalocyanine Polymers, J. Org. Chem., 2006, 71, 3345–3356 CrossRef CAS PubMed.
- M. M. Ayhan, A. Singh, C. Hirel, A. G. Gürek, V. Ahsen, E. Jeanneau, I. Ledoux-Rak, J. Zyss, C. Andraud and Y. Bretonnière, ABAB Homoleptic Bis(Phthalocyaninato) Lutetium(III) Complex: Toward the Real Octupolar Cube and Giant Quadratic Hyperpolarizability, J. Am. Chem. Soc., 2012, 134, 3655–3658 CrossRef CAS.
- T. Furuyama, Development of Controlled Reactions using an Element-Based design of Azaporphyrinoid Materials, J. Porphyrins Phthalocyanines, 2022, 26, 792–806 CrossRef.
- N. Kobayashi, Y. Kobayashi and T. Osa, Optically Active Phthalocyanines and their Circular Dichroism, J. Am. Chem. Soc., 1993, 115, 10994–10995 CrossRef CAS.
- N. Kobayashi, R. Higashi, B. C. Titeca, F. Lamote and A. Ceulemans, Substituent-Induced Circular Dichroism in Phthalocyanines, J. Am. Chem. Soc., 1999, 121, 12018–12028 CrossRef CAS.
- Y. Okada, T. Hoshi and N. Kobayashi, Recent Progress in Optically Active Phthalocyanines and their Related Azamacrocycles, Front. Chem., 2020, 8, 595998 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details and spectroscopic characterization of the compounds. See DOI: https://doi.org/10.1039/d3qo01630g |
| ‡ Note that when the reaction is carried out only with B-2 and 0.5 eq. of Zn(OAc)2, the formation of the B4 tetranitro-Zn(II)Pc takes place with 60% yield. Act-B-2 reacts easily with B-2 when they are the only two species in solution. |
| § In fact, when the reaction was carried out with 0.2 eq. of B-2 and 0.5 eq. of Zn(OAc)2, the yield of (AA)2-2 dropped to 3% since all the possible activation pathways can take place, which is unfavourable for the formation of the desired product. |
| This journal is © the Partner Organisations 2024 |