Chemical biology for target identification and validation
The struggles faced by the pharmaceutical industry in the high risk pursuit of new medicines, particularly through clinical development, have been highlighted recently. The main reason for high attrition rates is a lack of clinical efficacy demonstrated by the candidate drug and by successfully addressing this issue the industry could significantly improve its productivity.1,2 This has led many to argue that an increase in preclinical phenotypic screening (rather than target-based approaches) is required to deliver molecules that possess the desired efficacy and ‘horsepower’ in the cell or tissue of interest, and that this will result in an improvement in candidate drug survival.3This is an attractive paradigm, but it is important to appreciate that a lack of knowledge of the target protein is a significant obstacle to project progression, since many phenotypic screens are low throughput and technically challenging. Therefore, a recommended approach to expedite progression of a phenotypic screen-based project is to follow-up efficacious molecules with target identification and mode of action research to facilitate the drug discovery programme. If successful, not only may high throughput screening and synthesis be employed, but the biochemical insights provided will enable biomarker development and safety windows to be further understood.4 Biophysical methods such as structure-based drug design can be leveraged to significantly increase the speed of the optimization process. Target identification may also be employed to understand off-target effects for a drug that demonstrates a toxicological phenotype, thus helping address the other major cause of drug attrition.
Target elucidation for small molecule drugs is a time consuming and expensive task – there still exists a certain amount of naivety to these facts, which can be dangerous if the focus is on short term deliverables (for example, it is rare to read of failed target identification efforts in the literature). Firstly, there are a number of different technologies that can be applied to identify putative targets. Then each technology may require several years optimization to translate data output into meaningful information and hypothesis generation. And finally, each technology has its own advantages and limitations, and therefore amalgamating these different read-outs into a single irrefutable ‘answer’ is extremely challenging. These endeavours will no doubt increasingly rely on sophisticated computational techniques to decipher trends and inform future experiment design.
However, it should also be pointed out that the elucidation of pathways perturbed by a drug candidate may significantly advance a therapeutic approach and enable biomarker discovery, even if the exact target is not identified. It may also be that a molecule exerts a phenotypic effect through the binding of several protein targets, and some may argue that it is even more likely to be the case than not since many successful drugs discovered this way have been shown to be polypharmacological.
This special issue highlights recent advances in the application of chemical biology to drug discovery, with a particular focus on the development of new technologies that enable target identification and validation, or that further our understanding of molecular pharmacology. In particular, the synergy between chemical biology, biophysics and medicinal chemistry is a key facet of the research presented. Another overarching theme is the importance of molecular design and a deep appreciation of many of the factors that are important in medicinal chemistry, such as physicochemistry, molecular interactions and biodistribution, are also essential to the development of effective chemical probes and technologies (Fig. 1).
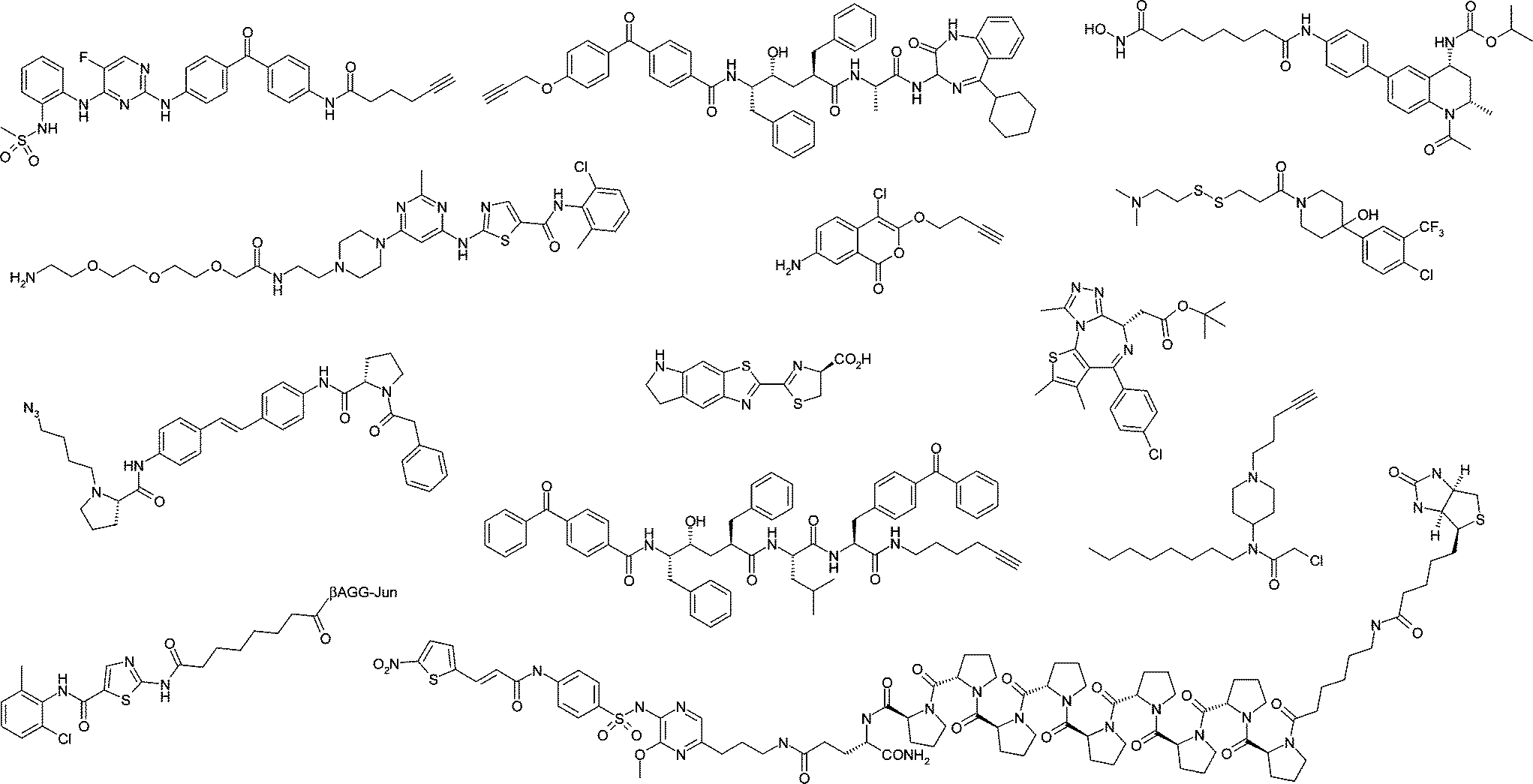 | ||
| Fig. 1 Some of the chemical biology probes highlighted in this issue. | ||
Ghosh and Jones (DOI:10.1039/c3md00277b) review the use of small molecule imaging probes that rely on what is termed a ‘silent’ reporter (a click handle) onto which a fluorescent dye can be appended intracellularly. These probes thus provide a more accurate picture of subcellular distribution and target engagement of the small molecule since the physicochemistry of a fluorometric dye can perturb the function of a chemical tool.
Another imaging technique, bioluminescence, is reviewed by Paley and Prescher (DOI:10.1039/c3md00288h). Applications of bioluminescence to visualizing biological processes in vivo are highlighted and the relevance of the technology to the medicinal chemistry community is described. It is apparent from the review that there are many exciting opportunities for the molecular design of novel luciferins and luciferase probes with unique functions and improved photostability.
Davda and Martin (DOI:10.1039/c3md00333g) review the design and development of inhibitors of acyl protein thioesterase (APT) enzymes, which process certain palmitoylated proteins. These tools have been used to interrogate the biochemistry and mechanisms of protein palmitoylation. A highlight in this area has been the integration of activity-based protein profiling (ABPP) methods with medicinal chemistry to optimise the discovery of selective inhibitors in complex proteomes. Another important application of the covalent probes has been the determination of target engagement ex vivo, thus providing an important component of translational pharmacology.
Affinity-based technologies have played essential roles in target identification and validation, and several examples are showcased in this issue. Kawatani and Osada (DOI:10.1039/c3md00276d) nicely review this important area, covering recent successful approaches using affinity purification with quantitative proteomics, array-based methods, photoaffinity labelling (often used in conjunction with bioorthogonal click chemistry) and label free methods. The authors also highlight the need for using these techniques in combination with other -omic approaches and imaging to more confidently define true binding partners, an important theme in this issue.
An affinity-based chemoproteomic approach was originally used to identify the BET bromodomains as targets of a phenotypic screening hit bearing the benzodiazepine unit. Müller and Knapp (DOI:10.1039/c3md00291h) review this discovery and the subsequent development and application of BET inhibitors (facilitated by their accessibility through the Structural Genomics Consortium) to help elucidate bromodomain biology, particularly in the areas of oncology and inflammation. Another family of important epigenetic regulators, the histone demethylase classes JMJD2 and JARID1, are reviewed by Zheng and Huang (DOI:10.1039/c3md00325f). This account explores the recent contributions made to further our pharmacological and mechanistic understanding of these enzymes.
Atkinson and co-workers (DOI:10.1039/c3md00285c) leverage the wealth of structural information for the BET and histone deacetylase (HDAC) proteins to design bifunctional HDAC/BET inhibitors as unique epigenetic modulators. A tetrahydroquinoline core (BET motif) was functionalised with hydroxamic acid (HDAC motif) to achieve efficient dual pharmacology, as confirmed by chemoproteomic techniques. These tools will provide further insights into the interplay of these epigenetic mechanisms.
The development of a useful chemical tool to inhibit the ubiquitin–proteasome system (UPS) is described by Linder and co-workers (DOI:10.1039/c3md00386h). Through a classic mechanistic investigation into the biochemical effects of this inhibitor, further pharmacological insight of this modality was gleaned.
Pan and co-workers (DOI:10.1039/c3md0000286a) describe the use of photoaffinity labelling to delineate the ‘off-targets’ of a LRRK2 inhibitor. A clickable 2,4-diaminopyrimidine photoprobe isolated glyoxalase I (GLO-1) from lysate and intact cells, and the chemical tools thus afforded a novel approach to profile GLO-1 biology in live cells. The authors also apply the techniques reviewed by Ghosh and Jones to determine the cellular localisation of probe targets using in-cell photolabeling and subsequent click chemistry-enabled imaging.
γ-Secretase is an aspartyl protease that plays important roles in Alzheimer's disease and cancer, and has therefore been the focus of significant therapeutic research. Two papers in this issue explore the pharmacology and mechanisms of action of γ-secretase modulators using clickable photoaffinity labelling probes. Li and co-workers (DOI:10.1029/c3md281k) developed a benzophenone-alkyne tethered version of a known inhibitor (CBAP-BPyne) to selectively label the presenilin N-terminal fragment of γ-secretase that should enable further understanding of the mechanisms of endoproteolysis of the target.
Ballard, Johnson and co-workers (DOI:10.1039/c3md283g) explore the protein interactions of γ-secretase using clickable chemical probes bearing two photoaffinity labels. The first photohandle captures the target protein, whilst the second is remote from the active site, so enabling accessory protein isolation. The bis-benzophenone probe was able to label the N- and C-terminal fragments of presinilin and nicastrin, and further refinement of the probe design would likely enable capture of the exo- γ-secretase complex.
Lei and co-workers (DOI:10.1039/c3md00278k) describe an impressive example of target identification using affinity pull-down experiments. SAR optimization of a hit from a phenotypic screen for necroptosis led to the development of a molecule dubbed ‘necrosulfonamide’ (NSA). Immobilization of this inhibitor using a rigid polyproline linker, which improved isolation of low abundance proteins, identified Mixed Lineage Kinase Domain-Like Protein (MLKL) as a direct target for NSA.
Maly, Ong and co-workers (DOI:10.1039/c3md00315a) further developed and optimised a technology that utilised quantitative mass spectrometry proteomics in combination with small molecule affinity matrices to accurately and rapidly measure direct targets of kinase inhibitors, including dasatinib and a panel of type II inhibitors. Additionally, phosphorylation sites for some kinases were elucidated without the need for further sample processing. This powerful technique illustrates the value of chemoproteomics in the drug discovery arena as it provides an unbiased assessment of selectivity and potential off-targets in whole cell lysates.
The split-luciferase based three-hybrid system is another approach that has been used previously to determine kinase inhibitor selectivity. The original method used the promiscuous kinase inhibitor staurosporine as a warhead to design chemical inducers of dimerisation (CID) in the three-hybrid system, but Ghosh and co-workers (DOI:10.1039/c3md00275f) describe in this issue the development of a novel CID, using a dasatinib derivative, to deliver a new screening technology to target tyrosine kinases specifically. Interestingly, this work suggests that lower affinity analogues of known inhibitors may be required to optimise displacement-based screening methods.
Couvertier and Weerapana (DOI:10.1039/c3md00289f) generated a small library of 4-aminopiperidine, cysteine-reactive probes primed with bioorthogonal clickable tags to enable subsequent target visualization and identification. In this remarkably simple and synthetically-enabled library, probes that reacted with GSTO1 and AKT1 were identified which could be used to explore cellular activity of these enzymes, similar to ABPP. This proof-of-principle experiment nicely illustrates the potential of larger ‘chemical biology’ libraries, where SARs for novel functions can be generated alongside more traditional ‘medicinal chemistry’ SAR generation on projects.
Wells, Pomerantz, Mapp and co-workers (DOI:10.1039/c3md356f) describe the development of a new technique using fluorescence polarization tethering (FP Tethering) to identify small molecules that perturb protein–protein interactions, in this case, between the KIX domain of CBP and the transcriptional activator peptide pKID. This fragment-based method relies on the equilibrium established between a library of disulfide-containing fragments and a target protein bearing a native or engineered cysteine proximal to the binding site. Fragments that interact with the protein will favour the formation of a mixed disulfide with the target, which in this work results in the displacement of a fluorometric peptide binder from the protein. This technology is an important innovation in the development of drugs that modulate protein–protein interactions.
And finally, Bender and co-workers (DOI:10.1039/c3md00313b) present a fascinating study of the importance of phenotype classification on subsequent in silico mode of action analyses. This work illustrates the importance of accurate and consistent phenotype annotations, which is particularly important when assigning a number of molecules the same phenotype for mechanistic elucidation
We are extremely grateful to the contributors of this special issue. They have pioneered the application of chemical biology to drug discovery and highlighted the value of research at the interface of chemistry and biology.
Lyn H. Jones and Nathanael S. Gray
Guest Editors
References
- D. C. Swinney and J. Anthony, Nat. Rev. Drug Discovery, 2011, 10, 507–519 CrossRef CAS PubMed.
- M. E. Bunnage, Nat. Chem. Biol., 2011, 7, 335–339 CrossRef CAS PubMed.
- G. C. Terstappen, C. Schlüpen, R. Raggiaschi and G. Gaviraghi, Nat. Rev. Drug Discovery, 2007, 6, 891–903 CrossRef CAS PubMed.
- M. E. Bunnage, E. L. P. Chekler and L. H. Jones, Nat. Chem. Biol., 2013, 9, 195–199 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |