
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deep representation learning determines drug mechanism of action from cell painting images†
Daniel R.
Wong
 *a,
David J.
Logan
b,
Santosh
Hariharan
c,
Robert
Stanton
a,
Djork-Arné
Clevert
a and
Andrew
Kiruluta
a
*a,
David J.
Logan
b,
Santosh
Hariharan
c,
Robert
Stanton
a,
Djork-Arné
Clevert
a and
Andrew
Kiruluta
a
aMachine Learning and Computational Sciences, Pfizer Worldwide Research Development and Medical, 610 Main Street, Cambridge, Massachusetts 02139, USA. E-mail: daniel.wong@pfizer.com
bInternal Medicine Research Unit, Pfizer Worldwide Research Development and Medical, 610 Main Street, Cambridge, Massachusetts 02139, USA
cDiscovery Sciences, Pfizer Global Research and Development, Groton Laboratories, 280 Shennecossett Rd, Groton, CT 06340, USA
First published on 16th August 2023
Abstract
Fluorescent-based microscopy screens carry a broad range of phenotypic information about how compounds affect cellular biology. From changes in cellular morphology observed in these screens, one key area of medicinal interest is determining a compound's mechanism of action. However, much of this phenotypic information is subtle and difficult to quantify. Hence, creating quantitative embeddings that can measure cellular response to compound perturbation has been a key area of research. Here we present a deep learning enabled encoder called MOAProfiler that captures phenotypic features for determining mechanism of action from Cell Painting images. We compared our method with both the traditional gold-standard means of feature encoding via CellProfiler and a deep learning encoder called DeepProfiler. The results, on two independent and biologically different datasets, indicated that MOAProfiler encoded MOA-specific features that allowed for more accurate clustering and classification of compounds over hundreds of different MOAs.
Introduction
High-content screening (HCS) produces diverse phenotypic information that is of great interest to the drug discovery process.1,2 One such procedure known as Cell Painting3 allows for broad profiling of cellular phenotypes in response to different compounds. Much effort has gone into quantitatively characterizing these phenotypes,4–6 with the goal of creating representations for describing how different compounds affect biology. Quantitatively profiling cellular response to compound perturbation has many use cases, such as target identification, concentration optimization, and mechanism of action (MOA) determination (e.g. heat shock protein inhibitor, CDK inhibitor, histamine receptor antagonist).7,8Traditional computer vision methods such as CellProfiler (CP),9 which is the gold standard in cellular profiling, rely on extracting preset human-selected features from image data. Methods like this have proven useful for encoding subtle changes in cellular phenotype to derive biological insight, with a variety of applications such as object detection,10 cell viability assessment,11 transcriptomic querying,12 and MOA determination.7 Although traditional computer vision techniques have also been used, they often need substantial fine-tuning and require human intelligence and intuition for deciding which phenotypic features and parameters are important to measure. In contrast, deep learning has emerged as a tool for learning and encoding meaningful representations4 (i.e. embeddings) without requiring humans to know beforehand what features may be useful for the task of interest. Indeed, deep representation learning13,14 has facilitated improved understanding in both biology14–16 and medicine.17–19
One goal of phenotypic HCS is to determine the MOAs of compounds.20 Ascertaining MOA is a challenging endeavor, especially given its multi-faceted and complex nature.21 But the task is worthwhile and can provide insight into drug efficacy, side effects, dosing, and possible success in clinical trials.22,23 Some success has been seen outside of phenotypic screening endeavors, such as through analyzing a compound's structure and effect on transcriptomic profiles.24–26 For phenotypic screening, deep learning has performed better than traditional techniques for MOA determination.27–33 However, these studies only served as a proof-of-concept, encompassing a small set of about a dozen MOAs.
Here, we present a MOA determination method spanning hundreds of MOAs that showed efficacy on two independent datasets: (1) the Joint Undertaking in Morphological Profiling (JUMP1) pilot dataset34 (2) the Library of Integrated Network-Based Cellular Signatures (LINCS) dataset.35,36 We compared our method called MOAProfiler (MP) with CP as well as a deep learning based method called DeepProfiler (DP).37,38 We present MP as an open-source and readily available tool for deep phenotypic profiling of Cell Painting images.
Results
JUMP1 performance
To assess whether we could determine a broad range of MOAs from phenotypic information alone, we developed a deep learning method across two datasets differing in cell type, concentration, and time-points (Methods). For the first, we turned to the JUMP1 pilot dataset which had MOA metadata information provided by the Clue Connectivity Map.39 To simplify the compound space and limit the intricacies associated with polypharmacology,40 we took a subset of the compounds that had at most one known MOA. The resulting dataset spanned 266 compounds, with 176 MOAs used for training and a subset of 59 (multi-compound MOAs) used for testing (Fig. 1a). Most compounds were plated at 23 replicate wells (Fig. 2a) and most MOAs were represented by one compound (Fig. 2b). There were either 9 or 16 image fields per well (Fig. 2c). Cell Painting images consisted of five channels capturing different areas of cellular morphology (Fig. 1b).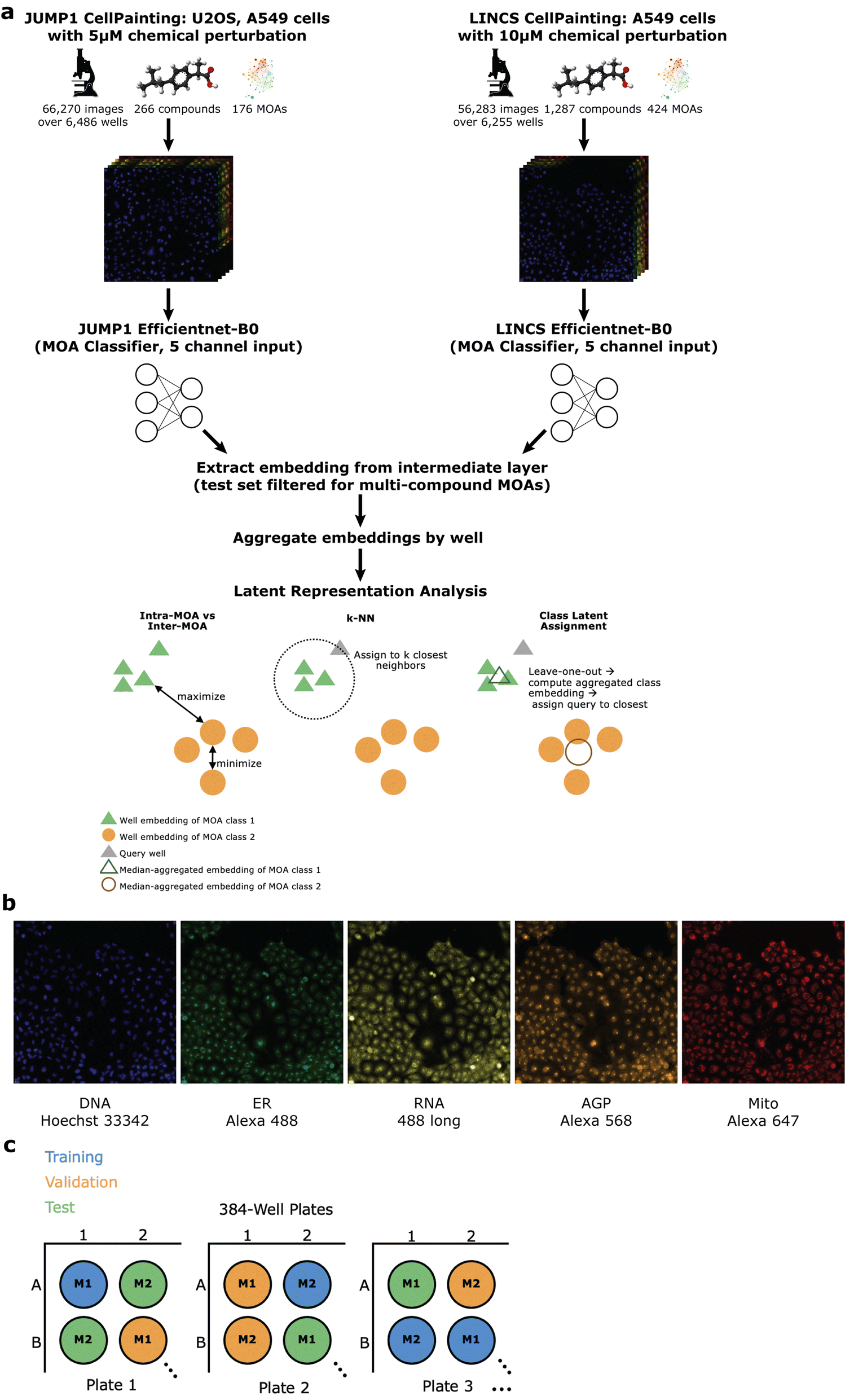 | ||
| Fig. 1 Study overview. (a) Study overview for well-holdout scheme. We applied the same method to two datasets independently: the JUMP1 pilot dataset as well as the LINCS dataset. Training and analyses were performed independently for each study. Below: pictorial visualizations of the various metrics assessing embedding viability and clustering. (b) Example cell painting images from the JUMP1 dataset. Color added for visualization. DNA = deoxyribonucleic acid, ER = endoplasmic reticulum, RNA = ribonucleic acid, AGP = actin cytoskeleton, Golgi, plasma membrane, Mito = mitochondria. (c) Schematic of training (blue), validation (orange), test (green) split. M1 indicates MOA class one and M2 indicates MOA class two. Each circle is a well. The schematic is illustrative and not the actual location splits used in the study. | ||
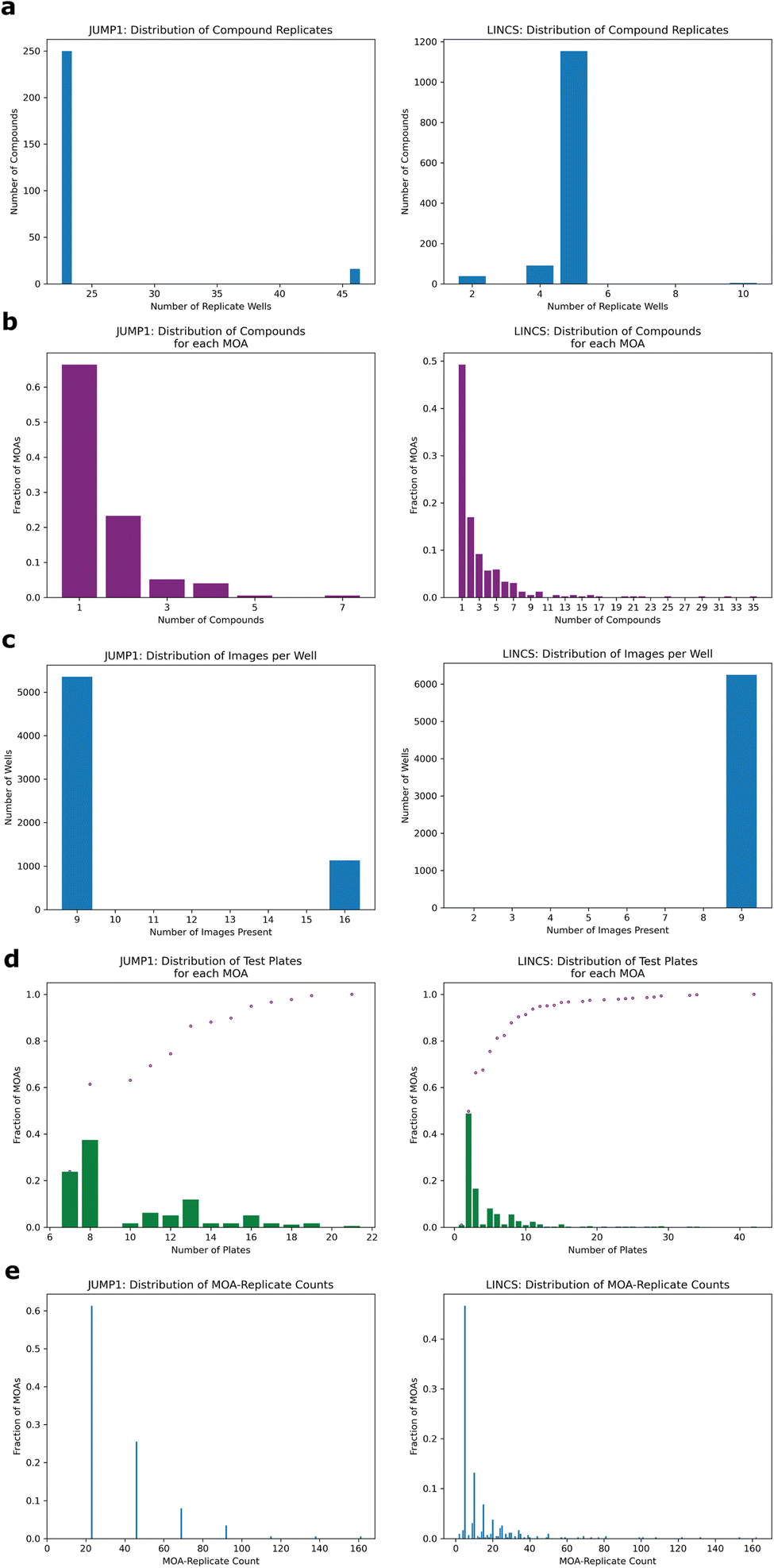 | ||
| Fig. 2 Distributions of study metadata. All panels exclude the negative control DMSO condition. JUMP1 (left), LINCS (right). (a) Distribution of replicate well count per compound. E.g. left panel: Most compounds were each plated in 23 wells. (b) Distribution of compounds for each MOA. E.g. left panel: 66% of JUMP1 MOAs were represented by one compound each. (c) Distribution of images per well. Most wells had 9 non-overlapping image fields. (d) Distribution of number of plates in the test set for each MOA. Example from left figure leftmost bar: 24% of MOAs were each represented on 7 plates in the held-out test set. Purple dots show the cumulative frequencies at each plate count. (e) Distribution of MOA-replicate counts. “MOA-replicates” is defined as any wells with any compounds with the same MOA. E.g. left panel: 62% of MOAs were each represented by 23 different wells in the entire dataset. | ||
To develop a model capable of creating MOA-specific embeddings from cellular phenotypes, we trained an EfficientNet model41 to classify an image field's MOA, with the motivation of directly learning which features are important for segregation. We chose EfficientNet because of its high performance on the ImageNet dataset and relatively few number of parameters. Only images were provided as input to the model, with no information on compound, concentration, or any other form of metadata (Methods). To minimize the model learning confounding experimental features like well-artifacts, we split the dataset such that each well was randomly assigned to only one of three sets: training, validation, or test (Fig. 1c). We divided the dataset such that 60% of the wells were assigned to training, 10% to validation, and 30% to test (Methods). We also ensured each MOA's test wells spanned multiple plates (at least seven for JUMP1, Fig. 2d, left) to assess the model's performance across potential variation of plate-specific conditions. Since the MOA-replicate count was imbalanced (Fig. 2e, left), we measured the model's precision recall characteristics.
Since most MOAs were only represented by one compound each (Fig. 2b), we filtered the held-out test set to only include MOAs that were each represented by multiple compounds, resulting in 59 MOAs. This way we could assess whether the model had simply learned to group compound replicates, which could be the case if the dataset was heavily over-represented by single-compound MOAs. In this well-holdout scheme, the compounds present in the test set were also present in the training set.
On the held-out test set, the model achieved an area under the precision recall curve (AUPRC) of 0.46 (random AUPRC = 0.006) for image field classification (Fig. 3a). This equated to an accuracy of 0.51 (random accuracy = 0.017). To assess the influence of well-location confounders common in microscopy,42,43 we compared the model's performance on edge wells versus non-edge wells. We found minor differences in classification accuracy (0.54 vs. 0.50) suggesting that the model was not leveraging much confounding edge-specific features for its learning (ESI Fig. 1A†). We also found differences in classification accuracy stratified by timepoint (ESI Fig. 1B†) and cell type (ESI Fig. 1C†).
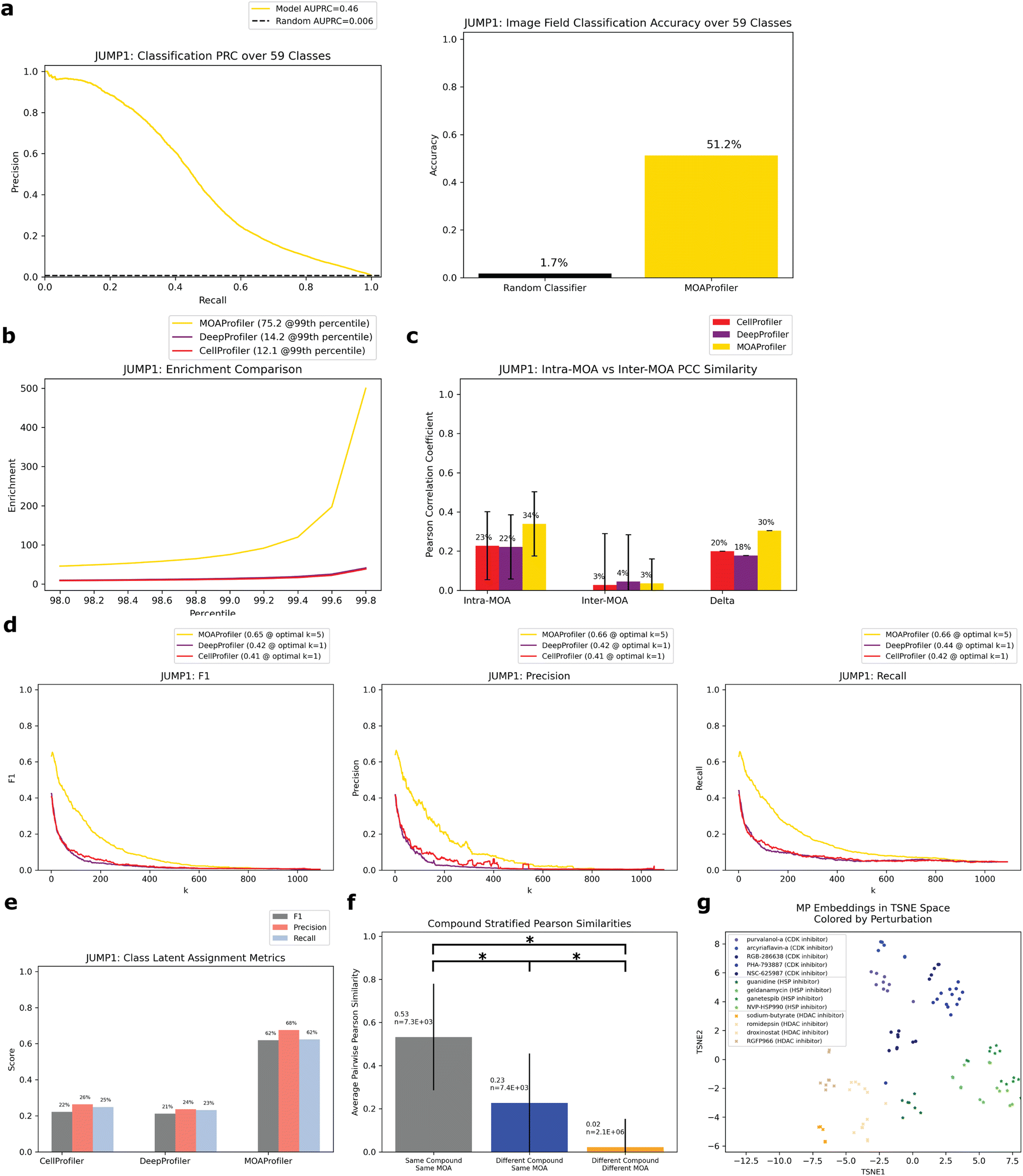 | ||
| Fig. 3 Performance of model trained and evaluated on the JUMP1 dataset. All evaluation was over the held-out test set filtered for multiple-compound MOAs. (a) Left: PRC of the classifier, random baseline = positive prevalence of binarized labels. Right: classification accuracy, random baseline = 1/59. Classification was over image fields (not embeddings as in panels d and e). (b) Enrichment scores for the three methods from the 98th percentile to the 99.8th percentile with step sizes of 0.2. Enrichment at the 99th percentile (rounded to the nearest tenth) is shown in the figure legend. (c) Average of pairwise PCCs for two groups of well-level embeddings: intra-MOA (embeddings with the same MOA class), inter-MOA (different MOA classes). Delta = intra-MOA average − inter-MOA average. Error bars span one standard deviation in each direction. (d) k-NN metrics for embedding classification calculated for all values of k. F1 (left), precision (middle), and recall (right). The highest score for each method and corresponding k are shown in the legend (rounded to the nearest hundredth). (e) F1, precision, and recall values for the class latent assignment metric. Scores rounded to the nearest hundredth. (f) Average of pairwise PCCs for three groups of embeddings. For each group, we calculated PCCs for each possible pair of wells. Significance (*) indicates p ≪ 0.0001 for a two-sided z test. Error bars span one standard deviation in each direction. (g) TSNE visualization of well embeddings of three example MOAs (chosen because they were each represented by four or more compounds). Circles = CDK inhibitor, stars = HSP inhibitor, x marks = HDAC inhibitor. Different compounds with the same MOA were given similar but different colors. | ||
With the trained classification model, we measured how well the model created embeddings13 that were meaningful for MOA classification. Hence, we extracted image embeddings from an intermediate layer in the network,14 median-aggregated them by well, standardized them with respect to DMSO control, and assessed how valuable these well-level embeddings were for the task of MOA classification compared to CP and DP (Methods). We performed all analyses on the held-out test set which spanned 59 MOAs.
Ideally, embeddings with the same MOA (intra-MOA) should be more similar than embeddings with different MOAs (inter-MOA). Hence, we measured how likely it was to observe intra-MOA embeddings in strongly correlated versus weakly correlated embedding pairs (pairs of well-level embeddings). We used different thresholds of correlation for defining strong and weak correlation (Methods). Through a Fisher's exact test, we found that intra-MOA embedding pairs were more likely to be found in strongly correlated (by Pearson correlation coefficient (PCC), Methods) versus weakly correlated embedding pairs, with greater enrichment of MP-derived embeddings vs. CP-derived and DP-derived embeddings (enrichment at the 99th percentile = 12.1 CP, 14.2 DP, 75.2 MP, Fig. 3b). Similarly, we asked which of the methods could better generate embeddings that captured phenotypic differences between the MOAs. There was greater difference between intra-MOA embeddings and inter-MOA embeddings when constructed by MP instead of CP or DP (delta = 0.30 for MP, Fig. 3c). For both CP and DP, the difference was smaller (delta = 0.20 for CP, 0.18 for DP). This indicates that MP encoded different MOAs in different and distinguishable phenotypic spaces, which may be advantageous if a new compound were to be queried for its MOA.
To simulate this situation of predicting the MOA of a query compound using its phenotypic embedding, we performed two analyses using the embeddings of the held-out test set: k-nearest-neighbors (k-NN) and an analysis we constructed called the “class latent assignment” (Fig. 1a for a pictorial visualization, Methods). We used each well in the test set as a held-out query. For k-NN, we predicted the query well's MOA as the majority MOA of its k-nearest neighbors (Methods). For all values of k, we calculated F1, precision, and recall values. We found that for all metrics, MP outperformed CP (percent improvement: 60.8% F1, 60.9% precision, and 57.5% recall). MP also outperformed DP (percent improvement: 54.1% F1, 58.8% precision, and 49% recall, Fig. 3d).
The “class latent assignment” method was a parallel way to classify a query compound's MOA by instead using similarity to aggregated MOA-level embeddings (MLEs) rather than to well-level embeddings for predicting a query well's class (Methods). This metric has the advantage of being less sensitive to single-well outliers and reduces the impact of the immediate closest neighbors so that embeddings can be queried against more representative class-wide embeddings. We computed an aggregated MLE for each MOA by taking the median of all the same-MOA well embeddings in the test set with the query well's embedding left out. We then predicted the query well's MOA as the MOA of the most similar MLE. We calculated F1, precision, and recall scores for the resulting predictions. By this metric, MP was more performant than CP (percent improvement: 179% F1, 157% precision, and 151% recall). MP also exceeded DP's metrics (percent improvement: 192% F1, 186% precision, and 170% recall, Fig. 3e).
As an additional metric to assess the viability of each methods' embeddings for MOA prediction, we also trained a logistic regression model on the training set embeddings and assessed the model's MOA-prediction performance on the test set embeddings (ESI Fig. 2†). We found the same trends, with MP-derived embeddings being the most informative for MOA classification.
Since different compounds shared the same MOA, we wanted to know whether the model was learning MOA-specific phenotypes consistent through different compounds with the same MOA as opposed to simply learning each compound's phenotypic effect. Hence, we calculated the average PCC of three groups of well pairs with: (1) the same compound (and hence same MOA) (2) different compounds with the same MOA and (3) different compounds with different MOAs. We found that of the embedding pairs with different compounds, the pairs with the same MOA class were more similar than those with different MOA classes (average PCC = 0.23 vs. 0.02, Fig. 3f). The three populations' PCC averages were all significantly different (p ≪ 0.0001 for all two-sided z-tests). This suggests that the phenotypic embeddings that the model encoded were MOA-specific rather than compound-specific. From a low-dimensional t-distributed stochastic neighbor embedding (TSNE) visualization of embeddings from three example MOAs, we could see that different compounds with the same MOA were clustered together with different MOAs inhabiting different areas in latent space (Fig. 3g). CP and DP did not display the same degree of clustering for these MOAs (ESI Fig. 3†). Additionally, when we compared baseline MP to heavily optimized CP embeddings, which underwent many post-processing steps such as normalization by plate, feature selection, and spherization to DMSO31 (Methods), MP was still more performant than CP, but performance gains were smaller (ESI Fig. 4†).
To simulate the real-world use case of identifying MOAs of unknown held-out compounds, we performed an analysis where we split the dataset by compound instead of by wells (Fig. 4a, Methods). In this scheme, we randomly selected and held out one compound for each of the MOA classes that were represented by at least two distinct compounds. We chose a threshold of two compounds so that each held-out compound would have at least one other same-MOA compound in the training set to facilitate learning. This resulted in 59 compounds that we used as a test set, with the remaining 207 compounds (plus negative control DMSO) used for training and validating a new model (Methods). Despite the model never being exposed to these held-out compounds during training, it was able to correctly predict MOAs for 20.3–22% of the compounds in a space of 59 possible MOAs (Fig. 4b and c). Compared to a random baseline of 1/59 and an expected value of (1/59) × 59 = 1 compound discovered, this was a 12–13× improvement. Performance varied depending upon whether we predicted MOA by an individual well-level vote (Fig. 4b) or by similarity of an aggregated compound-level embedding (CLE) to an aggregated MLE (Fig. 4c, Methods). CP, DP, and MP had variable performance across the different MOAs (ESI Table 1†). There was no clear trend between MP performance and number of available training compounds (ESI Fig. 5A†). Held-out compound replicate embeddings had greater similarity to each other than to other embeddings (ESI Fig. 5B and C†).
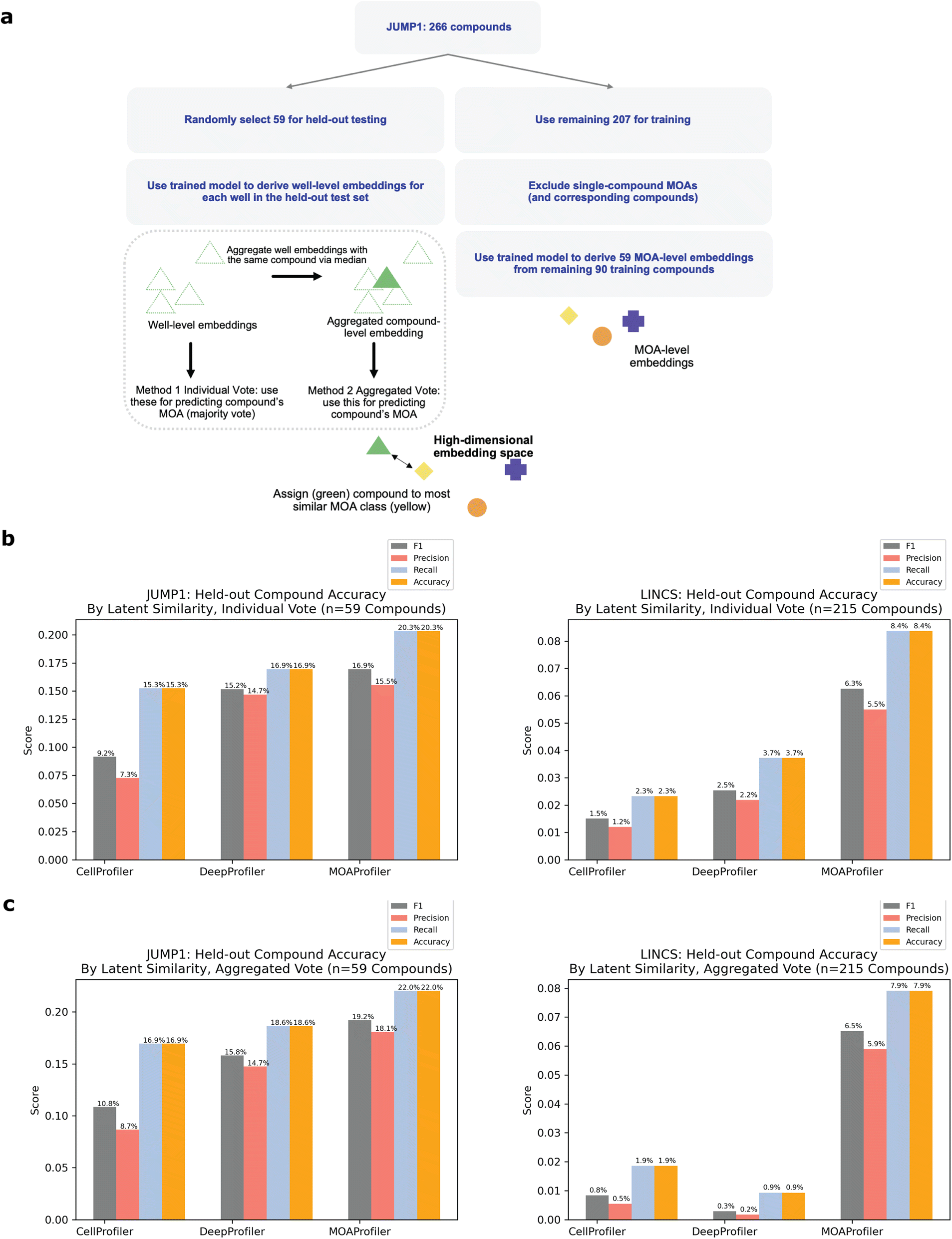 | ||
| Fig. 4 Performance on held-out compounds via two methods. (a) Learning schematic for compound-holdout. JUMP1 is pictured here as an example, but we also used the same process for the LINCS dataset (1287 compounds total: 1072 for training, 215 for test). Similarity was determined via PCC (Methods). (b) Individual vote is the scheme in which the held-out compound's MOA is predicted by taking the majority MOA over each well-level prediction (Methods). Y-axis: different scores for MOA identification. Left: JUMP1, right: LINCS. (c) Aggregated vote is the scheme in which we aggregated the held-out compound's well-level embeddings into a CLE and predicted its MOA as the MOA of the most similar MLE derived from the training set (Methods). Y-axis: different scores for MOA identification. Left: JUMP1, right: LINCS. | ||
LINCS performance
To assess the method on a second dataset, we turned to the LINCS Cell Painting Dataset.36 Like the JUMP1 dataset, we took a subset of the LINCS data that only had compounds with one known MOA. Furthermore, we took a subset of the data at a fixed concentration of 10 μM since this dose produced the strongest phenotype across compounds.43 The resulting dataset spanned 1287 compounds, with 424 MOAs used for training and a subset of 215 (multi-compound MOAs) used for testing (Fig. 1a). Most compounds were plated in five replicate wells (Fig. 2a). There were 9 images per well (Fig. 2c).We trained a separate EfficientNet on this dataset with the same task of classifying MOAs from solely image data. We used the same training-validation-test split scheme by well as we did for JUMP1 along with the same hyperparameter choices (Methods). Like the JUMP1 study, most MOAs were only represented by one compound each (Fig. 2b). Hence, we also assessed the performance metrics with a test dataset that only included MOAs that were each represented by multiple compounds, resulting in 215 MOAs. The model achieved an AUPRC of 0.34 (vs. 0.002 random) and an accuracy of 0.43 (vs. 0.005 random) for image field classification (Fig. 5a). Like the JUMP1 dataset, we did not detect much edge-location confounders affecting classification performance (0.47 edge accuracy vs. 0.42 non-edge accuracy, ESI Fig. 1D†).
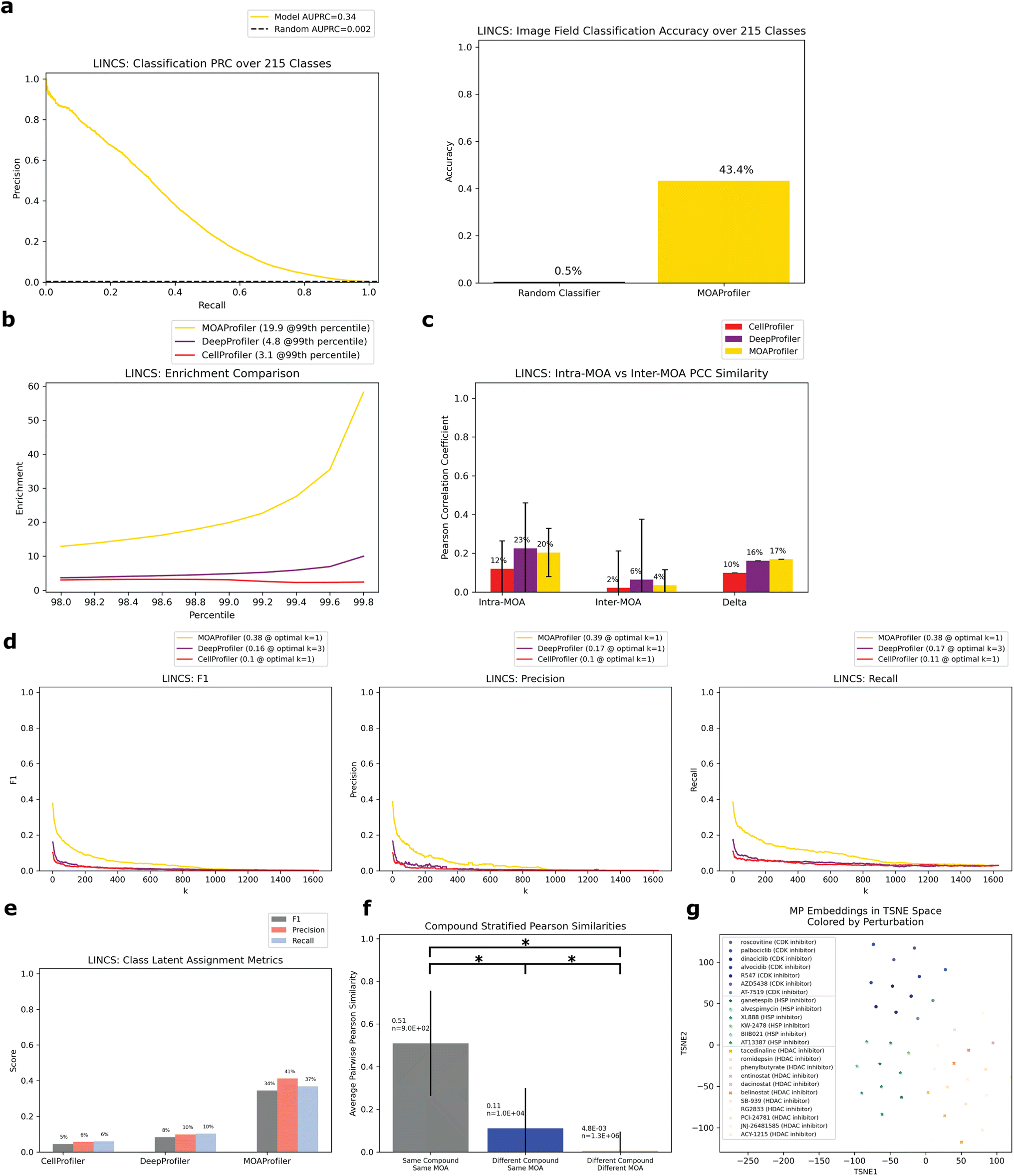 | ||
| Fig. 5 Performance of model trained and evaluated on the LINCS Dataset. Same analysis as Fig. 3. All evaluation was over the test set filtered for multiple-compound MOAs. (a) PRC and accuracy of image classification. Random baseline for PRC is 1/215. (b) Enrichment comparison at different percentiles. (c) Intra-MOA vs. inter-MOA average pairwise PCC similarity. (d) k-NN embedding metrics. (e) Class latent assignment metrics. (f) Average pairwise PCCs of three different groups stratified by perturbation. (g) TSNE visualization of well embeddings from three example MOAs. Circles = CDK inhibitor, stars = HSP inhibitor, x marks = HDAC inhibitor. Different compounds with the same MOA were given similar colors. | ||
We derived well-level embedding metrics for the held-out test set and determined that MP was advantageous over CP and DP for MOA determination. MP enabled greater enrichment for intra-MOA pairs among strongly correlated embeddings, with 6.4× fold increa se in enrichment over CP and 4.1× fold increase over DP (Fig. 5b). MP also had greater intra-MOA and inter-MOA delta compared to CP and DP (delta = 0.10 CP, 0.16 DP, 0.17 MP, Fig. 5c). MP facilitated greater k-NN metrics than CP (percent improvement: 270% F1, 288% precision, and 252% recall Fig. 5d). We also observed sizeable performance gains compared with DP (percent improvement: 135% F1, 133% precision, and 120% recall). For the class latent assignment metrics, MP outperformed CP (percent improvement: 665% F1, 623% precision, and 516% recall) and DP (percent improvement: 317% F1, 317% precision, and 259% recall, Fig. 5e). When we trained a logistic regression model on the training set embeddings derived from the different models, we likewise saw sizeable performance gains of a model trained on MP-derived embeddings instead of CP and DP-derived embeddings (ESI Fig. 2†). Like the JUMP1 dataset, we also observed significantly greater embedding similarity among different compounds with the same MOA (0.11 average PCC) versus different compounds with different MOAs (4.8 × 10−3 average PCC) (Fig. 5f). This was a smaller differential than what we observed in the JUMP1 dataset, with greater PCC profile variability in the LINCS dataset between different compounds and lower PCC similarity on average between different compounds having the same MOA. From a TSNE visualization of three example MOAs colored by compound, we can see both MOA separability in latent space and clustering between different compounds with the same MOA (Fig. 5g). When we compared baseline MP to heavily optimized and post-processed CP embeddings, MP was still more performant but performance gains were smaller (ESI Fig. 6†).
Like the JUMP1 study, we also performed an analysis where we split the dataset by compound instead of by wells, resulting in 215 compounds that we used as a held-out test set (Fig. 4a). Despite the model never being exposed to these compounds during training, it was able to correctly predict MOAs for 7.9–8.4% of the compounds in a space of 215 possible MOAs (Fig. 4b and c). Compared to a random baseline of 1/215 and expected value of (1/215) × 215 = 1 compounds discovered), this was a 17–18× improvement. Performance varied depending upon whether we predicted MOA by an individual well-level vote or by CLE similarity to a MLE (Methods). Performance differed across the MOAs (ESI Table 2†), with no clear trend between performance and number of training compounds available (ESI Fig. 5A†). Like JUMP1, held-out compound replicate embeddings had greater similarity to each other than to other embeddings (ESI Fig. 5B and C†).
Discussion
Our findings are consistent with the growing body of literature suggesting deep learning can encode broad phenotypic changes captured by Cell Painting. Two points merit emphasis: (1) We developed an embedding encoder for MOA identification of compounds, (2) The approach outperformed both a traditional (via CP) and a deep learning enabled method (via DP) for this task on two independent datasets. Higher similarity of intra-MOA embeddings (even across different compounds) than inter-MOA embeddings suggests that the model was capturing MOA-specific phenotypic features vs. simply learning an individual compound's phenotypic effect. This specificity towards MOA is important because in drug discovery campaigns many compounds could have the same MOA. Furthermore, a compound's MOA is often unknown. Hence, when trying to ascertain an unknown compound's MOA, a reasonable hypothesis is to predict its MOA as the MOA of its most phenotypically similar compounds. Since the embedder can encode similar embeddings for different compounds with the same MOA (Fig. 3f and 5f), perhaps it can likewise be useful for MOA discovery across a diverse compound space. Indeed, the held-out compound analysis (Fig. 4) suggests that the method can be used to identify the MOAs of new compounds the model has never used for training, which is an important use case for HCS.The ranking of PCC similarity for the three pairwise-embedding groups (Fig. 3f and 5f) fit with our expectation: Same-compound same-MOA > different-compound same-MOA > different-compound different-MOA. However, the average pairwise PCC for the same-compound same-MOA group was lower than expected (average PCC = 0.53 for JUMP1, 0.51 for LINCS), indicating embedding variation even between compound replicates. This could be due to several factors, such as natural experimental variation within compound replicates, or the presence of features that are not important for discriminating MOAs (and hence room for model improvement). The average pairwise PCC score of 0.23 (and 0.11 for LINCS) for the different-compound same-MOA pairings was also similar to other recent findings.43 This indicated that although the model correctly grouped different compounds that had the same MOA into the same class, same-MOA embeddings had diversity. Perhaps this is because different compounds in the same MOA class may have differing degrees to which they induce phenotypic changes, with some compounds exerting more evident changes while others were perhaps more modest, which would then lead to phenotypic embeddings with higher variance. Still, the fact that the average PCC for the different-compound same-MOA group was 12 times higher (and 26 times higher for LINCS) than the different-compound different-MOA average PCC indicates that the embeddings were capturing MOA-specific phenotypes despite the diversity within same-MOA groupings. This reinforces the notion that the model had indeed learned shared MOA phenotypes that persisted across different compounds.
Accurate performance on test wells never seen during the model's training suggests that the model was not leveraging intra-well specific features for its learning, which can be a confounder in microscopy.38 Furthermore, we ensured that each MOA's test set wells were spread across multiple plates (Fig. 2d). MP-derived embeddings for JUMP1 data did not seem to encode large batch effects44 (difference in average PCC of same-MOA embeddings was 0.01 between different plates, ESI Fig. 7†). However, this effect was more prevalent for the LINCS data (average PCC difference was 0.10). As a future avenue to explore, perhaps preprocessing images to remediate batch effects in the LINCS dataset prior to learning would enhance performance.
The model encoded MOA-associated phenotypes for two independent Cell Painting datasets and exceeded performance of two other methods in generating MOA-specific embeddings. As an alternative to hand-engineered feature extraction, deep learning did not require biological expertise for deciding which features to extract, yet both DP and MP consistently yielded better results than CP. In comparison to CP, MP was more performant for both class latent assignment and k-NN (Fig. 3d, e and 5d, e). Since we demonstrated that features specific to the biological domain of interest can be learned, we advocate for deep representation learning over traditional feature engineering for phenotypic MOA determination.
Although DP is another deep learning approach to phenotypic profiling that also uses an EfficientNet backbone architecture, we observed larger performance gains with MP. This indicates that architecture alone is not sufficient for better MOA profiling, but rather attention to parameters such as learning objective and image scale is necessary (ESI Fig. 8A†). It is also interesting to see MP's advantage even when compared to a separate DP model that was trained directly on the LINCS dataset instead of on ImageNet (ESI Fig. 8B–E†). This DP model was trained with the objective of classifying compounds as an auxiliary task for the primary task of MOA classification (i.e. weak supervision). When both models were allowed to train on Cell Painting images, MP still yielded improvement in enrichment (2.6× improvement over DP), k-NN metrics (percent improvement: 194% F1, 188% precision, 182% recall), and the class latent assignment metric (percent improvement: 256% F1, 300% precision, 212% recall). DP trained on LINCS had higher performance in enrichment and class latent assignment than DP trained on ImageNet, indicating that feature extraction is most performant when the model is trained on the same data types. Other than the quantitative advantages in performance, MP has a practical advantage over DP of not requiring single cell locations as input, extraction of which can be cumbersome.45,46 MP's standing as compared to DP for biological areas of interest other than MOA have not been determined. Indeed DP (or CP) may be a better choice for other biological domains or other datasets.
It is important to note that metrics of success were confined mainly to the task of identifying MOAs from learned representations. The reason we emphasize embedding generation as opposed to accurate image classification is because a classification model is constrained to the single task of MOA classification, and because the classifier will be of little value if a compound's MOA is not part of the training set. In contrast, an embedder can generate embeddings for any Cell Painting images regardless of MOA (even MOAs not included in the training set). These feature embeddings can then be used for a variety of exploratory downstream tasks other than simply identifying MOAs (e.g. understanding relationships between perturbed MOAs and toxicity, or training additional classifiers on these embeddings for other biological tasks related to MOA). Tests for other biological areas of interest, such as cellular viability, drug toxicity, or protein target identification, have yet to be studied. In such cases, deep learning can provide the advantage of learning features that are directly relevant to the biological area of interest (vs. undirected and unsupervised feature extraction such as CP). We trained the model specifically to identify MOA, but the feature-extraction method can apply directly to any other discrete biological label of interest. However, since we used strong supervision for MOA determination, our trained models are likely not viable for other domains outside of studying MOA-related questions.
Certain considerations focus the scope of this study. Although valuable as a proof-of-concept for assessing MP's ability to embed unseen data in the right MOA spaces (Fig. 3f and g), the well-holdout scheme cannot assess profiling of unseen compounds, which is the more pressing pharmaceutical need. In contrast, the more rigorous compound holdout (Fig. 4) is closer to a real-world use case. Although performance differentials with CP and DP are overall lower for the compound-holdout case than the well-holdout, we demonstrate that MP-derived embeddings can be used to determine MOAs of unseen compounds at a higher rate than CP and DP. For the compound-holdout scheme, our study encompassed relatively few compounds (and only one held-out compound per MOA). Therefore, greater generalizability and application to a larger compound space has yet to be determined. Furthermore, MP performed poorly on certain MOAs (ESI Fig. 9†). This could be due to many reasons such as non-optimal experimental setups (e.g. with plate maps, drug concentration, cell type) or ineffective model architecture and training. Or, since Cell Painting only captured five channels, perhaps certain MOAs do not affect any of the biological markers used in the Cell Painting assay, and hence were phenotypically indistinguishable from control. Or perhaps the limited dataset size for both JUMP and LINCS did not provide adequate sample sizes for robust learning. Additionally, all the analyses were performed on compounds with just one known MOA. Understanding drugs that are associated with multiple MOAs is an important task, but our study did not address this question. Moreover, our study spanned just two concentrations: 5 μM (for JUMP1) and 10 μM (for LINCS). Generalizability to other concentrations, particularly more clinically relevant lower concentrations, has yet to be explored. It is also possible that these concentrations were too low for certain drugs to affect the designated MOA—understanding of a drug's mechanistic effect at specific concentrations remains an open question. Moreover, some MOA labels were at a broad pathway level (e.g. DNA synthesis inhibitor) rather than specific to a target (e.g. CDK inhibitor). Additionally, when we evaluated the method on completely held-out compounds (Fig. 4a), a minority of compounds had their MOAs correctly identified (at most 22% for JUMP1 and 8.4% for LINCS depending on the prediction method). Perhaps this is a limitation of Cell Painting's ability to have distinguishable phenotypes for certain MOAs. Or perhaps compounds even within the same MOA class exerted unique phenotypic changes. For certain compounds, these changes may have been too different from what the model was exposed to during training. Despite identifying a minority of held-out compounds, performance was still higher on average than both CP and DP (and multiple folds above a random classification). MOA identification is an innately difficult problem, but since MP performed above the industry gold-standard (CP) for phenotypic profiling, we advocate for MP as a means of directed hypothesis generation. Furthermore, as with most deep representation learning approaches, interpretability is lacking and falls behind traditional computer vision approaches like CP, which is more interpretable. The latent feature embeddings that MP creates, although more performant in this context, are largely uninterpretable. Lastly, practical considerations may also inform the application of the method. EfficientNets require high memory consumption and graphic processing unit (GPU) hardware for tractable training. However, if adopters of the method simply apply MP without any training on their datasets, then memory and compute constraints are much less limiting. Furthermore, we found the model did not need all technical replicates for training for near-maximal performance, especially with the LINCS dataset, indicating that identifying MOAs is possible in smaller datasets (ESI Fig. 10†).
Here we provide a tool for creating quantitative representations relevant to the task of MOA identification. With a MOA-specific embedder, we can query a drug's phenotypic effect on cells and determine its MOA by similarity. We can then follow up with these predictions via traditional target-based screens. This strategy of broad profiling followed with target-based experiments can potentially be a powerful and cost-effective means of searching for new therapeutics. Moreover, if the hypothesis holds true that biologically similar MOAs would be similar in phenotypic latent space, then a future direction can be using tools like MP to query biological similarity between different MOAs. Although our study included hundreds of MOAs, including more MOAs would be another useful direction that will likely increase biological inference power. Another future endeavor can be understanding how concentration affects MOA. Since MP relies solely on phenotypic data, we can query images taken at different concentrations and generate hypotheses about MOA concentration dependence. We hope that the method will be readily applicable to archival Cell Painting datasets and of broad use to future phenotypic screening endeavors. To facilitate open sharing, the model and source code are freely available at https://github.com/pfizer-opensource/moa-profiler/.
Methods
Dataset preparation
The JUMP1 dataset is described in Chandrasekaran et al.34 Download instructions can be found here: https://github.com/jump-cellpainting/2021_Chandrasekaran_submitted. We downloaded the data on February 14, 2022. Briefly, they conducted the Cell Painting protocol with compound perturbation plated at 5 μM. We kept only compounds that had no more than one known MOA according to the CLUE Connectivity Map,39 which can be found at https://clue.io/. Experimentalists used both U2OS and A549 cells, which were subjected to both 24 and 48 hours of compound treatment before being imaged. The resulting dataset after filtering single-MOA compounds consisted of 81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 310 images (66270 excluding DMSO) and 7958 wells (6486 excluding DMSO) coming from 23384-well plates.
310 images (66270 excluding DMSO) and 7958 wells (6486 excluding DMSO) coming from 23384-well plates.
The LINCS Cell Painting dataset36,43 can be found at doi: 10.5281/zenodo.5008187. We downloaded the data on March 12, 2022, and sub-selected the data as follows. We chose batch 1 due to its larger cell count. For batch 1, authors only used A549 cells, which were subjected to 48 hours of compound treatment before being imaged. Like the JUMP1 dataset, we kept only compound data that had no more than one known MOA according to the CLUE Connectivity Map. We also sub-selected compounds at 10 μM concentration because this dose produced the strongest phenotype across compounds.43 The resulting dataset after filtering for single-MOA compounds at 10 μM concentration consisted of 87729 images (56283 excluding DMSO) and 9749 wells (6255 excluding DMSO) coming from 136384-well plates. All compounds had replicates plated at the same well locations.
Data preprocessing and model training
Each well was shuffled and partitioned into only one of three sets, following a 60% training, 10%, validation, and 30% test split, such that each well and all its image fields were assigned to only one of the three sets. The 60/10/30 dataset splits were constructed independently for each MOA. Each MOA was present in each of the three sets. We then class-balanced the training set so that each MOA had equal representation (by duplicating minority-class image examples shown to the model). We also included the negative DMSO as a class to learn but excluded it from all performance metrics because of its overrepresentation in the dataset. All MOAs (including MOAs with only one compound) were included in training to maximize the dataset size and to direct the model to differentiate between as many MOAs as possible. However, these single-compound MOAs were excluded in all test analyses to better assess inter-compound MOA profiling versus replicate profiling.For both datasets, images were captured in five channels at a resolution of 1080 × 1080px. The five channels came from five different biomarkers: Hoechst 33342 for DNA, Alexa 488 for the endoplasmic reticulum, Alexa 488 long for RNA, Alexa 568 for the actin cytoskeleton, golgi, and plasma membrane, and Alexa 647 for the mitochondria. We first scaled each image to the range zero to one, and then standardized them to a mean of zero and standard deviation of one. We augmented training by permuting each channel's brightness and contrast independently by a random factor in the range of 0 to 0.30 (just for the LINCS dataset because the image type for JUMP1 was not supported by PyTorch's transforms.ColorJitter function). As a final training augmentation step for both datasets, we performed random 90-degree rotations on each image, along with random horizontal flips. For a given field of view, we stacked each of the five augmented image channels into a single tensor.
We then fed this tensor (5 × 1080 × 1080) into a modified EfficientNet-B0 architecture with the task of classifying the image's compound's MOA (multi-class classification). The only modification to EfficientNet was adjusting the input layer to receive five channels instead of three. We trained for 100 epochs and selected the model that had the highest accuracy on the validation set (epoch 76 for JUMP1 and epoch 82 for LINCS). We used a learning rate of 0.1, a weight decay of 0.0001, a dropout rate of 0.2, a learning momentum of 0.9, a learning rate scheduler with a gamma decay of 0.1 at epoch 50 and 75, and batch size of 56 for training. Hyperparameters were chosen based on the default recommendations of the EfficientNet package. We trained using four NVIDIA A-100 GPUs.
For training on smaller subsets of data (ESI Fig. 10†), all hyperparameter choices were kept the same as the training scheme on the full training set. The compound-replicate wells excluded from training were chosen randomly for each training run. The maximal allotted compound-replicates values went up to the maximal number of compound replicates in the full training set.
For the logistic regression models (ESI Fig. 2†), we trained models to classify an image embedding's MOA. We used sklearn's LogisticRegression package with a one-vs.-rest scheme, L2 regularization, the Broyden–Fletcher–Goldfarb–Shanno algorithm solver, and a max-iteration count of 10000. These were the standard default parameters except for the max-iteration count, which required more iterations before convergence. To generate the MP training set embeddings, we fed each image in the training set through the trained EfficientNet, extracted the last convolutional layer, performed an average pool operation, and flattened the result. For CP and DP training set embeddings, we DMSO-standardized the existing embeddings (see CellProfiler and DeepProfiler Extraction) before feeding them to the logistic regression model. We then applied the trained logistic regression models (one for each of MP, CP, and DP) to the held-out test set filtered for multiple-compound MOAs.
CellProfiler and DeepProfiler extraction
All CP embeddings were provided at the well-level (in this case, average embeddings of all single cells within a well). We downloaded CP embeddings for the JUMP1 dataset from https://github.com/jump-cellpainting/2021_Chandrasekaran_submitted. The CP pipeline used for extraction can be found at https://github.com/jump-cellpainting/2021_Chandrasekaran_submitted/tree/main/pipelines/2020_11_04_CPJUMP1. We downloaded CP embeddings for LINCS from: https://github.com/broadinstitute/lincs-cell-painting/tree/master/profiles. The CP pipeline used for extraction can be found at https://github.com/broadinstitute/lincs-cell-painting/blob/master/profiles/README.md and https://github.com/broadinstitute/lincs-cell-painting/blob/master/profiles/profiling_pipeline.py.To compare CP and MP embeddings independent of the many possible downstream post-processing transformations, we used the raw provided CP embeddings and performed a simple standardization of features with respect to the DMSO control wells on the same plate (DMSO-standardization). For each feature f on plate p, we subtracted the mean of the DMSO wells of plate p of feature f and divided by the standard deviation of the DMSO wells of plate p of feature f. This was done to prevent features with wider ranges from disproportionately affecting similarity metrics, and to normalize features to the plate controls. To compare baseline MP performance to more optimized CP embeddings (robustizing by plate via median absolute deviation, selecting features via variance and feature correlation thresholding, and spherizing to DMSO), we also downloaded optimized profiles for JUMP1 from https://github.com/jump-cellpainting/2021_Chandrasekaran_submitted/tree/main/profiles and for LINCS from https://github.com/broadinstitute/lincs-cell-painting/tree/master/spherized_profiles/profiles. We compared these hyper-optimized profiles with baseline MP embeddings that underwent DMSO-standardization (ESI Fig. 4 and 6†).
The repository for DP can be found at https://github.com/cytomining/DeepProfiler. DP leverages an EfficientNet trained on the ImageNet dataset for profiling. DP employed a weak-supervision approach (i.e. train on one task like ImageNet or compound identification, evaluate on a different task like MOA similarity). We derived DP embeddings using the “profile” function of DP with the default configurations and pre-trained weights automatically downloaded by DP on April 12, 2022. We aggregated single-cell DP embeddings into well-level embeddings by taking the median over all single-cell embeddings within a well, and then DMSO-standardized the features.
When comparing MP and DP when both were allowed to train on the LINCS dataset (ESI Fig. 8†), we extracted and DMSO-standardized all available DP embeddings from https://github.com/broadinstitute/neural-profiling/tree/main/training/runs/1102. We chose these embeddings because the author reported the highest validation accuracy on this set. Outlier control was not applied to any of the CP, DP, or MP embeddings.
Classification analysis
For all classifier evaluation metrics (Fig. 3a and 5a), we binarized the classes using sklearn's label_binarize function, and plotted using sklearn's precision_recall_curve function. Random baseline was set to the positive prevalence of the binarized labels. We did not include the negative control DMSO condition for analysis of raw-image classification due to its large class over-representation.Embedding extraction and analysis
Once the MP models were fully trained, we applied them to the held-out test set of wells never exposed to the model's training. Since a single-compound MOA cannot be disentangled from its compound, we removed these MOAs and corresponding wells from the test set. To extract MP embeddings, we fed each image within a well through the EfficientNet, extracted the last convolutional layer, performed an average pool operation, flattened the result, and DMSO-standardized the embedding. The image-field level embeddings had a size of 1280. We then took the median of all image-field level embeddings that belonged to the same well and assigned this as the well-level embedding.Once we had well-level embeddings, we performed four analyses to assess how well the embeddings captured MOA-specific features. We excluded negative control DMSO wells from all embedding analyses. For assessing the similarity between two embeddings, we used PCC (centered cosine similarity).
First, we calculated the enrichment factor, which was the odds ratio in a one-sided Fisher's exact test (Fig. 3b and 5b). This test assessed whether high PCC similarity for an embedding pair is independent of the embeddings sharing the same MOA. We used a range of PCC percentiles (from the 98th to the 99.8th percentile) as the thresholds for determining which embedding pairs were considered strongly correlated (versus weakly correlated). The odds ratio was calculated as (a/b)/(c/d) for the following 2 × 2 frequency table:
| MOA replica | Non-MOA replica | |
|---|---|---|
| Strongly correlated | a | b |
| Weakly correlated | c | d |
Second, we calculated the average pairwise PCCs for two groupings of well-level embeddings: (1) those that had the same MOA, (2) those that had different MOA. Within each grouping, we calculated the PCC for each pair of wells and averaged the result. Delta values were the differences between the two groups' averages (Fig. 3c and 5c).
Third, we calculated k-NN metrics by finding the k closest neighbors for each well based on PCC similarity between the well embeddings and taking the majority MOA of those neighbors as the predicted MOA (Fig. 3d and 5d). We evaluated all possible values of k from one to the total number of embeddings minus one. For each k, we used sklearn's f1_score, precision_score, and recall_score functions. Averages were weighted by support (the number of true instances for each label).
Fourth, we derived a MLE by grouping all the wells of that MOA and taking the median (Fig. 3e and 5e). We treated each well in the test set as a query well. For each query well, we calculated the class embeddings with the query well excluded, and then assigned a prediction for the query well's MOA based on which MLE was the most similar (by PCC) to the query well's embedding. We calculate weighted-average F1, precision, and recall scores for these predictions with a one-vs.-rest scheme using the sklearn's f1_score, precision_score and average_recall_score functions.
As a last metric of embedding integrity and MOA-specificity, we calculated pairwise PCC averages of three groups: (1) embeddings with the same compound (and hence same MOA), (2) embeddings with different compound but same MOA, and (3) embeddings with different compound and different MOA (Fig. 3f and 5f). All pairs were unique and an embedding was never paired with itself. We determined statistical significance of difference of means with a two-sided z-test (see “Statistical Tests”).
Analysis for held-out compounds
For the held-out compound analyses (Fig. 4), we split the dataset by compound instead of by well. We held out one randomly selected compound for each MOA class that was represented by at least two unique compounds. All other compounds were used for training and validation, with 70% of the wells used for training, and 30% for validation (wells were assigned randomly). We trained a new model based on this dataset split using the same hyperparameter choices as the analysis for the well-split scheme. We made MOA predictions for the held-out compounds using the model's generated embeddings. For the training set, each MOA was assigned a MLE by aggregating (via median) over all well-level embeddings belonging to the MOA. For determining the final MOA of a held-out compound, we evaluated two methods:(1) Individual vote: For each well-level embedding in the held-out test set, we assigned its MOA prediction as the MOA of the most similar MLE via PCC. Finally, we assigned a compound's MOA prediction as the majority MOA over these well-level predictions.
(2) Aggregated vote: For each held-out compound, we derived an aggregated CLE by taking the median over all well-level embeddings of the compound. Finally, we assigned the compound's MOA prediction as the MOA of the MLE most similar to CLE by PCC.
Statistical tests
For all statistical test of significance, we performed a two-sided z-test for difference of means. We used a null hypothesis stating that the means were equal, and an alternative hypothesis stating that the means were different. Significance was set at p < 0.05. Each sample corresponded to a well-level embedding.Data availability
The code and datasets used in this study are public and can be found at https://github.com/pfizer-opensource/moa-profiler.Conflicts of interest
There are no conflicts of interest to declare.References
- J. A. Lee, M. T. Uhlik, C. M. Moxham, D. Tomandl and D. J. Sall, Modern phenotypic drug discovery is a viable, neoclassic pharma strategy, J. Med. Chem., 2012, 55, 4527–4538 CrossRef CAS PubMed.
- Z. Li, M. E. Cvijic and L. Zhang, Cellular imaging in drug discovery: Imaging and informatics for complex cell biology, in Comprehensive Medicinal Chemistry III, eds., Chackalamannil, S., Rotella, D. and Ward, S. E., Elsevier, 2017, pp. 362–387 Search PubMed.
- M.-A. Bray, et al., Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes, Nat. Protoc., 2016, 11, 1757–1774 CrossRef CAS PubMed.
- J. C. Caicedo, C. McQuin, A. Goodman, S. Singh and A. E. Carpenter, Weakly supervised learning of single-cell feature embeddings, Proc. IEEE Comput. Soc. Conf. Comput. Vis. Pattern Recognit., 2018, 9309–9318 Search PubMed.
- S. J. Warchal, et al., High content phenotypic screening identifies serotonin receptor modulators with selective activity upon breast cancer cell cycle and cytokine signaling pathways, Bioorg. Med. Chem., 2020, 28, 115209 CrossRef CAS PubMed.
- R. E. Hughes, et al., Multiparametric high-content Cell Painting identifies copper ionophores as selective modulators of esophageal cancer phenotypes, ACS Chem. Biol., 2022, 17, 1876–1889 CrossRef CAS PubMed.
- V. Ljosa, et al., Comparison of methods for image-based profiling of cellular morphological responses to small-molecule treatment, J. Biomol. Screening, 2013, 18, 1321–1329 CrossRef CAS PubMed.
- M. Hofmarcher, E. Rumetshofer, D.-A. Clevert, S. Hochreiter and G. Klambauer, Accurate prediction of biological assays with high-throughput microscopy images and convolutional networks, J. Chem. Inf. Model., 2019, 59, 1163–1171 CrossRef CAS PubMed.
- A. E. Carpenter, et al., CellProfiler: image analysis software for identifying and quantifying cell phenotypes, Genome Biol., 2006, 7, R100 CrossRef PubMed.
- M.-A. Bray, M. S. Vokes and A. E. Carpenter, Using CellProfiler for automatic identification and measurement of biological objects in images, Curr. Protoc. Mol. Biol., 2015, 109, 14.17.1–14.17.13 Search PubMed.
- L. S. Gasparini, et al., In vitro cell viability by CellProfiler® software as equivalent to MTT assay, Pharmacogn. Mag., 2017, 13, S365–S369 CrossRef PubMed.
- I. Nassiri and M. N. McCall, Systematic exploration of cell morphological phenotypes associated with a transcriptomic query, Nucleic Acids Res., 2018, 46, e116 CrossRef PubMed.
- Y. Bengio, A. Courville and P. Vincent, Representation learning: A review and new perspectives, arXiv [cs.LG], 2012 Search PubMed.
- A. Kopf and M. Claassen, Latent representation learning in biology and translational medicine, Patterns, 2021, 2, 100198 CrossRef CAS PubMed.
- A. S. Garruss, K. M. Collins and G. M. Church, Deep representation learning improves prediction of LacI-mediated transcriptional repression, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022838118 CrossRef CAS PubMed.
- E. C. Alley, G. Khimulya, S. Biswas, M. AlQuraishi and G. M. Church, Unified rational protein engineering with sequence-based deep representation learning, Nat. Methods, 2019, 16, 1315–1322 CrossRef CAS PubMed.
- I. Landi, et al., Deep representation learning of electronic health records to unlock patient stratification at scale, NPJ Digit. Med., 2020, 3, 96 CrossRef PubMed.
- Y. Si, et al., Deep representation learning of patient data from Electronic Health Records (EHR): A systematic review, J. Biomed. Inform., 2021, 115, 103671 CrossRef PubMed.
- C. G. S. I. A. R. Sur, gene-subcategory interaction-based improved deep representation learning for breast cancer subcategorical analysis using gene expression, applicable for precision medicine, Med. Biol. Eng. Comput., 2019, 57, 2483–2515 CrossRef PubMed.
- L. B. Tulloch, et al., Direct and indirect approaches to identify drug modes of action, IUBMB Life, 2018, 70, 9–22 CrossRef CAS PubMed.
- M.-A. Trapotsi, L. Hosseini-Gerami and A. Bender, Computational analyses of mechanism of action (MoA): data, methods and integration, RSC Chem. Biol., 2022, 3, 170–200 RSC.
- Mechanism matters, Nat. Med., 2010, 16, 347, https://www.nature.com/articles/nm0410-347 Search PubMed.
- Chemical biology for target identification and validation, MedChemComm, 2014, 5, 244–246, https://pubs.rsc.org/en/content/articlehtml/2014/md/c4md90004a Search PubMed.
- C. Liu, et al., Deep learning-driven prediction of drug mechanism of action from large-scale chemical-genetic interaction profiles, J. Cheminform., 2022, 14, 12 CrossRef PubMed.
- J. Dai and S. Latifi, A deep learning framework for prediction of the mechanism of action, Int. J. Comput. Appl., 2021, 183, 1–7 Search PubMed.
- G. Jang, et al., Predicting mechanism of action of novel compounds using compound structure and transcriptomic signature coembedding, Bioinformatics, 2021, 37, i376–i382 CrossRef CAS PubMed.
- C. Kandaswamy, L. M. Silva, L. A. Alexandre and J. M. Santos, High-content analysis of breast cancer using single-cell deep transfer learning, J. Biomol. Screening, 2016, 21, 252–259 CrossRef CAS PubMed.
- O. Z. Kraus, J. L. Ba and B. J. Frey, Classifying and segmenting microscopy images with deep multiple instance learning, Bioinformatics, 2016, 32, i52–i59 CrossRef CAS PubMed.
- W. J. Godinez, I. Hossain, S. E. Lazic, J. W. Davies and X. Zhang, A multi-scale convolutional neural network for phenotyping high-content cellular images, Bioinformatics, 2017, 33, 2010–2019 CrossRef CAS PubMed.
- N. Pawlowski, J. C. Caicedo, S. Singh, A. E. Carpenter and A. Storkey, Automating morphological profiling with generic deep convolutional networks, bioRxiv, 2016, DOI:10.1101/085118.
- D. M. Ando, C. Y. McLean and M. Berndl, Improving phenotypic measurements in high-content imaging screens, bioRxiv, 2017, DOI:10.1101/161422.
- A. Kensert, P. J. Harrison and O. Spjuth, Transfer learning with deep convolutional neural networks for classifying cellular morphological changes, SLAS Discov., 2019, 24, 466–475 CrossRef PubMed.
- G. Tian, P. J. Harrison, A. P. Sreenivasan, J. Carreras-Puigvert and O. Spjuth, Combining molecular and cell painting image data for mechanism of action prediction, Artif. Intell. Life Sci., 2023, 100060 CAS.
- S. N. Chandrasekaran, et al., Three million images and morphological profiles of cells treated with matched chemical and genetic perturbations, bioRxiv, 2022, DOI:10.1101/2022.01.05.475090.
- A. B. Keenan, et al., The library of integrated network-based cellular signatures NIH program: System-level cataloging of human cells response to perturbations, Cell Syst., 2018, 6, 13–24 CrossRef CAS PubMed.
- T. Natoli, et al., Broadinstitute/Lincs-Cell-Painting: Full Release of LINCS Cell Painting Dataset, 2021, DOI:10.5281/ZENODO.5008187.
- DeepProfiler: Morphological profiling using deep learning, https://github.com/cytomining/DeepProfiler.
- N. Moshkov, et al., Learning representations for image-based profiling of perturbations, bioRxiv 2022.08.12.503783, 2022, DOI:10.1101/2022.08.12.503783.
- A. Subramanian, et al., A next generation connectivity map: L1000 platform and the first 1,000,000 profiles, Cell, 2017, 171, 1437–1452.e17 CrossRef CAS PubMed.
- A. Anighoro, J. Bajorath and G. Rastelli, Polypharmacology: challenges and opportunities in drug discovery, J. Med. Chem., 2014, 57, 7874–7887 CrossRef CAS PubMed.
- M. Tan and Q. V. Le, EfficientNet: Rethinking model scaling for convolutional Neural Networks, arXiv [cs.LG], 2019 Search PubMed.
- B. K. Lundholt, K. M. Scudder and L. Pagliaro, A simple technique for reducing edge effect in cell-based assays, J. Biomol. Screening, 2003, 8, 566–570 CrossRef CAS PubMed.
- G. P. Way, et al., Morphology and gene expression profiling provide complementary information for mapping cell state, bioRxiv, 2021, DOI:10.1101/2021.10.21.465335.
- C. A. Glastonbury, M. Ferlaino, C. Nellåker and C. M. Lindgren, Adjusting for confounding in unsupervised latent representations of images, arXiv [cs.CV], 2018 Search PubMed.
- N. F. Greenwald, et al., Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning, Nat. Biotechnol., 2022, 40, 555–565 CrossRef CAS PubMed.
- J. C. Caicedo, et al., Evaluation of deep learning strategies for nucleus segmentation in fluorescence images, Cytometry, Part A, 2019, 95, 952–965 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00060e |
| This journal is © The Royal Society of Chemistry 2023 |