
Synthesis, self-assembly and optical properties of some rigid π-bridged triphenylene dimers†
Hang
Lin
a,
Ke-Xiao
Zhao
a,
Min
Jing
a,
Xiu-Hai
Long
a,
Ke-Qing
Zhao
 *a,
Ping
Hu
a,
Bi-Qin
Wang
a,
Peng
Lei
b,
Qing-Dao
Zeng
b and
Bertrand
Donnio
*c
*a,
Ping
Hu
a,
Bi-Qin
Wang
a,
Peng
Lei
b,
Qing-Dao
Zeng
b and
Bertrand
Donnio
*c
aCollege of Chemistry and Materials Science, Sichuan Normal University, Chengdu 610066, China. E-mail: kqzhao@sicnu.edu.cn
bKey Laboratory of Standardization and Measurement for Nanotechnology, Centre for Excellence in Nanoscience, National Centre for Nanoscience and Technology (NCNST), Beijing 100190, China
cInstitut de Physique et Chimie des Matériaux de Strasbourg (IPCMS), CNRS-Université de Strasbourg (UMR7504), Strasbourg, 67034, France. E-mail: bertrand.donnio@ipcms.unistra.fr
First published on 2nd September 2022
Abstract
The synthesis of an exclusive family of liquid-crystalline dimers consisting of two protomesogenic triphenylene moieties connected via a rigid, π-conjugated bridge, and the study of their mesomorphous, gel self-assembly and optical properties as a function of the nature of the bridge are reported. Various triphenylene bridged dimers were successfully prepared by either palladium-catalyzed Suzuki–Miyaura cross-coupling between pentakis(alkoxy)triphenylene nonaflates and various commercially accessible aryldiboronic acids (as bridges) or by FeCl3-promoted Scholl oxidative homo-coupling of thiophene/thienothiophene/furan-containing pentakis(alkoxy)triphenylene derivatives. All linear dimers are mesomorphous, and self-organize into large multi-columnar rectangular superlattices, with columnar pairs arranged according to a chevron-like pattern, as deduced from SWAXS and supported by STM. At high temperature, most compounds also display a nematic columnar mesophase (NCol), easily recognized by characteristic optical textures. The transition temperature and mesophase ranges show a high dependency on the bridge nature and, to a lesser extent, on the chain-lengths around the triphenylene moieties. The sole kinked bridged-dimeric homolog of this series exhibits a single, room-temperature hexagonal mesophase (Colhex), as do the monofunctionalized triphenylene precursors, whereas the dimeric triphenylene with no bridge is not mesomorphous. UV-visible absorption and fluorescence emission spectra were measured in both solvents and thin films. The π-conjugated bridged-dimers exhibit emission spanning from 400–700 nm, thus covering the full visible-light range, whose emission maxima are obviously influenced by the chemical nature of the bridge. Furthermore, fluorescence quantum yields as high as 64% were measured for some of them. DFT theoretical computing calculations fully support the experimental measurements. Most dimers also form gels in cyclohexane and emit blue-to-orange light when irradiated by UV light; the corresponding xerogels revealed that their gelation ability results from the 3D morphology of entangled ultra-long and thin microfibers. The facile synthesis of these unique, multifunctional rigid discotic dimers, their rich mesomorphism, strong gelation ability and fine-tuned photophysical properties make these materials very attractive for the active field of organic electronics.
Introduction
The active search for new π-conjugated polycyclic aromatic hydrocarbons (PAHs) able to meet the insatiable demand and numerous requirements for their implementation in organic electroluminescent devices (OLEDs), organic photovoltaics (OPVs) and organic field-effect transistors (OFETs) has resulted in significant progress in synthetic methodologies, molecular design and, none the least, molecular diversity.1–3 For the synthesis of PAHs, transition metal-catalyzed Suzuki–Miyaura cross-coupling4 and intra-/inter-molecular FeCl3-catalyzed Scholl oxidative cyclodehydrogenation5–11 are among the most used chemical methods. The latter chemical reactions are also of prime importance as they are not only cost-effective and robust synthetic routes, but also possess environmentally friendly characteristics and produce small quantities of toxic wastes with recycling possibilities, features that are widely sought after for the sustainable development and carbon footprint reduction of the next generation of organic optoelectronic materials.The combination of electronic (photophysical and semiconducting) and anisotropic fluid properties within single compounds has also attracted much attention in these advanced fields of research too.12–14 Liquid crystalline materials possessing π-conjugated polycyclic aromatic cores and peripheral flexible aliphatic chains to improve solubility in organic solvents and promote their spontaneous self-organization into ordered self-healing mesophases further are interesting assets for their potential integration into various optoelectronic-based applications. Their use also permits substantial cost reduction in device fabrication, by, for instance, ink-jet printing or coating existing techniques. π-Extended conjugated discotic liquid crystals (π-DLCs)15–18 spontaneously self-organize into supramolecular columns promoted by π–π stacking of their polycyclic cores, and these columns further self-assemble into various long-range order nanostructures, i.e., 1D lamello-columnar (LamCol), 2D columnar hexagonal/rectangular (Colhex/Colrec), or 3D helicoidal columnar19–24 mesophases, propitious to the creation of potentially soft, unidimensional molecular pathways for the transport of electrons and holes.
The relationship between structures and property functions is one of the central topics in materials science to improve systems, but our predictive ability still remains elusive and challenging, hence a systematic exploration of molecular diversity is important. Although the emergence of liquid crystalline phases is quite easily guaranteed by adapted molecular design, some specific geometrical considerations and the association of various molecular species between them make it difficult to predict the types of organizations, as well as their stability and resilience towards thermal/optical/chemical stresses or the characteristics of their intrinsic physical properties.25 By using the versatile synthetic methods mentioned earlier, a great diversity of π-conjugated heterocyclic molecules has thus emerged, with molecules having cross-,26–31 board-32–34 and butterfly-like35,36 shapes, integrating various types of electro-active molecular units within their structure such as fluorene,36 fluorenone,36–38 carbazole,36 benzothienobenzothiophene,39,40 or other fused-thiophene41–50 building blocks. These systems show, as expected by design, mainly columnar mesophases with high thermal stability, as well as, depending on their intrinsic molecular structure, interesting photoluminescence, solvent gelation ability and/or charge carrier mobilities, of relevance for electronics-based applications.
In our search for the development of new and highly-functional molecular systems efficiently synthesized from simple and accessible basic components, we report on an exclusive library of dimeric disk-like mesogenic molecules consisting of two triphenylene moieties connected together by a conjugated, rigid molecular bridge, the liquid crystalline properties being conferred by the two triphenylene units. Surprisingly, only a few of such rigid systems have been reported so far,51–58 despite both fundamental and practical interests, such as for instance the possibility of numerous chemical alterations of the bridge to tune the physical properties precisely, or the induction of new types of columnar-like and nematic mesophases due to the specific constraints imposed by the inflexible bridge, of particular interest for some of the targeted applications mentioned above. In this work, using a diverse collection of small molecules, several new π-bridged discotic dimers (Scheme 1) were synthesized in good-to-high yields and good purity either by Suzuki–Miyaura cross-coupling of lipophilic monononaflate triphenylenes with appropriate aryl-bisboronic acids (bithiophene-, phenylene-, biphenylene-, thienothiophene- and benzodithiophene-bisboronic acids, respectively, Scheme 1), or by Scholl oxidative homo-coupling of thiophene-/thienothiophene-/furan-monosubstituted triphenylene derivatives (M1–M5, Scheme 1) to yield bithiophene-, bifuran- and bithienothiophene-bridged triphenylene dimers, respectively. One dimer with no bridge (Tp6Tp6, Scheme 1) was also synthesized to demonstrate the importance of the bridge in the induction of mesophases. In addition, two other, non-symmetrical σ-bridged dimeric compounds (Tp6T6 and Tp6Dt6, Scheme 1) were also synthesized by the versatile annulative π-extension (APEX) reaction from some of the monomeric precursors.59,60 These σ-bridged dimers were found to self-organize into Colrec whereas all the linear π-dimers showed NCol and multi-columnar mesophases with superlattices. Some of the compounds also displayed gelation behavior and interesting optical properties.
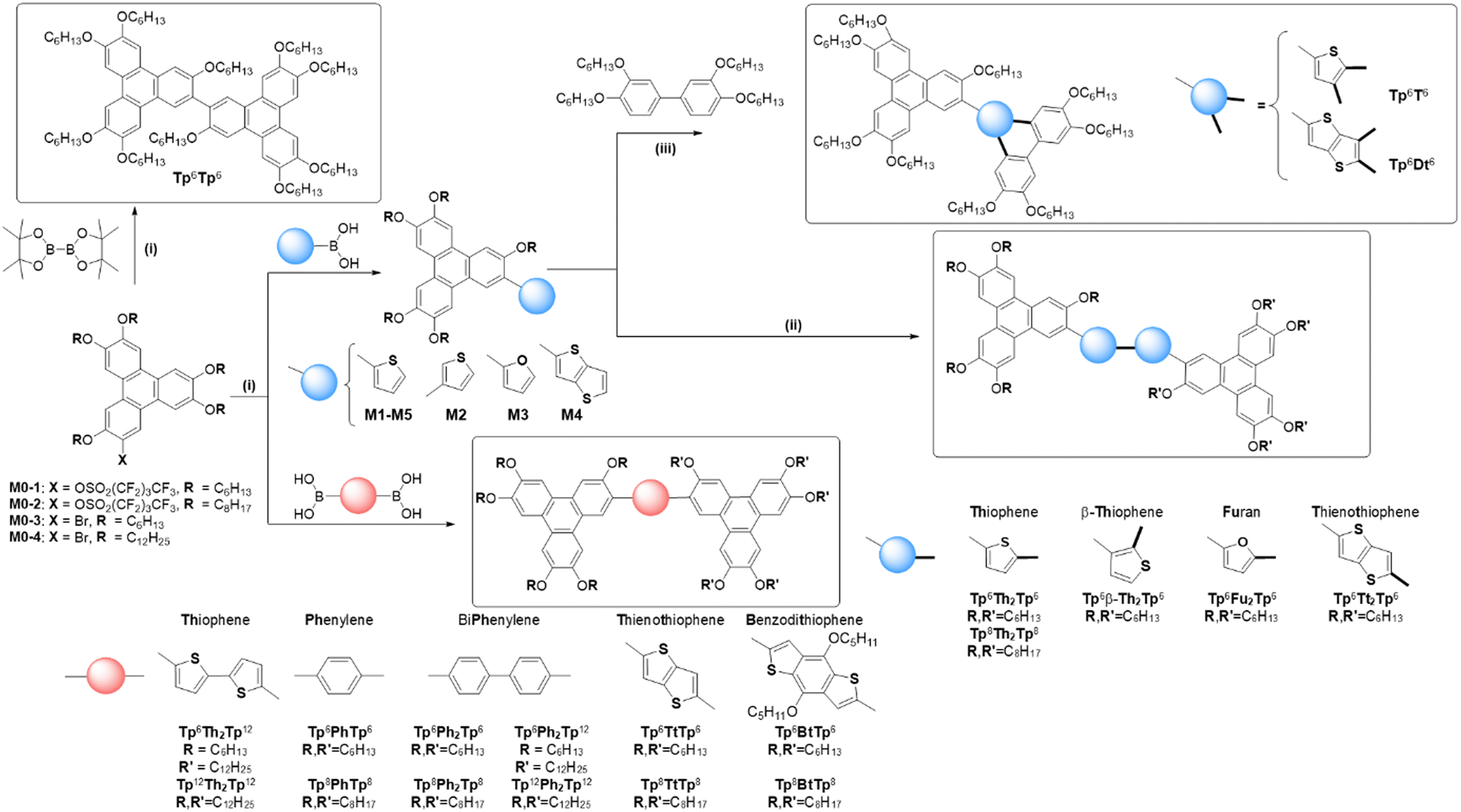 | ||
| Scheme 1 General synthetic scheme and nomenclature of the various monomeric and dimeric compounds (displayed in the frames): (i) Suzuki coupling; (ii) Scholl oxidative cyclodehydrogenation; and (iii) annulative π-extension (APEX) reactions. Nomenclature: Tpi,j-π-Tpi,j, where Tpi,j is the triphenylene moiety substituted by 5 alkoxy chains, OCnH2n+1, of length n = i and/or j, with i, j = 6, 8, 12, and π for the rigid, conjugated bridge: π = Th, Th2 for (bi)thiophene-, Fu, Fu2 for (bi)furan-, Tt, Tt2 for (bi)thienothiophene-, Ph, Ph2 for (bi)phenylene-, and Bt for benzodithiophene-based bridge. | ||
Results and discussion
Molecular designing, synthesis and characterization
Lipophilic triphenylene derivatives are workhorses of DLCs,61–64 and they display outstanding self-organizational behaviors and semi-conductive properties, which can be modulated by the nature of the peripheral substitution.62–64 The chemistry of functionalized triphenylene derivatives bearing various peripheral molecular moieties, i.e. polar (–CN, –SCN, –NC, –NO2, –CO2H, –NH2, N3, Aryl, …), halide and hydroxyl groups,65–69 has been widely explored and well developed to yield appealing functional building blocks for the construction of more complex molecules, dendrimers, polymers, coordination metal complexes, etc.65–69 The corresponding mesogenic triphenylene dimers and oligomers69–72 connected together by flexible aliphatic spacers tend to exhibit mainly Colhex mesophases, which is explained by the strong decoupling between the discotic units. In contrast, the less-reported rigid discotic dimers, when the two discrete discotic units are directly and physically bonded, and thus inter-dependent on each other, seem to preferentially dispense different types of mesophases, including also the elusive nematic mesophases (ND or NCol), due to some strong geometrical and conformational constraints.53,55 Some of the latter systems have in fact proven to be valuable and have been commercially applied as optical films for widening the view-angle of liquid crystal displays.64,73 Triphenylene-based dimers, oligomers and polymers containing electron-active aromatic cores, such as thiophene, thienothiophene, dibenzothiophene, and furan moieties, are also expected to show interesting supramolecular architectures and superior optical and electrical properties due to the synergy between the different molecular species.Here, aryl-connected lipophilic triphenylenes (i.e. 2-aryl-3,6,7,10,11-pentakis(alkoxy)-triphenylenes, M1–M5, Scheme 1 and Scheme S5, ESI†) were designed and used as the main precursors for the synthesis of several novel π-bridged and σ-bonded discotic dimers (Scheme 1). The first set of symmetrical π-bridged dimers was thus obtained by the Scholl oxidative homo-coupling cyclodehydrogenation reaction of thiophene-/thienothiophene-/furan-substituted triphenylene derivatives (Scheme 1 and Scheme S8, S9, ESI†), whereas, another set of dimers, both symmetrical and non-symmetrical, was prepared by the Suzuki–Miyaura cross-coupling reaction (Scheme 1 and Scheme S10, S13, ESI†) between the appropriate aryldiboronic acids and triphenylene nonaflate or bromo derivatives, respectively (M0–1, M0–2, Scheme S3, and M0–3, M0–4, Scheme S4, ESI†). As for the σ-bonded dimers, they were obtained by the thiophene-biphenyl annulative π-extension (APEX) reaction (Scheme 1 and Scheme S6, S7, ESI†).
The key precursors of M0–1 and M0–2, i.e. 2-hydroxy-3,6,7,10,11-pentakis(alkoxy)-triphenylenes, were synthesized as previously reported by the one-pot FeCl3 oxidation of the corresponding 1,2-di(alkoxy)phenylene.74 Triphenylene triflates were previously successfully applied in the Suzuki–Miyaura cross-coupling reaction for the synthesis of aryl-triphenylenes and triphenylene-fluorene-triphenylene discotic triads.55,75 However, considering the availability and high cost of the reagents, triphenylene nonaflates76 were synthesized to replace the triflates and used for the synthesis of the above-mentioned aryl-triphenylenes (Scheme 1). The Suzuki–Miyaura cross-coupling reaction between these triphenylene nonaflates (M0–1, M0–2) and commercially available or synthesized arylboronic acids produced the five 2-aryl-triphenylenes bearing 2-thiophenyl (M1/M5), 3-thiophenyl (M2), 2-furyl (M3), and 2-thieno[3,2-b]thiophene (M4) moieties, respectively, in high yields of ca. 90% (Scheme 1).
Oxidative homo-coupling of these electron-rich triphenylenes (Scheme 1 and Scheme S8, S9, ESI†) gave the corresponding symmetric discotic bridged dimers in good yields (Tp6Th2Tp6 (76%), Tp6β-Th2Tp6 (64%), Tp6Fu2Tp6 (67%), Tp6Tt2Tp6 (71%), and Tp8Th2Tp8 (73%)). For the 2-substituted thiophene (M1/M5), the 5-position is the most electron-rich and active, and thus, FeCl3 oxidation resulted in the linear, symmetric dimers (Tp6Th2Tp6 and Tp8Th2Tp8). Similarly for 2-substituted furan M3 and thieno[3,2-b]thiophene M4, the reaction patterns are the same, and the corresponding π-extended linear dimers Tp6Fu2Tp6 and Tp6Tt2Tp6 were thus also easily obtained. For the 3-substituted thiophene M2, however, the reaction pattern is different: the oxidative homo-coupling occurred at the 2-position, yielding only Tp6β-Th2Tp6, and did not occur in the less bulky 5-position; thus other isomers were not produced. It is demonstrated here that the Scholl reaction is electronically-determined but not sterically-driven. Furthermore, dimer Tp6β-Th2Tp6, with a head-to-head coupling product, was the only product isolated, with the complete absence of head-to-tail or tail-to-tail coupled isomers.
In order to demonstrate the great versatility of the Scholl reaction, 2 unsymmetrical σ-bonded discotic-like dimers were also synthesized by oxidative hetero-cross-coupling (annulated π-extension, APEX) between TP-2-thiophene (M1) and TP-2-thieno[3,2-b]thiophene (M4) and 3,3′,4,4′-tetra(alkoxy)biphenyl, respectively. Indeed, FeCl3-promoted oxidation of 1,2-di(alkoxy)benzene easily produces symmetrical 2,3,6,7,10,11-hexakis(alkoxy)-triphenylene in high yields,77,78 and the cross-coupling of electron-rich tetra(n-alkoxy)biphenyl with o-di(m-alkoxy)benzene lends the corresponding unsymmetrical triphenylenes (with n ≠ m).79 Similarly, thiophene and thienothiophene, as electron-rich arenes, cross-coupled with tetra(alkoxy)biphenyl yield in one step π-extensive thiophene-fused heteroarenes.39,49 To further extend this reaction, we successfully applied the FeCl3-mediated cross-coupling oxidation of M1/M4 with electron-rich tetra(alkoxy)biphenyl to yield two additional unsymmetrical σ-bonded discotic liquid crystals, Tp6T6 and Tp6Dt6 (Scheme 1 and Scheme S6, S7, ESI†), isolated in yields of 24 and 64%, respectively, by modifying the addition of M1/M4 to the mixed solution of excess biphenyl and FeCl3 in CH2Cl2. The cross-coupling products of unsymmetrical discotic dimers Tp6T6/Tp6Dt6, as well as their homo-coupling symmetrical products Tp6Th2Tp6/Tp6Tt2Tp6, were collected and separated by silica column chromatography. It was noted that Tp6T6 was synthesized in much lower yields than Tp6Dt6. M4, with a thieno[3,2-b]thiophene unit and a larger π-system, is effectively π-conjugated with the triphenylene core; the effective conjugation can inhibit free rotation of thieno[3,2-b]thiophene block, which is advantageous to the approach of tetra(hexyloxy)biphenyl for annulation. While the thiophene ring in M1 is smaller and less π-conjugated with the triphenylene core and more freely to rotate, the approach of tetra(hexyloxy)biphenyl is more difficult and consequently the annulative reaction is less efficient. The other explanation for this result is that the M1 homo-coupling occurs at a faster rate than cross-coupling; while on the contrary, cross-coupling is faster than homo-coupling for M4. It can be anticipated that, in the future, various types of thiophene-containing intermediates as well as electron-rich furan and pyrrole derivatives can be synthesized by this one-step, effective cross-coupling (oxidative APEX reaction)59,60 for the construction of original organic semiconductors.
The Suzuki–Miyaura cross-coupling was applied for the synthesis of eight other symmetrical π-bridged triphenylene dimers (Scheme 1 and Scheme S10, ESI†): triphenylene nonaflates coupled with the two commercial aryldiboronic acids produced dimers in good yield, i.e.Tp6,8PhTp6,8 (80 and 72%) and Tp6,8Ph2Tp6,8 (78 and 72%), whilst coupled with the two synthesized aryldiboronic acids yielded dimers Tp6,8TtTp6,8 (68 and 67%) and Tp6,8BtTp6,8 (64 and 60%) in good-to-moderate yields. To complete this series, additional non-symmetrical derivatives with a biphenyl and bithiophene bridge were prepared by reacting 4,4'-biphenyldiboronic acid or 5,5′-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,2′-bithiophene, respectively, with a mixture of two 2-bromo-3,6,7,10,11-pentakis(alkoxy)-triphenylene derivatives bearing alkoxy chains with different lengths (M0–3 and M0–4) in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1:1.1 (Scheme S4, ESI†). For the preparation of Tp6Ph2Tp12 (Scheme S11, ESI†), M0–4 was mixed first with 4,4'-biphenyldiboronic acid, and then, after 6 hours and cooling, the other monomer M0–3 was added to the mixture, and allowed to react for 12 hours. For the synthesis of Tp6Th2Tp12 (Scheme S12, ESI†), both triphenylene monomers M0–3 and M0–4 were mixed in an equimolar ratio at the beginning of the procedure with 5,5′-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,2′-bithiophene, and allowed to react together for 24 hours. In each case, the main compounds formed (with same or mixed chain periphery) could be easily separated by chromatography techniques: the non-symmetrical systems (Tp6Ph2Tp12and Tp6Th2Tp12) and the symmetrical ones (Tp12Ph2Tp12 and Tp12Th2Tp12, along with the other two homologous compounds with hexyloxy chains) were isolated in overall yields of ca 20–30% each.
:1.1:1.1 (Scheme S4, ESI†). For the preparation of Tp6Ph2Tp12 (Scheme S11, ESI†), M0–4 was mixed first with 4,4'-biphenyldiboronic acid, and then, after 6 hours and cooling, the other monomer M0–3 was added to the mixture, and allowed to react for 12 hours. For the synthesis of Tp6Th2Tp12 (Scheme S12, ESI†), both triphenylene monomers M0–3 and M0–4 were mixed in an equimolar ratio at the beginning of the procedure with 5,5′-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,2′-bithiophene, and allowed to react together for 24 hours. In each case, the main compounds formed (with same or mixed chain periphery) could be easily separated by chromatography techniques: the non-symmetrical systems (Tp6Ph2Tp12and Tp6Th2Tp12) and the symmetrical ones (Tp12Ph2Tp12 and Tp12Th2Tp12, along with the other two homologous compounds with hexyloxy chains) were isolated in overall yields of ca 20–30% each.
Finally, a bitriphenylene derivative with no bridge, Tp6Tp6, as the test compound, was also easily prepared by the direct Suzuki coupling between 2-bromo-3,6,7,10,11-pentakis(hexyloxy)-triphenylene and commercial bis(pinacolato)diboron in 71% yield (Scheme 1 and Scheme S13, ESI†).80
The complete synthetic details are reported in the experimental section of the ESI.† All the compounds were fully characterized by 1H/13C/19F NMR and elemental analysis, and the final compounds by additional HRMS (See ESI,† Fig. S1–S49).
Thermal behavior and mesomorphism
The thermal and mesomorphic behaviors of the discotic dimers and their aryl-containing triphenylene precursors were studied by polarized optical microscopy (POM) and thermal analyses (differential scanning calorimetry – DSC, and thermal gravimetric analysis – TGA), and the structure of the mesophases by variable temperature small- and wide-angle X-ray scattering (SWAXS). The POM images (Fig. S61, ESI†), TGA (Fig. S62, S63 and Table S7, ESI†) and DSC (Fig. S64–S66 and Table S8, ESI†) curves and the corresponding tables, as well as the SWAXS data (Fig. S67–S70 and Tables S9–S13, ESI†) are available in the ESI.†The triphenylene structure is one of the simplest and most studied basic polyaromatic systems in discoid liquid crystals.12,61 Due to its flat and nearly perfect circular shape, and once appropriately substituted in the six lateral positions (e.g. 2,3,6,7,10,11-hexasubstitution) by alkyl/alkoxy/ethylene glycol/perfluorinated chains, triphenylene-containing compounds12,61 show a natural tendency to stack into cylindrical columns with a short stacking distance of ca. 3.5 Å (hTP, see below) and to then self-organize mainly into hexagonal columnar mesophases (Colhex), with the chains homogeneously distributed around the piles. The archetypical n-alkoxy-chain compounds, included here as reference for comparison, show a columnar hexagonal mesophase, though over a rather narrow temperature range (e.g. n = 6, Iso 98 Colhex 53 °C Cr; n = 8, Iso 84 Colhex 46 °C Cr).81
In this work, several σ- and π-bridged dimers were thus synthesized from the monosubstituted triphenylene monomers. Two sets of compounds were obtained by the Scholl reaction: i) the unsymmetrical σ-bonded dimers, Tp6T6 and Tp6Dt6, by oxidative hetero-cross-coupling of M1 and M4, respectively, with the electron-rich (tetrahexyloxy)biphenyl, and ii) the symmetrical dimers Tp6Th2Tp6, Tp6β-Th2Tp6, Tp6Fu2Tp6, Tp6Tt2Tp6, and Tp8Th2Tp8, from the oxidative homo-coupling of M1–M5, respectively. Additionally, 11 other symmetrical π-bridged dimers were synthesized by the Suzuki–Miyaura coupling using monomers M0–1, M0–2, M0–3 and M0–4 and appropriate diboronic acid derivatives, including Tp6,8PhTp6,8, Tp6,8,12Ph2Tp6,8,12, Tp6,8TtTp6,8, Tp6,8BtTp6,8, Tp12Th2Tp12, and Tp6Tp6, with no bridge between the triphenylenes, and two compounds with unsymmetrical chain substitution patterns, Tp6Ph2Tp12 and Tp6Th2Tp12. The thermal behavior of the monomers will be briefly discussed first, followed by the mesomorphous properties of the unsymmetrical σ-bridged dimers Tp6T6 and Tp6Dt6, and finally, the mesomorphism of the complete set of π-bridged dimers, Tpi-π-Tpj.
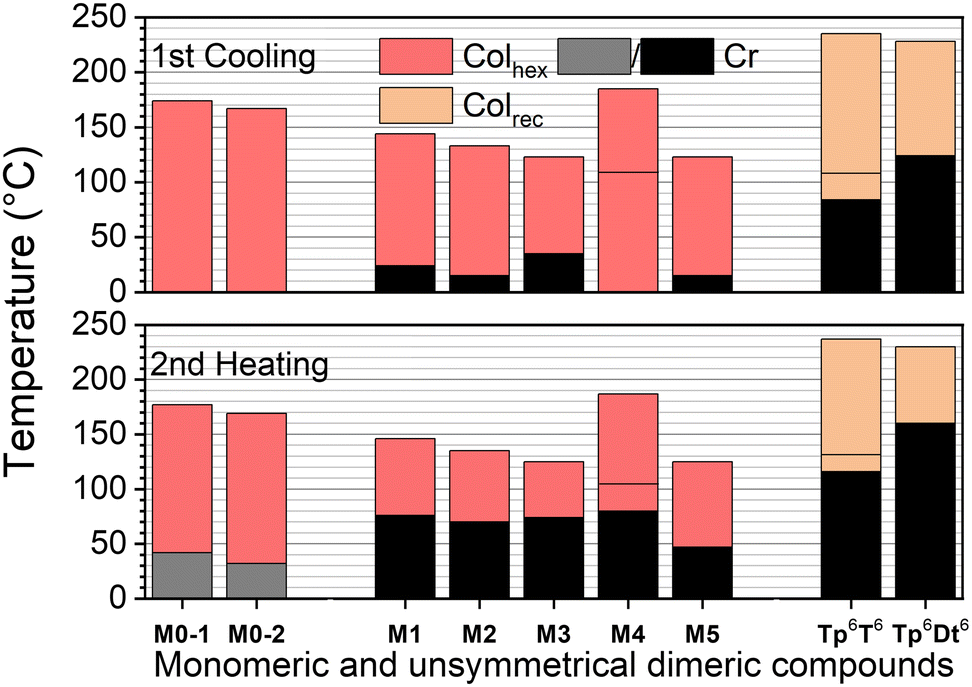 | ||
| Fig. 1 Phase diagrams (top: 1st cooling; bottom: 2nd heating) of the precursors, monomeric and unsymmetrical dimeric DLCs. For M4, a transition between two Colhex mesophases was detected by DSC and POM; For Tp6T6, a transition between two rectangular phases was detected by DSC and SWAXS. | ||
In all cases, the hexagonal symmetry of the mesophases was ultimately assigned by SWAXS measurements (Fig. S67 and Table S9, ESI†). Typical X-ray patterns display only one strong and sharp fundamental small-angle reflection assigned as (10) with, at best, an additional weak higher order, (21), along two sets of wide-angle scattering profiles, a broad scattering maximum at ca. 4.5–5.0 Å owing to the contribution of both the liquid-like lateral distances emerging from molten chains (hch) and nonaflate segments (hnf, for M0–1 and M0–2) or grafted rigid aromatic moieties (har, for M1–M5), respectively, and the wide-angle semi-diffuse peak at ca. 3.5 Å from the stacking of the triphenylene discs into columns (hTP). Therefore, these triphenylene molecules stack face-to-face into columns with the pending nonaflate segments or aromatic substituents randomly distributed in the aliphatic periphery thus without contributing to nano-segregation boundaries, but producing an irregular interface with the aliphatic periphery, in agreement with the quasi absence of high-order reflections in the SWAXS patterns. The average thicknesses of the molecules, hmol, deduced from the ratio between the molecular volumes and columnar cross-section areas (Table S10, ESI†), are close to the natural stacking distances between successive triphenylene cores, hTP, indicative of an almost orthogonal stacking of the TP cores into cylindrical columns, with a negligible tilt ψ of about 10–20° only with respect to the symmetry plane. This tilt likely emerges from the protruding nonaflate segment or pending aromatic moiety, respectively, that requires a larger cross-section area than an aliphatic chain (DFT, Fig. S55, ESI†). The Colhex mesophase therefore consists of these columns arranged at the nodes of a hexagonal lattice with a mixed and random periphery of chains and nonaflate/aromatic residues homogeneously distributed between the columns (Fig. S68, ESI†).
As expected, from the molecular shape change, the nature of the mesophase was modified accordingly. SWAXS patterns recorded at different temperatures confirmed the change of the mesophase nature, and the formation of columnar rectangular mesophases for both compounds (Fig. S69, ESI†). Although not so well resolved, X-ray patterns reveal nevertheless two very intense small-angle peaks, assigned to (11)/(20) of a rectangular lattice, plus, for the low-temperature phase of Tp6T6 (see below), a few other peaks of weaker intensity (Table S11, ESI†); the large-angle part reveals a broad band with two maxima (hch and hπ, from the chain and extended aromatic part, respectively). Furthermore, SWAXS confirmed the phase transition between two rectangular mesophases for Tp6T6 and the detailed peak analysis indicated the possibility of a symmetry change, i.e., c2mm and p2gg, between two Colrec mesophases; the high temperature mesophase was indexed, by convention, according to the highest centered symmetry (c2mm), whereas the low-temperature phase was logically indexed according to the herringbone-like symmetry (p2gg). Recall that these two planar symmetries correspond, respectively, to the specific orientations of the elliptical columnar cross-sections within the rectangular lattices, the long elliptical axis lying along the a-axis for the centro-symmetric c2mm lattice, or alternating its orientation with respect to the same a-axis (Fig. 2) to form a herringbone-like arrangement for the p2gg one. For Tp6Dt6, the rectangular phase was also confirmed by the presence in the SWAXS pattern of the two fundamental reflections (11) and (20) and the two broad wide-angle signals. Since no other reflections were present, the two reflections could not be differentiated, and the symmetry of the phase could not be solved either, as above the highest symmetry has been considered (Table S11, ESI†).
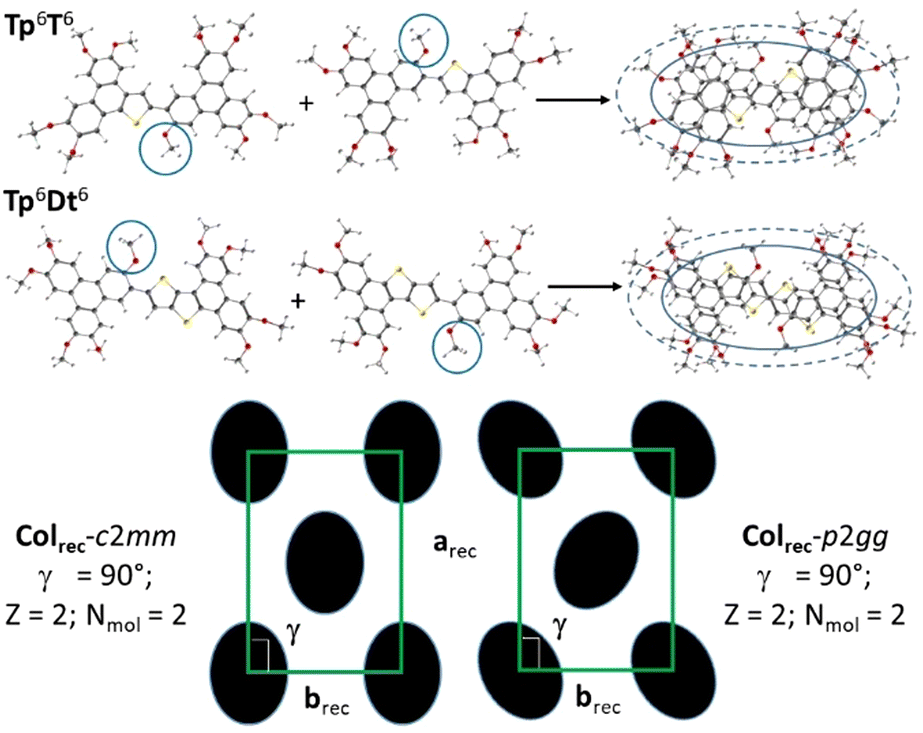 | ||
| Fig. 2 Schematic representations of molecular self-assemblies and supramolecular organizations in the various mesophases of the unsymmetrical σ-dimers, Tp6T6 (c2mm and p2gg) and Tp6Dt6 (c2mm); arec, brec, γ: lattice parameters and angle, Z: number of columns per lattice and Nmol: number of mesogens per lattice; the central chain is circled in blue (the chains are not represented for clarity). | ||
The average molecular thickness, hmol, in the mesophases diverges from the stacking distances between successive π-extended aromatic cores, hπ, regardless of the symmetry considered for Tp6T6, and indicative of tilted stacking of the rigid molecular cores, with respect to the symmetry plane (Table S12, ESI†). As confirmed by DFT simulations (Fig. S58, ESI†), these two molecules are not completely planar, with the triphenylene parts slightly tilted out of plane with respect to the thiophene-containing units, likely induced by the proximity of the aliphatic chain in the 3-position and the thiophene parts, thus affecting the stacking. Both the dimers have an average oval shape with 9 lateral chains, and their stacking somehow still preserves this shape and confers the elliptical cross-section to the columns, in agreement with the rectangular symmetry of the mesophases. This piling of the cores thus implies the periodic repartition of the central chain around the molecular cores that can be obtained by the alternated stacking of the molecules with respect to the columnar axis (i.e., point symmetry inversion, Fig. 2).
All of the linear dimers Tp6-π-Tp6, thus excluding the kinked dimer Tp6β-Th2Tp6 for now (see below), exhibit liquid crystalline properties over very broad temperature ranges (Fig. 3 and Fig. S66, ESI†). They all clear reversibly into the isotropic liquid at remarkably high temperatures from ca. 180 °C (Tp6PhTp6) and up to 310 °C (Tp6Tt2Tp6), that is at much higher temperatures than their monofunctionalized triphenylene precursors (Fig. 1). This trend more or less follows the length of the bridge binding the two triphenylene moieties, i.e., the longer the bridge, the higher the clearing. In contrast, the melting point does not follow this trend: some of the dimers are mesomorphous at room temperature (Tp6Ph2Tp6, Tp6Th2Tp6 and Tp6Tt2Tp6), Tp6PhTp6 melts at around 70 °C, whereas Tp6Fu2Tp6, Tp6TtTp6 and Tp6BtTp6 melt into liquid crystalline phases above 150 °C. Of interest, the simple ditriphenylene derivative, Tp6Tp6, in which both triphenylenes are directly linked, is not mesomorphous, and melts in the isotropic liquid at ca. 150 °C. As for the kinked dimer Tp6β-Th2Tp6, its clearing point is similar to that of its monomeric counterpart M2, and is also mesomorphous at room temperature. The effect of the chain-length (i = j = 6, 8, 12) and chain-repartition (i ≠ j) is also very effective over the control of the mesophases’ properties. Although the variation in the clearing temperatures mirrors the trend observed for the hexyloxy derivatives, these temperatures are significantly reduced with increasing chain-length for the octyloxy (clearing temperatures between ca. 145 and 250 °C) and for the dodecyloxy (132–136 °C) derivatives (Fig. 3). Similarly, compounds with non-symmetrical chain substitution patterns behave almost as their longer-chain (dodecyloxy) symmetrical homologs (125 °C for biphenylene and 167 °C for bithiophene). Thus, in these molecular systems, not only the bridge is essential in promoting mesomorphism, its chemical structure also strongly affects the transition temperatures of the dimers.
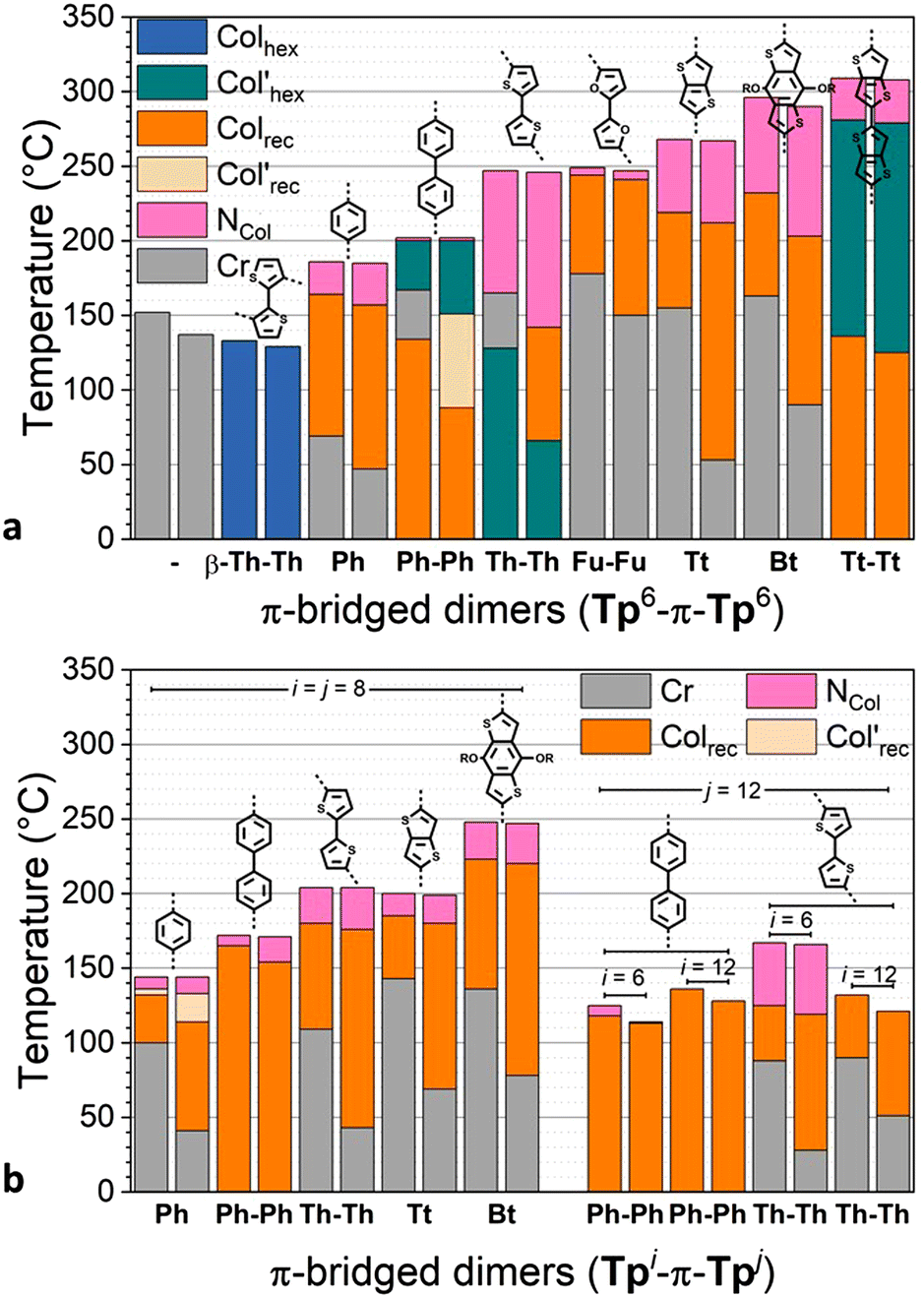 | ||
| Fig. 3 Phase diagram of π-bridged discotic dimers (both 2nd heating and 1st cooling are displayed in bar diagram for each compound): (a) Tp6-π-Tp6 and (b) Tpi-π-Tpj (i,j = 6, 8, 12). | ||
Furthermore, these dimers display a very rich and varied mesomorphism including nematic, rectangular and hexagonal columnar mesophases. On cooling from the isotropic liquid, a nematic phase is systematically induced for all the compounds, except for the kinked dimer (Tp6β-Th2Tp6) and for the two dodecyloxy derivatives (Tp12-π-Tp12). The nematic phase was recognized by the formation of Schlieren and/or thread-like textures, and in some case by the presence of large homeotropic areas (Fig. 4 and Fig. S61, ESI†). The domain of existence of the nematic phase is intimately entailed by the nature of the chemical bridge and the chain-length but no clear trend between the type of bridge and the extension of the nematic phase could be easily established. That said, this phase occurs only over a few degrees for the biphenylene and bifuran dimers, whereas is broad for all the thiophene-containing dimers. Of interest, the two central chains in the bridge of Tp6BtTp6 strongly promote this phase. Its occurrence was somehow not surprising as such a nematic phase has already been observed in other structurally related twin-like discotic systems.73 Increasing the chain-length reduces the range of existence of this mesophase, except for the octyloxy biphenylene term (Tp8Ph2Tp8). This phase is also observed for the two unsymmetrically substituted dimers, although for Tp6Ph2Tp12, the domain is larger on heating than on cooling, and this is probably as a consequence of the strong supercooling effect (of nearly 10 °C) observed for this compound.
 | ||
| Fig. 4 Selected POM textures of some representative π-bridged dimers. Top row: Tp6β-Th2Tp6 (100 °C, Colhex), Tp6Fu2Tp6 (228 °C, Colrec), and Tp6Tt2Tp6 (178 °C, Col'hex); bottom row: NCol of Tp8Th2Tp8 (195 °C), Tp6Ph2Tp6 (174 °C), and Tp8Ph2Tp8 (165 °C). Additional textures can be found in Fig. S57 (ESI†). | ||
The identification of the other mesophases was ultimately achieved by SWAXS experiments carried out at different temperatures, in the range defined by DSC measurements. SWAXS patterns recorded in the nematic phase of all compounds were similar in all cases (Fig. 5 and Fig. S70, ESI†). They consisted of a strong and slightly asymmetrical small-angle diffuse signal, D, at about 18–25 Å depending on the bridge type and chain-length, corresponding to the mean separation between residual short-range columnar stacks, which persisted over a few molecules. In addition, one single broad and diffuse signal combining the undifferentiated contributions of the molten chains, bridges and triphenylene stackings (h = hch + hbr + hTP) was observed in the wide-angle range. For some compounds, Tp6Th2Tp6, Tp6PhTp6, Tp6Ph2Tp6, and Tp8TtTp8, a very weak diffuse signal was also seen in the small-angle range, attributed to some reminiscent local 2D arrangement of columns in the low-orientationally ordered lattices (cybotactic groups). The maximum of the diffuse signal, observed at lower temperature, is no longer visible at the position of the hTP signal. However, the asymmetrical profile of the overall low-angle region signal is consistent with the persistence of this band around 3.5–4.0 Å, indicating that the piles would still correlate but to a few molecules only. This suggests that the dimers preserve some degree of stacking, and that the columnar nematic phases, NCol, are presumably made of short-range stacked twin-columns (Fig. 6).
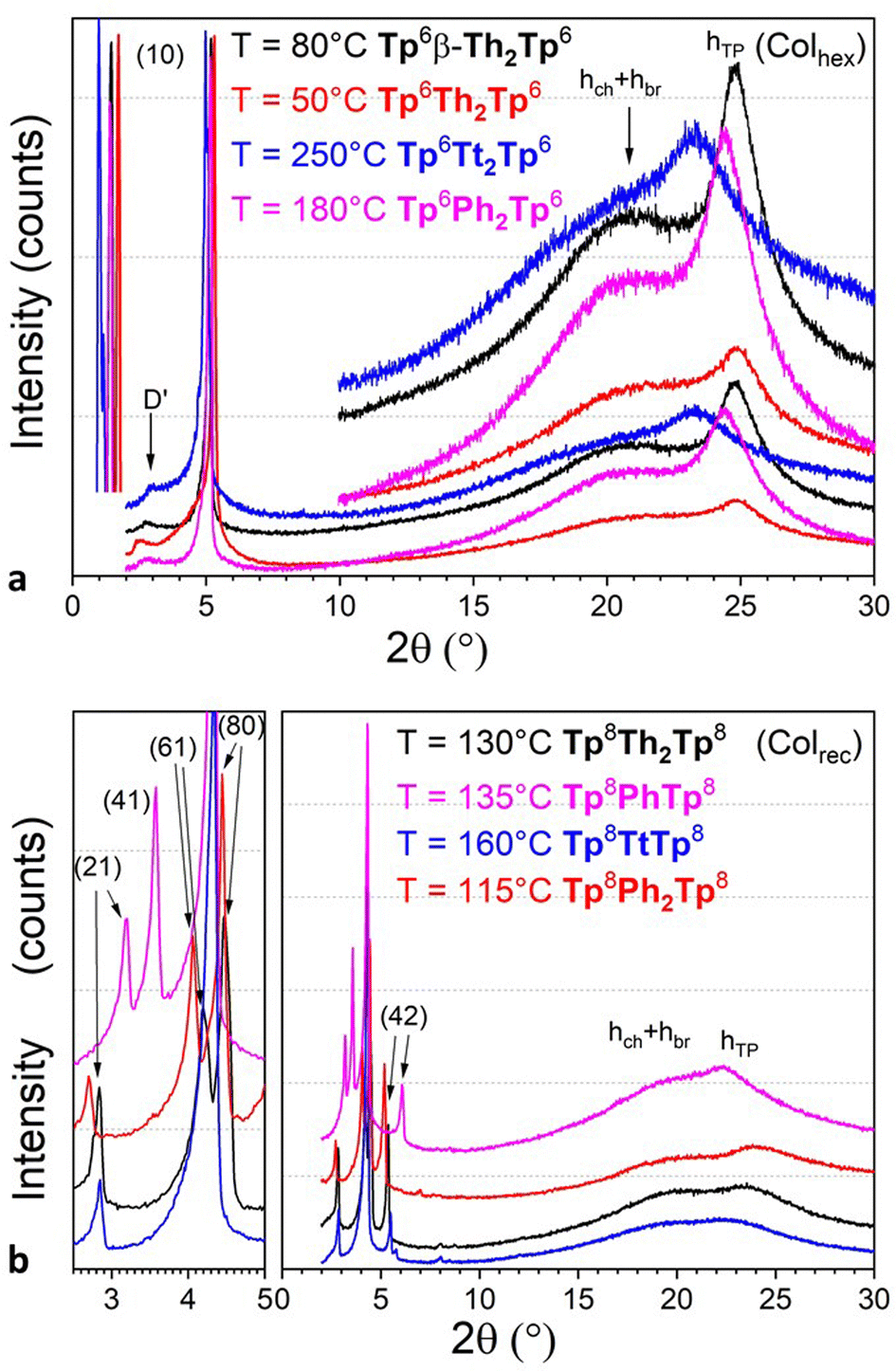 | ||
| Fig. 5 Representative SWAXS patterns of some representative dimeric compounds Tpi-π-Tpj (i,j = 6,8) recorded at various temperatures in (a) Colhex (Tp6-π-Tp6, top) and (b) Colrec (Tp8-π-Tp8, bottom) mesophases, respectively. Only the main small-angle reflections are indexed (see Fig. S66, ESI† for all SWAXS patterns). | ||
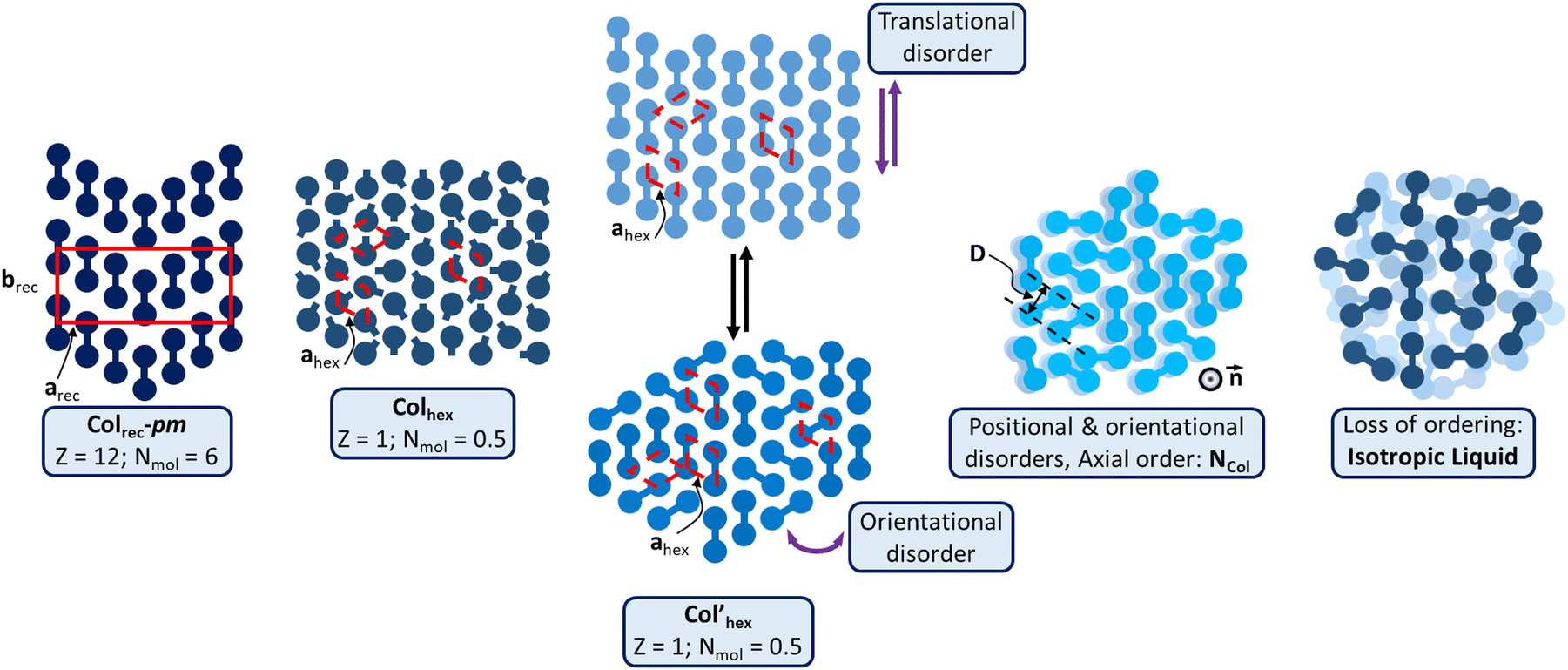 | ||
| Fig. 6 Schematic representation of the various supramolecular mesomorphous arrangements induced by the π-bridged dimeric mesogens: the Colrec-pm phase with chevron arrangements of the TP dimers; Col’hex phase, differentiated by translational or oriental order fluctuations; the Colhex phase of Tp6β-Th2Tp6 is similar to that of the monomeric precursors (Fig. S68, ESI†); the NCol phase characterized by loss of positional and orientational orders, but with short-range columns aligned along one main direction (axial order), the director n; isotropic liquid: no persisting molecular ordering. | ||
On decreasing the temperature, the texture of the nematic phase gives place to different, more structured textures, as observed by POM, consisting of bâtonnet-like, fern-like or mosaic-like defects (Fig. 4 and Fig. S61, ESI†), textures which are all reminiscent of columnar phases textures, as confirmed below by SWAXS measurements (Fig. S70, ESI†). As for the nematic phase, the thermodynamic stability of the more ordered mesophases was also found to strongly depend on the bridge nature (Fig. 3). Briefly, within the hexyloxy bridged dimers set, the phenylene, bifuran, thienothiophene and benzothiophene terms all display one single columnar phase below the NCol, over various temperature ranges, whilst the dithienothiophene shows two mesophases. The other terms behaved slightly differently. For the linear dithiophene compound, two mesophases were detected on cooling only (i.e. monotropic), with textures reminiscent of columnar mesophases (Fig. S70, ESI†), but with the low-temperature mesophase that remains stable at room temperature. On heating, crystallization occurs at ca 128°, and the nematic phase reforms at ca. 165 °C up to isotropisation. For the biphenylene derivative, a first mesophase is formed on cooling from the nematic phase, and on further cooling, two additional mesophases of different symmetry were also detected. As above, crystallization is induced upon re-heating the sample at ca. 134 °C, which melted at 165 °C in the columnar phase (Fig. 3); these two low-temperature mesophases are thus monotropic. The kinked dimer displays only one mesophase from room temperature onwards, i.e., a Colhex phase (Colhex 133 I) as recognized by its optical texture, and later confirmed by SWAXS, and is the only dimer that does not show the nematic phase. The behavior of the longer-chain homologs is simpler and monotonous as they exhibit a single columnar mesophase below the nematic phase, the exception being the octyloxy phenylene term, which displays two mesophases.
As just mentioned, only four compounds exhibit a Colhex mesophase, the kinked dimer, Tp6β-Th2Tp6, and three linear dimers. For the kinked dimer, the mesophase is present at room temperature and up to the isotropic liquid, whereas for the other dimers, the mesophase occurs either below the nematic phase (Tp6Ph2Tp6 and Tp6Tt2Tp6) or below the rectangular phase (Tp6Th2Tp6). The X-ray patterns for the Colhex mesophase of Tp6β-Th2Tp6 (Fig. 5) recorded on cooling from the isotropic liquid at different temperatures, display only one single but very intense small-angle reflection, indexed as the (10) reflection of a hexagonal lattice, along with the wide-angle scatterings corresponding to the mixed contribution of chains and bridges, hch + hbr, for the broad signal, and the semi-diffuse reflection characterizing the stacking of the triphenylenes, hTP, respectively (Fig. 5a and Fig. S70, ESI†). The correlation length for the intense intracolumnar signal was calculated to be ca. 23–33 Å, decreasing with temperature, that corresponds to about 6–10 stacked TP moieties on average (for the kinked dimer). Another weak scattering, D′ ≈ 30 Å, was also seen at a lower angle, likely the sign of some evanescent superlattice (see below). The SWAXS patterns of the Colhex phase for the other dimers are all very similar to that of Tp6β-Th2Tp6, and also consist of one major intense and sharp reflection, corresponding to the fundamental reflection (10) of a hexagonal lattice, and an almost extinct, broad small-angle signal, D′, as above (Fig. 5a). The wide-angle scattering was, also as for the kinked dimer, split into two distinct signals, corresponding respectively to the combined contributions of the molten chains and bridges (hch + hbr) and to the stacking of the TP cores (hTP). The lattice parameter of the Colhex phase of the different compounds, despite their different shapes and the nature of the bridge, is in the same range (ahex ≈ 20 ± 1 Å, Table 1), which corresponds to one triphenylene-based column per lattice for the four dimers. This hexagonal mesophase is not induced in the compounds with small bridges, such as phenylene and thienothiophene derivatives, nor in the benzodithiophene derivatives, probably due to some steric hindrance, and is no more present in the longer-chain homologues.
| Tpi,j-π-Tpi,j | T | Phaseb | V mol | ρ | a | b | a/bd | γ | A | N mol | h mol | Zcole | h TP (ξ)f/[h] | 〈ψ〉g | χTPh | 〈DTP〉i | 〈sch〉j | 〈q〉k |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Temperature of the experiment (°C).
b Mesophase type, square brackets refer to the monotropic phase.
c Molecular volume (Å3) and density (g cm−3) calculated by additivity of partial elementary volumes: Vmol = VTP + Vbr + Vch, the sum of the volume of the TP and bridge parts (from reference compounds) and volume of the chains, Vch, see ref. 82; ρ = MW/(NAVmol).
d Lattice parameters a, b and γ (Å, °) and cross-section areas A (Å2): Colhex, a = b = ahex, A = a2sinγ; for Colrec: a = arec, b = brec, A = a × b; a/b: anisotropic ratio.
e Number of molecules per lattice, Nmol, columnar slice thickness, hmol = Vmol × Nmol/A (Å), and number of columns per lattice, Zcol = Nmol × nTP, where nTP is the number of TP per molecules (i.e. nTP = 2).
f Face-to-face π–π stacking distance, hTP, (Å) from scattering maximum from the SWAXS pattern, and ζ, correlation length (Å) determined using the Debye–Scherrer formula; h: undifferentiated liquid-like contributions of the chains, bridges and triphenylene moieties.
g Out-of-plane tilt angle ψ(°) of TP mesogen cores inside columns: ψ = arcos(hTP/hmol).
h Triphenylene volume fraction (χTP = VTP/Vmol).
i Average TP diameter of equivalent circular columnar core cross-sectional area: 〈DTP〉 = [(4 × A × χTP)/(Zcol × π)]1/2.
j Average cross-section area per chain per TP unit: 〈sch〉 = π〈DTP〉 × hmol/6 (5 chains + 1 bridge).
k Chain packing ratio: 〈q〉 = 〈sch〉/σch, σch being the available cross-sectional area for a molten chain (σCH2 = VCH2/1.27), ref. 82.
|
||||||||||||||||||
| Tp6Th2Tp6 | 120 | [Colrec] | 2889 | 0.931 | 134.0 | 35.0 | 3.83 | 90 | 4691.3 | 6 | 3.69 | 12 | 3.72 (9) | ∼0 | 0.274 | 11.68 | 22.60 | 0.99 |
| 50 | [Col’hex] | 2720 | 0.989 | 19.3 | 19.3 | 1 | 120 | 334.0 | 0.5 | 4.07 | 1 | 3.58 (15) | 28 | 0.283 | 10.98 | 23.40 | 1.08 | |
| Tp6β-Th2Tp6 | 120 | Colhex | 2889 | 0.931 | 19.9 | 19.9 | 1 | 120 | 341.9 | 0.5 | 4.22 | 1 | 3.63 (23) | 26 | 0.274 | 10.92 | 24.14 | 1.06 |
| 80 | Colhex | 2788 | 0.965 | 19.8 | 19.8 | 1 | 120 | 339.9 | 0.5 | 4.10 | 1 | 3.58 (31) | 24 | 0.280 | 11.00 | 23.62 | 1.06 | |
| 40 | Colhex | 2561 | 1.051 | 19.7 | 19.7 | 1 | 120 | 337.1 | 0.5 | 3.80 | 1 | 3.54 (33) | 21 | 0.305 | 11.43 | 22.75 | 1.05 | |
| Tp6Fu2Tp6 | 200 | Colrec | 3111 | 0.848 | 139.1 | 32.1 | 4.34 | 90 | 4463.0 | 6 | 4.18 | 12 | 3.91 (−) | 21 | 0.262 | 11.14 | 24.39 | 1.01 |
| Tp6Tt2Tp6 | 250 | Col’hex | 3358 | 0.857 | 20.9 | 20.9 | 1 | 120 | 380.1 | 0.5 | 4.42 | 1 | 3.85 (13) | 29 | 0.247 | 10.94 | 25.32 | 1.02 |
| 100 | Colrec | 2923 | 1.036 | 141.3 | 34.0 | 4.16 | 90 | 4800.7 | 6 | 3.65 | 12 | 3.55 (35) | 14 | 0.269 | 11.70 | 22.37 | 0.99 | |
| Tp6PhTp6 | 120 | Colrec | 2273 | 0.917 | 144.6 | 30.5 | 4.74 | 90 | 4404.8 | 6 | 3.78 | 12 | 3.94 | — | 0.286 | 11.55 | 22.86 | 1.00 |
| Tp6Ph2Tp6 | 180 | Col’hex | 3098 | 0.862 | 19.9 | 19.9 | 1 | 120 | 343.6 | 0.5 | 4.51 | 1 | 3.66 (21) | 36 | 0.261 | 10.69 | 25.25 | 1.06 |
| 120 | [Col’rec] | 2923 | 0.914 | 137.7 | 37.5 | 3.67 | 90 | 5164.4 | 6 | 3.40 | 12 | 3.59 (42) | — | 0.271 | 12.18 | 21.69 | 0.95 | |
| 50 | [Colrec] | 2754 | 0.967 | 132.3 | 38.2 | 3.47 | 90 | 5053.3 | 6 | 3.27 | 12 | 3.57 (32) | — | 0.280 | 12.25 | 20.98 | 0.97 | |
| Tp6TtTp6 | 190 | Colrec | 3006 | 0.881 | 145.9 | 33.8 | 4.31 | 90 | 4939.4 | 6 | 3.65 | 12 | 3.92 (8) | — | 0.270 | 11.90 | 22.75 | 0.95 |
| 80 | Colrec | 2698 | 0.981 | 143.0 | 32.7 | 4.38 | 90 | 4673.1 | 6 | 3.46 | 12 | 3.81 | — | 0.289 | 11.97 | 21.69 | 0.98 | |
| Tp6BtTp6 | 190 | Colrec | 3475 | 0.868 | 143.0 | 34.4 | 4.16 | 90 | 4920.6 | 6 | 4.24 | 12 | [4.21] | — | 0.234 | 11.05 | 24.53 | 1.02 |
| Tp8Th2Tp8 | 130 | Colrec | 3598 | 0.877 | 157.7 | 36.1 | 4.36 | 90 | 5700.1 | 6 | 3.79 | 12 | 3.79 (−) | — | 0.221 | 11.56 | 22.94 | 1.00 |
| 50 | Colrec | 3271 | 0.965 | 155.6 | 35.9 | 4.33 | 90 | 5590.7 | 6 | 3.51 | 12 | 3.67 (67) | — | 0.236 | 11.82 | 21.73 | 1.00 | |
| Tp8PhTp8 | 135 | Col’rec | 3400 | 0.885 | 163.4 | 32.1 | 5.08 | 90 | 5254.6 | 6 | 3.88 | 12 | 3.99 | — | 0.234 | 11.43 | 23.22 | 1.00 |
| 100 | Colrec | 3302 | 0.912 | 164.2 | 31.6 | 5.20 | 90 | 5185.8 | 6 | 3.82 | 12 | 3.95 | — | 0.238 | 11.44 | 22.89 | 1.02 | |
| 50 | Colrec | 3155 | 0.954 | 163.9 | 31.0 | 5.29 | 90 | 5076.6 | 6 | 3.73 | 12 | 3.88 | — | 0.244 | 11.47 | 22.41 | 1.03 | |
| Tp8Ph2Tp8 | 115 | Colrec | 3488 | 0.899 | 159.0 | 38.0 | 4.18 | 90 | 6046.7 | 6 | 3.46 | 12 | 3.70 (13) | — | 0.227 | 12.06 | 21.85 | 0.96 |
| 50 | Colrec | 3305 | 0.949 | 156.2 | 37.6 | 4.16 | 90 | 5869.9 | 6 | 3.38 | 12 | 3.62 (6) | — | 0.233 | 12.05 | 21.33 | 0.98 | |
| Tp8TtTp8 | 160 | Colrec | 3509 | 0.887 | 170.5 | 34.8 | 4.90 | 90 | 5932.7 | 6 | 3.55 | 12 | 3.83 | — | 0.229 | 12.00 | 22.32 | 0.95 |
| Tp8BtTp8 | 200 | Colrec | 4048 | 0.860 | 163.8 | 34.8 | 4.71 | 90 | 5698.3 | 6 | 4.26 | 12 | [4.30] | — | 0.201 | 11.04 | 24.62 | 1.02 |
| 100 | Colrec | 3684 | 0.945 | 162.9 | 34.8 | 4.68 | 90 | 5669.8 | 6 | 3.90 | 12 | [4.28] | — | 0.13 | 11.33 | 23.13 | 1.03 | |
| Tp6Th2Tp12 | 105 | Colrec | 4099 | 0.827 | 182.8 | 34.2 | 5.34 | 90 | 6260.9 | 6 | 3.93 | 12 | 3.68 | 20 | 0.192 | 11.30 | 23.24 | 1.03 |
| 50 | Colrec | 3664 | 0.925 | 177.6 | 34.4 | 5.16 | 90 | 6107.7 | 6 | 3.60 | 12 | 3.62 | — | 0.210 | 11.68 | 22.01 | 1.01 | |
| Tp12Th2Tp12 | 110 | Colrec | 4590 | 0.891 | 187.3 | 34.4 | 5.44 | 90 | 6446.2 | 6 | 4.27 | 12 | 3.71 | 29 | 0.172 | 10.84 | 24.24 | 1.07 |
| Tp6Ph2Tp12 | 100 | Colrec | 4084 | 0.825 | 173.3 | 36.0 | 4.82 | 90 | 6231.1 | 6 | 3.93 | 12 | 3.72 | 19 | 0.192 | 11.21 | 23.06 | 1.02 |
| 50 | Colrec | 3698 | 0.911 | 173.1 | 35.1 | 4.93 | 90 | 6078.2 | 6 | 3.65 | 12 | 3.62 | 7 | 0.208 | 11.59 | 22.16 | 1.02 | |
| Tp12Ph2Tp12 | 110 | Colrec | 4624 | 0.880 | 194.4 | 37.3 | 5.21 | 90 | 7256.9 | 6 | 3.82 | 12 | 3.76 | 10 | 0.171 | 11.47 | 22.95 | 1.01 |
| 50 | Colrec | 4408 | 0.923 | 193.6 | 37.3 | 5.20 | 90 | 7213.5 | 6 | 3.67 | 12 | 3.66 | 0 | 0.175 | 11.57 | 22.23 | 1.02 | |
All compounds, except the kinked dimer, show one or more mesophases with a rectangular symmetry. X-ray patterns recorded at various temperatures unequivocally confirmed their liquid crystalline nature (Fig. 5b and Fig. S70, ESI†), resulting from the stacking of the TP rings and the segregation between hard cores and molten chains into 1D columns. The SWAXS patterns were rather unconventional and were composed of several sharp reflections in the small-angle range with a particularly intense one, confirming the formation of long-range ordered mesophases with large superlattices of reduced symmetry (Fig. 5b). The presence of a strong and extended wide-angle signal completed the patterns. This scattering was clearly split into the distinct molecular segments’ contributions; the broader scattering corresponding to the combined and undifferentiated contributions of the molten chains and bridges (hch + hbr), and the more-or-less semi-diffuse signal associated with the stacking of the TP cores (hTP), respectively. Only the dimers with central chains (Tp6,8BtTp6,8) showed one single diffuse signal, corresponding to the overlap of the various molecular contributions (h = hch + hbr + hTP), somehow expected since all the segments have almost identical cross-sections.
The sharp, low-angle reflections observed for most compounds were indexed within a planar rectangular symmetry (Table S13, ESI†). The overwhelming, mid-angle, single reflection, systematically present for all compounds, was ultimately assigned to the (80), whereas the other reflections were indexed as (21), (61) and (42), respectively, thus corresponding to strongly anisotropic, large two-dimensional rectangular superlattices (arec ≫ brec, Table 1). Such an indexation of the patterns for all the dimers is entirely satisfying, particularly for the high-order reflections and for the non-symmetrically substituted compounds (see Δ in Table S13, ESI,† and below). This indexation solution gives very large and anisotropic rectangular lattices with the a-parameter varying between 132 and 146 Å and the b-parameter between 30.5 and 38.2 Å for the Tp6-π-Tp6 compounds (Table 1). The a-parameters increase obviously with chain-lengths whereas the b-parameters remain quite constant throughout (arec ≈ 156–170 Å and brec ≈ 31–38 Å for the Tp8-π-Tp8, and arec ≈ 187–194 Å and brec ≈ 34–37 Å for the Tp12-π-Tp12). These large parameters imply several columns per lattice. The uneven distribution of the intensity of the reflections in this angular range and the partial or total extinction of the fundamental reflections (in this case, complete extinction of the fundamental reflections (11) and (20)), are the signature of important electronic density modulation emerging from periodic alternations between electron-rich zones (triphenylenes and bridges) and electron-poor areas (aliphatic continuum) within these large superlattices. The formation of such large superlattices of low symmetry strongly supports the formation of multicolumnar mesophases, somehow expected due to the intrinsic “dumbbell-like” shape of the mesogens. The absence of conditions on the reflections (hk) in most cases indicated also the formation of non-centered rectangular lattices.
The overall average intensity of the signal (hTP) indicates that the overlap between successive TP rings of the same dimer is not infinite but still predominates and extends to several stacks; this limitation is likely due to some distortion imposed by a slight twist of the bridges and/or their bulkiness with respect to the two adjacent triphenylene moieties (see DFT calculations, Fig. S58, ESI†). Although the stacking between the triphenylenes appears perturbed, as evidenced by the enlargement of the hTP stacking signal, the correlation length for the intracolumnar stacking could nevertheless be calculated for some compounds from the peak thickness at mid height using the Debye–Scherrer formula, providing that the reflection was sufficiently sharp. These calculations show that the correlation length was ca. 35 (Tp6Tt2Tp6), 42 (Tp6Ph2Tp6) and 67 Å (Tp8Th2Tp8), which correspond to about 10–20 stacked TP moieties for these most ordered systems. The persistence of this signal (hTP) for most compounds in the mesophase is also a further indication that this stacking is preferentially intracolumnar (the dimers would stack into discrete double cable columns in this case) rather than intercolumnar (undifferentiated stacking of TPs with connections between neighboring dimers), which would lead to unfavorable mixing between aromatic cores and lateral chains.
The two enantiotropic phases of Tp8PhTp8, detected below the nematic phase (Fig. 3), showed similar patterns to the rectangular phases described above, with an intense mid-angle reflection that could be indexed also as the (80), plus additional weaker reflections, and split wide-angle scattering. The only difference between these two mesophases was the emergence in the high-temperature phase of another reflection indexed as (81), likely corresponding to slight orientational changes of the dimers with respect to the lattice axes. As for the two monotropic phases of Tp6Ph2Tp6, detected below the high-temperature Colhex phase, slightly different X-ray patterns were obtained, but still preserving some of the common features already described above; differences mainly occur in the mid-angle range (Fig. S70 and Table S13, ESI† for details of the indexation).
Therefore, and not so surprisingly, all these dimeric compounds exhibit a rich mesomorphism, whose induction is strongly driven by the tendency of the triphenylene units to stack into one-dimensional twin-columns and, for the 2D phase, to further nanosegregate in space due to the peripheral alkyl chains. The relative arrangement of the twin-columns into rectangular (and single column for the hexagonal) lattices however is not straightforward, and is somehow controlled by the geometrical constraints imposed by the bridge. As previously developed,11,34,35 more intimate details about the respective organization of the columns within the superlattices can be revealed in part. The ratio between the molecular volume (Vmol) and the lattice area (A), scaled by the total number of dimers per lattice, Nmol, according to the formula, hmol = [Vmol × Nmol]/[A] (Table 1), simultaneously provides Nmol and a value for hmol, the molecular slice thickness, after successive trial and error loops, and keeping the most viable solution in agreement with the symmetry elements of the planar group and the various geometrical constraints (Table 1). Here, for the rectangular phases, Nmol was found to converge to 6 dimers, which correspond to columnar rectangular superlattices containing 12 TP-based single columns (Zcol = 2 × Nmol, for a molecular slice thickness value hmol that lies in the vicinity of the stacking periodicity along the TP-based columns, hTP). For the Colhex phases, these calculations give half a dimer that corresponds to the self-assembly of 1 TP-based columns (Table 1). As discussed above for the monomeric precursors, the average thickness of the molecules, hmol, is also close to the stacking distance between successive triphenylene cores, hTP, indicative of an almost orthogonal piling of the TP cores into cylindrical columns.
The striking feature commonly observed in all the SWAXS patterns of the Colrec mesophase is the systematic presence of the very intense, mid-angle reflection, (80), which is located at the same angle to that of the intense fundamental reflection (10) of the Colhex phase (of the triphenylene precursors and some dimers). As recalled from above, the natural tendency of TP mesogens is to stack into perfectly cylindrical columns that sit at the nodes of a hexagonal lattice, in order for the chains to homogeneously fill the available space. This suggests that an underlying similar local hexagonal arrangement of the almost cylindrical individual columns of each dimeric system is likely preserved in the rectangular phases, although at the local range, and that the reduction of the symmetry of the mesophases is imposed by the respective positional, translational and orientational orderings of the bridges connecting both TP units with respect to each other. Here, in a first simple approximation, it is assumed that the discs are located at the nodes of a pseudo hexagonal net, with the spacing being determined by the bridge and chains lengths, in order for the chains to fully fill the available space. This would then lead to a kind of staircase-like arrangement of the dimers, with one of the triphenylene part facing the bridge of its neighbor, arrangement that would be further facilitated by the absence of chains around the bridge. The self-assembling of the 6 dimers (i.e. 12 columns) according to a seamless zigzag chevron pattern is proposed that satisfies both lattice parameters and molecular dimensions. It is therefore possible to arrange the 6 dimers side-by-side in each chevron, with one TP unit interlocking into the free space left by the bare bridge of its close neighbors, and with all the TP units being equally spaced (i.e., as in a pseudo hexagonal net), and obviously depending on the bridge type. In other words, the molecules fit together like the pieces of a puzzle to maximize the density of the packing. The rectangular symmetry implies first the periodic change of the staircase direction at mid-lattice length (Fig. 6 and Fig. S72, ESI†), and second, that each chevron has to be separated from one another by a distance which is different from the TP–TP distance within the chevron. Under these conditions, this arrangement satisfies the symmetry and geometrical criteria outlined above, and gives a 2D tiling with the pm rectangular symmetry (no conditions on the hk reflections). This model is further supported by the packing chain ratio which is close to unity (Table 1). The other rectangular mesophases present in this system may involve tiny modifications, such as a slight tilt of the triphenylene discs with respect to the b-axis. Obviously, a deeper study, e.g. molecular modelling, correlating the separations between each chevron and between each neighboring dimer in the chevrons as a function of the length of the alkoxy chains irradiating from the TP cores and the length of the bridges, respectively, with the mesophase formed (Fig. 6 and Fig. S72–S74, ESI†), would support this model proposal and give other valuable information, but such investigations have not yet been realized.
DFT calculations (Fig. S58, ESI†) reveal that in the case of the kinked dimer, the bithiophene bridge favors the segregation between the aromatic core and chains by forcing the two TP units to almost lie on top of each other to form a pseudo cylindrical subunit likely at the origin of the induction of the Colhex mesophase. Structural and geometrical parameters agree with the formation of such types of single triphenylene-based columns (Table 1) and that half-twin Tp6β-Th2Tp6, i.e., one triphenylene core fits within the hexagonal lattice. The folded bridge however obviously protrudes out of the cylindrical core, thus reducing the free rotation of the overall dimer around the columnar axis and responsible for the irregular chain–core interface, explaining partly the absence of higher reflection orders. Tp6β-Th2Tp6 thus self-assembles in the same kind of Colhex mesophase as its monomeric precursors (Fig. S64, ESI†). The evanescent superlattice of reduced symmetry would then arise from the random orientation of the folded bridge (Fig. 6).
For linear dimers showing the hexagonal phase, the rigid bridges connecting the two triphenylenes do not bend and thus, the supramolecular organization in the Colhex phase is expected to be different from that of the kinked dimer. However, the hexagonal lattice parameters also agree with the formation of one triphenylene-based column as for the kinked dimer. The formation of the hexagonal columnar mesophases therefore necessarily results from the exact positioning of each triphenylene moiety at the nodes of a hexagonal network, with the basic unit being half of a twinned column. The formation of this mesophase stemming, on cooling, from the columnar nematic phase (for compounds Tp6Tt2Tp6 and Tp6Ph2Tp6) must somehow preserve some features of this phase, i.e., some of the orientational disorder of the twinned columnar piles relative to the bridge axis. Then, upon continuing cooling, the molecules probably continue to rearrange so that their major axes are parallel, which is the premise for the transition to the Colrec phase at lower temperatures. In this case, this intermediate phase would thus play the role of a template between these two mesophases (NCol and Colrec), and may be referred to as a transitory hexagonal (Col’hex, Fig. 6). However, for that stemming on cooling from the rectangular phase (compound Tp6Th2Tp6), the molecules likely retain some of the longitudinal ordering (see above), and would only differ by some degrees of translational disorder (along the bridge axis) to break the chevrons (Fig. 6). The formation of the low-temperature hexagonal mesophase of Tp6Th2Tp6 may be simply driven by the decrease in the chain fluidity. In both situations, the phase symmetry is increased and averaged to hexagonal (Fig. 6), but the core-chains interface is obviously not well defined, in agreement with the absence of high order reflections. It is also important to note that the TP stacking is preserved, suggesting persistent intercolumnar interactions. On further heating, the loss of translational order combined with the loss of orientational order of either Colrec or Colhex phases, respectively, actually prefigure the emergence of the nematic columnar phase which partially starts to lose the positional order of the columns upon chain dilation. The position of the broad scattering corresponding to the accumulation of the three main molecular contributions (chains, bridges and TP stacking) implies that some local intermolecular stacking is still present. The asymmetrical shape of the small-angle diffuse signal, D, also indicates that some local short-range ordering is persisting in the mesophase. Based on these observations, the nematic can be described as being made of twin-columns whose director is defined by the column's long axis (Fig. 6).
The chevron-like arrangement proposed above for the dimer organization within the rectangular superlattices was partially supported, at least the seeding of this arrangement, by scanning tunneling microscopy (STM), a powerful technique used to directly visualize single molecules on surfaces.83 The variation of the tunneling current in STM allows indeed visualizing the positions and distribution of the molecules directly at the atomic level without damaging the samples.84 The high-resolution STM images directly supply two-dimensional (2D) self-assembled patterns of molecules, and may support the self-organized patterns of the bulk, soft and disordered liquid crystalline materials in the mesophase, deduced by SWAXS. The mesomorphic character also further allows the spontaneous healing of self-organization defects and the development of long-range correlated segregated arrangements after thermal annealing and slow cooling of the samples. In addition, since most electronic devices are supported by a conductive substrate, the interfacial region between the substrate and the organic semiconductor is of significant importance, and although many favorable characteristics are reflected in the stacking direction constructed by π-conjugated systems, the behavior of these molecules on two-dimensional substrate should not be ignored.
Here, two samples, chosen as representative examples, namely Tp6Tt2Tp6 and Tp8Th2Tp8, were dissolved in 1-phenyloctane at concentrations less than 10−4 mol L−1. Then 2 μl of the solutions were placed on the fresh and clean highly oriented pyrolytic graphite (HOPG) surface for STM imaging. The STM tip (Pt/Ir, 80/20) was sharpened by mechanical cutting, and the detailed experiment conditions are labeled under the corresponding STM images (Fig. 7 and Fig. S71, ESI†). Detailed arrangements of the 2D self-assemblies of both π-dimers Tp6Tt2Tp6 and Tp8Th2Tp8, at the solid/liquid interface are presented in Fig. 7 and Fig. S71, ESI.† Large scale STM images reveal that these dimers are self-assembled into monolayer nanopatterns and arranged into long-range ordering structures. The shift or tilt of the molecules is essentially due to the crowded triphenylene units that are almost facing the bare bridge parts of the neighboring molecules to maximize the packing arrangement. Due to the large π-conjugated subunits constituting the dimers, each segment interacts with the HOPG substrate via very strong π–π interactions, while the peripheral alkyl side chains interact with the solvent 1-phenyloctane via van der Waals interactions. The images show that the thermodynamic stability of the patterns depends mainly on the interactions of the functional molecules/substrate, molecule/molecule, and molecule/solvent.
 | ||
| Fig. 7 Large-scale (a), medium range (b) and high-resolution (c) STM images of the self-assembly structure of Tp6Tt2Tp6 on the 1-phenyloctane/HOPG interface (imaging parameters: (a): Iset = 299.1 pA, Vbias = 699.8 mV; (b): Iset = 247.2 pA, Vbias = 847.2 mV; and (c): Iset = 299.1 pA, Vbias = 699.8 mV); large-scale (d), medium range (e) and high-resolution (f) STM images of the self-assembly structure of Tp8Th2Tp8 on the 1-phenyloctane/HOPG interface (imaging parameters: (d): Iset = 271.6 pA, Vbis = 770.0 mV; (e): Iset = 271.6 pA, Vbis = 770.0 mV; and (f): Iset = 271.6 pA, Vbis = 770.0 mV). | ||
Both dimeric molecules could form stable 2D nanostructures on the HOPG substrate and high-resolution STM images could then be easily obtained. These high-resolution STM images reveal that both the dimers Tp6Tt2Tp6 and Tp8Th2Tp8 are highly ordered, regularly aligned in parallel rows with well-organized 2D arrangements. In the high resolution STM image of the Tp6Tt2Tp6 structure (Fig. 7c), each brightened rod was assigned to one Tp6Tt2Tp6 molecule with the length of one molecule measured to be about 2.7 ± 0.1 nm. The oblique unit cell is highlighted in Fig. 7c and defined by the parameters, a = 2.3 ± 0.1 nm, b = 3.4 ± 0.1 nm, and α = 68 ± 1°. Similarly, high resolution STM images of Tp8Th2Tp8 dimers (Fig. 7f) also exhibit an oblique unit cell for the self-assembled 2D structure with similar parameters (a = 2.1 ± 0.1 nm, b = 3.6 ± 0.1 nm, α = 70 ± 1°, and with Tp8Th2Tp8 length of ca. 2.2 ± 0.1 nm). Thus, the above STM results agree well with the molecular lengths of both Tp6Tt2Tp6 and Tp8Th2Tp8, which possess either longer π-bridge (Tt2 > Th2) or shorter peripheral chains (C6H13versus C8H17), respectively. The molecular arrangement in the interfacial region is remarkably comparable to that of the chevron model proposed for the columnar sub-lattice of the bulk structure in the Colrec mesophase for both dimers Tp6Tt2Tp6 and Tp8Th2Tp8. In both cases, the molecules lie more or less parallel to each other, with a slight shift along their main axis in order to accommodate the hairy part (TP rings) near the naked bridges to optimize packing. The segregation pattern and the alternating row structure is thus very similar in both cases, except that both in-plane packing directions are long-range correlated in the film, whereas it is periodically alternated in the mesophase. Moreover, the lattice parameters (Table 1) are not excessively different: for Tp6Tt2Tp6, brec = 34 Å, and arec/6 ≈ 23.5 Å, and for Tp8Th2Tp8, brec = 36 Å, and arec/6 ≈ 26.0 Å; the 3D bulk organization is a fluid mesophase and involves dynamics with rotations and translations of the molecules, whereas chain-interdigitation occurs on the HOPG substrate. STM therefore kind of supports the self-organized patterns of the liquid crystalline mesophase deduced by SWAXS measurements and geometrical data analysis.
Photophysical properties and DFT results
The photophysical properties of these new compounds were studied by UV-vis absorption in solution and fluorescence emission in both solution and thin film states (Fig. 8), and the detailed parameters are provided in Table 2. The investigation of these photophysical properties of both isolated single molecules (molecular electronic structures, frontier orbitals and energy levels) and aggregated states is essential for the estimation of the potential performances and applications of organic electronic materials in OLEDs and OPVs.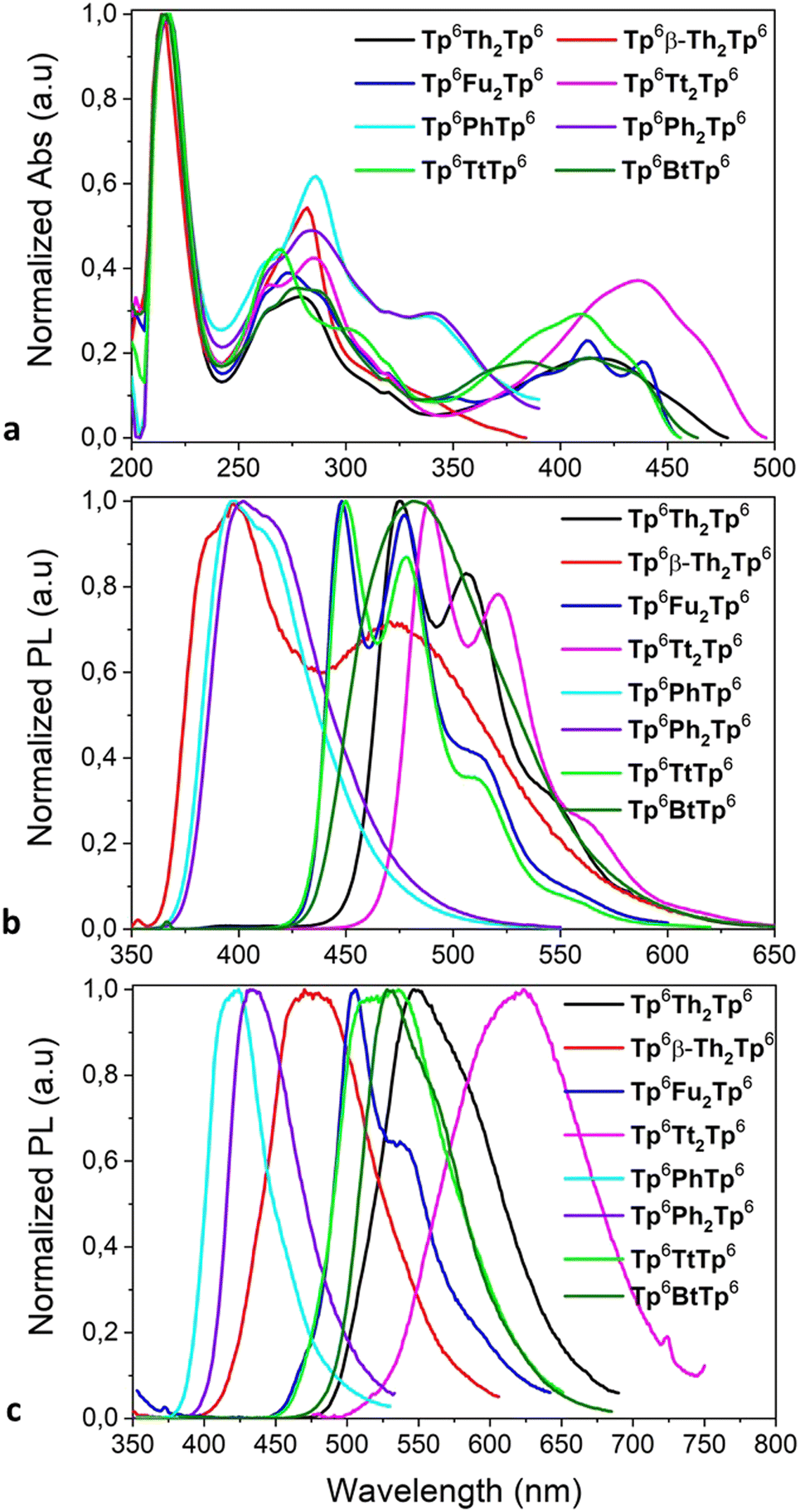 | ||
| Fig. 8 (a) UV-Vis absorption spectra, (b) fluorescence emission spectra in solution, and (c) thin film fluorescence emission spectra of various π-bridged dimers. | ||
| Compound | λ abs | ε | λ em | λ em | Φ |
|---|---|---|---|---|---|
| a UV-vis absorption measured in the THF solution at a concentration of 1 × 10−5 mol L−1 (maximum absorption, λabs in nm, ε, absorption coefficient, in ×104 L mol−1 cm−1). b Emission (λem in nm) in THF solutions at a solution concentration of 1 × 10−5 mol L−1. Excitation wavelength: 290 nm for Tp6,8Th2Tp6,8; 320 nm for Tp6β-Th2Tp6; 270 nm for Tp6Fu2Tp6; 271 nm for Tp6Tt2Tp6; 268 nm for Tp6,8PhTp6,8 and Tp6,8Ph2Tp6,8; 280 nm for Tp6,8TtTp6,8; and 367 nm for Tp6,8BtTp6,8. c Emission (λem in nm) in thin films. The thin films were obtained by drop-casting dilute THF solution and evaporation of solvent. Excitation wavelength: 350 nm for Tp6,8Th2Tp6,8; 320 nm for Tp6β-Th2Tp6; 340 nm for Tp6Fu2Tp6; 380 nm for Tp6Tt2Tp6; 268 nm for Tp6,8PhTp6,8 and Tp6,8Ph2Tp6,8; 330 nm for Tp6TtTp6; 340 nm for Tp8TtTp8; and 367 nm for Tp6,8BtTp6,8. d Quantum yields (Φ) in % were measured at a solution concentration of 1 × 10−5 mol L−1 in THF excited at different wavelengths (with the same excitation wavelengths used forb). Values in brackets are for the octyloxy Tp8-π-Tp8 homologs, only quoted when different. Data for unsymmetrical dimers (Tp6T6 and Tp6Dt6) are quoted in Table S2 (ESI). | |||||
| Tp6Th2Tp6 (Tp8Th2Tp8) | 214 | 28.73 (28.43) | 475 | 547 (545) | 30.47 (29.82) |
| 264 | 8.67 (11.39) | 506 (507) | |||
| 278 | 9.59 (12.57) | 545 | |||
| 320 | 3.12 (4.02) | ||||
| 416 | 5.43 (7.20) | ||||
| Tp6β-Th2Tp6 | 214 | 33.57 | 385 | 470 | 2.28 |
| 282 | 18.26 | 397 | |||
| 320 | 4.80 | 470 | |||
| Tp6Fu2Tp6 | 216 | 25.54 | 448 | 506 | 66.27 |
| 262 | 8.68 | 477 | 537 | ||
| 272 | 9.96 | 513 | |||
| 288 | 8.62 | ||||
| 320 | 3.92 | ||||
| 350 | 2.46 | ||||
| 390 | 3.80 | ||||
| 412 | 5.88 | ||||
| 438 | 4.62 | ||||
| Tp6Tt2Tp6 | 216 | 27.10 | 489 | 623 | 36.26 |
| 264 | 9.82 | 521 | |||
| 284 | 11.51 | 563 | |||
| 320 | 4.06 | ||||
| 436 | 10.09 | ||||
| Tp6PhTp6 (Tp8PhTp8) | 216 (218) | 27.38 (26.98) | 398 | 424 (422) | 44.81 (44.80) |
| 264 | 9.89 (11.24) | 410 | |||
| 286 | 15.89 (17.99) | ||||
| 338 | 5.93 (6.79) | ||||
| Tp6Ph2Tp6 (Tp8Ph2Tp8) | 218 | 26.78 (26.41) | 402 (401) | 434 (431) | 48.41 (48.93) |
| 266 | 9.74 (10.65) | 413 (412) | |||
| 284 | 12.11 (13.15) | ||||
| 340 | 6.50 (7.13) | ||||
| Tp6TtTp6 (Tp8TtTp8) | 218 | 26.28 (26.66) | 450 | 517 (−) | 53.80 (52.48) |
| 270 | 11.72 (11.69) | 478 | 536 (569) | ||
| 300 | 6.80 (6.80) | 510 (509) | |||
| 410 | 7.71 (7.51) | ||||
| Tp6BtTp6 (Tp8BtTp8) | 216 | 29.18 (29.11) | 481 | 532 (531) | 17.30 (17.82) |
| 264 | 8.94 (9.77) | 563 (562) | |||
| 276 | 10.37 (11.34) | ||||
| 386 | 5.28 (5.75) | ||||
| 414 | 5.58 (6.06) | ||||
In solution, the discotic monomers display absorption bands in the UV and deep-blue wavelength region (Fig. S50 and Table S1, ESI†). The absorption maxima (278–294 nm) are slightly redshifted in connection to the substituent moiety grafted onto the TP ring, increasing in the order M0 → M2 → M1(5)/M3 → M4; the chain-length has no effect on these values. Their fluorescence emission maxima in solution are centered in the narrow range of 350–450nm, with redshifts of the maxima following the same order of substitution as above (and no effect of chain-length either). The solid film emission spectra of the monomers are shifted to longer wavelength than the emission in solution, up to 490 nm, due to the enhancement of the intermolecular interactions attributed to the increase of the intramolecular π-conjugation system. For instance, thin film of M3 (containing the furan group) shows the most important red-shifted and broadest emission band, centered at nearly 500 nm, whilst M4 (with the 2-thieno[3,2-b]thiophene moiety) shows a broad emission band centered at ca. 460 nm. Isomerism (M1vs.M2) and chain-length (M1vs.M5) also affect the maximum emission wavelength of thin films. The emissions of the thiophene-containing compounds are found below 450 nm, with the isomer M2 even down to 400 nm. Among the functionalized discotic monomers, M3 and M4 also displayed the highest absolute quantum yields in solution: 25% and 30%, respectively.
As expected, the corresponding π-bridged dimers retain some of the absorption features of their parent compounds M1–M4 (Fig. 8a): the first main absorption band in the range between 250 and 350 nm does not change much upon dimerization, but the bands around 350 nm are shifted to above 400 nm for Tp6Th2Tp6, Tp6Fu2Tp6, Tp6Tt2Tp6 and Tp6TtTp6; the kinked dimer Tp6β-Th2Tp6 presents quasi identical features to its monomer parent M2. The same observations can be made for emission, for which the emission maximum is systematically redshifted in both solutions and films (Fig. 8b and c), with the larger shifts in the films (see for example M4versusTp6Tt2Tp6, the shift is about 200 nm).
Isomers Tp6Th2Tp6 and Tp6β-Th2Tp6 displayed different absorption and emission behavior due to their different connection patterns: in solution they show a combination of folded monomeric and extended dimeric conformation related emissions, while in the solid film only π-local monomeric emission is observed. The emission quantum yield (Φ) of Tp6Th2Tp6 is 30% with a peak at 547 nm, while for the isomer Tp6β-Th2Tp6, the Φ value is only 2% at 470 nm. It was noted that the highest Φ value of 66% was measured for the bifuran-bridged dimer Tp6Fu2Tp6, which showed almost identical absorption and emission spectra to that of Tp6Th2Tp6, probing that they have similar π-electron conjugation. Tp6Tt2Tp6, the longest π-bridged dimer, exhibits red-shifted wavelength of absorption and emission and an improved Φ value to 36%. As for Tp8Th2Tp8, homologue of Tp6Th2Tp6, it exhibits the same electronic spectra and Φ value, expected since the length of the peripheral alkyl chain has less or no effect on the photophysical properties. The absorption and fluorescence emission of the other dimers (Fig. 8a and b) showed a similar tendency: the thiophene-fused bridge systems Tp6,8TtTp6,8 exhibit red-shifted absorption and emission compared to the phenyl-/biphenyl-bridged dimers Tp6,8PhTp6,8/Tp6,8Ph2Tp6,8; their solid-state emission being further shifted to longer wavelength than that of their solution spectra. Unsymmetrical σ-bonded discotic dimers Tp6T6/Tp6Dt6 show red-shifted absorption and emission with Φ of 45%, due to the larger bithienothiophene-fused core (Fig. S51, ESI†).
The density functional theory (DFT, method: B3LYP, basis set: 6-31G*) method was used to calculate the frontier molecular orbital (FMO) distribution, energy levels and gaps for the methoxy homologs of these π-bridged dimers and precursors (Fig. 9 and Fig. S56, S57, S59, ESI†). Compared with the TP nonaflate M0, the discotic monomers M1–M4 show narrowed HOMO–LUMO energy gaps (ΔEgap) due to the obvious increase of the HOMO energy levels, caused by π-conjugation extension. The isomeric M1 and M2 display an obvious ΔEgap difference of 0.2 eV; for M1 with α-substitution, the thiophene moiety is better conjugated with the TP core, thus showing a narrowed energy gap than for M2. The furan monomer M3 shows a similar calculated electronic structure to the thiophene monomer M1. Among the discotic monomers, the fused dithiophene M4 has the most extended π-system and conjugation, showing lowered LUMO and most narrowed ΔEgap (3.76 eV) compared with the TP nonaflate (4.22 eV). The DFT results (Fig. S56 and S57, ESI†) are coincident with the photophysical properties measured for the discotic monomers at the single molecular state in diluted solution.
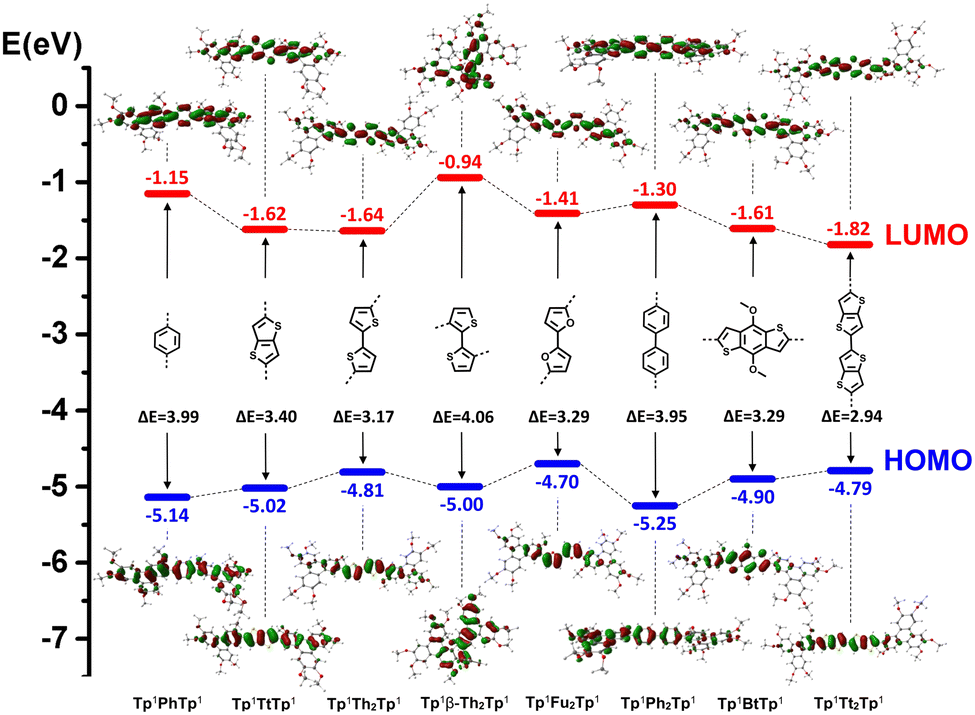 | ||
| Fig. 9 HOMO–LUMO and energy gaps of the discotic dimers. | ||
The DFT computational results for the discotic dimers (Fig. 9 and Fig. S59, ESI†) are also consistent with the experimental measurements. For the kinked molecular structure, Tp6β-Th2Tp6 exists as a twisted conformer with reduced π-conjugation, while all the other discotic dimers possess an extended conformation with a tendency for the aromatic cores and π-bridge to lie in the same plane. Tp6β-Th2Tp6 shows theoretically the largest ΔEgap (4.06 eV), just slightly narrower than that of its monomer M2 (4.19 eV). Then, the phenyl/biphenyl bridged dimers Tp6PhTp6 (3.99 eV) and Tp6Ph2Tp6 (3.95 eV) displayed partial π-conjugation in the single molecular state due to some degrees of rotation existing between the TP cores and the bridge. While the discotic dimer with the smallest ΔEgap (2.94 eV) was Tp6Tt2Tp6; this value is obviously smaller than that of the monomer M4 (3.76 eV). The other thiophene/furan fused/bridged dimers also displayed narrowed ΔEgap compared with that of their monomers (Fig. 9), due to the increase of the HOMO and lowering of the LUMO levels, respectively, upon the extension of π-conjugation. These DFT calculated electronic properties therefore agree fairly well with the measured photophysical behaviors of absorption and fluorescence emission.
In summary, the discotic monomers and dimers reported here show tailored π-conjugation systems and finely tuned HOMO–LUMO energy levels and gaps, color absorption and fluorescence emission; they are potential, highly valued electronic materials for further device fabrication.
Gelling behavior of discotic dimers
Gel is an interesting type of soft matter and has tremendous industrial applications.85 Organic π-gelators can be defined as polycyclic aromatic hydrocarbons (PAH) which dissolve first into an organic solvent when heated, then that self-assemble into one-dimensional supramolecular structures, entangle into fibers and cross-link into networks, immobilizing the solvent molecules on slow cooling. It was observed that the larger and the more planar the π-conjugated scaffold, the stronger the gelation ability. The extension of the π-conjugation and the strong intermolecular π–π interactions in these synthesized discotic dimers resulted in aggregation in solution as for other PAHs34–36 and in the formation of gels (Fig. 10 and Fig. S53, ESI†). | ||
| Fig. 10 Gels and frozen solutions of the dimers formed in cyclohexane on slow cooling, at room temperature and below 0 °C, in day light (top) and under UV irradiation (365 nm, bottom). G, S and PS indicate the state of the mixture corresponding to gel, solution and poor solution, respectively. The numbers in brackets correspond to the critical (minimum) gelation concentration (CGC/MGC) in mg mL−1; “o” indicates that the gel is opaque. | ||
The gelling properties of these π-conjugated discotic systems are determined by the nature of the bridge and the peripheral chain length. Heating the discotic dimers in cyclohexane and subsequently slow cooling down to room temperature yielded room temperature organic gels for the dimeric Tp6Th2Tp6, Tp6Fu2Tp6, Tp8Ph2Tp8, Tp8TtTp8 and Tp8BtTp8, with low critical (minimum) concentrations of 2–5 mg mL−1 (below 1%, they are termed supergelators), though the solutions started to gel at various temperatures on cooling. The other tested discotic dimers, i.e., Tp6β-Th2Tp6, Tp6Tt2Tp6, Tp6Dt6 and Tp8PhTp8, only form organo-gels when cooled down below 0 °C (Fig. S53, ESI†). Discotic dimers with short chains, Tp6Ph2Tp6, Tp6TtTp6 and Tp6BtTp6, exhibit inferior gelling behavior and show a tendency to crystallize from the solvents.
To gain some insights into the self-assembled gel micro-structures or crystalline aggregation from solvents, xerogels/crystalline fibers, the re-precipitated micro-particles were studied by scanning electron microscopy, and the morphologies of the xerogels are shown in Fig. 11 and Fig. S54 (ESI†).
 | ||
| Fig. 11 Self-assembled morphologies of some representative π-bridged dimers: SEM images of xerogels or solution-deposited micro-particles from organic solvents. | ||
As revealed by SEM, the thiophene-bridged/fused discotic dimers (Tp6Th2Tp6, Tp6Tt2Tp6, Tp6Dt6, Tp6,8TtTp6,8 and Tp6,8BtTp6,8) self-assemble into ultra-long net-shaped microwires with regular widths, demonstrating the balance of strong π–π core aggregations and the solubility of soft peripheral alkyl chains for these rigid discotic dimers. The gel formed from isomer Tp6β-Th2Tp6 shows a different morphology to that of Tp6Th2Tp6, assembling into rigid micro-plates with about 100 μm length and 10 μm width; this morphology further reflects the fact that Tp6β-Th2Tp6 has a rigid, twisted backbone and limited free-rotation conformation. The bifuran-bridged dimer Tp6Fu2Tp6, having a good solubility in most organic solvents but weaker π–π stacking interactions, did not form regular micro fibers. Tp6,8PhTp6,8 and Tp6,8Ph2Tp6,8, with a phenyl and biphenyl bridge, respectively, assemble into irregular micro particles. The SEM results here reveal the advantages of thiophene-containing building blocks for ordered self-assembling architectures. Compared with the fused π-conjugated polycyclic aromatic hydrocarbons (PAH) recently reported,34–36 these original discotic dimers show weaker gelling ability of organic solvent, due to the higher rotational freedom of both discogenic TPs and weaker π–π stacking interactions with the molecules of solvent. In polar organic solvents and aromatic hydrocarbon solvents, they show good solubility and organogels were not formed. Furthermore, the shorter-chain discotic dimers had better solubility in organic solvent and lower gelling ability. However, the discotic dimers with longer peripheral chains formed gels in cyclohexane.
Conclusions
Triphenylene nonaflates were used in Suzuki–Miyaura cross-coupling reactions for large scale synthesis of aryl-triphenylene and triphenylene discotic dimers. Bifuran-, bithiophene- and bithienothiophene-bridged dimers were synthesized by FeCl3 oxidative homo-coupling dimerization from the corresponding thiophene-, furan-, and thienothiophene-triphenylene. Moreover, FeCl3 oxidative cross-coupling of 2-thiophene- and 2-thieno[3,2-b]thiophene-triphenylene with electron-rich 3,3′,4,4′-tetra(alkoxy)biphenyl (annulated π-extension, APEX) afforded two original, unsymmetrical thiophene-fused dimers, the latter prepared in quite a good yield.All discotic dimers exhibited columnar (rectangular, hexagonal, and nematic) mesophases, over broad temperature ranges. From the SWAXS data and geometrical analysis, and supported by direct STM imaging, the colrec mesophase organization is thought to result from the disposition of 6 dimers into a chevron arrangement, whereas the formation of the hexagonal columnar mesophases necessarily results from the exact positioning of every triphenylene moieties (half a dimer) at the nodes of a hexagonal network (to yield single TP columns), with in-plane translational and/or orientational disorder. The nematic columnar phase results from short-range stacked twinned-columns, with a long-range axial order.
These rigid discotic dimers also show strong fluorescence in solution and in thin films, with emission color, ranging from blue to red, depending on the bridge π-conjugation length and the overall molecular conformation; therefore, they represent an original set of full-color tunable photoluminescent liquid crystalline materials. Furthermore, most of the dimers show moderate to high fluorescence quantum yields (Φ) in solution, and, in particular, the bifuran-bridged dimer shows the highest Φ value near 66.3%. These experimental measurements were fully supported by theoretical DFT computing calculation.
These dimeric compounds also form gels in cyclohexane and emit blue-to-orange light when irradiated by UV light. SEM revealed that their gelation ability results from the entanglements of, depending on the bridge and chain lengths, ultra-long or thin microfibers into intricate 3D morphologies.
The facile synthesis of these exclusive, potentially multifunctional rigid discotic dimers, their rich mesomorphism, strong gelation ability and fine-tuned photophysical properties make these materials very attractive in the active field of organic electronics. This methodology could be applicable to many other discotic and bridge system combinations (symmetrical and non-symmetrical), with the additional possibility to modify the nature of the terminal flexible chains in order to tame the electronic and organizational properties.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Contract No. 21772135, 51773140, and 51973143). BD thanks the CNRS and the University of Strasbourg for support.Notes and references
- T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and M. Funahashi, Nat. Rev. Mater., 2017, 2, 17001 CrossRef.
- A. C. Grimsdale and K. Müllen, Angew. Chem., Int. Ed., 2005, 44, 5592–5629 CrossRef CAS.
- R. E. Martin and F. Diederich, Angew. Chem., Int. Ed., 1999, 38, 1350–1377 CrossRef.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS.
- A. A. O. Sarhan and C. Bolm, Chem. Soc. Rev., 2009, 38, 2730–2744 RSC.
- Y. D. Yang, J. B. Lan and J. S. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- T. Doba, L. Ilies, W. Sato, R. Shang and E. Nakamura, Nat. Catalysis, 2021, 4, 631–638 CrossRef CAS.
- M. Grzybowski, B. Sadowski and H. Butenschön, Angew. Chem., Int. Ed., 2020, 59, 2998–3027 CrossRef CAS PubMed.
- K. Q. Zhao, J. Q. Du, X. H. Long, M. Jing, B. Q. Wang, P. Hu, H. Monobe, B. Henrich and B. Donnio, Dyes Pigm., 2017, 143, 252–260 CrossRef CAS.
- K. Q. Zhao, M. Jing, L. L. An, J. Q. Du, Y. H. Wang, P. Hu, B. Q. Wang, H. Monobe, B. Heinrich and B. Donnio, J. Mater. Chem. C, 2017, 5, 669–682 RSC.
- D. Myśliwiec, B. Donnio, P. J. Chmielewski, B. Heinrich and M. Stępień, J. Am. Chem. Soc., 2012, 134, 4822–4833 CrossRef PubMed.
- R. J. Bushby, S. M. Kelly and M. O’Neill, Liquid Crystalline Semiconductors, Springer, 2013 Search PubMed.
- R. J. Bushby and O. R. Lozman, Curr. Opin. Colloid Interface Sci., 2002, 7, 343–354 CrossRef CAS.
- R. J. Bushby and O. R. Lozman, Curr. Opin. Solid State Mater. Sci., 2002, 6, 569–578 CrossRef CAS.
- S. Chandrasekhar, B. K. Sadashiva and K. A. Suresh, Pramana, 1977, 9, 471–480 CrossRef CAS.
- T. Wöhrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 116, 1139–1241 CrossRef PubMed.
- S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- M. Lehmann, S. Gloza and S. Roth, Chem. Mater., 2015, 27, 8181–8184 CrossRef CAS.
- S. Park and B. K. Cho, Soft Matter, 2015, 11, 94–101 RSC.
- E. Beltrán, J. Barberá, J. L. Serrano, A. Elduque and R. Giménez, Eur. J. Inorg. Chem., 2014, 1165–1173 CrossRef.
- M. Masuda, M. Kohri and K. Kishikawa, Liq. Cryst., 2021, 48, 295–306 CrossRef CAS.
- M. A. Shcherbina, X. B. Zeng, T. Tadjiev, G. Ungar, S. H. Eichhorn, K. E. S. Phillips and T. J. Katz, Angew. Chem., Int. Ed., 2009, 48, 7837–7840 CrossRef CAS PubMed.
- Y. Cao, M. Alaasar, L. Zhang, C. H. Zhu, C. Tschierske and F. Liu, J. Am. Chem. Soc., 2022, 144, 6936–6945 CrossRef CAS PubMed.
- S. H. Eichhorn, A. O. El-Ballouli, A. Cassar and B. R. Kaafarani, ChemPlusChem, 2021, 86, 319–339 CrossRef CAS.
- M. Lehmann, M. Dechant, M. Lambov and T. Ghosh, Acc. Chem. Res., 2019, 52, 1653–1664 CrossRef CAS PubMed.
- M. Dechant, M. Lehmann, G. Uzurano, A. Fujii and M. Ozaki, J. Mater. Chem. C, 2021, 9, 5689–5698 RSC.
- T. Ghosh, L. Gerbig, M. Lambov, M. Dechant and M. Lehmann, J. Mater. Chem. C, 2020, 8, 5562–5571 RSC.
- M. Hügel, M. Dechant, N. Scheuring, T. Ghosh and M. Lehmann, Chem. – Eur. J., 2019, 25, 3352–3361 Search PubMed.
- M. Lehmann, Top. Curr. Chem., 2012, 318, 193–224 CrossRef CAS.
- N. Tober, J. Winter, M. Jochem, M. Lehmann and H. Detert, Eur. J. Org. Chem., 2021, 798–809 CrossRef CAS.
- H. Taing, A. M. Cassar, M. U. Ocheje, S. Rondeau-Gagné, T. H. El-Assaad, C. A. Sharabati, B. R. Kaafarani and S. H. Eichhorn, ChemPlusChem, 2019, 84, 103–106 CrossRef CAS PubMed.
- X. M. Zhu, X. Y. Bai, H. F. Wang, P. Hu, B. Q. Wang and K. Q. Zhao, Acta Chim. Sin., 2021, 79, 1486–1493 CrossRef.
- T. Ma, H. F. Wang, K. Q. Zhao, B. Q. Wang, P. Hu, H. Monobe, B. Heinrich and B. Donnio, ChemPlusChem, 2019, 84, 1439–1448 CrossRef CAS PubMed.
- T. Ma, Y. J. Zhong, H. F. Wang, K. Q. Zhao, B. Q. Wang, P. Hu, H. Monobe and B. Donnio, Chem. – Asian J., 2021, 16, 1106–1117 CrossRef CAS PubMed.
- J. F. Hang, H. Lin, K. Q. Zhao, P. Hu, B. Q. Wang, H. Monobe, C. H. Zhu and B. Donnio, Eur. J. Org. Chem., 2021, 1989–2002 CrossRef CAS.
- F. Lincker, A. J. Attias, F. Mathevet, B. Heinrich, B. Donnio, J. L. Fave, P. Rannou and R. Demadrille, Chem. Commun., 2012, 48, 3209–3211 RSC.
- F. Lincker, B. Heinrich, R. D. Bettignies, P. Rannou, J. Pécaut, B. Grévin, A. Pron, B. Donnio and R. Demadrille, J. Mater. Chem., 2011, 21, 5238–5247 RSC.
- C. X. Liu, H. Wang, J. Q. Du, K. Q. Zhao, P. Hu, B. Q. Wang, H. Monobe, B. Heinrich and B. Donnio, J. Mater. Chem. C, 2018, 6, 4471–4478 RSC.
- O. Kwon, X. Q. Cai, W. T. Qu, F. Liu, J. Szydłowska, E. Gorecka, M. J. Han, D. K. Yoon, S. Poppe and C. Tschierske, Adv. Funct. Mater., 2021, 31, 2102271 CrossRef CAS.
- T. Yasuda, T. Shimizu, F. Liu, G. Ungar and T. Kato, J. Am. Chem. Soc., 2011, 133, 13437–13444 CrossRef CAS PubMed.
- Y. F. Li, A. Concellon, C. J. Lin, N. A. Romero, S. B. Lin and T. M. Swager, Chem. Sci., 2020, 11, 4695–4701 RSC.
- Q. Xiao, T. Sakurai, T. Fukino, K. Akaike, Y. Honsho, A. Saeki, S. Seki, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2013, 135, 18268–18271 CrossRef CAS.
- I. Bala, J. De, S. P. Gupta, H. Singh, U. K. Pandey and S. K. Pal, Chem. Commun., 2020, 56, 5629–5632 RSC.
- L. X. Guo, Y. B. Xing, M. Wang, Y. Sun, X. Q. Zhang, B. P. Lin and H. Yang, J. Mater. Chem. C, 2019, 7, 4828–4837 RSC.
- N. Hu, R. F. Shao, Y. Q. Shen, D. Chen, N. A. Clark and D. M. Walba, Adv. Mater., 2014, 26, 2066–2071 CrossRef CAS PubMed.
- A. Demenev, S. H. Eichhorn, T. Taerum, D. F. Perepichka, S. Patwardhan, F. C. Grozema, L. D. A. Siebbeles and R. Klenkler, Chem. Mater., 2010, 22, 1420–1428 CrossRef CAS.
- D. M. Knawby and T. M. Swager, Chem. Mater., 1997, 9, 535–538 CrossRef CAS.
- K. C. Zhao, J. Q. Du, H. F. Wang, K. Q. Zhao, P. Hu, B. Q. Wang, H. Monobe, B. Heinrich and B. Donnio, Chem. – Asian J., 2019, 14, 462–470 CrossRef CAS PubMed.
- W. J. Deng, S. Liu, H. Lin, K. X. Zhao, X. Y. Bai, K. Q. Zhao, P. Hu, B. Q. Wang, H. Monobe and B. Donnio, New J. Chem., 2022, 46, 7936–7949 RSC.
- S. Kumar, Liq. Cryst., 2005, 32, 1089–1113 CrossRef CAS.
- S. Kumar and S. K. Varshney, Liq. Cryst., 2001, 28, 161–163 CrossRef CAS.
- S. Kumar and S. K. Varshney, Org. Lett., 2002, 4, 157–159 CrossRef CAS PubMed.
- K. Q. Zhao, H. Zhou, W. H. Yu, B. Q. Wang and P. Hu, Acta Chim. Sin., 2011, 69, 1895–1902 CAS.
- Y. Yang, H. Wang, H. F. Wang, C. X. Liu, K. Q. Zhao, B. Q. Wang, P. Hu, H. Monobe, B. Heinrich and B. Donnio, Cryst. Growth Des., 2018, 18, 4296–4305 CrossRef CAS.
- L. Zhang, H. Gopee, D. L. Hughes and A. N. Cammidge, Chem. Commun., 2010, 46, 4255–4257 RSC.
- L. Zhang, D. L. Hughes and A. N. Cammidge, J. Org. Chem., 2012, 77, 4288–4297 CrossRef CAS PubMed.
- W. K. Xiao, Z. Q. He, S. Remiro-Buenamañana, R. J. Turner, M. Xu, X. Yang, X. P. Jing and A. N. Cammidge, Org. Lett., 2015, 17, 3286–3289 CrossRef CAS PubMed.
- H. Ito, K. Ozaki and K. Itami, Angew. Chem., Int. Ed., 2017, 56, 11144–11164 CrossRef CAS PubMed.
- H. Ito, Y. Segawa, K. Murakami and K. Itami, J. Am. Chem. Soc., 2019, 141, 3–10 CrossRef CAS PubMed.
- S. Kumar, Liq. Cryst., 2004, 31, 1037–1059 CrossRef CAS.
- S. Kumar and J. J. Naidu, Liq. Cryst., 2002, 29, 899–906 CrossRef CAS.
- D. Adam, P. Schuhmacher, J. Simmerer, L. Häussling, K. Siemensmeyer, K. H. Etzbach, H. Ringsdorf and D. Haarer, Nature, 1994, 371, 141–143 CrossRef CAS.
- R. J. Bushby and K. Kawata, Liq. Cryst., 2011, 38, 1415–1426 CrossRef CAS.
- R. Chico, C. Domínguez, B. Donnio, B. Heinrich, S. Coco and P. Espinet, Cryst. Growth Des., 2016, 16, 6984–6991 CrossRef CAS.
- R. Chico, E. de Domingo, C. Domínguez, B. Donnio, B. Heinrich, R. Termine, A. Golemme, S. Coco and P. Espinet, Chem. Mater., 2017, 29, 7587–7595 CrossRef CAS.
- A. B. Miguel-Coello, M. Bardají, S. Coco, B. Donnio, B. Heinrich and P. Espinet, Inorg. Chem., 2018, 57, 4359–4369 CrossRef CAS PubMed.
- D. Zeng, I. Tahar-Djebbar, Y. Xiao, F. Kameche, N. Kayunkid, M. Brinkmann, D. Guillon, B. Heinrich, B. Donnio, D. A. Ivanov, E. Lacaze, D. Kreher, F. Mathevet and A. Attias, Macromol., 2014, 47, 1715–1731 CrossRef CAS.
- A. Zelcer, B. Donnio, C. Bourgogne, F. D. Cukiernik and D. Guillon, Chem. Mater., 2007, 19, 1992–2006 CrossRef CAS.
- K. Q. Zhao, L. L. An, X. B. Zhang, W. H. Yu, P. Hu, B. Q. Wang, J. Xu, Q. D. Zeng, H. Monobe, Y. Shimizu, B. Heinrich and B. Donnio, Chem. – Eur. J., 2015, 21, 10379–10390 CrossRef CAS PubMed.
- K. Q. Zhao, X. Y. Bai, B. Xiao, Y. Gao, P. Hu, B. Q. Wang, Q. D. Zeng, C. Wang, B. Heinrich and B. Donnio, J. Mater. Chem. C, 2015, 3, 11735–11746 RSC.
- H. Lin, Q. B. Lv, H. F. Wang, K. Q. Zhao, P. Hu, B. Q. Wang, B. Heinrich and B. Donnio, Dyes Pigm., 2022, 197, 109911 CrossRef CAS.
- H. K. Bisoyi and S. Kumar, Chem. Soc. Rev., 2010, 39, 264–285 RSC.
- S. Kumar and B. Lakshmi, Tetrahedron Lett., 2005, 46, 2603–2605 CrossRef CAS.
- K. Q. Zhao, Y. Gao, W. H. Yu, P. Hu, B. Q. Wang, B. Heinrich and B. Donnio, Eur. J. Org. Chem., 2016, 2802–2814 CrossRef CAS.
- S. J. Mahoney, M. M. Ahmida, H. Kayal, N. Fox, Y. Shimizu and S. H. Eichhorn, J. Mater. Chem., 2009, 19, 9221–9232 RSC.
- H. Bengs, O. Karthaus, H. Ringsdorf, C. Baehr, M. Ebert and J. H. Wendorff, Liq. Cryst., 1991, 10, 161–168 CrossRef CAS.
- N. Boden, R. C. Borner, R. J. Bushby, A. N. Cammidge and M. V. Jesudason, Liq. Cryst., 1993, 15, 851–858 CrossRef CAS.
- N. Boden, R. J. Bushby and A. N. Cammidge, J. Am. Chem. Soc., 1995, 117, 924–927 CrossRef CAS.
- C. F. Nising, U. K. Schmid, M. Nieger and S. Bräse, J. Org. Chem., 2004, 69, 6830–6833 CrossRef CAS PubMed.
- Y. Hirai, H. Monobe, N. Mizoshita, M. Moriyama, K. Hanabusa, Y. Shimizu and T. Kato, Adv. Funct. Mater., 2008, 18, 1668–1675 CrossRef CAS.
- B. Donnio, B. Heinrich, H. Allouchi, J. Kain, S. Dele, D. Guillon and D. W. Bruce, J. Am. Chem. Soc., 2004, 126, 15258–15268 CrossRef CAS PubMed.
- Y. L. Yang and C. Wang, Chem. Soc. Rev., 2009, 38, 2576–2589 RSC.
- K. S. Mali, J. Adisoejoso, E. Ghijsens, I. de Cat and S. de Feyter, Acc. Chem. Res., 2012, 45, 1309–1320 CrossRef CAS PubMed.
- S. S. Babu, V. K. Praveen and A. Ajajaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, experimental and synthetic procedures, 1H, 13C, and 19F NMR, HRMS, UV-vis and photoluminescence spectra, SEM images of xerogels, DFT calculations, TGA curves, POM photos, DSC curves, S/WAXS patterns, STM images, and model variations (111 pages). See DOI: https://doi.org/10.1039/d2tc02441a |
| This journal is © The Royal Society of Chemistry 2022 |