
Cu-catalyzed arylation of S-tosyl peptides with arylboronic acids†
Junjie Yinga,
Jingrong Huanga,
Chenguang Liua,
Fa-Jie Chen *a,
Chunfa Xu*ab and
Fen-Er Chen*acd
*a,
Chunfa Xu*ab and
Fen-Er Chen*acd
aInstitute of Pharmaceutical Science and Technology, College of Chemistry, Fuzhou University, Fuzhou, 350108, P. R. China. E-mail: fajie.chen@fzu.edu.cn
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, P. R. China. E-mail: xucf@fzu.edu.cn
cEngineering Center of Catalysis and Synthesis for Chiral Molecules, Department of Chemistry, Fudan University, 220 Handan Road, Shanghai, 200433, P. R. China. E-mail: rfchen@fudan.edu.cn
dShanghai Engineering Research Center of Industrial Asymmetric Catalysis of Chiral Drugs, Fudan University, 220 Handan Road, Shanghai, 200433, P. R. China
First published on 8th November 2023
Abstract
Transition metal-mediated S-arylation has emerged as a powerful tool for the synthesis of S-arylcysteine and S-arylpeptide, which are useful building blocks in pharmacophores and biomolecules. In contrast, the catalytic protocols for arylation remain unexplored, particularly methods employing abundant metal catalysts (e.g. Cu and Ni). Herein, we reported the copper-catalyzed arylation chemistry of S-tosyl peptides with readily available arylboronic acids. This method features excellent yields and a wide variety of aryl groups, enabling the efficient synthesis of S-arylated cysteines and peptides under mild reaction conditions (room temperature, weak base). The reaction can be carried out in both batch and flow, demonstrating its utility in organic synthesis.
Introduction
S-Arylcysteine is an important synthetic intermediate and building block for many pharmacophores and biologically active molecules, such as the potent HIV-1 protease inhibitor Viracept1,2 and the death-cap mushroom cytotoxin α-amanitin3 (Scheme 1). Compared to the naturally occurring cysteine residue, S-arylated cysteine can endow drug molecules with enhanced stability towards protease in the cellular environment, and potentially stronger binding affinity to the protein of interest (POI) due to the hydrophobic interaction with the aromatic cycle.1,2 Cysteine arylation chemistry forming S-arylcysteine has also been demonstrated as a powerful tool for bioconjugation of peptides, proteins, sugars, and nucleic acids.4 Efficient methods for the construction of S-arylcysteine have attracted enormous attention in the fields of organic chemistry and biological science.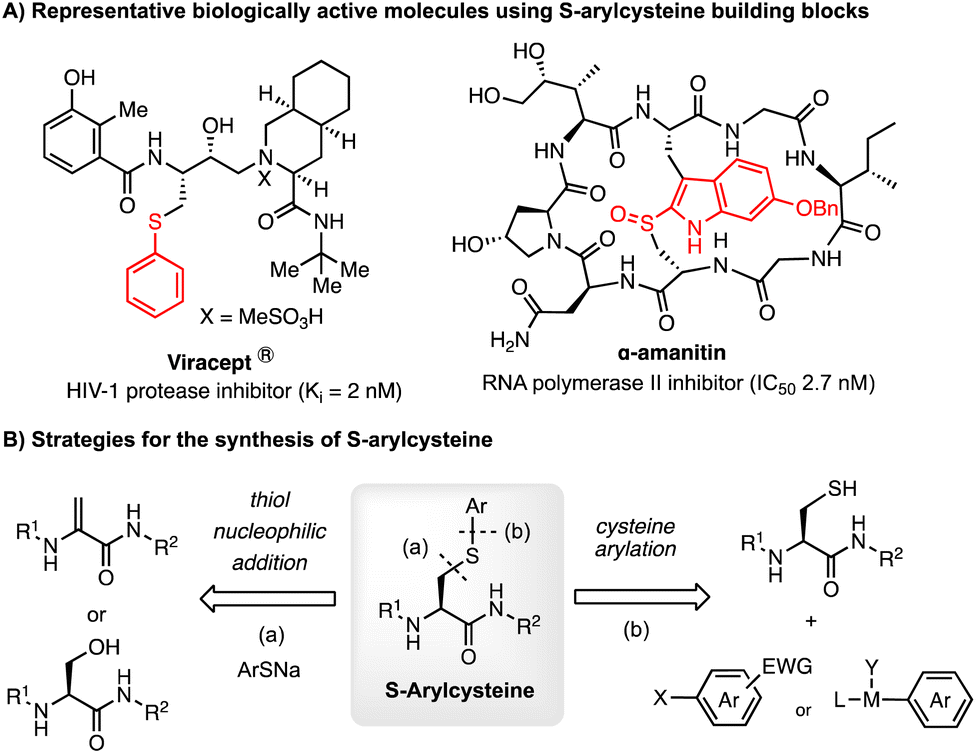 | ||
| Scheme 1 S-Arylcysteine as a useful building block. | ||
Traditionally, S-arylcysteines have been prepared by S–C(sp3) bond formation through Michael addition to dehydroalanine (Dha)5 or SN2 substitution of the serine hydroxyl group6 (Mitsunobu reaction) using arylthiolate as the aryl source.7 The limitation of thiol-addition to Dha is the production of a racemic arylcysteine mixture. Alternatively, S-arylcysteine can be accessed through direct arylation of a cysteine thiol through S–C(sp2) bond formation. Examples include cysteine arylation with electron-deficient aryl halides for the SNAr reaction (e.g. perfluoroarene,8–10 fluorinated pyridinium salts,11 and aryl thioethers12) and activated arene species (e.g. hypervalent iodine reagent13 and diazo compounds14,15). The methods using reactive arene species suffer from the limitation of substrate scope due to the oxidative or acidic reaction conditions as well as the complexity of reagent synthesis. Recently, organometallic reagents have emerged as a powerful tool for cysteine arylation. Selected reagents include organometallic complexes16 (e.g. palladium complex,17–21 gold complex,22–28 and nickel reagent29) (Scheme 2A). These metal-involved arylation strategies have been successfully applied to the bioconjugation of native peptides and proteins. The use of a stoichiometric amount of an organometallic reagent is acceptable for biological applications, considering the value of substrates like antibodies and the relatively low working concentration. However, these metal-mediated strategies are not ideal for organic synthesis considering the reagent cost and atom economy. Cysteine arylation protocols using catalytic amounts of transition metals and readily available arylation reagents are highly appealing, especially methods with abundant metals (such as Cu and Ni). However, this catalytic peptide arylation chemistry has been rarely reported,30 regardless of the extensive studies on the Chan–Lam type S-arylation,31–33 as well as copper-34 and nickel-promoted29 cysteine arylation.
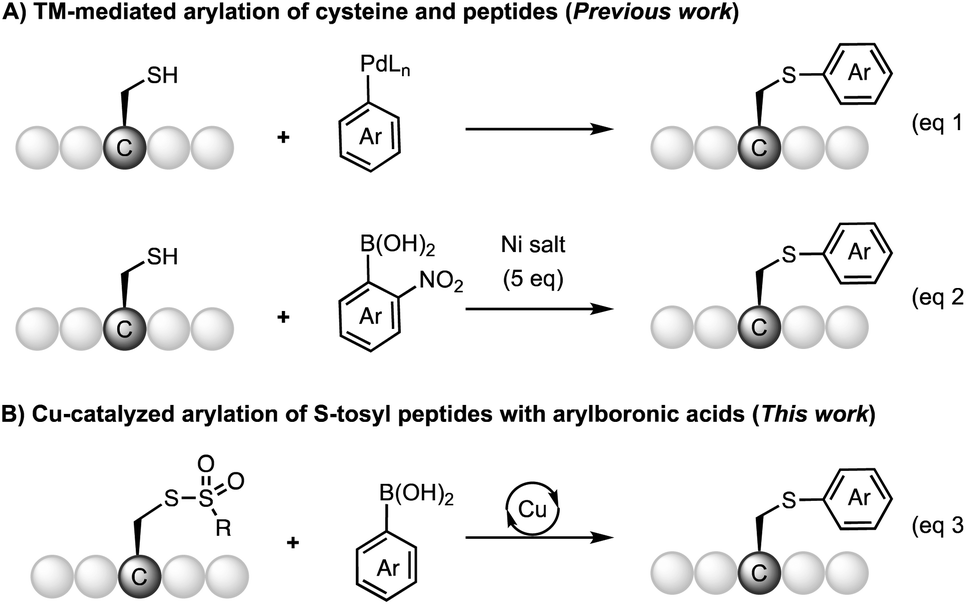 | ||
| Scheme 2 Transition-metal mediated S-arylation. | ||
In this work, we reported the copper-catalyzed arylation chemistry of S-tosyl cysteines and peptides with readily available arylboronic acids and a copper catalyst (Scheme 2B). The reaction can be carried out under mild reaction conditions (room temperature, weak base) with high efficiency. The method allows for the construction of a broad range of S-arylated dipeptides, tripeptides, and pentapeptides from the corresponding cysteine arylthiosulfonate derivatives.
Results and discussion
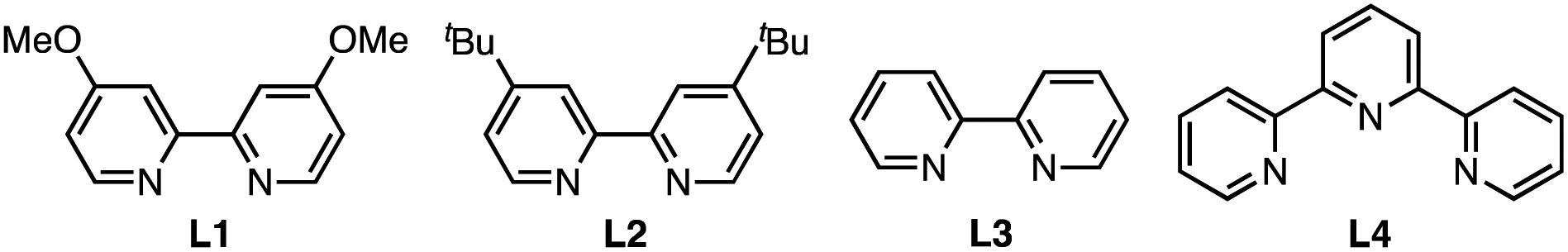
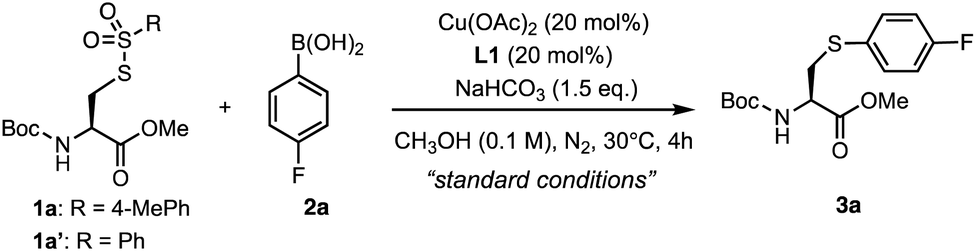
Inspired by the benzenesulfonothioate-mediated S–C(sp2) bond formation,35,36 we explored catalytic S-arylation by investigating the reaction of S-tosyl (Ts) protected cysteine 1a with 4-fluorophenyl boronic acid 2a in the presence of a copper catalyst (Table 1). Upon systematic optimization of reaction conditions, we were able to identify an efficient catalytic arylation protocol: 1a (1.0 eq.), 2a (2.0 eq.) in the presence of Cu(OAc)2 (20 mol%), 4,4′-dimethoxy-2,2′-bipyridine L1 (20 mol%), and NaHCO3 (1.5 eq.) in MeOH (0.1 M) at 30 °C for 4 hours. 98% yield of the desired S-arylcysteine 3a was obtained under the abovementioned “standard conditions” (Table 1, entry 1). In the absence of a copper catalyst, the reaction was completely shut down, demonstrating the crucial role of copper species in the catalytic procedure (Table 1, entry 2). Other copper(II) catalysts, such as Cu(OTf)2 and CuSO4, were also effective (Table 1, entries 7 and 8). Weak bases and ligands were beneficial but not essential for the arylation (Table 1, entries 3 and 4). Relatively strong bases (Na2CO3, K2CO3, and K3PO4) significantly decreased the yields. Only a moderate yield was obtained when the reaction was carried out under an ambient atmosphere (entry 5). EtOH or DMF as a solvent gave low yields, while CH3CN provided the product with acceptable efficiency (entries 15–17). Interestingly, 20% of water as the co-solvent was well tolerated, affording 3a in 88% yield (entry 6). In addition, benzenesulfonothioate derivative 1a′ was found to be as equally efficient as tosyl substrate 1a, generating 3a in 98% yield (entry 18).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield of 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (%) a (%) |
| Standard reaction conditions: 1a (0.1 mmol, 1.0 eq.), 2a (2.0 eq.), NaHCO3 (1.5 eq.), Cu(OAc)2 (20 mol%), L1 (20 mol%) in MeOH (0.1 M) under a N2 atmosphere at 30 °C for 4 h.a Yield determined by crude 19F NMR spectra analysis using trifluoromethyl benzene as an internal standard.b 2.0 equiv. of the base was used.c Isolated yield. | ||
| 1 | None | 98 (98c) |
| 2 | w/o Cu(OAc)2 | 0 |
| 3 | w/o ligand | 77 |
| 4 | w/o NaHCO3 | 74 |
| 5 | Ambient atmosphere | 42 |
| 6 | MeOH/H2O (80/20) | 88 |
| 7b | Cu(OTf)2 instead of Cu(OAc)2 | 82 |
| 8b | CuSO4 instead of Cu(OAc)2 | 77 |
| 9b | L2 instead of L1 | 91 |
| 10b | L3 instead of L1 | 55 |
| 11b | L4 instead of L1 | Trace |
| 12b | Na2CO3 instead of NaHCO3 | 64 |
| 13b | K2CO3 instead of NaHCO3 | 50 |
| 14b | K3PO4 instead of NaHCO3 | Trace |
| 15b | EtOH instead of MeOH | 14 |
| 16b | DMF instead of MeOH | Trace |
| 17b | CH3CN instead of MeOH | 68 |
| 18b | 1a′ instead of 1a | 98 |
 |
||
With the optimized reaction conditions in hand, we explored the substrate scope of arylboronic acids for the arylation of sulfonothioate 1a (Scheme 3). In general, arylboronic acids with an electron-withdrawing group or an electron-donating group were well tolerated, affording the corresponding arylcysteine 3 in excellent yields. For example, products with electron-withdrawing fluorine (3b), trifluoromethyl (3f), ester (3h), and nitro (3i) groups were prepared in excellent yields. 98% yield of 3c bearing an electron-donating methoxyl group was obtained. Surprisingly, this protocol is capable of tolerating arylboronic acids bearing an unprotected amino group (3d) and hydroxyl group (3e). These functionalities were found to deactivate the copper catalyst due to the strong coordination property, and could potentially lead to the N- or O-Chan–Lam coupling reaction.32 Interestingly, a coumarin motif could be installed into the cysteine smoothly, generating sulfide 3i in a quantitative yield. In addition, pyrene-conjugated cysteine derivative 3j was also synthesized with high efficiency. When a bifunctional phenyl boronic acid was used, the two-fold arylation provided sulfide 3k (66%) bearing two cysteine motifs. These results demonstrate the broad substrate scope for arylation reagents and potential application for payload installation in cysteine derivatives.
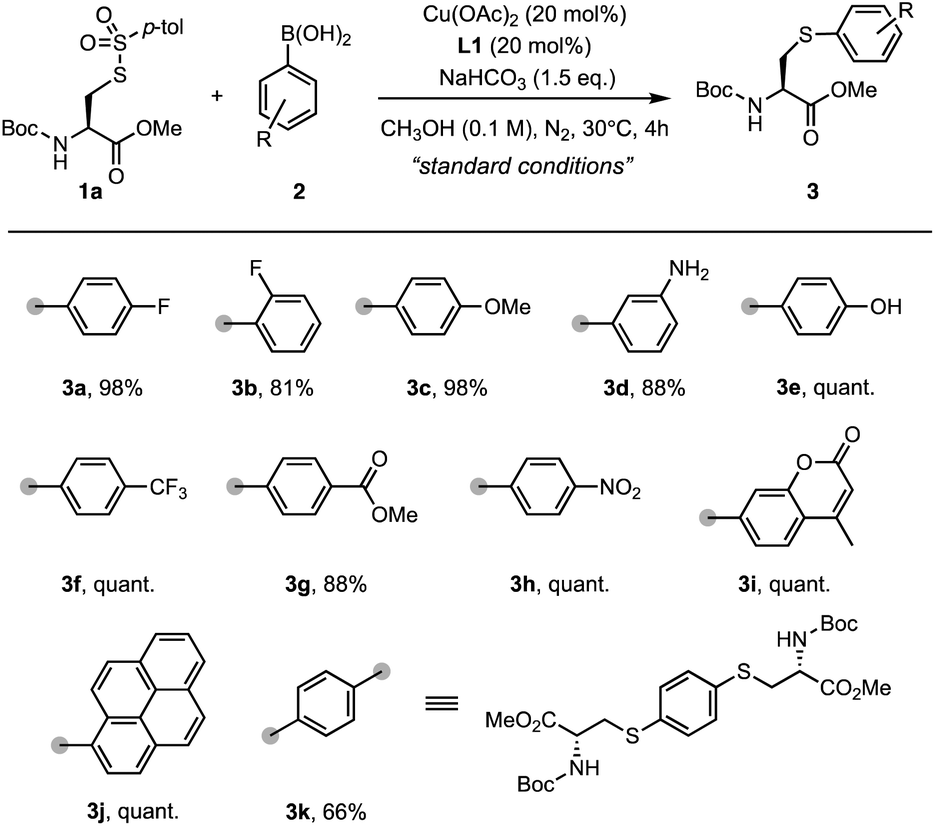 | ||
| Scheme 3 Substrate scope of arylboronic acids. Reaction conditions: 1a (0.1 mmol, 1.0 eq.), arylboronic acid 2 (2.0 eq.), NaHCO3 (1.5 eq.), Cu(OAc)2 (20 mol%), and L1 (20 mol%) in MeOH under a N2 atmosphere at 30 °C for 4 h. Isolated yield. | ||
Encouraged by the wide substrate scope of arylation reagents, we further explored the generality of the Cu-catalyzed arylation at the peptide level. As shown in Scheme 4, the arylation proceeds smoothly to afford cysteine-arylated dipeptides and tripeptides in high yields. The reaction shows good chemo-selectivity for cysteine-related side chains over other nucleophilic functionalities, including the hydroxyl group in tyrosine (4c) and threonine (5f), and the indole group in tryptophan (4h and 4i).
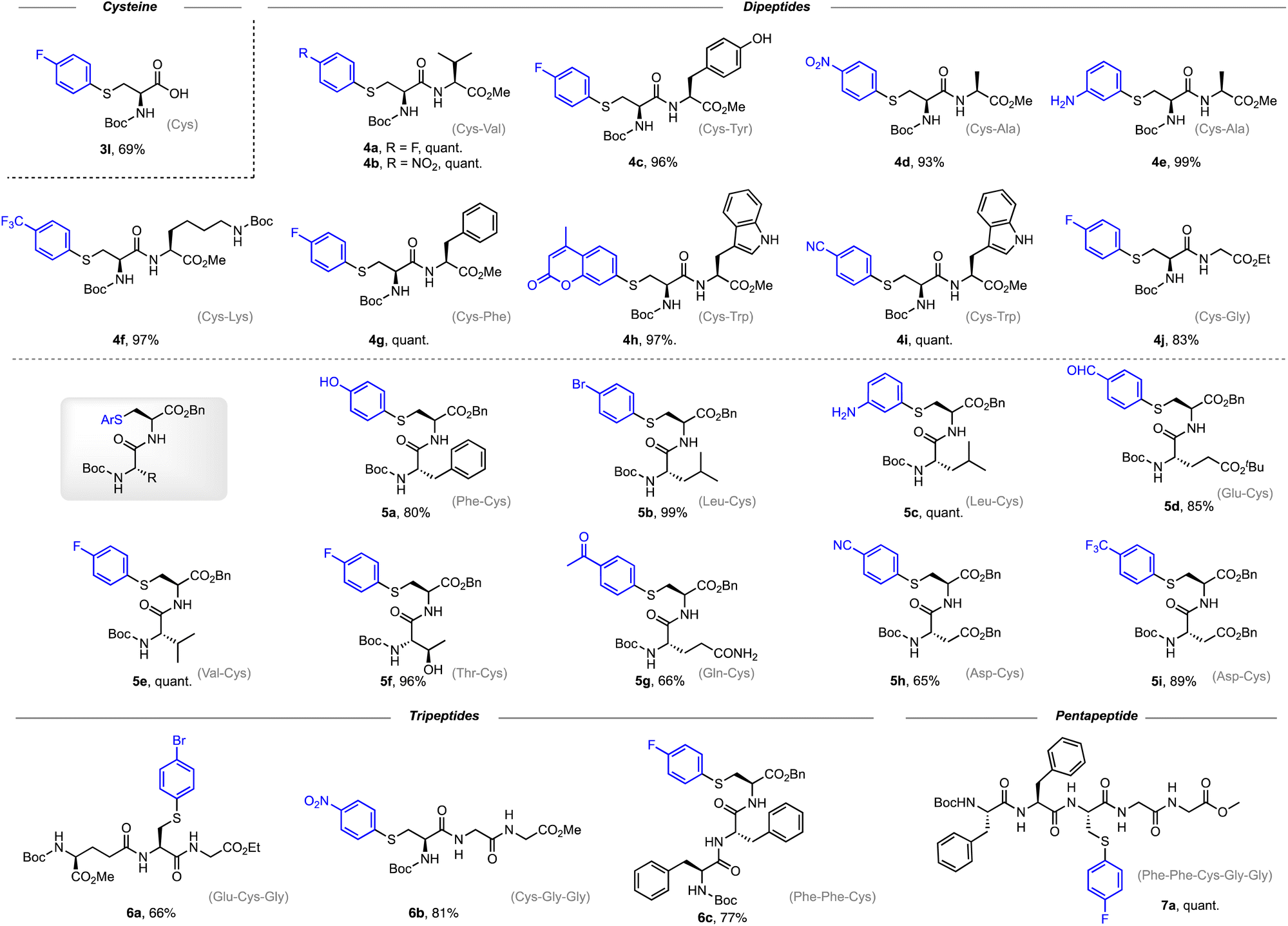 | ||
| Scheme 4 Substrate scope of peptides. Reaction conditions: benzenesulfonothioate derivatives of the peptide substrate (0.1 mmol, 1.0 eq.), arylboronic acid (2.0 eq.), NaHCO3 (1.5 eq.), Cu(OAc)2 (20 mol%), and L1 (20 mol%) in MeOH under a N2 atmosphere at 30 °C for 4 h. Isolated yield. | ||
It is worth noting that dipeptide 5d carrying an aldehyde motif was obtained in 85% yield, and the aldehyde functionality has been proved to be a useful crosslinker for bioconjugation of peptides and proteins.37–39 Arylated dipeptides 4e and 5c carrying an aniline motif were synthesized in 99% and quantitative yield, respectively. Interestingly, we were able to prepare a tripeptide 6a bearing a bromo substituent, providing an extra coupling handle for further conjugation and modification. Other tripeptide products such as 6b and 6c were also obtained in good yields. In addition, late-stage modification of the pentapeptide with arylboronic acid afforded arylated product 7a in an excellent yield. These results demonstrate the utility of the copper-catalyzed S-arylation reaction for the construction of S-arylated peptides.
To investigate the potential applications of the Cu-catalyzed arylation for unprotected peptide substrates, we studied the model arylation of S-tosyl cysteine 1a with arylboronic acid 2a under the standard conditions with an amino acid as the additive (Table 2). It was found that adding 1 equivalent of N-mono-protected amino acids, which bear a free carboxylic group, led to a decrease in the arylation efficiency. Only moderate yields (52–58%) of product 3a were generated in the presence of N-Boc-Pro, N-Boc-Arg and N-Boc-Met (entries 1–3). N-Acetyl amino acid methyl (or ethyl) esters were also studied. It was found that adding Ac-Phe-OMe did not change the yield, and the addition of Bz-Arg-OEt slightly decreased the yield. A moderate yield of arylation was obtained with Ac-His-OMe (entry 5). With both amino and carboxylic groups unprotected, the reaction affords a low yield (16%) of the arylated product (entry 4). The decreasing efficiency of the arylation could be the result of the coordination of the carboxylate or amine group to the copper catalyst under basic conditions, which blocks the metal center from binding with substrates and arylation reagents. These results imply that the current protocol is not ideal for modifications of unprotected native peptides at this stage. Alternatively, arylated and unprotected peptides could be obtained through sequential Cu-catalyzed arylation/global deprotection as the phenyl thiol ethers are stable towards acids and bases (section 7, ESI†).17
|
|
||
|---|---|---|
| Entry | Additive | Yield of 3aa (%) |
| Reaction conditions: 1a (0.1 mmol, 1.0 eq.) 2a (2.0 eq.), NaHCO3 (2.5 eq.), amino acid additive (1.0 eq.), Cu(OAc)2 (20 mol%), and L1 (20 mol%) in MeOH under a N2 atmosphere at 30 °C for 4 h.a Yield determined by crude 19F NMR spectra analysis using trifluoromethyl benzene as an internal standard.b 24 h reaction time. | ||
| 1 | 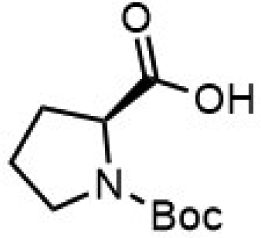 |
57 |
| 2 | 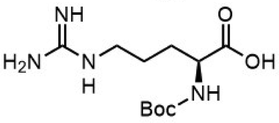 |
58 |
| 3 | 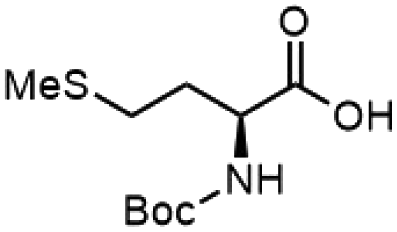 |
52 |
| 4 | 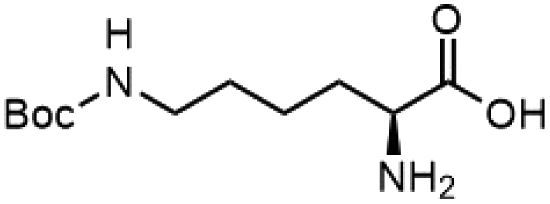 |
16 |
| 5 | 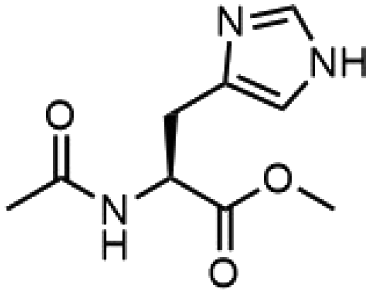 |
48b |
| 6 | 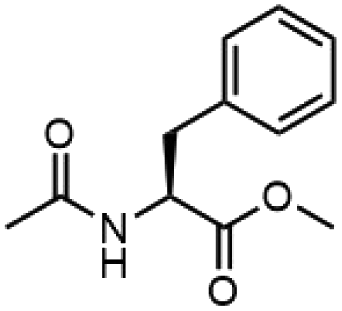 |
Quant. |
| 7 | 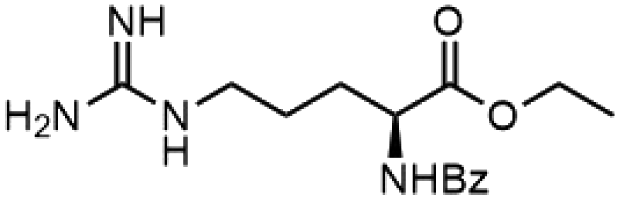 |
73 |

To demonstrate the utility of this method in organic synthesis, we carried out the synthesis of arylated cysteine 3a on a gram scale and in continuous flow (Scheme 5). Starting with the readily available material Boc-Cys-OMe, the gram-scale synthesis of 3a was conducted in batch through sequential oxidation, sulfonation, and arylation under the standard conditions (Scheme 5A). This three-step procedure was performed in a one-pot manner and only single column chromatography was required for the purification, affording 3a in 98% yield. The scalability of this method was further demonstrated by the development of a flow synthesis protocol (Scheme 5B). Water was used as the cosolvent to ensure a good solubility of NaHCO3. In contrast to the 4-hour reaction time in batch, a better mixing efficiency in the microreactor led to full conversion in 17.5 min, delivering the desired product in 91% yield. These results imply that this novel arylation chemistry could be a useful and scalable tool for the synthesis of valuable arylcysteinyl building blocks for organic synthesis and drug discovery.
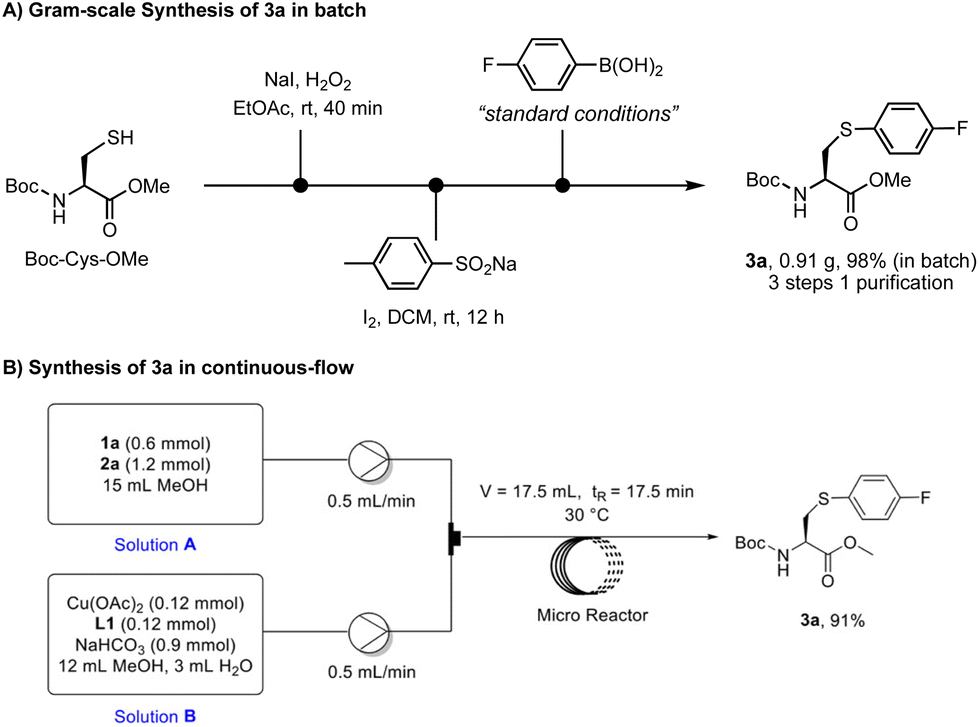 | ||
| Scheme 5 Synthesis of 3a on a gram scale and in continuous flow. | ||
Although the detailed mechanism of this catalytic arylation is still not clear at this stage, we proposed a plausible mechanism based on earlier studies on copper-catalyzed S–C(sp2) bond formation.35,40–44 As shown in Scheme 6, the reaction appears to initiate with disproportionation of the Cu(II) catalyst to afford a Cu(III) and a Cu(I) species.42,43 Subsequently, the oxidative addition of sulfonothionate to the Cu(I) species and the following transmetallation of arylboronic acid form a Cu(III) intermediate B.45,46 Alternatively, an anionic [ArB(OMe)2(OH)]− species could be the active arylation reagent.35 Finally, S–C reductive elimination leads to arylated product 3a, completing the catalytic cycle by regenerating the active Cu(I) catalyst. Experimentally, lower arylation efficiencies were observed on using substrates with lower oxidation states, including disulfide 1c and free cysteine thiol 1d. These results could be a support for the proposed mechanism with the Cu(I)/Cu(III) redox cycle.42
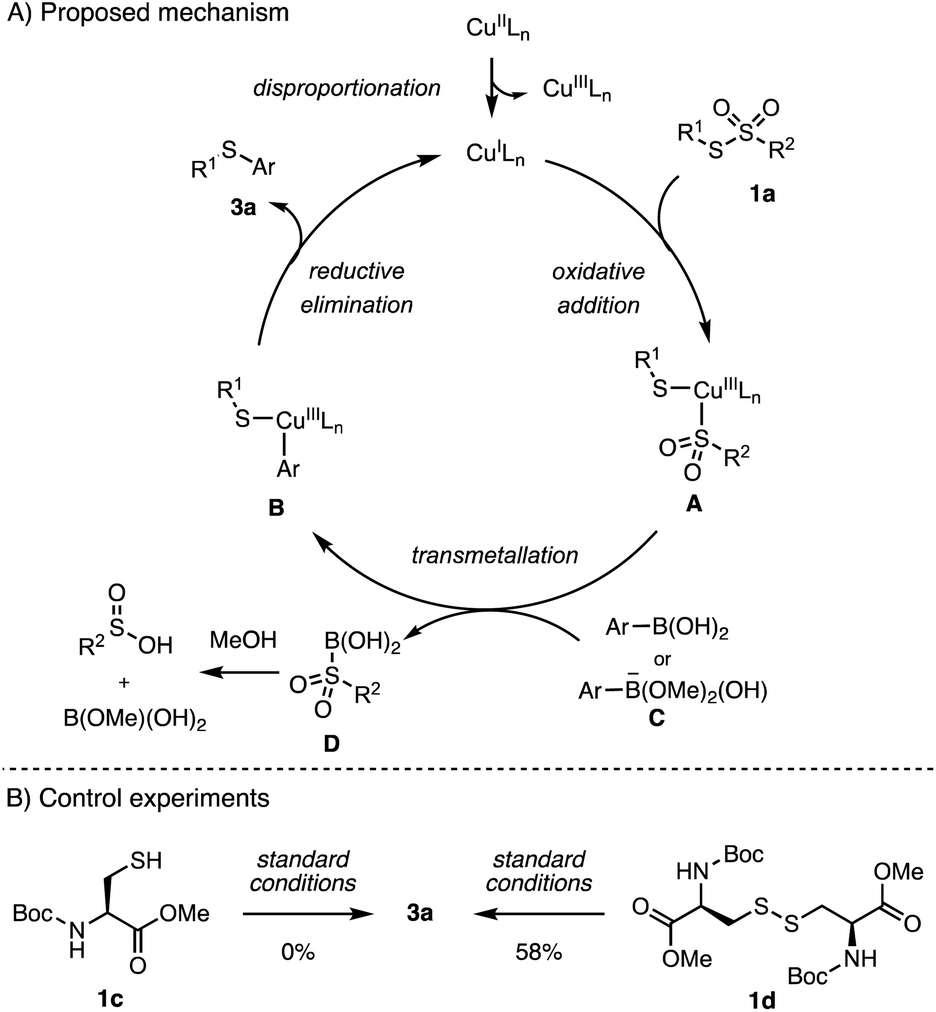 | ||
| Scheme 6 Proposed mechanism and control experiments. | ||
Conclusions
In conclusion, we have developed a catalytic arylation protocol for the synthesis of S-arylated cysteines and peptides. Readily available arylboronic acids were proved to be robust arylation reagents in the presence of an abundant and cheap copper catalyst. The reaction features mild reaction conditions (room temperature, weak base), broad substrate scope, and good functional group tolerance. We have successfully applied this chemistry to the synthesis of a wide range of dipeptides, tripeptides, and pentapeptides carrying an S-arylated cysteinyl scaffold, providing an efficient method for peptide modification. The development of catalytic arylation protocols for unprotected peptides under biocompatible conditions is going on in our labs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 83422104) and Fuzhou University (No. 511041 and 511264) is acknowledged.References
- S. W. Kaldor, V. J. Kalish, J. F. Davies, B. V. Shetty, J. E. Fritz, K. Appelt, J. A. Burgess, K. M. Campanale, N. Y. Chirgadze, D. K. Clawson, B. A. Dressman, S. D. Hatch, D. A. Khalil, M. B. Kosa, P. P. Lubbehusen, M. A. Muesing, A. K. Patick, S. H. Reich, K. S. Su and J. H. Tatlock, Viracept (Nelfinavir Mesylate, AG1343): A Potent, Orally Bioavailable Inhibitor of HIV-1 Protease, J. Med. Chem., 1997, 40, 3979–3985 CrossRef CAS PubMed.
- S. Nakatani, K. Hidaka, E. i. Ami, K. Nakahara, A. Sato, J.-T. Nguyen, Y. Hamada, Y. Hori, N. Ohnishi, A. Nagai, T. Kimura, Y. Hayashi and Y. Kiso, Combination of Non-natural d-Amino Acid Derivatives and Allophenylnorstatine–Dimethylthioproline Scaffold in HIV Protease Inhibitors Have High Efficacy in Mutant HIV, J. Med. Chem., 2008, 51, 2992–3004 CrossRef CAS.
- K. Matinkhoo, A. Pryyma, M. Todorovic, B. O. Patrick and D. M. Perrin, Synthesis of the Death-Cap Mushroom Toxin α-Amanitin, J. Am. Chem. Soc., 2018, 140, 6513–6517 CrossRef CAS.
- C. Zhang, E. V. Vinogradova, A. M. Spokoyny, S. L. Buchwald and B. L. Pentelute, Arylation Chemistry for Bioconjugation, Angew. Chem., Int. Ed., 2019, 58, 4810–4839 CrossRef CAS.
- J. Dadová, S. R. G. Galan and B. G. Davis, Synthesis of Modified Proteins via Functionalization of Dehydroalanine, Curr. Opin. Chem. Biol., 2018, 46, 71–81 CrossRef.
- L. D. Arnold, T. H. Kalantar and J. C. Vederas, Conversion of Serine to Stereochemically Pure .beta.-Substituted .alpha.-Amino Acids via .beta.-Lactones, J. Am. Chem. Soc., 1985, 107, 7105–7109 CrossRef CAS.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Methods for Converting Cysteine to Dehydroalanine on Peptides and Proteins, Chem. Sci., 2011, 2, 1666–1676 RSC.
- E. D. Evans and B. L. Pentelute, Discovery of a 29-Amino-Acid Reactive Abiotic Peptide for Selective Cysteine Arylation, ACS Chem. Biol., 2018, 13, 527–532 CrossRef CAS PubMed.
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, π-Clamp-Mediated Cysteine Conjugation, Nat. Chem., 2016, 8, 120–128 CrossRef CAS.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, A Perfluoroaryl-Cysteine SNAr Chemistry Approach to Unprotected Peptide Stapling, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS.
- B. M. Lipka, V. M. Betti, D. S. Honeycutt, D. L. Zelmanovich, M. Adamczyk, R. Wu, H. S. Blume, C. A. Mendina, J. M. Goldberg and F. Wang, Rapid Electrophilic Cysteine Arylation with Pyridinium Salts, Bioconjugate Chem., 2022, 33, 2189–2196 CrossRef CAS.
- J. Li, J.-J. Deng, Z. Yin, Q.-L. Hu, Y. Ge, Z. Song, Y. Zhang, A. S. C. Chan, H. Li and X.-F. Xiong, Cleavable and Tunable Cysteine-specific Arylation Modification with Aryl Thioethers, Chem. Sci., 2021, 12, 5209–5215 RSC.
- S. A. Byrne, M. J. Bedding, L. Corcilius, D. J. Ford, Y. Zhong, C. Franck, M. Larance, J. P. Mackay and R. J. Payne, Late-stage Modification of Peptides and Proteins at Cysteine with Diaryliodonium Salts, Chem. Sci., 2021, 12, 14159–14166 RSC.
- N. Naveen, S. Sengupta and S. Chandrasekaran, Metal-Free S-Arylation of Cysteine Using Arenediazonium Salts, J. Org. Chem., 2018, 83, 3562–3569 CrossRef CAS.
- C. Bottecchia, M. Rubens, S. B. Gunnoo, V. Hessel, A. Madder and T. Noël, Visible-Light-Mediated Selective Arylation of Cysteine in Batch and Flow, Angew. Chem., Int. Ed., 2017, 56, 12702–12707 CrossRef CAS.
- M. Jbara, Transition Metal Catalyzed Site-selective Cysteine Diversification of Proteins, Pure Appl. Chem., 2021, 93, 169–186 CrossRef CAS.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Organometallic Palladium Reagents for Cysteine Bioconjugation, Nature, 2015, 526, 687–691 CrossRef CAS.
- J. A. R. Tilden, A. T. Lubben, S. B. Reeksting, G. Kociok-Köhn and C. G. Frost, Pd(II)-Mediated C–H Activation for Cysteine Bioconjugation, Chem. – Eur. J., 2022, 28, e202104385 CrossRef CAS.
- A. J. Rojas, C. Zhang, E. V. Vinogradova, N. H. Buchwald, J. Reilly, B. L. Pentelute and S. L. Buchwald, Divergent Unprotected Peptide Macrocyclisation by Palladium-Mediated Cysteine Arylation, Chem. Sci., 2017, 8, 4257–4263 RSC.
- A. J. Rojas, B. L. Pentelute and S. L. Buchwald, Water-Soluble Palladium Reagents for Cysteine S-Arylation under Ambient Aqueous Conditions, Org. Lett., 2017, 19, 4263–4266 CrossRef CAS.
- J. Willwacher, R. Raj, S. Mohammed and B. G. Davis, Selective Metal-Site-Guided Arylation of Proteins, J. Am. Chem. Soc., 2016, 138, 8678–8681 CrossRef CAS PubMed.
- G. E. Mudd, S. J. Stanway, D. R. Witty, A. Thomas, S. Baldo, A. D. Bond, P. Beswick and A. Highton, Gold-Mediated Multiple Cysteine Arylation for the Construction of Highly Constrained Bicycle Peptides, Bioconjugate Chem., 2022, 33, 1441–1445 CrossRef CAS PubMed.
- S.-L. Zhang and J.-J. Dong, Mechanism and Chemoselectivity Origins of Bioconjugation of Cysteine with Au(iii)-aryl Reagents, Org. Biomol. Chem., 2019, 17, 1245–1253 RSC.
- M. S. Messina, J. M. Stauber, M. A. Waddington, A. L. Rheingold, H. D. Maynard and A. M. Spokoyny, Organometallic Gold(III) Reagents for Cysteine Arylation, J. Am. Chem. Soc., 2018, 140, 7065–7069 CrossRef CAS PubMed.
- H. R. Montgomery, M. S. Messina, E. A. Doud, A. M. Spokoyny and H. D. Maynard, Organometallic S-arylation Reagents for Rapid PEGylation of Biomolecules, Bioconjugate Chem., 2022, 33, 1536–1542 CrossRef CAS.
- J. M. Stauber, A. L. Rheingold and A. M. Spokoyny, Gold(III) Aryl Complexes as Reagents for Constructing Hybrid Peptide-Based Assemblies via Cysteine S-Arylation, Inorg. Chem., 2021, 60, 5054–5062 CrossRef CAS PubMed.
- S. Gukathasan, S. Parkin, E. P. Black and S. G. Awuah, Tuning Cyclometalated Gold(III) for Cysteine Arylation and Ligand-Directed Bioconjugation, Inorg. Chem., 2021, 60, 14582–14593 CrossRef CAS.
- M. N. Wenzel, R. Bonsignore, S. R. Thomas, D. Bourissou, G. Barone and A. Casini, Cyclometalated AuIII Complexes for Cysteine Arylation in Zinc Finger Protein Domains: towards Controlled Reductive Elimination, Chem. – Eur. J., 2019, 25, 7628–7634 CrossRef CAS.
- K. Hanaya, J. Ohata, M. K. Miller, A. E. Mangubat-Medina, M. J. Swierczynski, D. C. Yang, R. M. Rosenthal, B. V. Popp and Z. T. Ball, Rapid Nickel(II)-promoted Cysteine S-Arylation with Arylboronic Acids, Chem. Commun., 2019, 55, 2841–2844 RSC.
- R. A. A. Al-Shuaeeb, S. Kolodych, O. Koniev, S. Delacroix, S. Erb, S. Nicolaÿ, J.-C. Cintrat, J.-D. Brion, S. Cianférani, M. Alami, A. Wagner and S. Messaoudi, Palladium-Catalyzed Chemoselective and Biocompatible Functionalization of Cysteine-Containing Molecules at Room Temperature, Chem. – Eur. J., 2016, 22, 11365–11370 CrossRef CAS PubMed.
- J. X. Qiao and P. Y. S. Lam, Copper-Promoted Carbon-Heteroatom Bond Cross-Coupling with Boronic Acids and Derivatives, Synthesis, 2011, 829–856 CrossRef CAS.
- J.-Q. Chen, J.-H. Li and Z.-B. Dong, A Review on the Latest Progress of Chan-Lam Coupling Reaction, Adv. Synth. Catal., 2020, 362, 3311–3331 CrossRef CAS.
- H.-J. Xu, Y.-Q. Zhao, T. Feng and Y.-S. Feng, Chan–Lam-Type S-Arylation of Thiols with Boronic Acids at Room Temperature, J. Org. Chem., 2012, 77, 2878–2884 CrossRef CAS.
- P. S. Herradura, K. A. Pendola and R. K. Guy, Copper-Mediated Cross-Coupling of Aryl Boronic Acids and Alkyl Thiols, Org. Lett., 2000, 2, 2019–2022 CrossRef CAS.
- Q. Zhao, L. Lu and Q. Shen, Direct Monofluoromethylthiolation with S-(Fluoromethyl) Benzenesulfonothioate, Angew. Chem., Int. Ed., 2017, 56, 11575–11578 CrossRef CAS PubMed.
- Z. Sun, W. Yan, L. Xie, W. Liu, C. Xu and F.-E. Chen, A Robust Copper-Catalyzed Cross-Coupling of Glycosyl Thiosulfonate and Boronic Acids Enables the Construction of Thioglycosides, Org. Lett., 2023, 25, 5714–5718 CrossRef CAS PubMed.
- T. Sahu, M. Kumar, T. K. Sajeev, M. Joshi, R. K. Mishra and V. Rai, Residue-specific N-terminal Glycine to Aldehyde Transformation Renders Analytically Pure Single-site Labeled Proteins, Chem. Commun., 2022, 58, 12451–12454 RSC.
- Y. Song, F. Xiong, J. Peng, Y. M. E. Fung, Y. Huang and X. Li, Introducing Aldehyde Functionality to Proteins Using Ligand-directed Affinity Labeling, Chem. Commun., 2020, 56, 6134–6137 RSC.
- J. I. MacDonald, H. K. Munch, T. Moore and M. B. Francis, One-step Site-specific Modification of Native Proteins with 2-Pyridinecarboxyaldehydes, Nat. Chem. Biol., 2015, 11, 326–331 CrossRef CAS PubMed.
- F.-J. Chen, G. Liao, X. Li, J. Wu and B.-F. Shi, Cu(II)-Mediated C-S/N-S Bond Formation via C-H Activation: Access to Benzoisothiazolones Using Elemental Sulfur, Org. Lett., 2014, 16, 5644–5647 CrossRef CAS PubMed.
- Y. Guo, Z.-J. Quan, Y.-X. Da, Z. Zhang and X.-C. Wang, (2-Chlorobenzoyloxy)copper(I) Catalyzed C–S Cross-coupling of Di(hetero)aryl Disulfides with Aryl Boronic Acids under Base-free Conditions, RSC Adv., 2015, 5, 45479–45483 RSC.
- M. Font, T. Parella, M. Costas and X. Ribas, Catalytic C–S, C–Se, and C–P Cross-Coupling Reactions Mediated by a CuI/CuIII Redox Cycle, Organometallics, 2012, 31, 7976–7982 CrossRef CAS.
- N. Taniguchi, Convenient Synthesis of Unsymmetrical Organochalcogenides Using Organoboronic Acids with Dichalcogenides via Cleavage of the S–S, Se–Se, or Te–Te Bond by a Copper Catalyst, J. Org. Chem., 2007, 72, 1241–1245 CrossRef CAS.
- D. T. Cohen, C. Zhang, B. L. Pentelute and S. L. Buchwald, An Umpolung Approach for the Chemoselective Arylation of Selenocysteine in Unprotected Peptides, J. Am. Chem. Soc., 2015, 137, 9784–9787 CrossRef CAS PubMed.
- X.-Q. Chu, D. Ge, Y.-Y. Cui, Z.-L. Shen and C.-J. Li, Desulfonylation via Radical Process: Recent Developments in Organic Synthesis, Chem. Rev., 2021, 121, 12548–12680 CrossRef CAS.
- X. Xiao, Y.-Q. Huang, H.-Y. Tian, J. Bai, F. Cheng, X. Wang, M.-L. Ke and F.-E. Chen, Robust, Scalable Construction of an Electrophilic Deuterated Methylthiolating Reagent: Facile Access to SCD3-Containing Scaffolds, Chem. Commun., 2022, 58, 3015–3018 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qo01534c |
| This journal is © the Partner Organisations 2024 |