
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCustomising excitation properties of polycyclic aromatic hydrocarbons by rational positional heteroatom doping: the peri-xanthenoxanthene (PXX) case†
Cataldo
Valentini
 a,
Duncan
Gowland
b,
C. Grazia
Bezzu
a,
Deborah
Romito
c,
Nicola
Demitri
d,
Nicola
Bonini
*b and
Davide
Bonifazi
*ac
a,
Duncan
Gowland
b,
C. Grazia
Bezzu
a,
Deborah
Romito
c,
Nicola
Demitri
d,
Nicola
Bonini
*b and
Davide
Bonifazi
*ac
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK
bDepartment of Physics, King's College London, Strand, London, WC2R 2LS, UK. E-mail: nicola.bonini@kcl.ac.uk
cInstitute of Organic Chemistry, Faculty of Chemistry, University of Vienna, Währinger Str. 38, 1090, Vienna, Austria. E-mail: davide.bonifazi@univie.ac.at
dElettra – Sincrotrone Trieste, S.S. 14 Km 163.5 in Area Science Park, 34149 Basovizza, Trieste, Italy
First published on 12th May 2022
Abstract
In this paper we tackle the challenge of gaining control of the photophysical properties of PAHs through a site-specific N-doping within the structural aromatic framework. By developing a simple predictive tool that identifies C(sp2)-positions that if substituted with a heteroatom would tailor the changes in the absorption and emission spectral envelopes, we predict optimal substitutional patterns for the model peri-xanthenoxanthene (PXX) PAH. Specifically, TDDFT calculations of the electron density difference between the S1 excited state and S0 ground state of PXX allowed us to identify the subtleties in the role of sites i.e., electron donating or withdrawing character on excitation. The replacement of two C(sp2)-atoms with two N-atoms, in either electron donating or withdrawing positions, shifts the electronic transitions either to low or high energy, respectively. This consequently shifts the PXX absorption spectral envelop bathochromically or hypsochromically, as demonstrated by steady-state absorption spectroscopic measurements. Within the series of synthesised N-doped PXX, we tune the optical band gap within an interval of ∼0.4 eV, in full agreement with the theoretical predictions. Relatedly, measurements show the more blueshifted the absorption/emission energies, the greater the fluorescence quantum yield value (from ∼45% to ∼75%). On the other hand, electrochemical investigations suggested that the N-pattern has a limited influence on the redox properties. Lastly, depending on the N-pattern, different supramolecular organisations could be obtained at the solid-state, with the 1,7-pattern PXX molecule forming multi-layered, graphene-like, supramolecular sheets through a combination of weak H-bonding and π–π stacking interactions. Supramolecular striped patterned sheets could also be formed with the 3,9- and 4,10-congeners when co-crystallized with a halogen-bond donor molecule.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are attracting great interest in the recent years for preparing organic semiconductors that, absorbing in the UV-vis spectral range of solar irradiation, generate excited states of interest for light-harvesting applications1 and photocatalysis.2–5 Capitalising on the wealth of organic synthetic methodologies, chemists have tailored the photophysical properties of PAHs by: (i) changing the size and edge of the aromatic scaffold; (ii) varying the molecular planarity; (iii) changing the resonance energy of the constituting monomeric units; (iv) varying the peripheral functionalisation of the edges through the insertion of either electron-donating or -withdrawing substituents; (v) replacing C(sp2) atoms by isostructural analogues, i.e., heteroatom doping.6–9 Amid all approaches, the doping route can effectively tailor the photophysical properties of a PAH without the need to add peripheral groups while maintaining the structural topology.7,9 It is thus expected that one can encode the functionalization that defines the photochemistry of PAHs via a site-specific substitutional pattern. Owing to the possibilities of introducing the heteroatom in different ratios (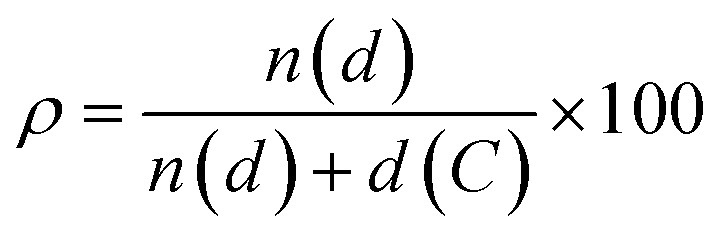 , where n(d) and d(C) are the numbers of doping heteroatom and carbons), positions, and symmetries, numerous configurations are possible even for small PAHs. While the general influence on the energy level of the frontier molecular orbitals of replacing a C(sp2) atom with a more electronegative heteroatom such as N is well established (i.e., lower-energy rigid shift of the HOMO and LUMO levels),10–21 it is more difficult to predict the influence of tailoring heteroatom position on optoelectronic properties.22,23 Herein, we raise the question of whether it is possible to rationally customise optical bandgap properties of a PAH by a specific heteroatom doping pattern and if this can be conceived a priori. In the absence of any formalized approach to conceive a tailored doping pattern, we would need synthesise and analyse each positional congeners to establish a structure–property relation. Considering the large number of possible doping patterns, this trial-and-error approach could result in a substantial amount of unnecessary synthetic efforts. To minimise these efforts, one needs a predictive tool that identifies those C(sp2)-positions in the PAH core that, if substituted with a heteroatom, would induce the desired changes in the absorption and emission spectral envelopes. It is with this challenge in mind that we have developed a simple conceptual approach that would allow organic chemists to devise pertinent heteroatom-doping patterns to rationally tune the absorption UV-vis envelope of a given PAH. Using peri-xanthenoxanthene (PXX) as a model PAH, herein we show that one can predictably tailor its UV-vis absorption and emission energies through specific substitutional patterns using N-atoms as dopants.
, where n(d) and d(C) are the numbers of doping heteroatom and carbons), positions, and symmetries, numerous configurations are possible even for small PAHs. While the general influence on the energy level of the frontier molecular orbitals of replacing a C(sp2) atom with a more electronegative heteroatom such as N is well established (i.e., lower-energy rigid shift of the HOMO and LUMO levels),10–21 it is more difficult to predict the influence of tailoring heteroatom position on optoelectronic properties.22,23 Herein, we raise the question of whether it is possible to rationally customise optical bandgap properties of a PAH by a specific heteroatom doping pattern and if this can be conceived a priori. In the absence of any formalized approach to conceive a tailored doping pattern, we would need synthesise and analyse each positional congeners to establish a structure–property relation. Considering the large number of possible doping patterns, this trial-and-error approach could result in a substantial amount of unnecessary synthetic efforts. To minimise these efforts, one needs a predictive tool that identifies those C(sp2)-positions in the PAH core that, if substituted with a heteroatom, would induce the desired changes in the absorption and emission spectral envelopes. It is with this challenge in mind that we have developed a simple conceptual approach that would allow organic chemists to devise pertinent heteroatom-doping patterns to rationally tune the absorption UV-vis envelope of a given PAH. Using peri-xanthenoxanthene (PXX) as a model PAH, herein we show that one can predictably tailor its UV-vis absorption and emission energies through specific substitutional patterns using N-atoms as dopants.
Results and discussions
The concept: use of charge-redistribution maps for rational heteroatom doping
We wish to develop a rational tool that allows organic chemists to tailor the optical gap of a given PAH through specific heteroatom doping patterns. Tailored absorption and emission properties could in principle be obtained by simply gaining control of the ground (PAH, S0) and excited (PAH*, S1, the first bright singlet excited state in this case) state energies and properties through chemical functionalisation (e.g., controlling the HOMO and LUMO energies and their overlaps). In a simplified picture, one can envisage that introducing a dopant which destabilises the electronic excited state (PAH*, S1) whilst keeping the ground state energy fixed leads to an opening of the optical gap, i.e., a hypsochromic shift in the spectral envelop. Of course, this primitive model is highly limited as all properties (both electronic and geometric) of a molecule are intimately related to the exact atomic composition and positions – the ground state energies and structural properties are not the same for different positional isomers.In the general case of a rigid molecule, it is reasonable to hypothesise a negligible structural distortion due to heteroatom doping and/or on excitation (this is discussed further in our results). This would allow one to more directly compare the electronic excitation properties of different doping patterns (i.e., a direct calculation and comparison of the S0 → S1 transition). The latter statement, that geometries vary little on excitation, is related to the Franck–Condon approximation, which in this case is assumed as we require that not only the ground state structures of the positional isomers are similar, but that their excited state dynamics are also similar. Following these approximations, we note the influence of the heteroatom position can be subtle but significant on both the ground and excited states. For example, visualisation or quantification of different regioisomer HOMO and LUMO orbitals can reveal little trend in their structure and one would have to understand the trends in both to make predictions about the optical gap. Therefore, we move to directly examining the properties of excitations rather than states, which more clearly would allow identification in trends on optical properties. More specifically, we calculate and utilise a charge redistribution ‘map’ (related to the TDDFT density difference) on excitation of the PAH structure to identify electronic withdrawing and donating domains, allowing prediction of whether replacing specific C(sp2) atoms would either favour or disfavour the charge redistribution and could induce an energy shift of the electronic transition. This methodology is powerful as it allows rich comparison and can incorporate changes to both ground and excited state electronic and structural properties between isomers. Thus, the idea is to map the charge redistribution between the S0 and S1 states and use it as the matrix to rationalise the choice of a given doping pattern.
Our case study: rational N-doping of PXX
Our group is involved in synthetic programs developing O-doped PAH chromophores.24–27 One of our targets is PXX (Fig. 1), the O-containing analogue of anthanthrene.3,28–33 Considering the vis-absorbing bandgap, with minor Stokes shift and good emission properties (ΦF = 60%) of PXX (Fig. 1), we envisioned that the replacement of one or more C(sp2)-atoms with N-atoms could affect its optical bandgap.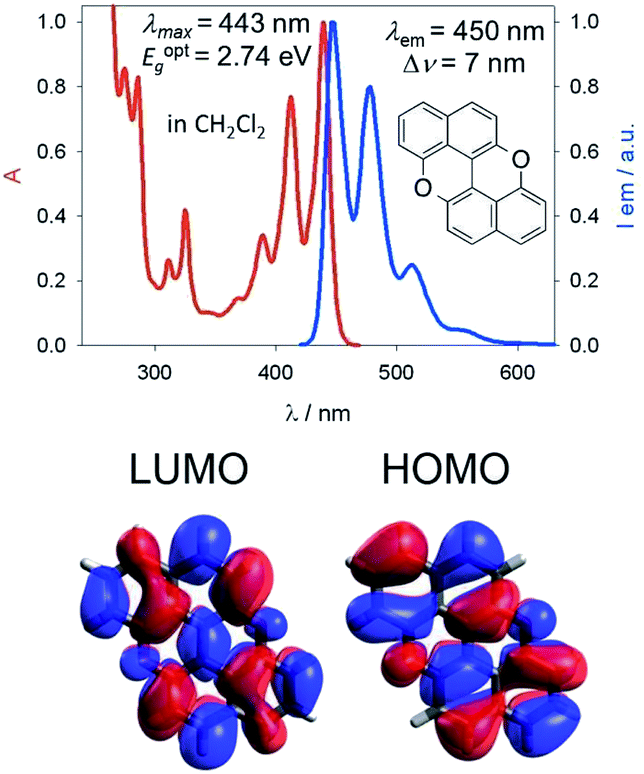 | ||
| Fig. 1 Structural framework of PXX along with its fundamental optical gap properties. | ||
This conjecture led us to design a series of N-doped PXX derivatives (N,N-PXX, featuring a ρ value of 9%), in which two N-atoms replace specific C(sp2)–H functions at the peripheries (Fig. 2). Given the large number of positional possibilities, here we focused our attention on N,N-PXX regioisomers bearing one N-atom for each naphthalenyl ring (Fig. 2). This provides a way to explore a reasonable variety of structures at fixed doping ratio (ρ). For the PXX molecule and the set of regioisomers, we performed ab initio calculations at the TD-/DFT/B3LYP/6-311+G* level of theory in Gaussian 09 (the accuracy of these and further computational details are discussed below and in the ESI†).34–40 For these systems we find that the first excited state (S1) is bright and energetically well separated from the second state (S2) that is consistently much darker. Our calculations also suggest that PXX and its N-doped congeners do not experience any significant conformational change in either the ground state or on electronic transition.
 | ||
| Fig. 2 Positional isomers of N,N-PXX, where the N-atoms are peripherally localised. | ||
Therefore, we moved to directly comparing excitations. The unique methodology we employ to compare excitations is based on calculating the set of atomic point charges (in the CHELPG formalism41) of the ground and excited state and examining the difference (Fig. 3). This difference is then interpolated across the space of the molecule to create the 2D maps which show how many electrons are gained or lost around an atom on excitation. The accuracy of these maps is examined in the ESI,† where we compare it to more physical quantities, such as the exact TDDFT density differences and molecular orbitals. As shown in the charge redistribution map of PXX (Fig. 3), the peripheral C-atoms display a significant and unequal electron redistribution upon the electronic transition. In particular, the C-atoms at positions 3 and 9 (marked as deeper red sites in Fig. 3) tend to donate electrons, whereas those at positions 4 and 10, or 5 and 11 (marked as deeper blue sites in Fig. 3) behave like electron-accepting sites during the electronic transition. Chemical intuition suggests that replacing a donor-like C(sp2) site with an atom with either more electronegative or electron withdrawing character on excitation might disrupt the PXX excitation pattern, possibly leading to poorer excitation properties (higher excitation energies, lower intensity, for example). For now we assume this to be the case, and this suggests that replacing the C-atoms with N-atoms in positions 3 and 9, i.e., the most electron depleting sites in the charge redistribution map, leads to a hypsochromic shift of the UV-vis absorption envelop when compared to its parent PXX.
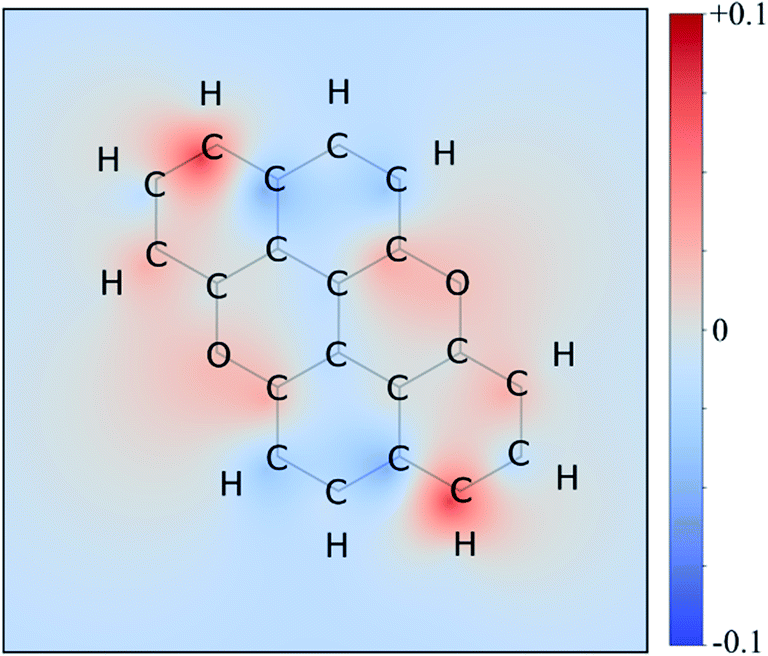 | ||
| Fig. 3 Excitation charge redistribution map for PXX. This is the excited state CHELPG charges minus ground state CHELPG charges of PXX interpolated over the atomic positions on the σ-scaffold, with the blue and red area indicating the electron depletion and accumulation regions respectively. The colour map ranges from +0.1 (red, donors) to −0.1 (blue, acceptors) electrons. | ||
On the other hand, if the N-replacement happens in electron-enriching positions, a bathochromic shift of the electronic transition is expected as the PXX charge redistribution should be enhanced. This is reflected by a shrinking of the optical gap. Consequently, this predicts the largest bathochromic shifts for the 4,11-, 4,10- and 5,11-doping patterns. Between the extreme cases of gap variation, there is also potential for fine-tuning the molecular optical gap. This would be achieved by combining the effects of the two N-dopants in sites that are characterised by a different role (withdrawing/donating/electronegative/electropositive). For the set of regioisomers of interest, the hypothesis that introducing N atoms at electron enriching positions leads to bathochromic shifts and the opposite for electron depleting sites is demonstrated in theory through jointly examining the regioisomer charge redistribution maps and TDDFT excitation energies (Fig. 4).
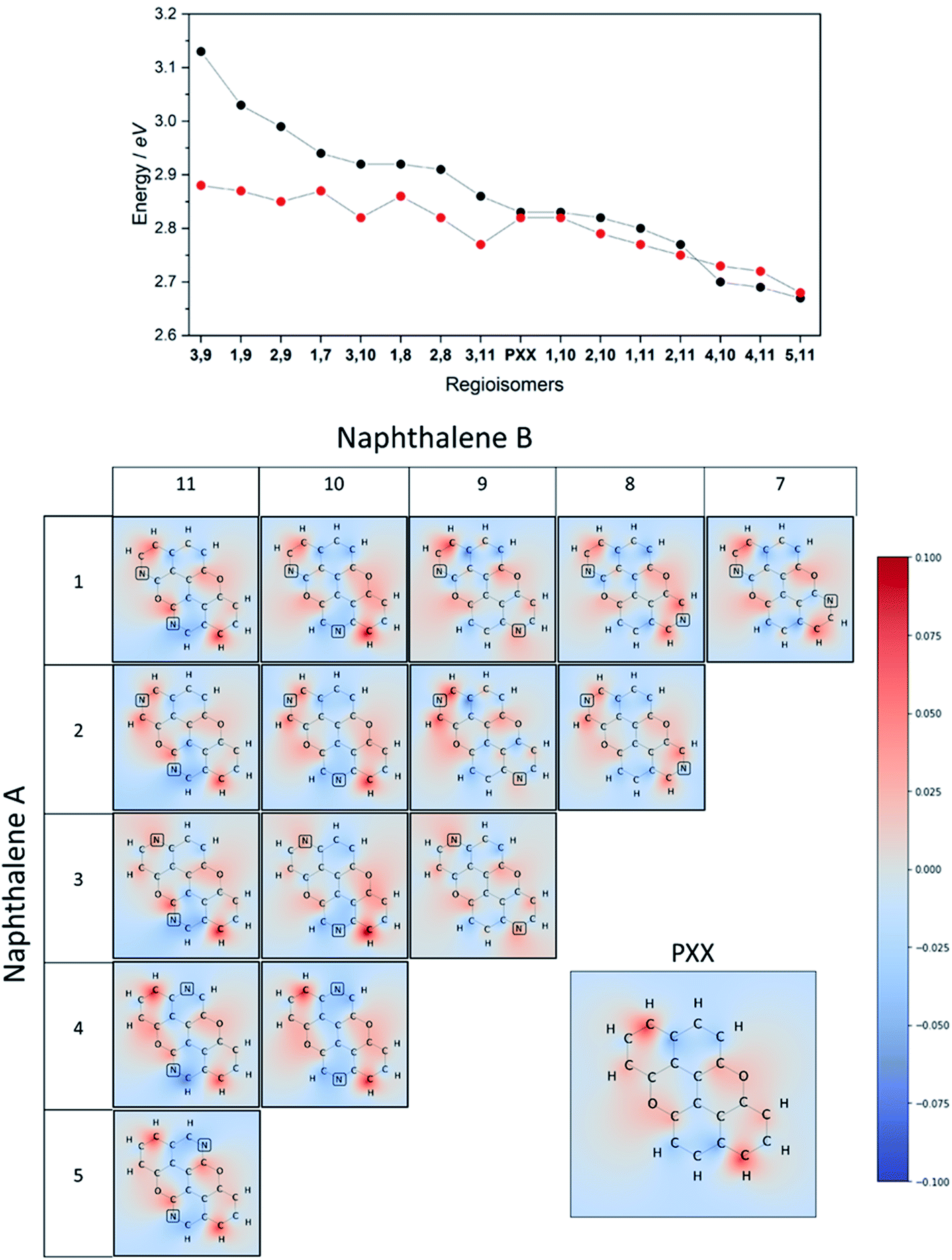 | ||
| Fig. 4 Top: predicted absorption energy for each positional isomers of N,N-PXX (DFT in black and Huckel in red). Centre: charge redistribution maps plotted on the σ-scaffold of positional isomers of N,N-PXX. The colour map ranges from +0.1 (red, donors) to −0.1 (blue, acceptors). | ||
Similarly, we predict an almost smooth control over the excitation energies depending on the doping configuration. Notably, the TDDFT results suggest that site-specific N-substitution allows a tuning of the optical gap by about 0.46 eV, going from +0.30 eV to −0.16 eV with respect to the gap of PXX. The final trend is that the ‘colour-bleached’ excitation maps (when contrasted to PXX) lead to hypsochromic shifts, implying that the reduced amount of electron transfer leads to a widening of the optical gap. The influence on transition intensity is more complex and is not discussed extensively here, however, we do highlight trends in the x and y directional components of the transition dipole moment in relation to the doping site. Later in this article, we present spectroscopic measurement of synthesised regioisomers, which validates our electronic structure methodology and approximations through closely matching calculated Franck–Condon lineshapes (see ESI†).
To juxtapose the TDDFT energies, we also present results from calculations using a Hückel tight-binding method (Fig. 4 top).42,43 This simplified Hamiltonian performs remarkably well in predicting the trend of excitation energies for regioisomers and gives reasonable accuracy when employing an empirical β parameter. The strength of these results is likely explained by examining the character of the TDDFT excitations, as the S0 → S1 transition of these π-conjugated molecules is primarily described (a contribution of more than 96%) by the HOMO → LUMO transfer. We advocate the use of the Hückel calculation to quickly (and exceptionally cheaply) screen the range of excitation energies possible due to doping configurations, however TDDFT results are preferable in this work due to the complete set of accurate ab initio properties without the use of the Hückel β.
Finally, it is important to note that the extent of the effect of tuning the optical gap of PXXvia N-substitution is enhanced by the presence of the O-atoms. Indeed, calculations and previous work on the N-doping of anthanthrene show a minor impact on the optical gap, although the trend is the same as that of its PXX congener, i.e., the N-substitution of electron depleting and enriching sites causes small blue and bathochromic shifts, respectively (Table S5 and Fig. S17†).
Synthesis
Capitalising on the Cu-mediated Pummerer44,45 oxidative cyclisation,31,46 we prepared N,N-PXX derivatives displayed in Scheme 1 following the (i) C–C and (ii) C–O bond formation strategy. Molecule 1 has been prepared following the reported protocol by Kwong.47 Quinolin-6-ol was dimerised via oxidative coupling in the presence of the mixture CuCl2/BnNH2 at room temperature. BIQOL 1 was obtained as a brown solid precipitate in 55% yield and reacted with CuO in PhNO2 under refluxing conditions (210 °C) to give 3,9-N,N-PXX in 23% yield. In a parallel avenue, isoquinolin-7-ol was dissolved in MeOH and treated with the CuCl2/BnNH2 complex at rt. BIQOL 2 was collected as brown precipitate and oxidised in the presence of CuO to form 1,7-N,N-PXX in 52% yield. When passing to quinolin-3-ol, no C–C coupling product could be formed under the CuCl2/BnNH2 reaction conditions and only starting material was recovered. Increasing the reaction temperature to 120 °C for 16 hours in DMSO led to full conversion, but only a mono O-annulated derivative could be isolated from the reaction mixture (molecule 3 in the ESI†). Thus, we turned our attention to the oxidative dimerization strategy developed by Milicevic and coworkers,48 who reported the synthesis of BINOLs through oxidative coupling of 1-lithiated naphthol derivatives obtained through ortho-metalation. Quinolin-3-ol was dissolved in dry pyridine and treated with carbamoyl chloride to yield carbamate derivative 4 in 95% yield. Treatment of 4 with LDA at −78 °C, followed by the addition of anhydrous FeCl3, gave carbamate-protected BIQOL 5 in 52% yield. Hydrolysis of the carbamoyl moieties with KOH in MeOH yielded BIQOL 6 in 98% that, under CuO-mediated oxidative cyclisation reaction, formed desired molecule 4,10-N,N-PXX in 20% yield.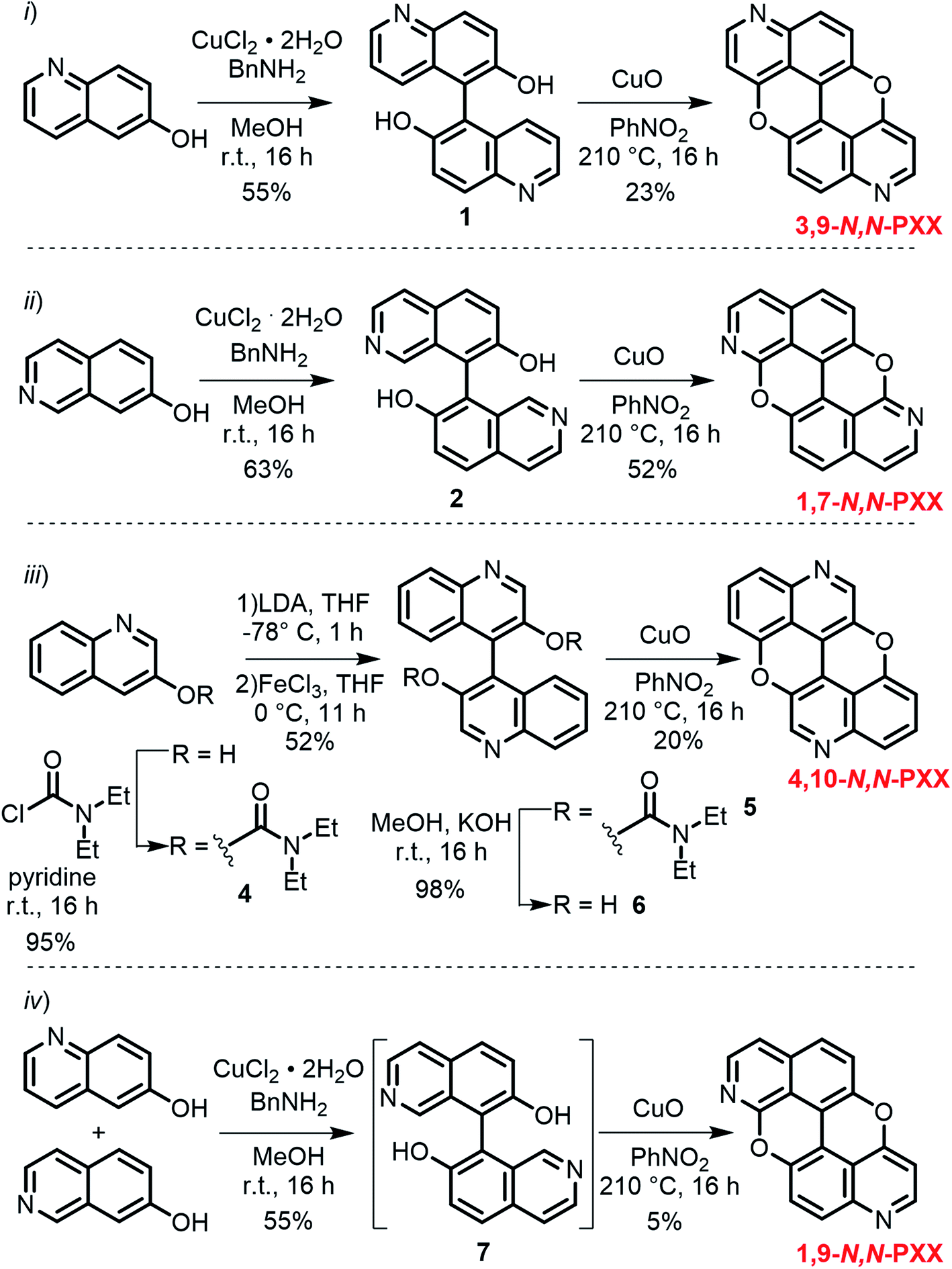 | ||
| Scheme 1 Synthetic routes to N,N-PXX: (i) 3,9-N,N-PXX, (ii) 1,7-N,N-PXX, (iii) 4,10-N,N-PXX and (iv) 1,9-N,N-PXX. | ||
Capitalising on the synthetic routes used to prepare derivatives 1,7-N,N-PXX and 3,9-N,N-PXX, reference molecule 1,9-N,N-PXX was also prepared.31,47 Isoquinolin-7-ol and quinolin-6-ol were dissolved in MeOH with CuCl2 and BnNH2 and coupled to form a statistical mixture of BIQOL derivatives. Subsequent oxidation with CuO yielded derivative 1,9-N,N-PXX as a yellow solid in 5%. Despite the numerous synthetic attempts, we could not prepare any of the N,N-PXX isomers depicting an N atom either at the 2 or 11 position (for further details see the ESI†). All molecules were fully characterised by NMR techniques, HR-MS spectrometry and single-crystal X-ray diffraction (see section below) analyses.
Photophysical investigations
Steady-state UV-vis absorption and emission measurements were carried out in toluene solutions (Fig. 5 and Table 1). The spectral envelopes of N,N-PXX molecules are similar to that of PXX, and are all characterized by π → π* transitions according to the TDDFT calculations. PXX depicts the lowest-energy electronic transition at 443 nm, whereas congeners 3,9-N,N-PXX, 1,9-N,N-PXX, and 1,7-N,N-PXX display blue-shifted bands centred at 413 nm, 427 nm and 437 nm, respectively (Table 1).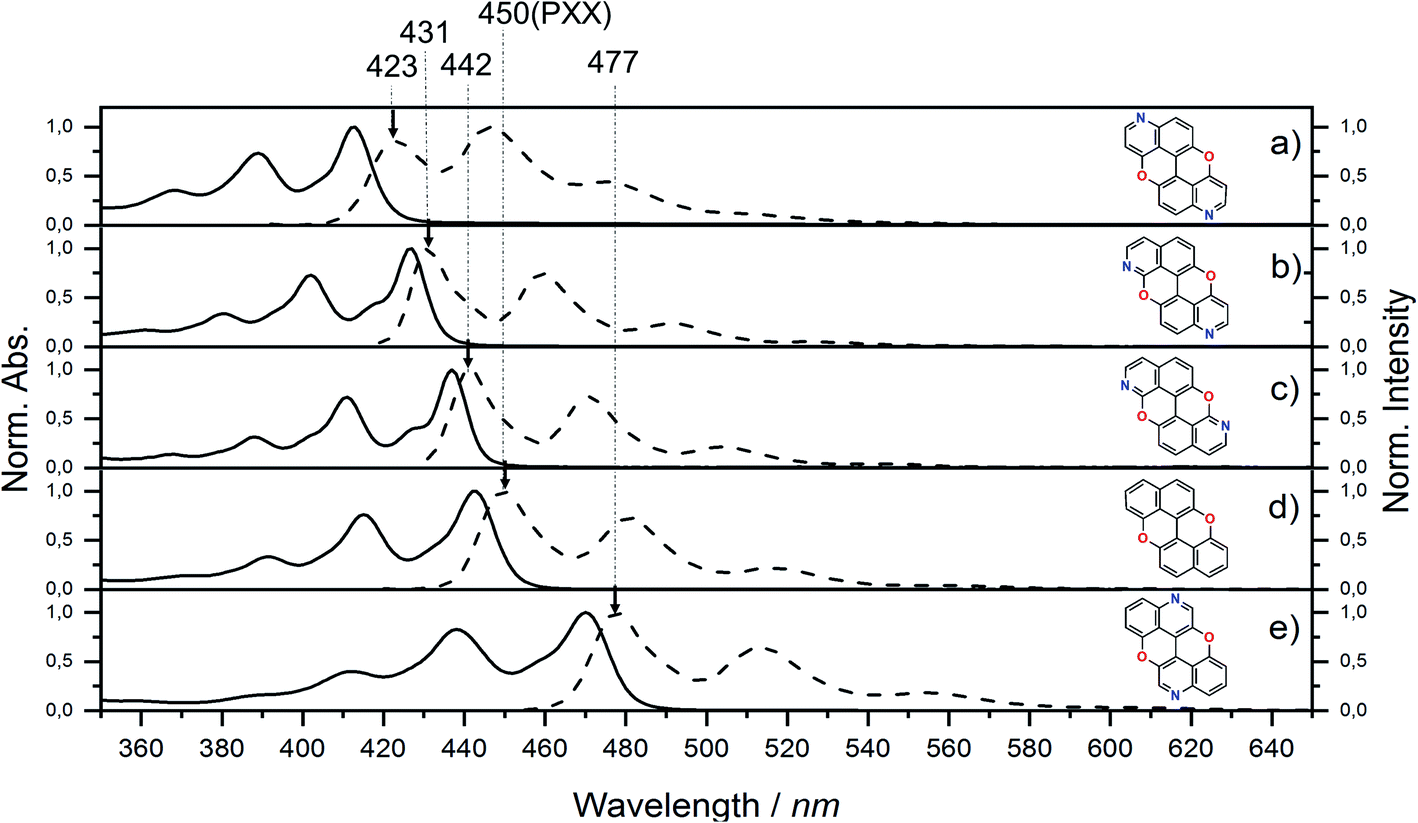 | ||
| Fig. 5 UV-vis absorption (full line) and emission (dashed line) spectra for N,N-PXX derivatives and PXX in toluene at rt. | ||
| Absorbance | Emission | Energy gaps | |||||||
|---|---|---|---|---|---|---|---|---|---|
| λ max (eV) [nm] | ε (M−1 cm−1) | E Texc (eV) | λ max (eV) [nm] | (ns) | Φ F (%) | Stokes shift (eV) [nm] | E optg (eV) | E Tg (eV) | |
| a Φ fl measured by optical dilute method and coumarin 153 as reference. b E optg = 1240/λmax (nm). c TDDFT calculated value. | |||||||||
| 3,9-N,N-PXX | 3.00 [413] | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 838 838 |
3.13 | 2.93 [423] | 6.6 | 75 | 7 × 10−2 [10] | 2.93 | 3.43 |
| 1,9-N,N-PXX | 2.90 [427] | 16667 |
3.03 | 2.88 [431] | 4.5 | 64 | 2 × 10−2 [4] | 2.84 | 3.37 |
| 1,7-N,N-PXX | 2.84 [437] | 26611 |
2.94 | 2.80 [442] | 3.8 | 61 | 4 × 10−2 [5] | 2.78 | 3.32 |
| PXX | 2.80 [443] | 17560 |
2.83 | 2.76 [450] | 5 | 61 | 4 × 10−2 [7] | 2.74 | 3.30 |
| 4,10-N,N-PXX | 2.63 [470] | 7448 | 2.70 | 2.60 [477] | 13.5 | 41 | 3 × 10−2 [7] | 2.57 | 3.02 |
Similarly, all emission spectral envelopes of 3,9-N,N-PXX, 1,9-N,N-PXX, and 1,7-N,N-PXX are blue-shifted compared to that of PXX (450 nm). In full agreement with our designing principle and theoretical predictions (see the experimental optical and theoretical energy gaps, Eoptg and ETg, Table 1), the 3,9-N-doping pattern of molecule 3,9-N,N-PXX induces the largest blueshift (ΔEoptg = +0.19 eV, Eoptg = 2.74 eV and 2.93 eV for PXX and 3,9-N,N-PXX, respectively), as the two most-depleting 3,9-sites in PXX have been replaced by two N-atoms. As anticipated by our model, a smaller blueshift of the optical gap was observed (ΔEoptg = +0.1 eV) with congener 1,7-N,N-PXX (Eoptg = 2.84 eV), as the 1,7-N-doping pattern is localised to positions in which minor electron redistribution occurs. Finally, a bathochromic shift (ΔEoptg = −0.17 eV) was observed with isomer 4,10-N,N-PXX, which depicts the π → π* transition centred at 470 nm (Eoptg = 2.57 eV). As anticipated by our predictive tool, the 4,10-N-doping pattern induces the strongest bathochromic shifts, being the N-atoms overlaying the most electron-enriching sites of PXX. While the fluorescence quantum yield (ΦF) values for 1,9-N,N-PXX, 1,7-N,N-PXX and PXX are virtually the same (∼60%, Table 1), a significant enhancement (∼75%) was observed for 3,9-N,N-PXX, the most blueshifted-absorbing derivative. In contrast, redshifted 4,10-N,N-PXX features the lowest ΦF value (41%). All lifetime values, τ, are within the same range (between 6 and 3 ns), but that for 4,10-N,N-PXX, which features the longest value (13.5 ns).
In light of this, the correlation between doping position and the variation of the absorption and emission maxima theoretically postulated was demonstrated since the optical bandgaps of the four positional isomers N,N-PXX follow perfectly the predicted values. Similarly, the TDDFT calculations are validated, showing good accuracy with respect to the optical gap and being only slightly red shifted and under sensitive to the doping pattern. These limitations may be addressed by choice of the exchange correlation functional, however the recreation of the UV-vis lineshapes by Franck–Condon (Fig. S14†) indicates perfectly adequate results here.
Electrochemical investigations
The effect of the N-substitution was also investigated against the redox behaviour of PXX (Table 2 and ESI†). Cyclic voltammetry (CV) studies of PXX and its N,N-PXX congeners were performed in orthodichlorobenzene (ODCB). As expected all N,N-PXX derivatives displayed higher (of ∼0.3–0.4 V) oxidation potentials (E1/2ox = 0.71 V, 0.71 V, 0.63 V and 0.65 V for 3,9-N,N-PXX, 1,9-N,N-PXX, 1,7-N,N-PXX and 4,10-N,N-PXX, respectively) when compared to that of PXX (E1/2ox = 0.30 V).| E 1/2ox (V) [mV] | E 1/2red1 (V) [mV] | E 1/2red2 (V) [mV] | E CVg (eV) |
|
|
HOMOb (eV) | LUMOb (eV) | |
|---|---|---|---|---|---|---|---|---|
| a Half-wave potentials are calculated as E1/2 = (Epa + Epc)/2 considering anodic (Epa) and cathodic (Epc) peak potentials, unless differently specified. Values in parentheses refer to the peak separation (Epa − Epc, in mV) of each process; “n.d.” stands for “not detected”. b Detected in CH3CN.3 c Values in parentheses are referred to the theoretical values calculated by DFT. | ||||||||
| 3,9-N,N-PXX | 0.71 [148] | −1.81 [116] | n.d. | 2.52 | −2.22 | 1.12 | −5.81 [−5.68] | −3.29 [−2.52] |
| 1,9-N,N-PXX | 0.71 [115] | −1.83 [125] | n.d. | 2.54 | −2.13 | 1.01 | −5.81 [−5.67] | −3.27 [−2.28] |
| 1,7-N,N-PXX | 0.63 [167] | −1.87 [97] | n.d. | 2.50 | −2.15 | 0.91 | −5.73 [−5.66] | −3.23 [−2.30] |
| PXX | 0.30 [119] | −2.59 [148]c | n.d. | 2.89 | −2.44 | 0.15c | −5.40 [−5.06] | −2.51 [−1.76] |
| 4,10-N,N-PXX | 0.65 [229] | −1.94 [108] | −2.35 [186] | 2.59 | −1.92 | 0.63 | −5.74 [−5.71] | −3.16 [−2.70] |
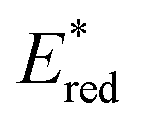
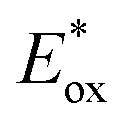
While PXX showed no reductive events within the investigated potential window, all N,N-PXX derivatives displayed reversible reduction events. For instance, reversible reduction peaks centred at E1/2red = −1.81 V, −1.83 V, and −1.87 V were observed for 3,9-N,N-PXX, 1,9-N,N-PXX and 1,7-N,N-PXX, respectively. Interestingly, the CVs of 4,10-N,N-PXX showed two reversible reduction peaks centred at −1.94 V (E1/2red1) and −2.35 V (E1/2red2), respectively. As expected, these data suggest that the N-doping causes a low-energy rigid shift of the energy levels of the HOMO and LUMO orbitals. When looking at the electrochemical gap (ECVg), calculated as E1/2ox − E1/2red1, the replacement of two C-atoms with N-atoms shrank the ECVg value of around 0.4 V when compared to that of PXX. However, the ECVg values of the N,N-PXX isomers are very similar, with the largest ΔECVg (0.09 eV) being that between isomers 1,7-N,N-PXX and 4,10-N,N-PXX (Table 2). These observations suggest that the doping pattern has a minor impact on the ECVg value when compared to that of the doping ratio ρ, which is a more influential parameter for the redox properties, i.e., higher oxidations and reduction potentials and a shrank ECVg, of this class of molecules.
From the combination of photophysical and redox properties, one can estimate the oxidation and reductions of the excited states and thus the photoredox properties of PXX and its N-containing congeners (Fig. 6).6 Consideration of the redox properties in both the excited and ground states is crucial when evaluating and/or tailoring the photochemical reactivity of a chromophore in the context of light-triggered applications (e.g., light-harvesting systems, photosynthesis and photocatalysis to name a few).6,49–51 From Fig. 6 it is evident that, when comparing the reductive and oxidation properties of the excited states, the N,N-PXX derivatives are respectively stronger oxidisers and weaker reducers at their S1 states than PXX. While the reduction potentials of the S1 state 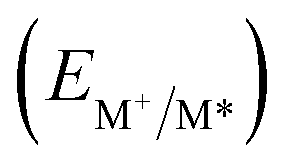 within the N,N-PXX series are virtually the same within the experimental errors, the S1-centered oxidation potential
within the N,N-PXX series are virtually the same within the experimental errors, the S1-centered oxidation potential 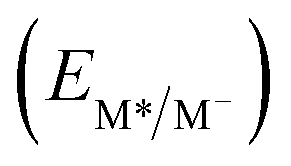 increases in the order 4,10-N,N-PXX, 1,7-N,N-PXX, 1,9-N,N-PXX, and 3,9-N,N-PXX (Fig. 6). The N-pattern has a strong impact on the oxidation property of the S1 state, with the most hypsochromically- and bathochromically-shifted absorbers being the stronger and weaker oxidisers respectively.
increases in the order 4,10-N,N-PXX, 1,7-N,N-PXX, 1,9-N,N-PXX, and 3,9-N,N-PXX (Fig. 6). The N-pattern has a strong impact on the oxidation property of the S1 state, with the most hypsochromically- and bathochromically-shifted absorbers being the stronger and weaker oxidisers respectively.
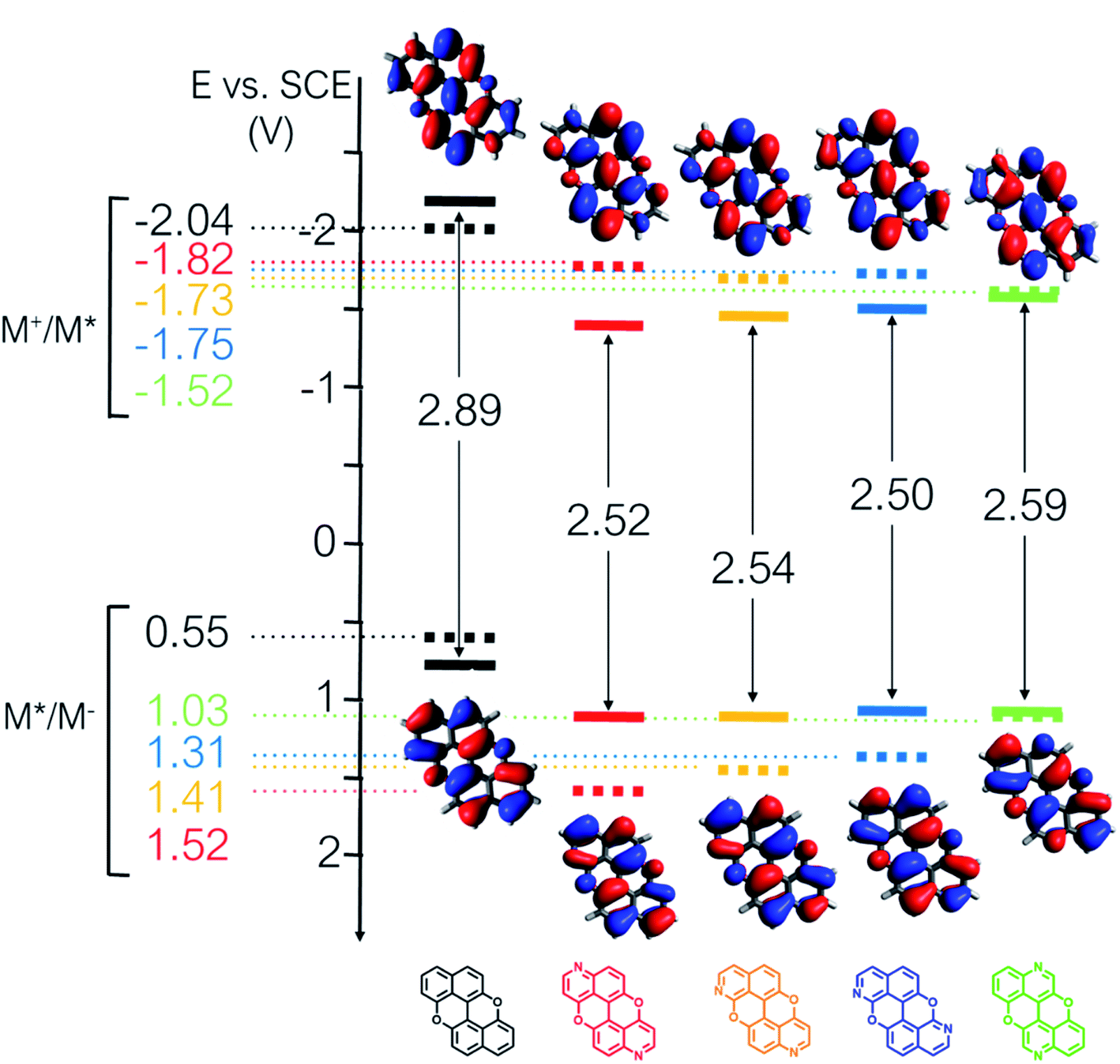 | ||
Fig. 6 Redox potentials vs. SCE of PXX, 3,9-N,N-PXX, 1,9-N,N-PXX, 1,7-N,N-PXX and 4,10-N,N-PXX estimated from the CV data obtained in ODCB. Dashed lines: reduction 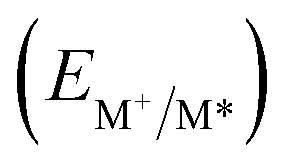 and oxidation and oxidation 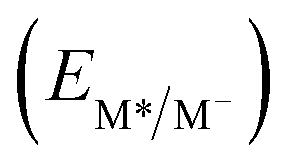 potentials of the singlet excited states estimated using the optical band gap measured in toluene.6 Inserts: DFT calculated HOMO (bottom) and LUMO (top) lobes.6 potentials of the singlet excited states estimated using the optical band gap measured in toluene.6 Inserts: DFT calculated HOMO (bottom) and LUMO (top) lobes.6 | ||
Solid-state self-assembly
At last, we studied the effect of the N-doping on the solid-state organisation of PXX. PXX undergoes the typical herringbone arrangement of PAHs (Fig. 7),52 where the molecules organise through edge-to-face and face-to-face (dπ–π = 3.381 Å) along the a and c axes, respectively.52 The molecule featuring the N-atoms along the 1,7-pattern undertakes a different organisation (Fig. 8). In particular, X-ray diffraction analysis of crystals obtained from slow vapour diffusion of Et2O into a toluene solution of 1,7-N,N-PXX revealed the formation of a non-covalent lamellar architecture, in which the molecules arrange into supramolecular sheets (i.e., 2D polymeric architecture (1,7-N,N-PXX)n) and are held together by four pairs of weak DD⋯AA-type H-bonds.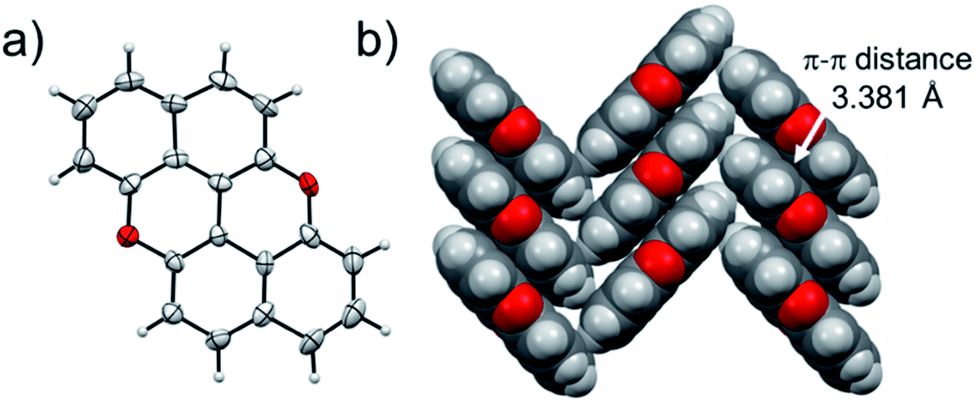 | ||
| Fig. 7 Single-crystal X-ray structure of PXX. ORTEP representation (a) and (b) herringbone-type arrangement. Space groups: P21/c. Data taken from literature.52 Atom colours: red O, grey C, white H. | ||
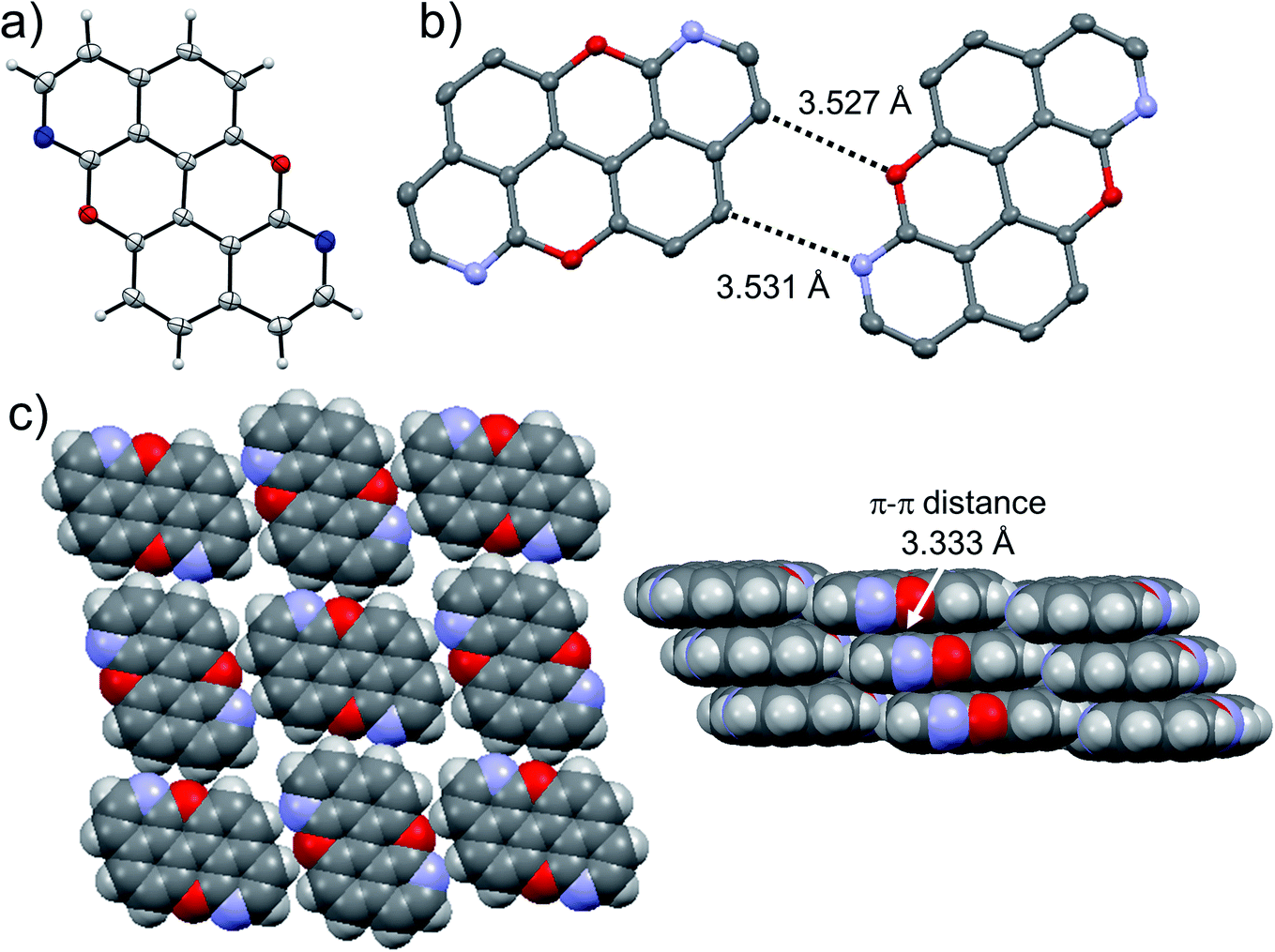 | ||
| Fig. 8 Single-crystal X-ray structure of 1,7-N,N-PXX. ORTEP representation (a) and structure of two isolated molecules engaging into H-bonds (b) depicting a multilayer-type organisation of π–π stacked supramolecular H-bonded sheets (c). Space groups: P21/c. Atom colours: red O, blue N, grey C, white H. | ||
These are established between the side-by-side O and N heteroatoms and the pseudo–peri H atoms of a neighbouring molecule. In total, each molecule establishes four H-bonds as donor and four as acceptor. Within the non-covalent sheet, N⋯C and O⋯O distances are equal to 3.531 Å and 3.527 Å. At the molecular level, the supramolecular sheets organise in an off-set multilayered, graphite-like, fashion displaying an averaged interplanar π–π stacking distance of 3.333 Å (Fig. 8).
Suitable crystals for X-ray analysis for 3,9-N,N-PXX were obtained from slow evaporation of a CHCl3 solution. The analysis revealed the formation of a co-crystal with water in a 1:2 ratio with 3,9-N,N-PXX (Fig. 9). Interestingly, a supramolecular polymer (3,9-N,N-PXX·2H2O)n is formed through a 1:2 association of both components. One can easily discern dimeric associates of the water molecules through an O⋯H bond (dO⋯O = 2.552 Å). Each water dimer sandwiches a molecule of 3,9-N,N-PXX through two H-bonds established with the quinoline-like N-atoms (dN⋯O = 2.858 Å). Notably, 3,9-N,N-PXX arranges in columnar π–π stacks (estimated dπ–π = 3.4 Å) separated by a layer of dimeric water molecules (Fig. 9). The solid-state arrangement led to a segregation between the hydrophilic and non-hydrophilic moieties.
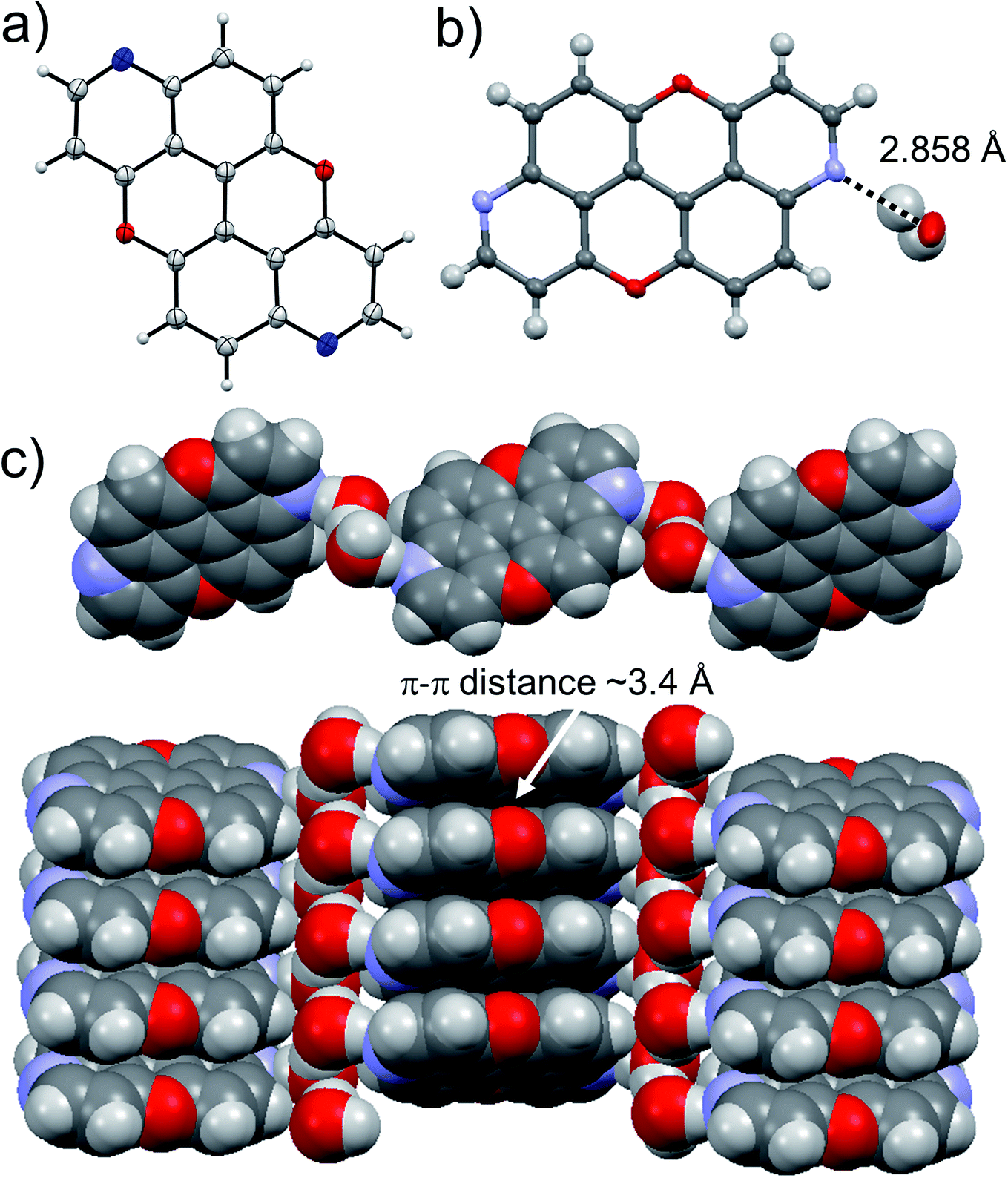 | ||
| Fig. 9 Single-crystal X-ray structure of 3,9-N,N-PXX. ORTEP representation (a) and structure of the water and 3,9-N,N-PXX molecules engaging into H-bonds (b) depicting the supramolecular polymer organisation (c). Space groups: P21/c. Atom colours: red O, blue N, grey C, white H. | ||
At last, we studied the solid-state organisation of isomer 4,10-N,N-PXX. The crystals were grown by slow diffusion of hexane vapours into a CHCl3 solution of 4,10-N,N-PXX. Only a columnar arrangement was observed (Fig. 10), with the molecule undergoing off-set face-to-face π–π stacking (estimated dπ–π = 3.4 Å). The columnar stacks (Fig. 10) lies each other through very weak C–H⋯O bonds (dC–H⋯O = 3.530 Å).
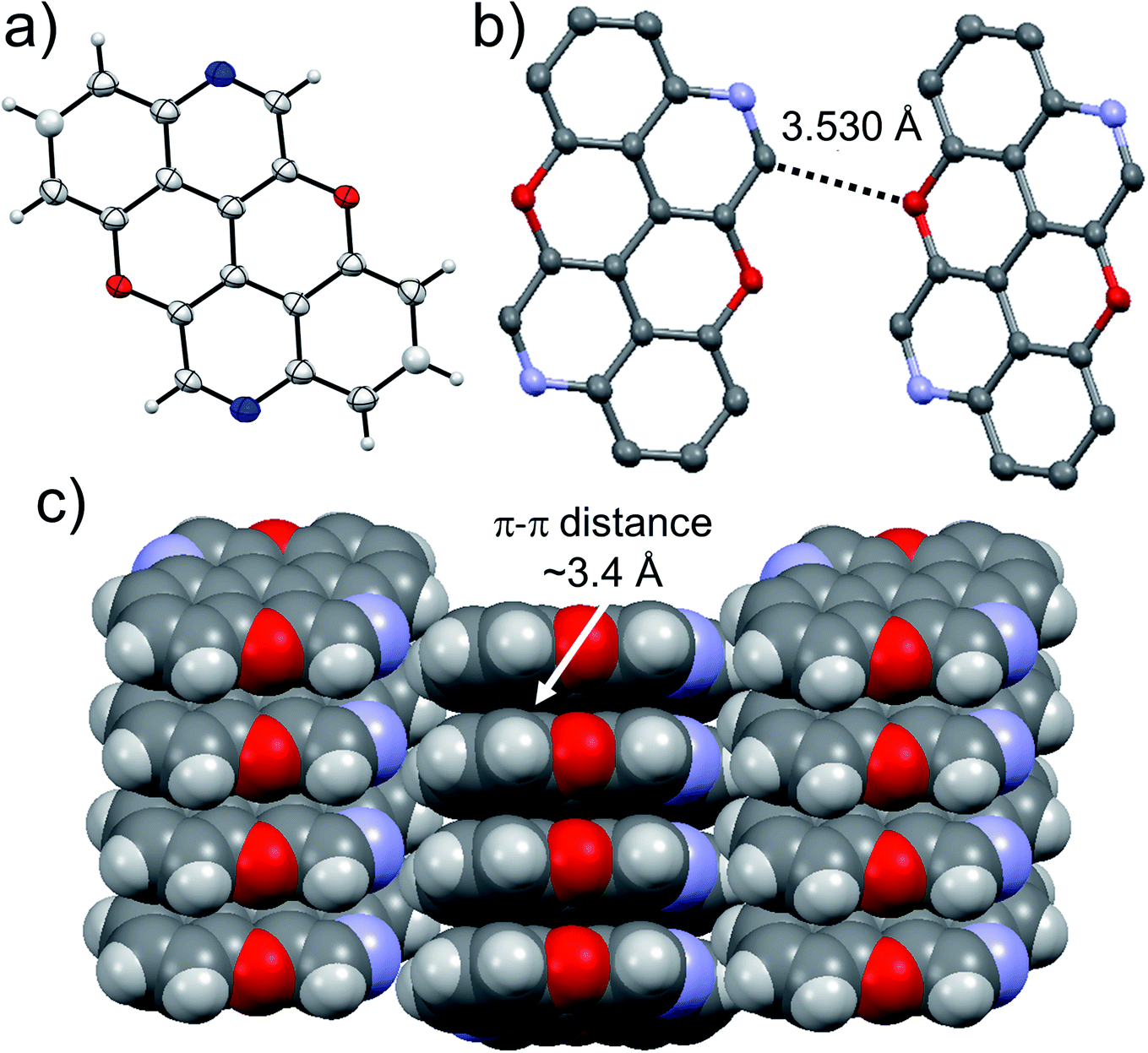 | ||
| Fig. 10 Single-crystal X-ray structure of 4,10-N,N-PXX. ORTEP representation (a) and structure of two molecules engaging into H-bonds (b) forming a columnar organization (c). Space groups: P21/n. Atom colours: red O, blue N, grey C, white H. | ||
The presence of the nucleophilic quinoline-like N-atoms offered us the possibility to exploit halogen-bonding interactions (XBIs) to govern the solid-state organisation. Thus, we independently co-crystallized molecules 3,9-N,N-PXX and 4,10-N,N-PXX with 1,4-diiodo-2,3,5,6-tetrafluorobenzene (DITFB) in a 1:1 ratio, and grew crystals from slow evaporation of a CHCl3 solution. As expected, in both cases the given N,N-PXX derivatives undergo XBIs with DITFB, giving rise to halogen-bonded complexes 3,9-N,N-PXX·DITFB and 4,10-N,N-PXX·DITFB, respectively. In the case of 3,9-N,N-PXX·DITFB, a supramolecular polymeric ribbon (3,9-N,N-PXX·DITFB)n was formed at the solid state (Fig. 11). The structure displays the quinoline-like moieties establishing XBIs (dN⋯I = 2.871 Å) with DITFB, with its tetrafluoroaryl core being coplanar with the polycyclic ring of 3,9-N,N-PXX. The halogen-bonded ribbons arrange through lateral weak H-bonds (dO⋯C = 3.476 Å) in striped patterned sheets. The sheets arrange in multilayers through π–π stacking interactions, where the fluorinated and non-fluorinated moieties are segregated and the 3,9-N,N-PXX and DITFB modules engage with homomolecular off-set π–π stacking interactions (dπ–π = 3.357 Å and 3.540 Å, off-set of 0.85 Å and 1.43 Å, Fig. 11). Also 4,10-N,N-PXX·DITFB organises in supramolecular polymeric ribbons (4,10-N,N-PXX·DITFB)n at the solid state (Fig. 12). In this case, longer-distance XBIs (dN⋯I = 2.949 Å) are established between the halogen acceptor and donor molecules, which are also segregated in fluorinated and non-fluorinated domains. In contrast to (3,9-N,N-PXX·DITFB)n, the monomeric units in polymer (4,10-N,N-PXX·DITFB)n are not coplanar (i.e., the aromatic cores of 4,10-N,N-PXX and DITFB are not coplanar) and terraced layers are formed (Fig. 12). Neighbouring supramolecular polymers are held together by lateral very weak H-bonds (dO⋯C = 3.582 Å). Similarly to the case of co-crystal 3,9-N,N-PXX·DITFB, molecules 4,10-N,N-PXX and DITFB engage with homomolecular π–π stacking interactions (dπ–π = 3.393 Å and 3.732 Å, off-set of 1.04 Å and 1.42 Å, Fig. 12).
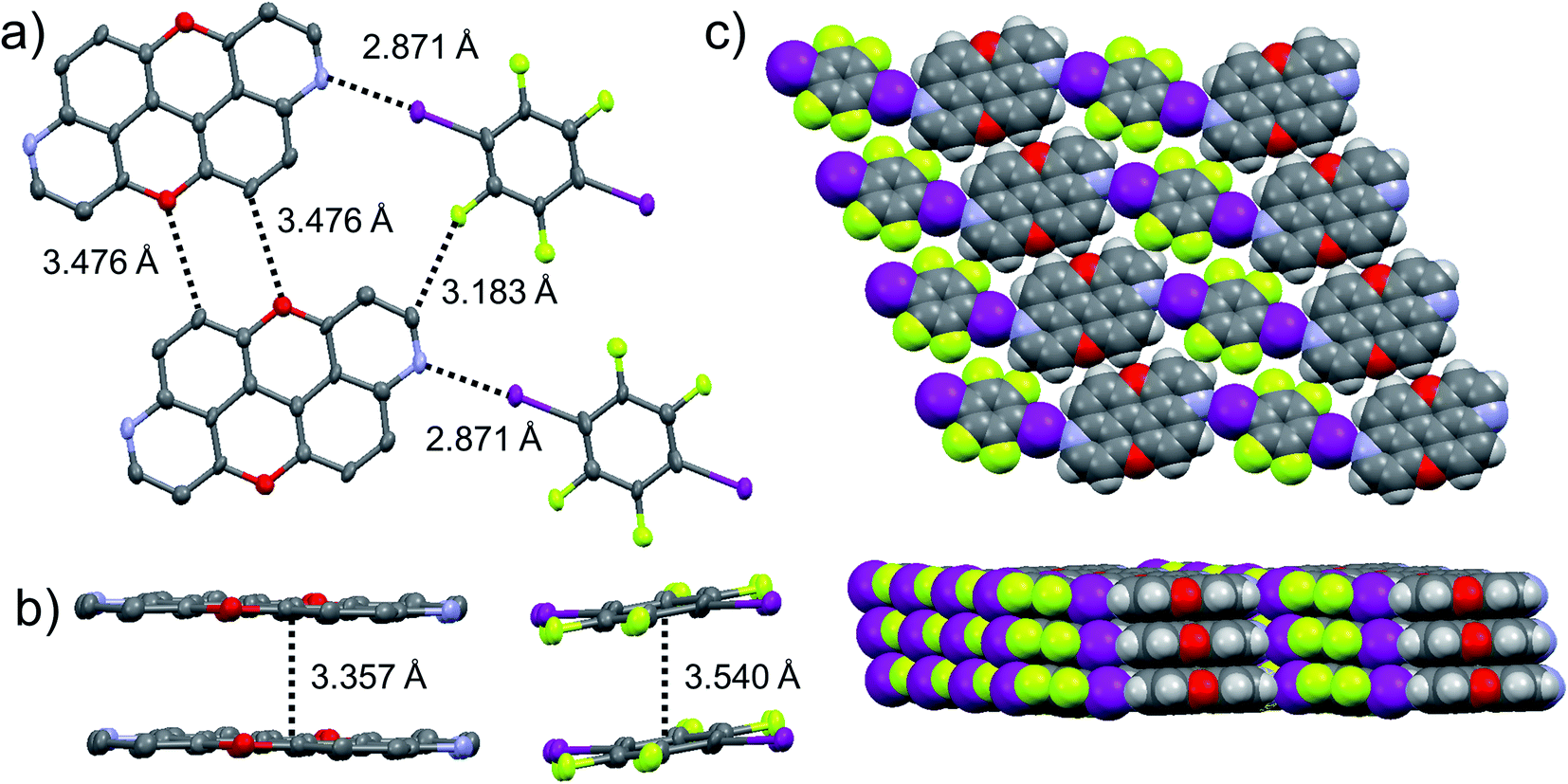 | ||
Fig. 11 Single-crystal X-ray structure of co-crystal 3,9-N,N-PXX·DITFB. ORTEP representation and structure of four molecules engaging into H-bonds and XBIs (a) organised in supramolecular multi-layered π–π stacked (b) sheets (c). Space groups: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Atom colours: red O, blue N, purple I, green F, grey C, white H. . Atom colours: red O, blue N, purple I, green F, grey C, white H. | ||
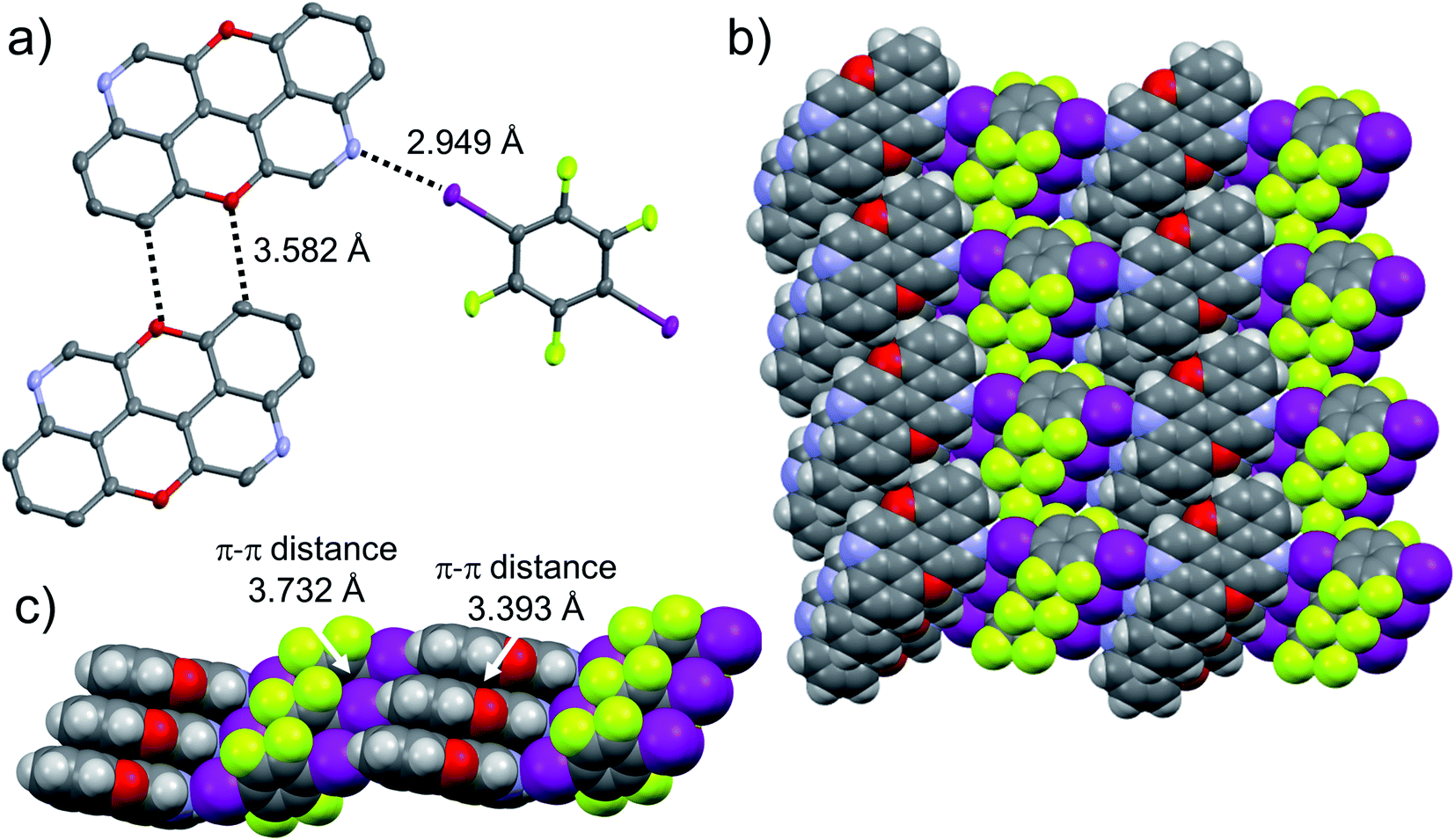 | ||
| Fig. 12 Single-crystal X-ray structure of co-crystal 4,10-N,N-PXX·DITFB. ORTEP representation and structure of three molecules engaging into H-bonds and XBIs (a) organised in supramolecular π–π stacked (b) supramolecular polymers (c). Space groups: P. Atom colours: red O, blue N, purple I, green F, grey C, white H. | ||
Conclusion
In this paper we have raised the question of whether (i) we can rationally tailor the optical bandgap properties of a PAH by a specific heteroatom doping pattern and (ii) if this can be predicted a priori to guide organic chemists in the structural design. Thus, we have developed a predictive tool that identifies C(sp2)-positions in the π-conjugated core of a given PAH that, if substituted with a heteroatom, would cause the desired changes in the UV-vis absorption and emission spectral envelopes. Using peri-xanthenoxanthene (PXX) as a model polyaromatic hydrocarbon, we showed that TDDFT calculations of the electron density difference between the S1 excited state and S0 ground state of PXX allowed us to identify the subtleties in the role of atomic sites, i.e., electron withdrawing or donating abilities, on the excitation energy. Specifically, we have found that the replacement of two C(sp2)-atoms with two N-atoms in either electron donating or withdrawing positions, shift the electronic transitions either bathochromically or hypsochromically, respectively. To validate the predictive tool, we synthesised a series of N-doped PXX positional isomers peripherally bearing one N-atom for each naphthalenyl ring and characterized their electronic transitions by means of steady-state absorption and emission UV-vis spectroscopies. In full agreement with theoretical predictions, the spectral envelops of the N-doped PXX derivatives depicting the N atoms in the most-depleting sites are blueshifted when compared to that of PXX (with 3,9-N,N-PXX displaying the largest shift: +0.19 eV). On the other hand, when overlaying the N-pattern on the most electron-enriching sites of PXX, a bathochromic shift of the absorption bands was observed (−0.17 eV for 4,10-N,N-PXX), as anticipated by our predictive tool. Fluorescence quantum yield measurements show the more blueshifted the absorption/emission energies, the greater the ΦF value (from ∼41% to ∼75%). Surprisingly, CV studies suggested that the redox properties are only marginally affected by the N-pattern and all N,N-PXX depict very similar redox properties at the ground state. Lastly, we have studied the solid-state organisation of the N-doped PXX, and showed that the N-pattern has a strong impact on the supramolecular architecture. While the N,N-PXX derivative depicting the 1,7-pattern forms multi-layered, graphene-like supramolecular sheets, the 3,9- and 4,10-congeners self-assemble into wire-like polymeric architectures. When the 3,9- and 4,10-N,N-PXX derivatives are co-crystallized with a halogen-bond donor molecule, supramolecular striped patterned sheets are formed.To the best of our knowledge, this work describes the first design of heteroatom-doped PAHs in which charge-redistribution maps of singlet-state excitations are used to rationally conceive the doping patterns tailoring a given electronic energy transition. This simple approach can be generalised and assists organic chemists to conceive tailored chromophores via specific doping patterns, thus minimising synthetic efforts.
Conflicts of interest
There are no conflicts to declare.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
CV carried out all organic synthetic work, structural characterisation, electrochemical studies and UV-vis steady-state, spectroscopic absorption and emission studies and interpreted the data with DB. DG performed all the DFT calculations, computed the charge-redistribution maps and interpreted the data with NB. GB prepared and characterised molecule 1,9-N,N-PXX. DR performed the X-ray diffraction analysis and solved the X-ray structure for all molecules but derivative 3, which was performed by ND. DB conceived and coordinated the project and conceptualised the content of the manuscript together with NB and DG. All authors participated in discussing the data and wrote the manuscript.Acknowledgements
D. B. gratefully acknowledge the EU through the MC-RISE “INFUSION” and H2020-IA “DecoChrom” projects, the School of Chemistry at Cardiff University (CA) and the University of Vienna for financial support. CV thanks the School of Chemistry at CA for his doctoral fellowship. D. G. and N. B. acknowledge the supportive research environment of the EPSRC Centre for Doctoral Training in Cross-Disciplinary Approaches to Non-Equilibrium Systems (CANES, No. EP/L015854/1) and the U.K. Materials and Molecular Modelling Hub (MMM Hub) for compute resources, which is partially funded by EPSRC (EP/P020194/1).References
- O. Kulyk, L. Rocard, L. Maggini and D. Bonifazi, Chem. Soc. Rev., 2020, 49, 8400–8424 RSC.
- C. Pezzetta, A. Folli, O. Matuszewska, D. Murphy, R. W. M. Davidson and D. Bonifazi, Adv. Synth. Catal., 2021, 363, 4740–4753 CrossRef CAS.
- A. Sciutto, A. Fermi, A. Folli, T. Battisti, J. M. Beames, D. M. Murphy and D. Bonifazi, Chem.–Eur. J., 2018, 24, 4382–4389 CrossRef CAS PubMed.
- A. L. Trifonov, L. I. Panferova, V. V. Levin, V. A. Kokorekin and A. D. Dilman, Org. Lett., 2020, 22, 2409–2413 CrossRef CAS PubMed.
- Q. Ma, J. Song, X. Zhang, J. Yu, L. Ji and S. Liao, Nat. Commun., 2021, 12, 429 CrossRef CAS PubMed.
- A. Fermi and D. Bonifazi, Photochemistry, 2020, 47, 293–325 Search PubMed.
- A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Zyła-Karwowska and M. Stępien, Chem. Rev., 2021, 122, 565–788 CrossRef PubMed.
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed.
- A. Narita, X. Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- D. B. Granger, Y. Mei, K. J. Thorley, S. R. Parkin, O. D. Jurchescu and J. E. Anthony, Org. Lett., 2016, 18, 6050–6053 CrossRef CAS PubMed.
- Y. S. Park, D. J. Dibble, J. Kim, R. C. Lopez, E. Vargas and A. A. Gorodetsky, Angew. Chem., Int. Ed., 2016, 55, 3352–3355 CrossRef CAS PubMed.
- A. Riaño, K. Strutyński, M. Liu, C. T. Stoppiello, B. Lerma-Berlanga, A. Saeki, C. Martí-Gastaldo, A. N. Khlobystov, G. Valenti, F. Paolucci, M. Mell and A. Mateo-Alonso, Angew. Chem., Int. Ed., 2021, 61, e202113657 Search PubMed.
- R. K. Dubey, M. Melle-Franco and A. Mateo-Alonso, J. Am. Chem. Soc., 2021, 143, 6593–6600 CrossRef CAS PubMed.
- D. Lehnherr, J. M. Alzola, C. R. Mulzer, S. J. Hein and W. R. Dichtel, J. Org. Chem., 2017, 82, 2004–2010 CrossRef CAS PubMed.
- L. Ji, A. Friedrich, I. Krummenacher, A. Eichhorn, H. Braunschweig, M. Moos, S. Hahn, F. L. Geyer, O. Tverskoy, J. Han, C. Lambert, A. Dreuw, T. B. Marder and U. H. F. Bunz, J. Am. Chem. Soc., 2017, 139, 15968–15976 CrossRef CAS PubMed.
- Z. Zeng, H. Jin, K. Sekine, M. Rudolph, F. Rominger, A. Stephen and K. Hashmi, Angew. Chem., Int. Ed., 2018, 57, 6935–6939 CrossRef CAS PubMed.
- P. Karak, C. Dutta, T. Dutta, A. L. Koner and J. Choudhury, Chem. Commun., 2019, 55, 6791–6794 RSC.
- U. H. F. Bunz and J. Freudenberg, Acc. Chem. Res., 2019, 52, 1575–1587 CrossRef CAS PubMed.
- C. L. Deng, J. P. Bard, L. N. Zakharov, D. W. Johnson and M. M. Harley, Org. Lett., 2019, 21, 6427–6431 CrossRef CAS PubMed.
- B. L. Hu, C. An, M. Wagner, G. Ivanova, A. Ivanova and M. Baumgarten, J. Am. Chem. Soc., 2019, 141, 5130–5134 CrossRef CAS PubMed.
- J. P. Mora-Fuentes, A. Riaño, D. Cortizo-Lacalle, A. Saeki, M. Melle-Franco and A. Mateo-Alonso, Angew. Chem., Int. Ed., 2018, 58, 552–556 CrossRef PubMed.
- K. Jug and G. Hahn, J. Comput. Chem., 1983, 4, 410–418 CrossRef CAS.
- R. M. Hochstrasser and J. W. Michaluk, J. Chem. Phys., 1971, 55, 4668 CrossRef CAS.
- D. Stassen, N. Demitri and D. Bonifazi, Angew. Chem., Int. Ed., 2016, 55, 5947–5951 CrossRef CAS PubMed.
- A. Berezin, N. Biot, T. Battisti and D. Bonifazi, Angew. Chem., Int. Ed., 2018, 57, 8942–8946 CrossRef CAS PubMed.
- A. Sciutto, A. Berezin, M. Lo Cicero, T. Miletić, A. Stopin and D. Bonifazi, J. Org. Chem., 2018, 83, 13787–13798 CrossRef CAS PubMed.
- L. Đorđević, C. Valentini, N. Demitri, C. Mézière, M. Allain, M. Sallé, A. Folli, D. Murphy, S. Mañas-Valero, E. Coronado and D. Bonifazi, Angew. Chem., Int. Ed., 2019, 59, 4106–4114 CrossRef PubMed.
- J. A. Christensen, J. Zhang, J. Zhou, J. N. Nelson and M. R. Wasielewski, J. Phys. Chem. C, 2018, 122, 23364–23370 CrossRef CAS.
- C. Song and T. M. Swager, Macromolecules, 2009, 42, 1472–1475 CrossRef CAS.
- N. Lv, M. Xie, W. Gu, H. Ruan and S. Qiu, Org. Lett., 2013, 15, 2382–2385 CrossRef CAS PubMed.
- T. Miletić, A. Fermi, I. Orfanos, A. Avramopoulos, F. De Leo, N. Demitri, G. Bergamini, P. Ceroni, M. G. Papadopoulos, S. Couris and D. Bonifazi, Chem.–Eur. J., 2017, 23, 2363–2378 CrossRef PubMed.
- N. Kobayashi, M. Sasaki and K. Nomoto, Chem. Mater., 2009, 21, 552–556 CrossRef CAS.
- M. Noda, N. Kobayashi, M. Katsuhara, A. Yumoto, S. Ushikura, R. Yasuda, N. Hirai, G. Yukawa, I. Yagi, K. Nomoto and T. Urabe, SID Symp. Dig. Tech. Pap., 2010, 41, 710–713 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 2002, 98, 11623–11627 CrossRef.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, K. J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, 2016 Search PubMed.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- Y. Carissan, D. Hagebaum-Reignier, N. Goudard and S. Humbel, J. Phys. Chem. A, 2008, 112, 13256–13262 CrossRef CAS PubMed.
- Y. Carissan, D. Hagebaum-Reignier, D. N. Goudard and S. Humbel, 2008, http://www.hulis.free.fr.
- R. Pummerer and A. Rieche, Ber. Dtsch. Chem. Ges., 1926, 59, 2161–2175 CrossRef.
- R. Pummerer and F. Frankfurter, Ber. Dtsch. Chem. Ges., 1914, 47, 1472–1493 CrossRef CAS.
- T. Kamei, M. Uryu and T. Shimada, Org. Lett., 2017, 19, 2714–2717 CrossRef CAS PubMed.
- Y. X. Chen, L. W. Yang, Y. M. Li, Z. Y. Zhou, K. H. Lam, a. S. Chan and H. L. Kwong, Chirality, 2000, 12, 510–513 CrossRef CAS PubMed.
- P. R. Blakemore, C. Kilner and S. D. Milicevic, J. Org. Chem., 2005, 70, 373–376 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- T. Asari, N. Kobayashi, T. Naito and T. Inabe, Bull. Chem. Soc. Jpn., 2001, 74, 53–58 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional transient absorption data, synthetic and computational details, and X-ray crystallographic data. CCDC 1985339 (1,7-N,N-PXX), 1985342 (3,9-N,N-PXX), 1985344 (4,10-N,N-PXX), 2133474 (4,10-N,N-PXX·DITFB), 2133475 (3,9-N,N-PXX·DITFB) and 2151478 (3). For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01038k |
| This journal is © The Royal Society of Chemistry 2022 |