
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity of oxidants towards phenyl and benzyl substituted 5-selanylpentanoic acids: radiolytic and theoretical insights†
Beena G.
Singh
 *ab,
Kavanal P.
Prasanthkumar
c,
Francesca
Mangiavacchi
de,
Francesca
Marini
d and
Claudio
Santi
*d
*ab,
Kavanal P.
Prasanthkumar
c,
Francesca
Mangiavacchi
de,
Francesca
Marini
d and
Claudio
Santi
*d
aRadiation & Photochemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai – 4000851, India. E-mail: beenam@barc.gov.in
bHomi Bhabha National Institute, Anushaktinagar, Mumbai – 400 094, India
cPost Graduate and Research Department of Chemistry, Maharaja's College, Ernakulam, Kerala 682011, India. E-mail: prasanthkumarkp@gmail.com
dGroup of Catalysis, Synthesis and Organic Green Chemistry, Department of Pharmaceutical Sciences, University of Perugia, Via del Liceo 1, 06123 Perugia, Italy. E-mail: Francesca.marini@unipg.it; claudio.santi@unipg.it
eDepartment of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 13, Sesto Fiorentino, I-50019 Florence, Italy. E-mail: francesca.mangiavacchi@unifi.it
First published on 13th November 2023
Abstract
Two selanyl compounds, 5-(phenylselanyl)pentanoic acid (1) and 5-(benzylselanyl)pentanoic acid (2), were investigated for their reactivity towards one-electron oxidants, particularly the radiolytically generated hydroxyl radical (˙OH). The reactions of compounds 1 and 2 with ˙OH led to the formation of transient absorptions, characterized by peak wavelengths (λmax) at 630 nm and 560 nm, respectively. Concentration-dependent absorption studies revealed that the transient from 1 with λmax of 630 nm corresponds to a dimer species featuring a two-centre–three-electron (2c–3e) bond. Conversely, the species exhibiting λmax of 560 nm for 2 was identified as a monomer radical cation. To validate the identities of these transients, compounds 1 and 2 underwent reactions with specific one-electron oxidants, namely the dibromide radical (Br2˙−) and carbonate radical anion (CO3˙−). The differences in the reactivities of the transients derived from compounds 1 and 2 were distinguished by measuring their ability to oxidize glutathione, a biologically important thiol. Interestingly, the transient from compound 2 exhibited a relatively lower oxidizing ability than the transient from compound 1. Compound 2 also demonstrated higher scavenging rate constants in reactions with Br2˙−, CO3˙−, and peroxyl radicals (derived from thermal degradation of 2,2′-azobis(2-amidinopropane) hydrochloride, AAPH). Computational calculations were carried out employing the M05-2X/6-311+G(d,p)/SMD method to offer additional proof in favor of dimer radical species during the interactions of compound 1 with ˙OH. The calculations revealed a competitive formation of both σ- and π-type dimer radicals from compound 1. However, the experimental spectrum primarily corresponded to the σ-type dimer, as indicated by the predicted λmax (630 nm) using the TDDFT method. This study shows that the benzyl-containing selanyl system is a better free radical scavenger than the phenyl-containing selanyl system, highlighting its potential as a potent free radical scavenger.
1. Introduction
Selenium, as one of the chalcogens, exhibits unique redox properties and plays a significant role in various biological systems; moreover, it finds distinctive applications in numerous fields of materials sciences.1–6 Like its congeners, its ability to exhibit variable oxidation states allows elemental selenium and its compounds to be used as versatile catalysts in synthetic organic chemistry.2,6 This property has also been utilized in nature in various selenium-containing enzymes. For instance, enzymes such as glutathione peroxidase, thioredoxin reductase, and iodothyronine deiodinase utilize the proteogenic amino acid selenocysteine to perform their underlying biological functions involving redox reactions.1,4,5 The radical scavenging ability of selenium-containing enzymes has greatly inspired the development of several organoselenium antioxidants.7,8It is considered that the physicochemical properties of selenium are quite similar to sulfur; nevertheless, their radical-mediated redox chemistry differs significantly.1,9–12 Radiation chemical studies on certain water-soluble aliphatic selenium compounds from one of the authors' group have shown that selenium-centered radicals exhibit extra stability and lower reactivity than analogous sulfur-centered radicals.12–16 Previous antioxidant studies on selenium-bearing carboxylic acids have shown that they have higher activity, but the underlying reaction mechanism and product distribution vary considerably depending on the chain length.13,14 Furthermore, it is worth noting that the carboxylic group, found in various naturally occurring antioxidants such as lipoic acid, plays a significant role in their activity.17–19 Recent investigations have revealed that compounds derived from pentanoic acid demonstrate heightened neuroprotective capabilities, attributed to their ability to mitigate oxidative stress.20 While earlier studies have extensively explored the one-electron oxidations of organosulfur compounds, both aliphatic and aromatic, there is a scarcity of reports on organoselenium compounds.21 It is intriguing to mention that studies conducted on various biological models have shown that aromatic selenides exhibit relatively lower toxicity compared to their aliphatic counterparts.22 Interestingly, there is a lack of a detailed discussion regarding the one-electron oxidations of aromatic organoselenium compounds, particularly in an aqueous environment.23
The hydroxyl radical (˙OH) is an extremely reactive oxidant that plays a significant role in biological and environmental systems.24 Consequently, the development of effective and compatible scavengers or antioxidants for ˙OH and similar oxidants is crucial in the fields of biology and the environment. In this in vitro study, we have examined the radical scavenging abilities, specifically towards ˙OH, of two organoselenium compounds containing phenyl and benzyl groups attached to 5-selanylpentanoic acid. Notably, the use of Density Functional Theory (DFT) methods has been previously reported for studying the energetics and mechanisms of ˙OH reactions with various compound types.25–27 In this study, DFT calculations have been employed to support and validate the formation of a key transient species proposed experimentally in the reactions between the phenyl-containing selanyl system and ˙OH. The findings of this study have importance for the design of selenium-based antioxidants with potential applications in therapeutics and industry.
2. Material and methods
Synthesis of selanyl compounds
5-(Phenylselanyl)pentanoic acid (1) and 5-(benzylselanyl)pentanoic acid (2) (Scheme 1) were prepared according to a procedure reported in the literature.28,29 Diphenyl diselenide or dibenzyl diselenide is reduced by treatment with NaBH4 in EtOH to give a selenate which reacts with the ethyl ester of the 5-chloropentenoic acid. The resulting selenide is subsequently hydrolyzed by treatment with sodium hydroxide in refluxing ethanol to obtain the corresponding carboxylic acids 1 and 2. | ||
| Scheme 1 Structure of selenium compounds. | ||
Pulse radiolysis studies
Radiolytic studies were carried out using a linear accelerator delivering 7 MeV electron pulses (with a duration of 500 ns). Transient species were monitored using the optical absorption detection method. To determine the dose per pulse, an aerated KSCN solution (0.01 M) was used to monitor the formation of (SCN)2˙− at 475 nm, where the G × ε at 475 nm is 2.59 × 10−4 m2 J−1. The radiation chemical yield (G) is the amount of product formed upon absorption of 1 joule of energy (mol J−1), while ε, is the molar absorption coefficient (m2 mol−1). The dose was maintained at a range of 10–12 Gy per pulse, where 1 Gy is equal to 1 J kg−1. Upon electron pulse irradiation of N2 saturated aqueous solutions, radiolysis of water takes place, leading to the production of radical, neutral, and charged species, as described by eqn (1). Notably, among the radical species generated, the G of hydroxyl radical (˙OH) and hydrated electron (e−aq) are significantly higher compared to that of hydrogen atom (˙H).| H2O → ˙H, ˙OH, e−aq, H2, H2O2, H3O+ | (1) |
The hydroxyl radical (˙OH) exhibits oxidizing properties. To ensure reactions of ˙OH solely with substrate molecules, it is preferable to remove the e−aq generated during radiolysis. In N2O-saturated solutions, e−aq can be quantitatively converted to ˙OH, as depicted in eqn (2). Under these reaction conditions, the G of ˙OH is approximately 0.60 μmol J−1.30
| N2O + e−aq → N2 + ˙OH + OH− | (2) |
In addition to the ˙OH, oxidations of compounds 1 and 2 have been conducted using radiolytically generated dibromide radical anion (Br2˙−) and carbonate radical anion (CO3˙−). These specific one-electron oxidants are formed through the reactions of ˙OH with 0.1 M Br− and 10 mM CO32−/HCO3−, as illustrated in eqn (3), where X and Y represent Br and CO3, respectively.
| ˙OH + X− or Y2− → ½X2˙− or Y˙− + OH− | (3) |
The bimolecular rate constants for the reactions involving Br2˙− and CO3˙− with the selenium compounds were determined by observing the decay of Br2˙− and CO3˙− at 365 nm and 600 nm respectively while varying the concentrations of selenium compounds.31
The yield of oxidizing transients (G(oxidizing)) derived from 1 and 2 was determined by following the oxidation of the reducing agent, 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS2−) to produce ABTS˙− radicals, which absorb at 415 nm. The G(oxidizing) using a molar absorption coefficient (ε) of 3.4 × 103 m2 mol−1 for ABTS˙− at 415 nm, as described previously.14,32 To study the kinetics of the reaction between oxidizing transients and ABTS2−, the N2O-saturated aqueous solutions at pH 7 containing 1 mM of compounds 1 or 2 and 10–120 μM ABTS2− was pulse irradiated and determined the rate constant by tracking the formation of ABTS˙−. The yield of the reducing transient G(reducing) was determined by studying the reduction of the oxidizing agent methyl viologen (MV2+). When MV2+ reacts with reducing transients, it produces the MV˙+, which absorb at 605 nm (ε = 1.37 × 103 m2 mol−1).27
We evaluated the in vitro antioxidant abilities of compounds 1 and 2 by measuring their capacity to protect the organic dye fluorescein from degradation. The degradation of fluorescein was induced by peroxyl radicals (ROO˙) derived from the decomposition of AAPH at 37 °C.33 Reagents were prepared in 75 mM phosphate buffer (pH 7.4). In each microplate well, 20 μL of selenium compound 1 or 2 (ranging from 0.3 mM to 0.9 mM) and 120 μL of fluorescein (resulting in a final concentration of 120 nM) were added, and the total reaction volume in each well was adjusted to 200 μL with the addition of buffer solution. The mixture was incubated for 15 minutes at 37 °C, and then 50 μL of AAPH solution (to give a final concentration of 12 μM) was rapidly added. The microplate was immediately placed in a plate reader equipped with a constant-temperature cell holder (Hybrid Synergy). The fluorescence intensity decay of fluorescein was automatically recorded at 1 minute intervals over a period of 90 minutes using the multi-well microplate reader. The fluorescent probe (fluorescein) was excited at 485 nm, and the emission wavelength was set at 512 nm. The microplate was automatically shaken prior to each measurement. Control experiments were conducted in a similar manner, but without the addition of selenium compounds. All analyses were performed in triplicate.
Cyclic voltammetry
Cyclic voltammograms of 1 and 2 were recorded with a potentiostat–galvanostat PGSTAT20 system (Autolab, EcoChemie, The Netherlands) using a three-electrode setup, which included a glassy carbon working electrode, a platinum wire counter electrode, and an Ag/AgCl (3 M KCl) reference electrode. Compound 1 or 2 at pH 7 was dissolved in 0.1 M phosphate buffer and purged with N2 to remove dissolved oxygen. The supporting electrolyte used was also a 0.1 M phosphate buffer.16Computational details
All geometry optimizations and subsequent frequency calculations were performed using the M05-2X method34 in combination with the 6-311+G(d,p) basis set, as implemented in the Gaussian 16 suite of programs.35 The simulations were augmented with the influence of the solvent (water) environment using the SMD method.36 The TDDFT method was employed to estimate the electronic spectra of the transients.373. Results and discussion
Radiation chemical studies
Compound 1 is characterized by a phenyl substituent on the Se atom, while compound 2 has a benzyl moiety connected to the Se atom. Correspondingly, it can be inferred that the phenyl/benzyl substituent plays a significant role in the formation and stabilization of a radical centered on the Se atom, which would be generated upon one-electron oxidations of compounds 1 and 2 by ˙OH, Br2˙−, and CO3˙− radicals. It is presumed that the anionic forms of 1 and 2 are involved in reactions with oxidizing radicals based on the literature-reported pKa value of ∼5 to 5-phenylvaleric acid.38The transient absorption spectra obtained from the reaction of ˙OH with compound 1 (500 μM) showed two distinct bands in the UV region, centered around 320 nm and 380 nm, respectively, and a broad absorption extending beyond 700 nm, with a maximum at 630 nm (Fig. 1). The reactions of ˙OH with organic compounds are generally non-specific.31 The ˙OH has the ability to undergo electrophilic addition reactions with C–C double bonds as well as polarizable heteroatoms such as S or Se.21,31 It can also undergo H-atom abstraction (RH + ˙OH → R˙ + H2O) or simple one-electron oxidation (RH + ˙OH → RH˙+ + OH−).31 In the case of compound 1, it appears that ˙OH can undergo electrophilic addition either to the phenyl ring or to the Se atom. H-atom abstractions can occur from the alkyl moiety, while one-electron oxidation can produce a Se-centered radical cation of 1 (denoted as 1˙), which is an overall charge-neutral species. Additionally, species 1˙ can also be obtained via the dehydroxylation of the initially formed Se–OH˙ adduct. In several organoselenium compounds, it has been reported that the radical induced one-electron oxidations can lead to the formation of relatively stable radical cations.11–14 These radical cations can achieve further stabilization through intramolecular or intermolecular two-centre–three-electron (2c–3e) bond formations.11–14,39 The intramolecular 2c–3e bond can be conceptualized as the interaction between the oxidized selenium center and the lone pair electron on a suitably positioned heteroatom. The intermolecular dimer radical formation can be visualized as a combination of the initially formed radical cation with a neutral parent compound characterized by a >Se∴Se< bond.11–16,39 In both instances, the formation of the 2c–3e bond is attributed to the high intrinsic covalent radius of selenium.
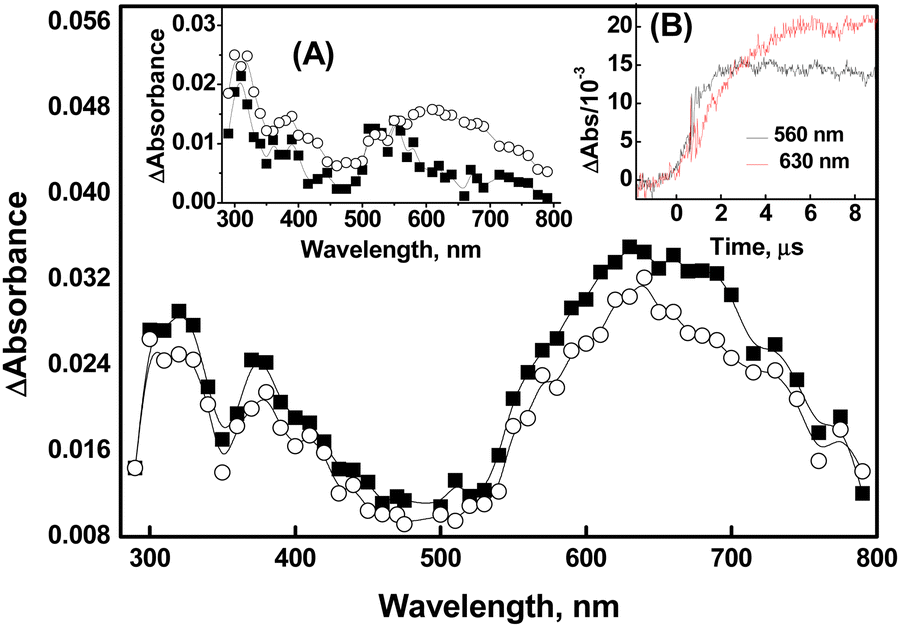 | ||
| Fig. 1 Transient absorption spectra generated by pulse radiolysis in an N2O-saturated aqueous solution containing 500 μM of compound 1 (■ = 2 μs and ○ = 10 μs after the pulse) at pH 7. Insets: (A) shows transient spectra from 50 μM of 1 at 2 μs (■) and 10 μs (○) after the pulse, generated under similar experimental conditions. (B) Shows the absorbance-time plot obtained from the reaction of ˙OH with 1 (50 μM) at 560 nm (black line) and 630 nm (red line). | ||
It is important to consider the changes associated with two distinct regions in the spectra in the ˙OH reactions of compound 1. The transient absorption spectrum obtained from the reaction of ˙OH with 50 μM of compound 1 exhibited λmax at 320 nm, 380 nm, and 560 nm at 2 μs after the pulse irradiation. However, the band with λmax at 560 nm completely disappeared, and a new band with λmax at 630 nm appeared at 10 μs after the irradiation (Inset A of Fig. 1). Therefore, it can be inferred that the emergence of the λmax 630 nm band is a consequence of the simultaneous transformation of a transient with λmax at 560 nm. This inference is supported by the absorbance vs. time plot acquired at 560 nm and 630 nm as shown in Inset B of Fig. 1. The formation of dimer radicals in the reactions of ˙OH with compound 1 was explored by observing changes in transient absorption spectra and their decay kinetics at varying initial concentrations of 1 (ranging from 50 μM to 500 μM). As shown in Fig. 1, the transient absorptions at 320 nm, 380 nm, and 630 nm increased with increasing concentrations of compound 1. However, the 630 nm peak exhibited a substantial increase in intensity relative to the other peaks, suggesting the formation of dimer radicals. The 10 μs spectra showed that the peak intensity at 630 nm increased 2.3-fold upon increasing the compound 1 concentration from 50 μM to 500 μM, further supporting this conclusion. This finding is consistent with previous reports of characteristic concentration-dependent transient absorptions in certain organoselenium compounds.11–14 Therefore, the species absorbing at 560 nm can be ascribed to the radical cation formed either by direct one-electron oxidation of compound 1 by ˙OH or by rapid dehydroxylation of the initial Se–˙OH adduct.
Compelling evidence for the generation of a radical cation (peaking at 560 nm) through direct one-electron oxidation was obtained from the reaction of compound 1 (50 μM) with ˙OH in a phosphate buffer. Previous studies have reported that the phosphate ion (H2PO4−) serves as a proton donor, enabling the dehydroxylation of ˙OH adducts of organic molecules.40 In Fig. 2, it is evident that in the absence of phosphate buffer, transient absorptions with peak wavelengths at 320 nm, 380 nm, and 560 nm were observed in the reactions of compound 1 (50 μM) with ˙OH at 2 μs after the pulse (spectrum a of Fig. 2). Additionally, broad absorption in the range of 600–760 nm appeared at a later time point (10 μs) (spectrum b in Fig. 2). However, when the reactions of compound 1 with ˙OH were conducted in the presence of 5 mM phosphate buffer, the peak at 560 nm was not observed. Instead, the formation of absorption band at 630 nm, corresponding to the dimer species, was directly noticed (spectrum c of Fig. 2). This suggests that the presence of phosphate enhances the mechanism that generates radical cation formation and its subsequent combination with the parent molecule to produce dimer species (spectrum d of Fig. 2). Therefore, it can be inferred that the existence of the radical cation in the medium is strongly influenced by the concentration of compound 1 and the kinetics of its combination with compound 1.
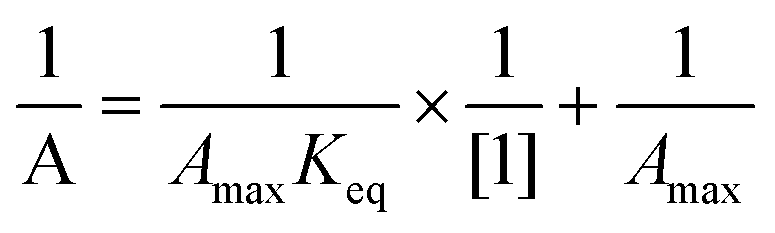 | (4) |
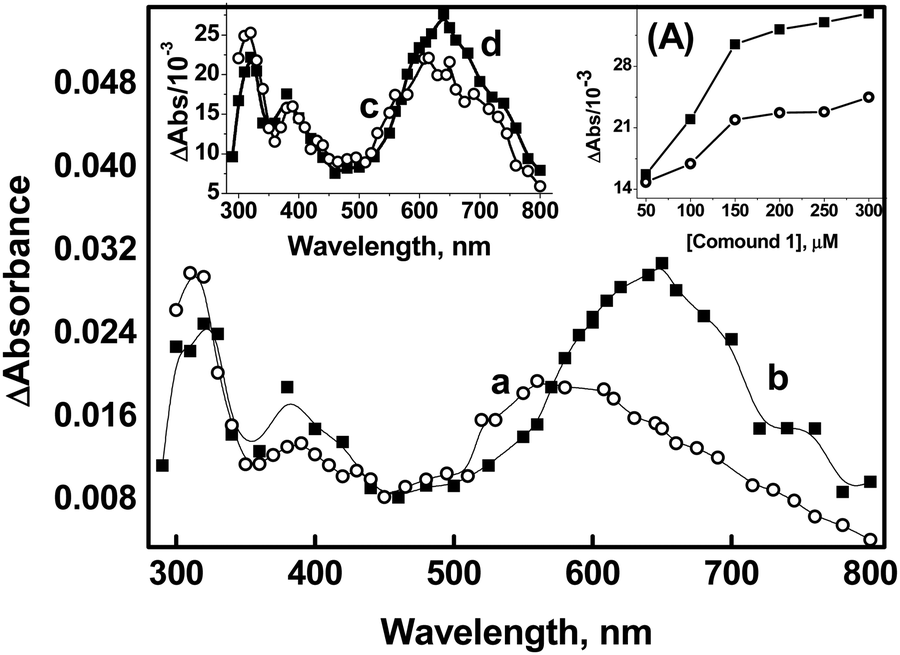 | ||
| Fig. 2 Transient absorption spectra generated from pulse radiolysis of N2O-saturated solution containing compound 1 (50 μM) in water (a = 2 μs, b = 10 μs) and in the presence of 5 mM phosphate buffer at pH 7 (c = 2 μs, d = 10 μs). Inset (A) shows the variation of absorbance at 630 nm (■) and 380 nm (○) as a function of the concentration of 1. | ||
The transient absorbing at 630 nm decayed by first order kinetics (1.0 × 104 s−1). The stability of the dimer radical can be determined by analyzing the equilibrium constant (Keq), which can be estimated using eqn (4). In this equation, A represents the absorbance at 630 nm at concentrations ranging from 50 to 300 μM; Amax corresponds to the maximum absorbance when all primary species resulting from the ˙OH reaction are converted to the dimer radical. Plotting a double reciprocal graph yields a straight line, as shown in Fig. S1 (ESI†), which is consistent with eqn (4). From the intercept and slope of the line, the estimated value of Keq is 9.9 × 103 M−1. This value is reasonably consistent with the reported Keq values for some aromatic selenium compounds, but higher in magnitude compared to several aromatic sulfur systems.12,23 The disparity in Keq values between aromatic selenium and sulfur compounds can be attributed to the stronger stabilization of the dimer species due to the larger covalent radius of selenium compared to sulfur. The larger covalent radius reduces steric hindrance and enhances the strength of the >Se∴Se< bond formed between the constituent species in the dimer radical.
Additional confirmation of the presence of the radical cation, characterized by its peak wavelength at 560 nm, has been acquired through the reactions of Br2˙− with compound 1. The kinetics of the reaction between Br2˙− and 1 were investigated by monitoring the decay of Br2˙− (which exhibits strong absorption at 365 nm) at various initial concentrations of compound 1. The bimolecular rate constant for the reaction was determined to be 3.5 ± 0.2 × 108 M−1 s−1. Transient absorption with λmax at 560 nm was observed in the reaction of Br2˙− with 1. Notably, the spectral features remain unchanged even at higher time scales, which is in contrast to the ˙OH reactions of 1 described earlier. This provides further support for the formation of the radical cation in the reaction of Br2˙− with compound 1 (Fig. 3A). Herein, we did not observe a concentration-dependent increase in transient absorption with a λmax at 630 nm even when tested up to the permissible solubility of compound 1, which clearly indicates the absence of dimer formation. This suggests that the radical cation formed in the reaction (1 + Br2˙−→ radical cation + 2Br−) may be stabilized through hemi-bond formation (>Se∴Br−) between the Br− and Se center. This assumption is supported by the observation that the radical adducts absorbing at 560 nm decayed by following first-order kinetics (k = 2.2 × 104 s−1).
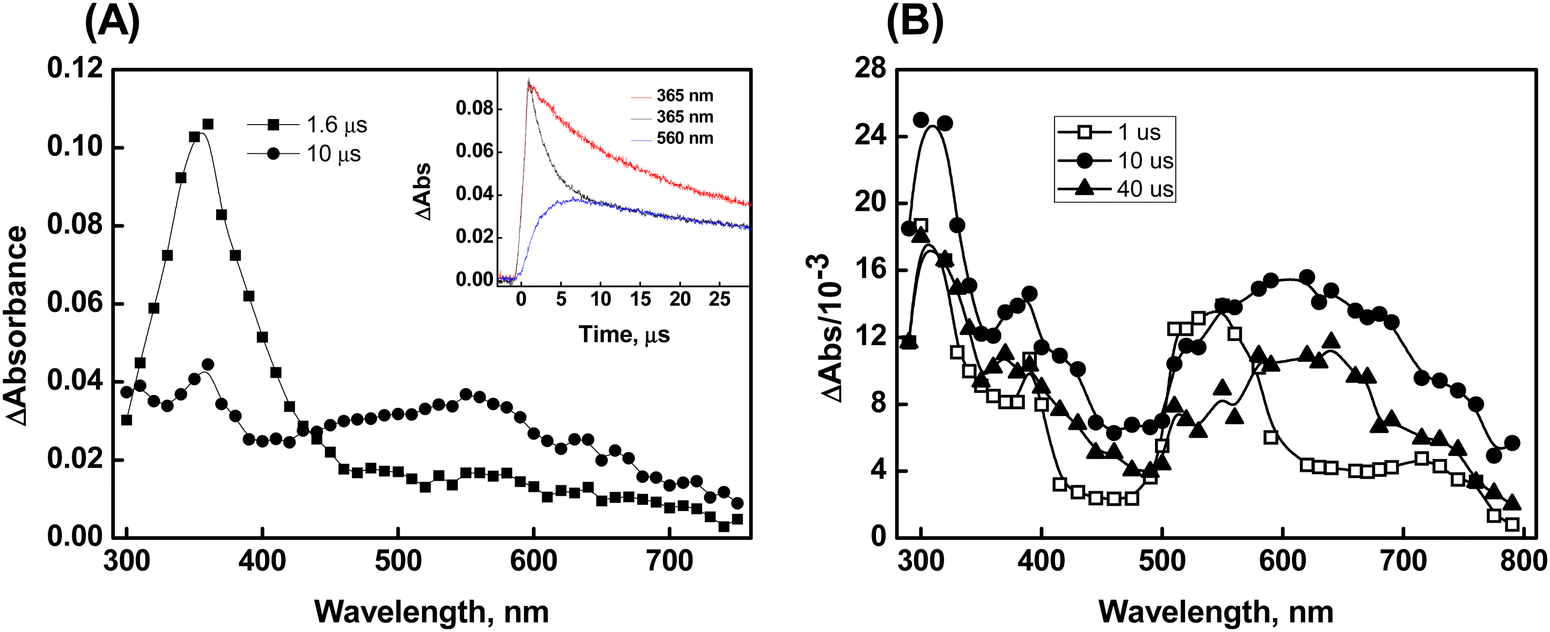 | ||
| Fig. 3 Plot (A) corresponds to the transient spectrum of compound 1 generated on reaction with Br2˙− radical at pH 7. The inset shows the time absorption plot at 365 nm obtained on radiolyzing aqueous solution containing 0.1 M KBr in the absence (red line) and presence of compound 1 (black line). The blue plot corresponds to the absorbance-time plot obtained at 560 nm by radiolyzing N2O saturated aqueous solution containing 0.1 M KBr and compound 1. Plot (B) corresponds to the transient spectrum obtained on reaction of CO3˙− radical with compound 1. | ||
The spectral features obtained from the reactions of compound 1 with CO3˙− (with a standard potential of 1.5 V vs. NHE) are comparable to those observed in the ˙OH reactions. The transient absorption spectrum recorded at 1 μs after the pulse in the CO3˙− reactions of 1 showed λmax at 320 nm, 380 nm, and 560 nm. Subsequently, the formation of dimer species was observed at 10 μs, as indicated by the emergence of the 630 nm peak and the simultaneous disappearance of the 560 nm band (Plot B of Fig. 3). The second-order rate constant for the CO3˙− reaction with 1 was estimated to be 1.1 ± 0.2 × 108 M−1 s−1.
The reaction of compound 2 with ˙OH yields a transient absorption spectrum with one band in the UV region with a λmax of 320 nm, a shoulder at 400 nm, and a broad absorption extending beyond 700 nm with a maximum at 550 nm (Fig. 4). Herein, it was found that, apart from the decay of the initial transient(s), no additional bands were observed at higher time scales. Furthermore, no rise in the absorbance of any bands was observed as the concentration of compound 2 increased. This suggests that the observed absorptions are solely attributed to monomer-type radical species. Interestingly, the phenyl ring in compound 1 promotes the stabilization of the dimer radical through its electron-withdrawing inductive effect, which delocalizes the odd spin. However, due to the presence of an intervening methylene group on the benzyl moiety, if a dimer radical had formed, this spin delocalization would have been less effective in compound 2. Thus, we postulate that the 550 nm peak corresponds to the radical cation, similar to the observation made in the case of compound 1. The transient spectra obtained from the reactions of compound 2 with ˙OH, both in the absence and presence of a phosphate buffer, displayed comparable spectral characteristics, with identical λmax, except for an elevated absorbance at 550 nm in the presence of the buffer. The increased absorbance at 550 nm in the presence of the phosphate buffer is ascribed to the enhanced acid-catalyzed dehydroxylation of the Se–OH˙ adduct, which is presumed to have formed initially.40
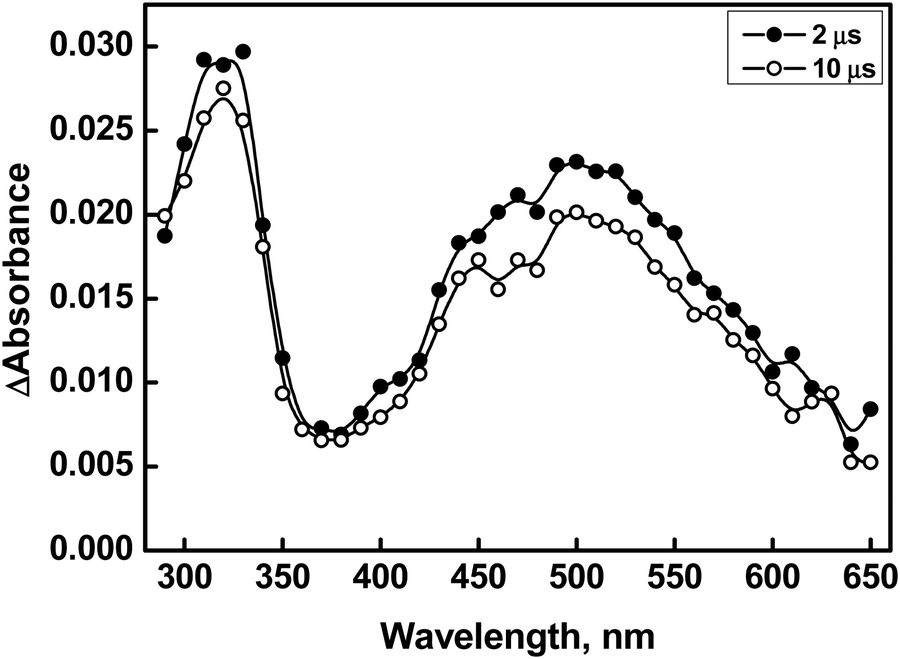 | ||
| Fig. 4 Transient absorption spectra generated from pulse radiolysis of N2O-saturated solution containing compound 2 (500 μM) at pH 7 (○ = 2 μs, ● = 10 μs). | ||
The reactions of compound 2 with CO3˙− exhibited similar spectral features to those observed for the ˙OH reactions, with λmax at 320 nm and 550 nm, and a shoulder at 400 nm (Fig. 5A). The second-order rate constant for the reaction between compound 2 and CO3˙− was determined to be 7.6 ± 0.2 × 108 M−1 s−1. Almost identical spectra have been observed for the reactions of compound 2 with Br2˙− (Fig. 5B). However, it is important to note that the significant absorbance at 360 nm, as observed in the 2 μs spectrum, resulting from Br2˙−, obscures the absorptions of the transients generated from the reactions of compound 2 with Br2˙−. At higher time scales, a significant loss of absorbance due to the radical cation at 550 nm is observed. This loss can be attributed to the formation of a hemi-bond (>Se∴Br) between Br− and the Se center, similar to the concept applied in the case of Br2˙− reactions of compound 1. The second-order rate constant for the reaction between compound 2 and Br2˙− was determined to be 2.4 ± 0.2 × 109 M−1s−1.
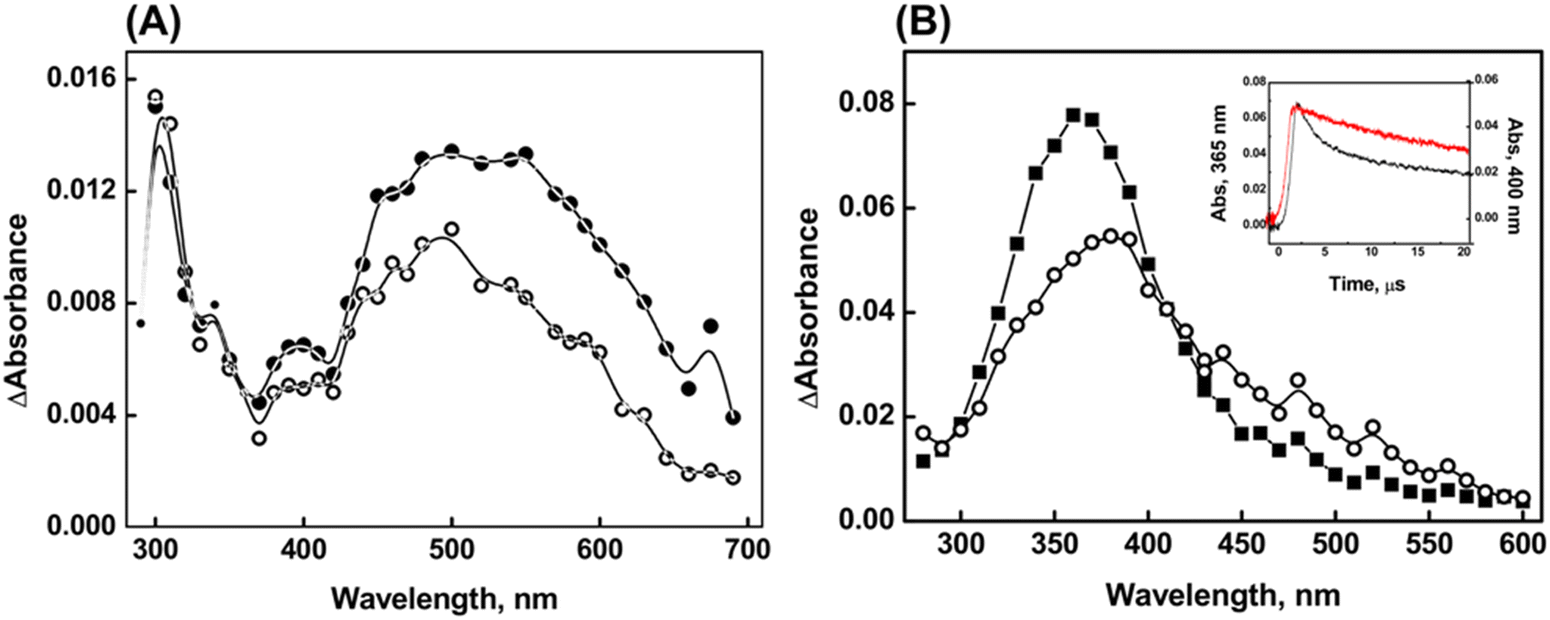 | ||
| Fig. 5 Transient absorption spectra obtained on reaction between 100 μM compound 2 and (A) CO3●−(○ = 4 μs, ● = 20 μs) and (B) Br2●− radical (■ = 2 μs, ○ = 5 μs) at pH 7. The inset in plot (B) is the absorbance-time plot obtained on reaction of compound 2 with Br2●− radical (black line = 365 nm, red line = 400 nm). | ||
The stabilities of the radical cations formed from the reaction of compounds 1 and 2 with the CO3˙− radical can be assessed by investigating their reactivity towards glutathione (GSH), a biologically significant tripeptide. It is crucial to highlight that, under the specified reaction conditions, the direct oxidation of GSH (GS˙, H+/GSH 0.92 V vs. NHE at pH 7) by CO3˙− does not take place.41 The second-order rate constants for the electron transfer reaction between the radical cation and GSH were determined by monitoring the decay of the radical cation at their respective λmax, while systematically varying the concentration of GSH from 0.5 to 8 mM. The results indicate that the rate of electron transfer between 1˙+ and GSH is higher (3.8 ± 0.4 × 107 M−1 s−1) compared to the rate between 2˙+ and GSH (4.5 ± 0.8 × 106 M−1 s−1) (Fig. 6A). This experiment suggests that the lower oxidizing ability of the radical cations derived from compound 2 is attributed to its higher stability.16
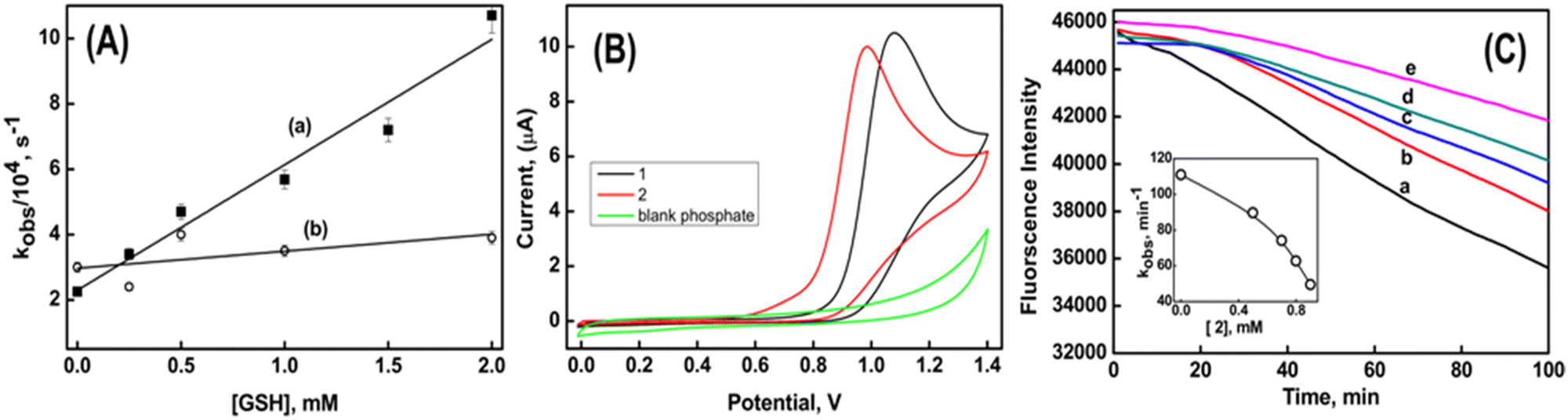 | ||
| Fig. 6 Plot (A) is the observed decay rate of the radical cation with varying concentrations of GSH, where (a) corresponds to compound 1 and (b) corresponds to compound 2. Plot (B) is the cyclic voltammogram of compound 1 and 2 in phosphate buffer at pH 7. Plot (C) shows the decay of fluorescein due to degradation by peroxyl radical in the absence (trace a) and in the presence of compound 2 (b = 0.5 mM, c = 0.7 mM, d = 0.8 mM and e = 0.9 mM) at pH 7. | ||
The radical scavenging abilities of compounds 1 and 2 have been assessed through various analyses. It was observed that both compounds did not react with azide radical (N3˙) and trichloromethyl peroxyl radical (Cl3COO˙). This observation indicates that their reduction potentials fall within the range of greater than 1.3 V but less than 1.5 V vs. NHE. Furthermore, previous studies in the radiation chemical section revealed that the reaction rates of Br2˙−and CO3˙− with compound 2 were higher compared to compound 1. This suggests that compound 2 is more susceptible to oxidation than compound 1. Moreover, the anodic peak potential determined via cyclic voltammetry showed that compound 2 exhibited a potential of 0.98 V, while compound 1 had a potential of 1.08 V. This indicates that compound 2 is more readily oxidized compared to compound 1 (Fig. 6B).
The antioxidant activity of compounds 1 and 2 was evaluated by their capacity to shield against the degradation of fluorescein. The degradation of fluorescein was induced by the generation of peroxyl radicals (ROO˙) through the thermal decomposition of AAPH at 37 °C. The fluorescence emitted by fluorescein alone displayed a linear decrease over time, as depicted by trace a in Fig. 6C. The rate of peroxidation (kobs) was determined to be 111 min−1 from the slope of the time-dependent fluorescence decay of fluorescein. In the presence of compound 1, there was no observed change in the time-dependent decay of fluorescein. However, in the presence of compound 2, a time delay of 20 minutes was observed before the fluorescence began to decrease. Notably, this time delay remained constant even with an increase in the concentration of compound 2. However, the respective kobs values decreased to 90 min−1 and 50 min−1 in the presence of 0.5 mM and 0.9 mM of compound 2, respectively. From Fig. 6C, it can be seen that compound 2 exhibits better inhibition efficiency in protecting against the degradation of fluorescein. This indicates that the benzyl group in compound 2 assists in free radical scavenging, similar to what is observed in natural compounds like glaucine and capsaicin. These compounds are known to retain their antioxidant activity due to the presence of the radical stabilizing effect of the benzyl group.42,43
Computational studies
The feasibility of dimer species formation in the one-electron oxidation of compound 1 by ˙OH has been explored through computational calculations. These calculations were driven by the experimental results obtained from the competitive reaction kinetics method and concentration-dependent absorption measurements, which provide compelling evidence for the existence of dimer species. Herein, it is proposed that the first step in the one-electron oxidation of compound 1 by ˙OH involves the formation of its corresponding radical cation, which is clearly a neutral radical (symbolically represented as 1˙). We determined that the free energy change (ΔG) related to the formation of 1˙ (1− + ˙OH →1˙ + OH−) is −0.29 kcal mol−1 using the M05-2X/6-311+G(d,p)/SMD level of theory. Additionally, we observed that the Mulliken spin density is maximum on Se (0.83 au) in the 1˙ species. It can be inferred that the localization of spin on the Se atom of 1˙ plays a vital role in the formation of dimer species, characterized by a >Se∴Se< bond. In practical terms, an accurate representation of the dimer species necessitates the consideration of both σ- and π-type dimer formations. Therefore, we modeled the σ- and π-type dimers in a manner where the aromatic rings are positioned apart from each other in the case of the σ-type dimer, while the aromatic rings are arranged in a face-to-face configuration in the π-type dimer. The σ- and π-type dimers resulting from the combination of 1˙ and 1− are labeled as D1 and D1′, respectively, as depicted in eqn (5).| 1˙ + 1− → D1 and/or D1′ | (5) |
The optimized geometries of D1 and D1′ obtained at the M05-2X/6-311 + G(d,p)/SMD level of theory are depicted in Fig. 7. The formations of D1 and D1′ were determined to be thermodynamically feasible processes, as evidenced by the ΔGr values for their formations (−0.13 kcal mol−1 and −1.91 kcal mol−1, respectively). Thermodynamically, the π-stacking interactions stabilize the D1′ by 1.73 kcal mol−1 relative to D1. As observed in Fig. 7, the odd spin density is nearly equally distributed over the two Se atoms in D1, whereas this is not the case for the D1′ species. The characteristic >Se∴Se< bond distance in D1 is measured at 3.009 Å, while the corresponding distance in D1′ is 2.997 Å. Additionally, we determined that the distance between the centers of the phenyl rings in the D1′ species is 3.775 Å.
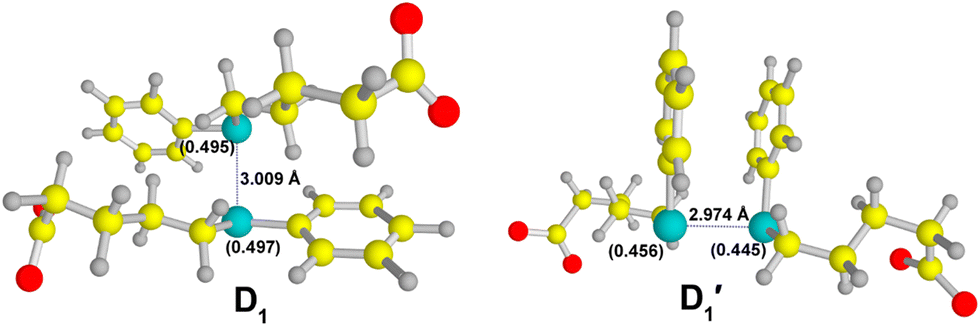 | ||
| Fig. 7 Optimized geometries of σ-(D1) and π-type (D1) dimers obtained at the M05-2X/6-311 + G(d,p)/SMD level of theory. Spin densities on Se are indicated in parentheses. Atom color code: oxygen (red), carbon (yellow), selenium (cyan), and hydrogen (grey). | ||
TDDFT calculations yielded a λmax of 630 nm for the D1 species, which is in excellent agreement with the experimental value. The calculated λmax for the D1′ species is 566 nm. Therefore, the σ-type dimer (D1) predominantly contributes to the observed experimental spectrum, despite the equal likelihood of formation of both σ- and π-type dimers. This observation represents a unique finding and has not been previously reported in the context of one-electron oxidations of organoselenium compounds. Indeed, both the D1 and D1′ species are characterized by a 2c–3e bond, with the odd electron occupying an anti-bonding orbital, leading to a half bond order for >Se∴Se< bond. In the case of hemi-bonded species, the presence of an unpaired electron in the anti-bonding orbital results in longer bond distances between the participating atoms, effectively reducing the repulsive interaction between them.12 As previously mentioned, the bond distance between the two Se atoms in the σ- and π-type dimer of compound 1 is 3.009 Å and 2.974 Å, respectively. The observed Se–Se bond distance in the D1 species suggests a higher level of stability compared to the D1′ species.
4. Conclusions
The pulse radiolytic studies of 5-(phenylselanyl)pentanoic acid (1) and 5-(benzylselanyl)pentanoic acid (2) demonstrated that the phenyl/benzyl group plays a significant role in determining the transients generated from their reactions with oxidizing radicals such as ˙OH, Br2˙− and CO3˙−. Theoretical calculations at the M05-2X/6-311+G(d,p)/SMD level explicitly established that formations of both σ- and π-type dimer anions characterized by >Se∴Se< bonds are thermodynamically feasible processes in the one electron oxidations of compound 1 by ˙OH. The λmax value calculated for the σ-type dimer species using the TDDFT method is in excellent agreement with the experimental value and hence considered as the prime contributor towards the transient absorption spectrum obtained in the experimental ˙OH reaction. Compound 2 exhibited superior antioxidant properties, as evidenced by its higher scavenging abilities towards Br2˙−, CO3˙−, and peroxyl (ROO˙) radicals compared to compound 1. These results gain significance in the design of new functionalized organoselenium systems as antioxidants.Conflicts of interest
There are no conflicts to declare.Acknowledgements
BGS acknowledge Dr Awadhesh Kumar, Head, RPC Division BARC, and Dr A. K. Tyagi, Director, Chemistry Group, BARC for their support and encouragement. CS, FM and FMan acknowledge University of Perugia for the financial support under the frames “Ricerca di Base”. FMan was supported by a grant from MIUR (Dipartimenti di Eccellenza). This study was performed as a collaborative action under the umbrella of the SeSRedCat Network (Selenium, Sulfur, Redox and Catalysts).References
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821 CrossRef CAS PubMed.
- T. Guo, Z. Li, L. Bi, L. Fan and P. Zhang, Tetrahedron, 2022, 112, 132752 CrossRef CAS.
- N. Bisht, P. Phalswal and P. K. Khanna, Mater. Adv., 2022, 3, 1415 RSC.
- F. Zhang, X. Li and Y. Wei, Biomolecules, 2023, 13, 799 CrossRef CAS.
- G. Bjørklund, M. Shanaida, R. Lysiuk, H. Antonyak, I. Klishch, V. Shanaida and M. Peana, Molecules, 2022, 27, 6613 CrossRef PubMed.
- Ö. Pehlivan, M. Waliczek, M. Kijewska and P. Stefanowicz, Molecules, 2023, 28, 3198 CrossRef.
- M. Chen, W. Cao, J. Wang, F. Cai, L. Zhu, L. Ma and T. Chen, J. Am. Chem. Soc., 2022, 144, 20825 CrossRef CAS PubMed.
- E. J. Ste. Marie, R. J. Wehrle, D. J. Haupt, N. B. Wood, A. van der Vliet, M. J. Previs, D. S. Masterson and R. J. Hondal, Biochem., 2020, 59, 3300 CrossRef CAS PubMed.
- K. Skotnicki, I. Janik, K. Sadowska, G. Leszczynska and K. Bobrowski, Molecules, 2021, 27, 133 CrossRef PubMed.
- N. C. Payne, A. Geissler, A. Button, A. R. Sasuclark, A. L. Schroll, E. L. Ruggles, V. N. Gladyshev and R. J. Hondal, Free Radical Biol. Med., 2017, 104, 249 CrossRef CAS.
- B. Mishra, A. Sharma, S. Naumov and K. I. Priyadarsini, J. Phys. Chem. B, 2009, 113, 7709 CrossRef CAS PubMed.
- P. V. Kumar, B. G. Singh, P. P. Phadnis, V. K. Jain and K. I. Priyadarsini, Chem. - Eur. J., 2016, 22, 12189 CrossRef CAS.
- B. G. Singh, E. Thomas, F. Kumakura, K. Dedachi, M. Iwaoka and K. I. Priyadarsini, J. Phys. Chem. A, 2010, 114, 8271 CrossRef CAS.
- B. G. Singh, S. A. Nadkarni, V. K. Jain and K. I. Priyadarsini, RSC Adv., 2015, 82, 66621 RSC.
- P. Prabhu, P. P. Bag, B. G. Singh, A. Hodage, V. K. Jain, M. Iwaoka and K. I. Priyadarsini, Free Radical Res., 2011, 45, 461 CrossRef CAS PubMed.
- B. G. Singh, E. Thomas, S. N. Sawant, K. Takahashi, K. Dedachi, M. Iwaoka and K. I. Priyadarsini, J. Phys. Chem. A, 2013, 117, 9259 CrossRef CAS.
- A. K. Tripathi, A. K. Ray, S. K. Mishra, S. M. Bishen, H. Mishra and A. Khurana, Rev. Bras. Farmacogn., 2023, 33, 272 CrossRef CAS.
- J. Chen, J. Yang, L. Ma, J. Li, N. Shahzad and C. K. Kim, Sci. Rep., 2020, 10, 2611 CrossRef CAS.
- B. Godlewska-Żyłkiewicz, R. Świsłocka, M. Kalinowska, A. Golonko, G. Świderski, Ż. Arciszewska, E. Nalewajko-Sieliwoniuk, M. Naumowicz and W. Lewandowski, Materials, 2020, 13, 4454 CrossRef.
- R. L. Jayaraj, R. Beiram, S. Azimullah, N. Meeran Mf, S. K. Ojha, A. Adem and F. Y. Jalal, Int. J. Mol. Sci., 2020, 21, 7670 CrossRef CAS PubMed.
- R. S. Glass, Top. Curr. Chem., 2018, 376, 22 CrossRef.
- C. W. Nogueira, N. V. Barbosa and J. B. T. Rocha, Arch. Toxicol., 2021, 95, 1179 CrossRef CAS PubMed.
- L. M. Bouchet and J. E. Argüello, J. Org. Chem., 2018, 83, 5674 CrossRef CAS.
- B. Halliwell, A. Adhikary, M. Dingfelder and M. Dizdaroglu, Chem. Soc. Rev., 2021, 50, 8355 RSC.
- K. P. Prasanthkumar, M. P. Rayaroth and J. R. Alvarez-Idaboy, J. Phys. Chem. B, 2020, 124, 6245 CrossRef CAS.
- K. P. Prasanthkumar, P. K. Sajith and B. G. Singh, New J. Chem., 2020, 44, 18858 RSC.
- K. P. Prasanthkumar, J. R. Alvarez-Idaboy, P. V. Kumar, B. G. Singh and K. I. Priyadarsini, Phys. Chem. Chem. Phys., 2016, 18, 28781 RSC.
- C. H. Schiesser and K. Sutej, J. Chem. Soc., Chem. Commun., 1992, 57 RSC.
- W. R. Bowman, P. T. Stephenson, N. K. Terrett and A. R. Young, Tetrahedron, 1995, 51, 7959 CrossRef CAS.
- R. H. Schuler, L. K. Patterson and E. Janata, J. Phys. Chem., 1980, 84, 2088 CrossRef CAS.
- G. V. Buxton and Q. G. Mulazzani, in Radiation-Chemical Techniques, Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, Germany, 1st edn, 2001, vol. 1, pp. 503–557 Search PubMed.
- S. L. Scott, W. J. Chen, A. Bakac and J. H. Espenson, J. Phys. Chem., 1993, 97, 6710 CrossRef CAS.
- E. Dorta, E. Atala, A. Aspee, H. Speisky, E. Lissi and C. Lopez-Alarcon, Free Radical Biol. Med., 2014, 75, S38 CrossRef PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Jr. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS.
- https://pubchem.ncbi.nlm.nih.gov/compound/5-Phenylvaleric-acid .
- S. Zhang, X. Wang, Y. Su, Y. Qiu, Z. Zhang and X. Wang, Nat. Commun., 2014, 5, 4127 CrossRef CAS PubMed.
- M. Bonifačić, I. Štefanić, G. L. Hug, D. A. Armstrong and K. Asmus, J. Am. Chem. Soc., 1998, 38, 9930 CrossRef.
- E. Madej and P. Wardman, Arch. Biochem. Biophys., 2007, 462, 94, DOI:10.1016/j.abb.2007.03.002.
- P. O’Brien, C. Carrasco-Pozo and H. Speisky, Chem. – Biol. Interact., 2006, 159, 1 CrossRef.
- A. Rosa, M. Deiana, V. Casu, S. Paccagnini, G. Appendino, M. Ballero and M. A. Dessí, J. Agric. Food Chem., 2002, 50, 7396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj04487d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |