
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid and column-free syntheses of acyl fluorides and peptides using ex situ generated thionyl fluoride†
Cayo
Lee
 ,
Brodie J.
Thomson
and
Glenn M.
Sammis
*
,
Brodie J.
Thomson
and
Glenn M.
Sammis
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: gsammis@chem.ubc.ca
First published on 29th November 2021
Abstract
Thionyl fluoride (SOF2) was first isolated in 1896, but there have been less than 10 subsequent reports of its use as a reagent for organic synthesis. This is partly due to a lack of facile, lab-scale methods for its generation. Herein we report a novel protocol for the ex situ generation of SOF2 and subsequent demonstration of its ability to access both aliphatic and aromatic acyl fluorides in 55–98% isolated yields under mild conditions and short reaction times. We further demonstrate its aptitude in amino acid couplings, with a one-pot, column-free strategy that affords the corresponding dipeptides in 65–97% isolated yields with minimal to no epimerization. The broad scope allows for a wide range of protecting groups and both natural and unnatural amino acids. Finally, we demonstrated that this new method can be used in sequential liquid phase peptide synthesis (LPPS) to afford tri-, tetra-, penta-, and decapeptides in 14–88% yields without the need for column chromatography. We also demonstrated that this new method is amenable to solid phase peptide synthesis (SPPS), affording di- and pentapeptides in 80–98% yields.
The past decade has witnessed a resurgence in the application of sulfur(VI) fluorides to organic synthesis. The majority of these studies have focused on the commodity chemical sulfuryl fluoride (SO2F2)1 and its derivatives,2 which readily react with a wide variety of oxygen-containing functional groups, such as alcohols (1),3 oximes (2),4 and carboxylates (3),5 to form activated fluorosulfate intermediates (5). Fluorosulfates behave like triflate surrogates and have been used in a wide variety of subsequent transformations.6 Due to the mild reaction conditions and stable sulfate byproducts, many of these transformations can be carried out in a single reaction vessel and often do not require flash column chromatography for purification.7 While fluorosulfate derivatives are powerful for some transformations, they are highly reactive and often undergo undesired side reactions. This problem is exemplified in peptide couplings, where epimerization is observed alpha to the initially formed acyl fluorosulfate (5, R5 = R′C(O), Fig. 1).5a Despite the importance of a liquid phase, column-free method for peptide coupling, it remains an unsolved challenge for S–F based reagents8 and other non-sulfur based deoxyfluorinating agents.9
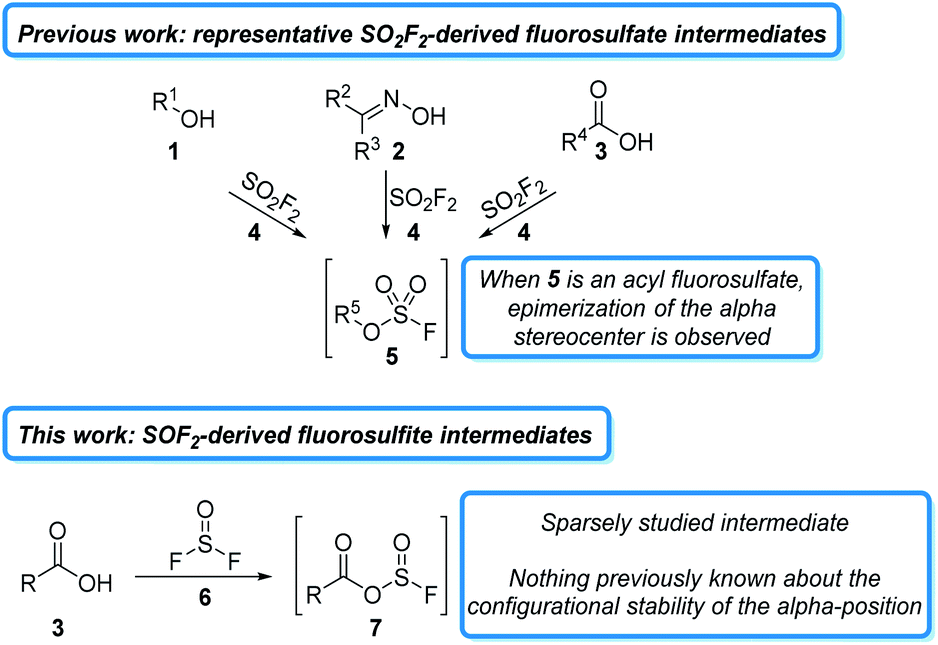 | ||
| Fig. 1 Activation of carboxylic acids for peptide coupling using sulfur fluoride gasses. | ||
An alternative, and largely unexplored strategy for carboxylic acid activation is to access the analogous acylfluorosulfite intermediate (7). These intermediates are less reactive at the acyl carbon than the analogous fluorosulfate derivates and should, therefore, be less susceptible to epimerization.10 One reagent that could be used to access these sulfite intermediates is thionyl fluoride (6). Thionyl fluoride is more reactive than sulfuryl fluoride, which should increase the rate of carboxylate activation.11 Intriguingly, thionyl fluoride has received very little attention as a reagent. The synthesis of thionyl fluoride was first reported in 1896,12 but it was not until 1985 when Shreeve and coworkers reported on its use to react with phosphorous derivatives, amines, and alkanes.13 Since then, only four manuscripts and three patents have detailed its use as a reagent.14 These studies indicate that thionyl fluoride forms activated fluorosulfite intermediates in the presence of oxygen nucleophiles, but the reactivity of these intermediates has not been extensively studied.
Key to further investigations into the synthetic potential of thionyl fluoride is the development of a facile and direct method for SOF2 formation. Thionyl fluoride is typically generated from thionyl chloride and a fluoride salt followed by isolation via condensation of the resulting gas.15 These methods are effective but isolation of the condensed gas is a significant practical impediment, and has likely limited studies of its reactivity. A more practical strategy is to obviate the need for isolation through ex situ generation and direct use of thionyl fluoride.16 This has been a powerful strategy for sulfuryl fluoride-based methodologies16a but it has not yet been applied to SOF2. As thionyl fluoride has a similar safety profile as SO2F2,17 an on-demand generation approach also minimizes the safety risk associated with its handling.
Ex situ SOF2 gas generation was examined using an analogous set-up as the recent ex situ generation of sulfuryl fluoride.16 Thionyl chloride and fluoride salts18 were added to one reaction vessel and the resulting SOF2 gas was bubbled through an organic solvent in a second vessel. Unlike SO2F2 generation, we found that an imidazole trap inserted between the two reaction chambers was necessary to remove any unwanted SOClF and HCl. Our final optimized conditions19 were effective for creating thionyl fluoride solutions using a wide variety of solvents as determined by 19F NMR spectroscopy (Table 1, arranged by descending dielectric constants). While dimethyl sulfoxide (DMSO, entry 1) reacts with SOF2, acetonitrile (ACN) is a viable solvent and affords comparable concentrations (entry 2) as the analogous reaction with SO2F2.20 Lower concentrations were observed in both N,N-dimethylformamide (DMF, entry 3)21 and dichloromethane (DCM, entry 4). Tetrahydrofuran (THF), 1,2-dimethoxyethane (DME), and ethyl acetate (EtOAc) afforded 0.13 M, 0.11 M, and 0.15 M solutions, respectively (entries 5–7). Chloroform and toluene (Tol) performed equivalently, both yielding 0.10 M solutions (entries 8–9), but SOF2 was poorly soluble in the least polar solvent that was screened, petroleum ether (Pet ether, entry 10).
|
|
||
|---|---|---|
| Entry | Solvent | SOF2 (M) |
| a Reaction conditions: 8 (3.0 mmol), KHF2 (3 equiv), solvent (6 mL), imidazole trap, 30 min. The molarity of SOF2 in the solvent was determined by 19F NMR spectroscopy using trifluorotoluene as an internal standard. | ||
| 1 | DMSO | 0 |
| 2 | ACN | 0.14 |
| 3 | DMF | 0.08 |
| 4 | DCM | 0.07 |
| 5 | THF | 0.13 |
| 6 | DME | 0.11 |
| 7 | EtOAc | 0.15 |
| 8 | Chloroform | 0.10 |
| 9 | Tol | 0.10 |
| 10 | Pet ether | 0.03 |

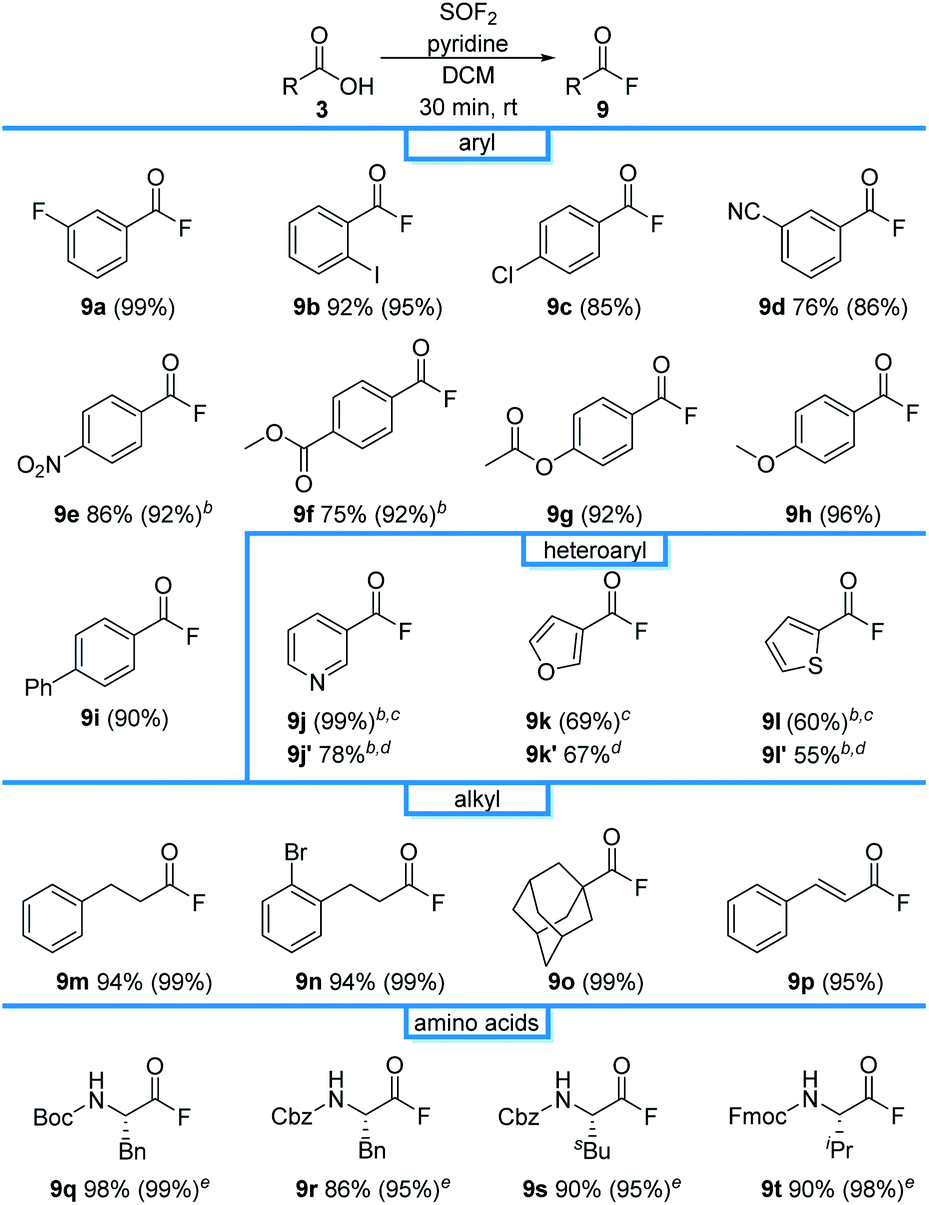
With a protocol for the ex situ generation of thionyl fluoride in hand, we then focused on the syntheses of peptides as it could not be readily accomplished using sulfuryl fluoride. We started our investigations by exploring the first step of this process, the direct conversion of carboxylic acids to the corresponding acyl fluoride. meta-Fluorobenzoic acid (1a) was selected as an initial substrate due to its simplicity and because we could use the aryl fluoride as a handle to track the reaction by 19F NMR spectroscopy. An initial screen found that acid fluoride 9a can be accessed using the SOF2-containing stock solutions from entries 2–10 (Table 1).22 DCM was particularly effective; treatment of 1a with a stock solution of thionyl fluoride in DCM afforded the desired product (9a) in 99% conversion after only 30 minutes at room temperature (Table 2), which is more effective than the analogous reaction using SO2F2.23 A direct comparison was performed under the same conditions (Fig. 2), which found that SOF2 promotes a higher reaction rate relative to SO2F2. This enhanced reaction rate compared to SO2F2 is consistent with literature reports describing the higher reactivity of SOF2.11 As acyl fluorides can be volatile, DCM was selected for further studies due to its low boiling point. The acyl fluorides can be isolated after extractive work-up by diluting with DCM and washing with 0.1 M NaHCO3 solution and brine.
| a Reaction conditions: 3 (0.6 mmol), SOF2 in DCM (1 equiv, approximately 0.07 M), pyridine (1 equiv), 30 min. Isolated yields for the one-pot reaction are reported, with 19F NMR yields using trifluorotoluene as the internal standard provided in parentheses. b SOF2 in ACN was used. c The reaction time was 1 h. d Yields of subsequent derivatization to the corresponding N-hydroxyphthalimide ester. See ESI for reaction details. e The reaction time was 20 min. |
|---|
|
 | ||
| Fig. 2 19F NMR spectroscopy kinetic study of the formation of 9a from 3-fluorobenzoic acid, with SOF2 or SO2F2. Blue = with SOF2; red = with SO2F2. Reactions were carried out in parallel on a 0.6 mmol scale. | ||
The reaction was effective for a wide range of benzoic acid derivatives, affording 9b–9i in 85–96% NMR yields (Table 2). Investigations next turned to the preparation of heteroaryl acyl fluorides. Pyridine (1j), furan (1k) and thiophene (1l) were effective substrates, affording 9j–9l in 99%, 69%, and 60% NMR yields, respectively. As substrates have low boiling points and have previously been documented to be unstable out of solution,23 they were derivatized to the corresponding N-hydroxyphthalimide esters 9j′–9l′ in 78%, 67%, and 55% overall isolated yields, respectively. The reaction was also compatible with alkyl carboxylic acids, affording 9m–9p in near quantitative conversion. Boc, Cbz, and Fmoc-protecting amino acids were also viable substrates, affording 9q–9t in excellent isolated yields without the need for flash column chromatography.
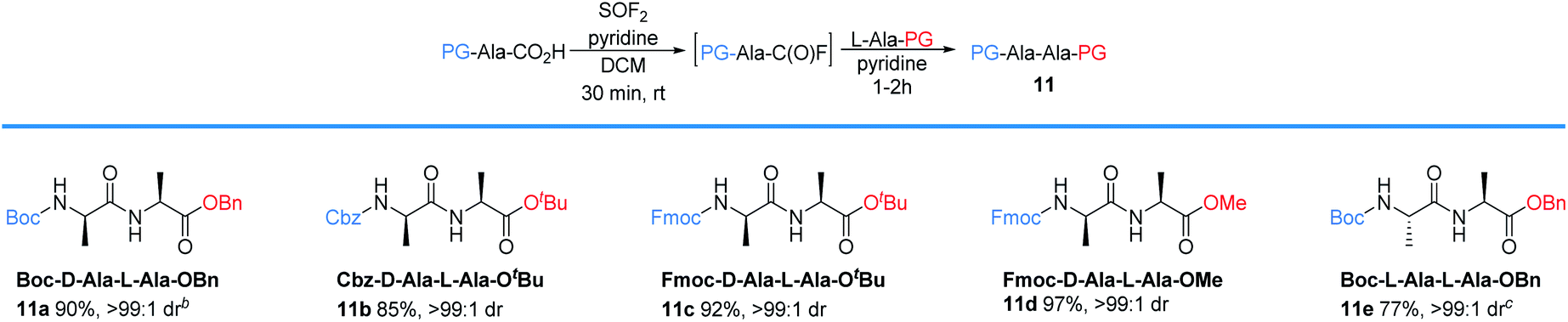
Investigations next focused on one-pot peptide couplings directly from Boc protected amino acids (Table 3). Subjecting Boc-protected glycine to our optimized thionyl fluoride reaction conditions, followed by sparging with nitrogen and addition of L-Ala-OtBu produced the desired dipeptide (10a) in 87% yield and >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er, as determined by HPLC. Notably, the side products are readily removed by extraction and the dipeptide could be isolated pure with no column chromatography required. Alanine (Ala) was compatible with the reaction conditions to deliver 10b in 92% yield with >99:1 dr. For comparison, we also conducted experiments using conventional synthetic methodologies including DCC/HOBt, PyBOP, and HBTU. In all cases, the reactions took 4 hours to afford 10b in 42%, 77%, and 76% yield, respectively all with >99:1 dr. Notably, column chromatography was required for each of these established coupling methods.
:1 er, as determined by HPLC. Notably, the side products are readily removed by extraction and the dipeptide could be isolated pure with no column chromatography required. Alanine (Ala) was compatible with the reaction conditions to deliver 10b in 92% yield with >99:1 dr. For comparison, we also conducted experiments using conventional synthetic methodologies including DCC/HOBt, PyBOP, and HBTU. In all cases, the reactions took 4 hours to afford 10b in 42%, 77%, and 76% yield, respectively all with >99:1 dr. Notably, column chromatography was required for each of these established coupling methods.
| a Reaction conditions: Boc-AA-CO2H (0.6 mmol), SOF2 in DCM or ACN (1 equiv.), pyridine (1 equiv.), 30 min. Followed by L-Ala-OtBu (1 equiv.), pyridine (1 equiv.), 1–2 h. Isolated yields are reported. Unless otherwise noted, the drs were determined by 1H NMR. b The drs and ers were determined by HPLC. |
|---|

|
Amino acids with other hydrophobic alkyl and aryl side chains, such as leucine (Leu), isoleucine (Ile), valine (Val), phenylalanine (Phe), tryptophan (Trp), and methionine (Met) were successfully coupled to produce dipeptides 10c–10h in good to excellent yields with minimal to no epimerization. This is in contrast to SO2F2-mediated amidation of amino acids, where substantial epimerization was observed.24 No epimerization was observed when excess SOF2 stock solution (1.5 equiv.) was used, suggesting that the issue of epimerization arises from the use of SO2F2 rather than the number of equivalents of SOF2 that were utilized. Coupling with cystine (Cys), proline (Pro), and tyrosine (Tyr) proceeded successfully to afford the desired dipeptides 10i–10k in 84%, 80%, and 88% yields, respectively. O-Protected amino acid serine (Ser) was also effective, and afforded 10l in 90% yield without epimerization. Threonine (Thr) coupling afforded a slightly lower yield (83% for 10m) than serine (Ser), likely due to increased sterics. Asparagine (Asn), glutamine (Gln), lysine (Lys), arginine (Arg), histidine (His), aspartic acid (Asp), and glutamic acid (Glu) were effective in this methodology, giving excellent yields of 10n–10t with >99:1 dr. No evidence of cyclization was detected for any of these substrates. Compared to the current state-of-the-art for peptide coupling methodologies, our method provides comparable yields and diastereoselectivities, but with improved reaction times and simpler purification protocols (Table 4).25
| a Reaction conditions: PG-AA-CO2H (0.6 mmol), SOF2 in DCM or ACN (1 equiv.), pyridine (1 equiv.), 30 min. Followed by L-Ala-PG (1 equiv.), pyridine (1 equiv.), 1–2 h. Isolated yields are reported. Unless otherwise noted, the drs were determined by 1H NMR. b The drs and ers were determined by HPLC. c 2 gram-scale (8 mmol). |
|---|
|
The method can also be applied for unnatural amino acids. Ornithine (Orn) was an efficient substrate, affording 10u in 82% yield. Phenylglycine (Phg) is recognized as one of the most easily racemized amino acids.26 Our method successfully coupled phenylglycine (Phg) to give 10v in 79% yield with 98:2 dr, in 2 h.
We next examined the protecting group tolerance of this new dipeptide coupling reaction (Table 4).27 Amino acids with N-Boc, N-Cbz, or N-Fmoc protecting groups effectively coupled with OBn, OtBu, or OMe amino esters to form the corresponding dipeptides 11a–11d in excellent yields with >99:1 dr. Our protocol can be performed on a 2 gram-scale to generate 11e safely and with similar efficacy.
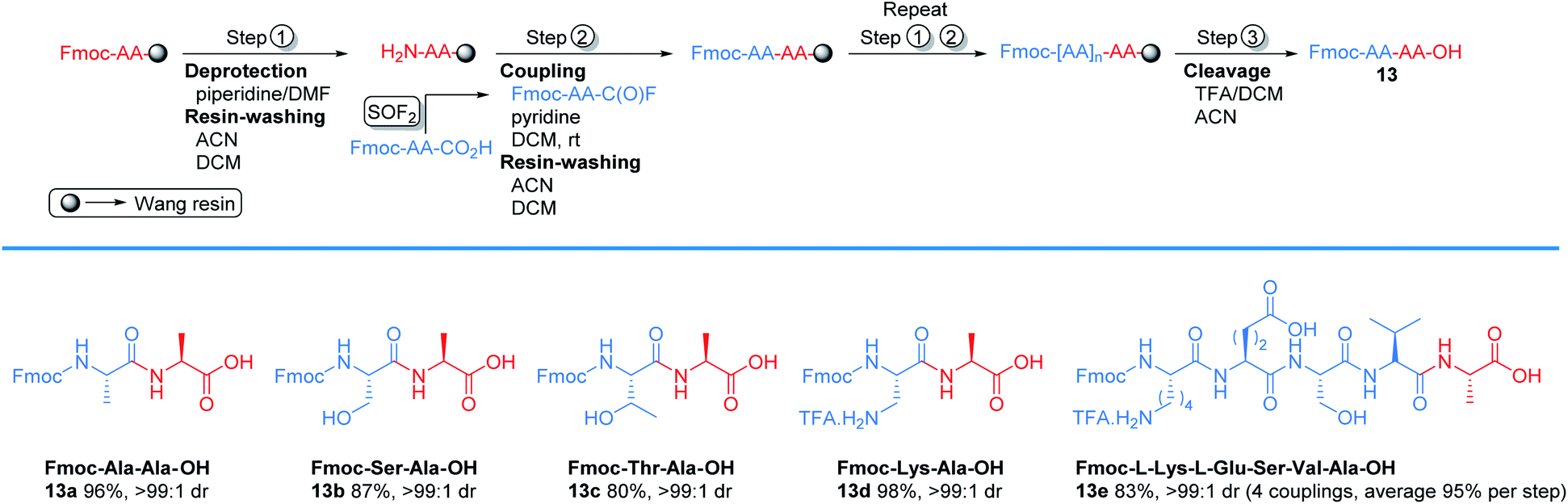
To explore the column-free, liquid phase syntheses of tri-, tetra-, penta-, and decapeptides, we designed the protocol outlined in Scheme 1. The protocol begins by first treating an N-terminal amino acid with Boc-protected amino acid fluoride, which was synthesized by our new method (step 1). Boc-protected dipeptides were obtained in 1–2 h after a simple aqueous work up. Subsequent deprotection of the Boc group was achieved with 4.0 M HCl in dioxane or TFA/DCM (1:1) in 1 h (step 2).28 Concentration in vacuo and neutralization afforded N-terminal peptides. Steps 1 and 2 were repeated, as necessary, for subsequent amino acid incorporation. The final coupling with the Boc-protected amino acid fluorides (step 3) afforded the desired polypeptides. This strategy was effective for the synthesis of tripeptides 12a and 12b, which were obtained in 84% and 88% yields over the three-step sequence. Tetrapeptide 12c and pentapeptide 12d were synthesized in 80% and 51% isolated yields using an analogous method as the tripeptides, except the dipeptide N-Boc-Leu-Gly-CO2H was used. Similary, a decapeptide (12e) was produced in 14% isolated yield over 8 couplings using the dipeptide N-Boc-Leu-Gly-CO2H. Notably, all of the tri-, tetra-, and pentapeptides could be assembled in a single day without the use of column chromatography.
 | ||
| Scheme 1 Representative examples of liquid phase peptide synthesis through acyl fluoride intermediates. | ||
 | ||
| Scheme 2 Representative example of solid phase peptide synthesis through acyl fluoride intermediate. | ||
To explore the potential of using SOF2 generated amino acid fluoride in solid phase peptide synthesis (SPPS),29 we examined the new protocol in couplings with Wang resin (Scheme 2). We started with Fmoc-deprotection of Fmoc-Ala-Wang resin using 20% piperidine in DMF to afford free amine. The free amine was subjected to coupling with Fmoc-Ala-C(O)F to generate Fmoc-Ala-Ala-Wang resin in 1 h. After coupling, the resin was cleaved with TFA/DCM and the target Fmoc protected dipeptide 13a was obtained in 96% yield. Serine (Ser), threonine (Thr), and lysine (Lys) were compatible with this solid phase peptide synthesis and afforded the corresponding products 13b–13c with excellent yields. Pentapeptide 13e was also synthesized in 83% isolated yield.
Conclusions
In conclusion, we have developed a novel acid activation peptide coupling strategy utilizing SOF2 to access acyl fluorides via acyl fluorosulfite intermediates. The ex situ generation of thionyl fluoride was achieved using inexpensive and readily available commodity chemicals, and displayed an expedited, column-free preparation of alkyl, aryl, and amino acid fluorides. Dipeptides were afforded in a one-pot, column-free protocol, effective across natural and unnatural amino acid substrates with a wide range of protecting groups and retention of optical purity. Our approach was applied to the syntheses of tri-, tetra-, and decapeptides, providing a competitive method for liquid phase, iterative peptide couplings. The new approach was also amenable to solid phase peptide synthesis.Data availability
The datasets supporting this article have been uploaded as part of the ESI† material.Author contributions
C. L. and G. M. S. conceived the project. C. L. and B. J. T. conducted and analyzed the experiments. C. L., B. J. T., and G. M. S. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. M. S., C. L., and B. J. T. thank the University of British Columbia (UBC), and the Natural Sciences and Engineering Research Council of Canada (NSERC) for funding.Notes and references
- (a) For representative reviews and books, see: J. Dong, K. B. Sharpless, L. Kwisnek, J. S. Oakdale and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53, 9466–9470 CrossRef CAS; (b) T. Abdul Fattah, A. Saeed and F. Albericio, J. Fluorine Chem., 2018, 213, 87–112 CrossRef CAS; (c) P. Martín-Gago and C. A. Olsen, Angew. Chem., Int. Ed., 2019, 58, 957–966 CrossRef; (d) R. Lekkala, R. Lekkala, B. Moku, K. P. Rakesh and H.-L. Qin, Org. Chem. Front., 2019, 6, 3490–3516 RSC; (e) A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731–4758 RSC; (f) N. D. Ball, in Emerging Fluorinated Motifs: Synthesis, Properties, and Applications, ed. J.-A. Ma and D. Cahard, Wiley-VCH, Weinheim, 1st edn, 2020, ch. 21, vol. 2, pp. 621–674 Search PubMed; (g) C. Lee, A. J. Cook, J. E. Elisabeth, N. C. Friede, G. M. Sammis and N. D. Ball, ACS Catal., 2021, 11, 6578–6589 CrossRef CAS PubMed.
- (a) M. K. Nielsen, C. R. Ugaz, W. Li and A. G. Doyle, J. Am. Chem. Soc., 2015, 137, 9571–9574 CrossRef CAS PubMed; (b) H. Zhou, P. Mukherjee, R. Liu, E. Evrard, D. Wang, J. M. Humphrey, T. W. Butler, L. R. Hoth, J. B. Sperry, S. K. Sakata, C. J. Helal and C. W. am Ende, Org. Lett., 2018, 20, 812–815 CrossRef CAS; (c) T. Guo, G. Meng, X. Zhan, Q. Yang, T. Ma, L. Xu, K. B. Sharpless and J. Dong, Angew. Chem., Int. Ed., 2018, 57, 2605–2610 CrossRef CAS PubMed.
- (a) W. Chen, J. Dong, S. Li, Y. Liu, Y. Wang, L. Yoon, P. Wu, K. B. Sharpless and J. W. Kelly, Angew. Chem., Int. Ed., 2016, 55, 1835–1838 CrossRef CAS; (b) S. D. Schimler, M. A. Cismesia, P. S. Hanley, R. D. J. Froese, M. J. Jansma, D. C. Bland and M. S. Sanford, J. Am. Chem. Soc., 2017, 139, 1452–1455 CrossRef CAS; (c) M. Epifanov, P. J. Foth, F. Gu, C. Barrillon, S. S. Kanani, C. S. Higman, J. E. Hein and G. M. Sammis, J. Am. Chem. Soc., 2018, 140, 16464–16468 CrossRef CAS; (d) Z. Liu, J. Li, S. Li, G. Li, K. B. Sharpless and P. Wu, J. Am. Chem. Soc., 2018, 140, 2919–2925 CrossRef CAS; (e) G.-F. Zha, W.-Y. Fang, Y.-G. Li, J. Leng, X. Chen and H.-L. Qin, J. Am. Chem. Soc., 2018, 140, 17666–17673 CrossRef CAS; (f) P. J. Foth, F. Gu, T. G. Bolduc, S. S. Kanani and G. M. Sammis, Chem. Sci., 2019, 10, 10331–10335 RSC; (g) M. Epifanov, J. Y. Mo, R. Dubois, H. Yu and G. M. Sammis, J. Org. Chem., 2021, 86, 3768–3777 CrossRef CAS PubMed.
- (a) J. Gurjar, J. Bater and V. V Fokin, Chem. – Eur. J., 2019, 25, 1906–1909 CrossRef CAS; (b) G. Zhang, Y. Zhao, L. Xuan and C. Ding, Eur. J. Org. Chem., 2019, 2019, 4911–4915 CrossRef CAS; (c) W.-Y. Fang and H.-L. Qin, J. Org. Chem., 2019, 84, 5803–5812 CrossRef CAS; (d) G. Zhang, Y. Zhao and C. Ding, Org. Biomol. Chem., 2019, 17, 7684–7688 RSC.
- (a) S.-M. Wang, C. Zhao, X. Zhang and H.-L. Qin, Org. Biomol. Chem., 2019, 17, 4087–4101 RSC; (b) S.-M. Wang, N. S. Alharbi and H.-L. Qin, Synthesis, 2019, 51, 3901–3907 CrossRef CAS; (c) J. Liu, S.-M. Wang and H.-L. Qin, Tetrahedron, 2020, 76, 131724 CrossRef CAS.
- (a) G. P. Roth and C. E. Fuller, J. Org. Chem., 1991, 56, 3493–3496 CrossRef CAS; (b) P. S. Hanley, M. S. Ober, A. L. Krasovskiy, G. T. Whiteker and W. J. Kruper, ACS Catal., 2015, 5, 5041–5046 CrossRef CAS; (c) Q. Liang, P. Xing, Z. Huang, J. Dong, K. B. Sharpless, X. Li and B. Jiang, Org. Lett., 2015, 17, 1942–1945 CrossRef CAS PubMed; (d) P. S. Hanley, T. P. Clark, A. L. Krasovskiy, M. S. Ober, J. P. O'Brien and T. S. Staton, ACS Catal., 2016, 6, 3515–3519 CrossRef CAS; (e) E. Zhang, J. Tang, S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Chem. – Eur. J., 2016, 22, 5692–5697 CrossRef CAS PubMed; (f) K. Domino, C. Veryser, B. A. Wahlqvist, C. Gaardbo, K. T. Neumann, K. Daasbjerg, W. M. DeBorggraeve and T. Skrydstrup, Angew. Chem., Int. Ed., 2018, 57, 6858–6862 CrossRef CAS; (g) R. Lekkala, R. Lekkala, L. Jing, R. K. Puttaswamy and Q. Hua-Li, Asian J. Org. Chem., 2018, 7, 662–682 CrossRef; (h) H. Xu, F. Ma, N. Wang, W. Hou, H. Xiong, F. Lu, J. Li, S. Wang, P. Ma, G. Yang and R. A. Lerner, Adv. Sci., 2019, 6, 1901551 CrossRef CAS; (i) V. Bieliūnas and W. M. De Borggraeve, J. Org. Chem., 2019, 84, 15706–15717 CrossRef; (j) M. Mendel, I. Kalvet, D. Hupperich, G. Magnin and F. Schoenebeck, Angew. Chem., Int. Ed., 2020, 59, 2115–2119 CrossRef CAS; (k) G. Kundu, T. Sperger, K. Rissanen and F. Schoenebeck, Angew. Chem., Int. Ed., 2020, 59, 21930–21934 CrossRef CAS.
- (a) G.-F. Zha, W.-Y. Fang, J. Leng and H.-L. Qin, Adv. Synth. Catal., 2019, 361, 2262–2267 CrossRef CAS; (b) C. Zhao, G.-F. Zha, W.-Y. Fang, N. S. Alharbi and H.-L. Qin, Tetrahedron, 2019, 75, 4648–4656 CrossRef CAS; (c) J. Y. Mo, M. Epifanov, J. W. Hodgson, R. Dubois and G. M. Sammis, Chem. – Eur. J., 2020, 26, 4958–4962 CrossRef CAS.
- (a) G. S. Lal, G. P. Pez, R. J. Pesaresi, F. M. Prozonic and H. Cheng, J. Org. Chem., 1999, 64, 7048–7054 CrossRef; (b) J. M. White, A. R. Tunoori, B. J. Turunen and G. I. Georg, J. Org. Chem., 2004, 69, 2573–2576 CrossRef CAS; (c) C. O. Kangani and D. E. Kelley, Tetrahedron Lett., 2005, 46, 8917–8920 CrossRef CAS PubMed; (d) F. Beaulieu, L.-P. Beauregard, G. Courchesne, M. Couturier, F. LaFlamme and A. L'Heureux, Org. Lett., 2009, 11, 5050–5053 CrossRef CAS.
- (a) S. Gross, S. Laabs, A. Scherrmann, A. Sudau, N. Zhang and U. Nubbemeyer, J. Prakt. Chem., 2000, 342, 711–714 CrossRef CAS; (b) C. Chen, C.-T. Chien and C.-H. Su, J. Fluorine Chem., 2002, 115, 75–77 CrossRef CAS; (c) Y. Liang, Z. Zhao, A. Taya and N. Shibata, Org. Lett., 2021, 23, 847–852 CrossRef CAS.
- Fluorosulfate intermediates display a similar reactivity to triflates for nucleophilic substitutions (ref. 3f), whereas fluorosulfites are not as electron-withdrawing and therefore should be less susceptible to SN2 reactions..
- E. R. Falardeau and D. D. DesMarteau, J. Chem. Eng. Data, 1976, 21, 386–387 CrossRef.
- M. Meslans, Bull. Soc. Chim. Fr., 1896, 15, 391–392 Search PubMed.
- T. Mahmood and J. M. Shreeve, Inorg. Chem., 1985, 24, 1395–1398 CrossRef CAS.
- (a) H. Grützmacher and H. W. Roesky, J. Fluorine Chem., 1987, 35, 295–306 CrossRef; (b) W. T. Konieczko, A. Łopusiński and J. Michalski, Phosphorus, Sulfur Silicon Relat. Elem., 1989, 42, 103–104 CrossRef CAS; (c) N. R. Patel and R. L. Kirchmeier, Inorg. Chem., 1992, 31, 2537–2540 CrossRef CAS; (d) N. R. Patel, J. Chen, Y. F. Zhang, R. L. Kirchmeier and J. M. Shreeve, Inorg. Chem., 1994, 33, 5463–5470 CrossRef CAS; (e) J. Lopez and G. Rajoharison, WO Pat. 037887, 2006; (f) Axichem AB, BE Pat., 1019713, 2012; (g) J. Dong, S. Ke and L. Xu, CN Pat., 112079755, 2020.
- (a) H. S. Booth and F. C. Mericola, J. Am. Chem. Soc., 1940, 62, 640–642 CrossRef CAS; (b) F. Seel and L. Riehl, Z. Anorg. Allg. Chem., 1955, 282, 293–306 CrossRef CAS; (c) C. W. Tullock and D. D. Coffman, J. Org. Chem., 1960, 25, 2016–2019 CrossRef CAS.
- (a) C. Veryser, J. Demaerel, V. Bieliūnas, P. Gilles and W. M. De Borggraeve, Org. Lett., 2017, 19, 5244–5247 CrossRef CAS PubMed; (b) J. Demaerel, C. Veryser and W. M. De Borggraeve, React. Chem. Eng., 2020, 5, 615–631 RSC.
- (a) Sulfuryl fluoride; CAS RN: 2699-79-8; M016-2-04; rev 1; Synquest Laboratories: Alachua, FL, September 01, 2016, http://www.synquestlabs.com/product/id/51619.html, accessed 2021-09-23 Search PubMed; (b) Thionyl fluoride; CAS RN: 7783-42-8; M016-2-03; rev 1; Synquest Laboratories: Alachua, FL, January 12, 2017, http://www.synquestlabs.com/product/id/25936.html, accessed 2021-09-23 Search PubMed.
- NaF, KF, and KHF2 were used for the sources of fluoride, see the ESI† for the details..
- See the ESI† for further details on optimization of SOF2..
- In comparison, 1,1′-Sulfonyldiimidazole (5.5 mmol) using De Borggraeve's method from ref. 12, resulted in 0.12 M SO2F2 in ACN..
- DMF reacts slowly with SOF2 to form a white precipitate..
- See the ESI† for details of the solvent screen..
- The analogous reaction with SO2F2 took 1 h at 40 ˚C. P. J. Foth, T. C. Malig, H. Yu, T. G. Bolduc, J. E. Hein and G. M. Sammis, Org. Lett., 2020, 22, 6682–6686 CrossRef CAS PubMed.
- As an example, amide bond formation starting from Boc-Ala-CO2H afforded 50% ee, and Boc-Met-CO2H afforded 64% ee. 2 examples of alpha-stereocenters also present in dipetide coupling without ee values reported. See ref. 5a for details..
- (a) L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397–4398 CrossRef CAS; (b) H. Wenschuh, M. Beyermann, H. Haber, J. K. Seydel, E. Krause, M. Bienert, L. A. Carpino, A. El-Faham and F. Albericio, J. Org. Chem., 1995, 60, 405–410 CrossRef CAS; (c) L. A. Carpino and A. El-Faham, Tetrahedron, 1999, 55, 6813–6830 CrossRef CAS; (d) M. Koshizuka, K. Makino and N. Shimada, Org. Lett., 2020, 22, 8658–8664 CrossRef CAS PubMed; (e) W. Muramatsu, T. Hattori and H. Yamamoto, J. Am. Chem. Soc., 2019, 141, 12288–12295 CrossRef CAS PubMed; (f) W. Muramatsu, C. Manthena, E. Nakashima and H. Yamamoto, ACS Catal., 2020, 10, 9594–9603 CrossRef CAS; (g) F. Albericio and A. El-Faham, Org. Process Res. Dev., 2018, 22, 760–772 CrossRef CAS; (h) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS.
- (a) G. G. Smith and T. Sivakua, J. Org. Chem., 1983, 48, 627–634 CrossRef CAS; (b) E. D. Stroud, D. J. Fife and G. G. Smith, J. Org. Chem., 1983, 48, 5368–5369 CrossRef CAS.
- A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS.
- G. Han, M. Tamaki and V. J. Hruby, J. Pept. Res., 2001, 58, 338–341 CrossRef CAS PubMed.
- S. L. Pedersen, A. P. Tofteng, L. Malik and K. J. Jensen, Chem. Soc. Rev., 2012, 41, 1826–1844 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, methods, and optimization data; NMR, IR, HPLC and MS data including 1H, 13C, and 19F NMR spectra. See DOI: 10.1039/d1sc05316g |
| This journal is © The Royal Society of Chemistry 2022 |