
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCavity catalysis: modifying linear free-energy relationship under cooperative vibrational strong coupling†
Jyoti
Lather‡
,
Ahammad N. K.
Thabassum‡
,
Jaibir
Singh
and
Jino
George
 *
*
Indian Institute of Science Education and Research (IISER), Mohali, Punjab-140306, India. E-mail: jgeorge@iisermohali.ac.in
First published on 25th November 2021
Abstract
Here, we used an unconventional idea of cooperative vibrational strong coupling of solute and solvent molecules to enhance the rate of an esterification reaction. Different derivatives of p-nitrophenyl benzoate (solute) and isopropyl acetate (solvent) are cooperatively coupled to an infrared Fabry–Perot cavity mode. The apparent rates are increased by more than six times at the ON resonance condition, and the rate enhancement follows the lineshape of the vibrational envelope. Very interestingly, a strongly coupled system doesn't obey the Hammett relations. Thermodynamics suggests that the reaction mechanism remains intact for cavity and non-cavity conditions. Temperature-dependent experiments show an entropy-driven process for the coupled molecules. Vacuum field coupling decreases the free energy of activation by 2–5 kJ mol−1, supporting a catalysis process. The non-linear rate enhancement can be due to the reshuffling of the energy distribution between the substituents and the reaction center across the aromatic ring. These findings underline the non-equilibrium behavior of cavity catalysis.
Introduction
The last decade witnessed progress in studying light–matter interaction in molecular systems.1–3 Earlier, semiconductor researchers used this concept to understand the interesting properties of these hybrid, half-photonic-half-matter states. Recent experiments suggest that chemical and physical properties can be controlled precisely by coupling to a vacuum electromagnetic field.4 For example, room temperature Bose–Einstein condensation,5 polariton lasing,6 enhanced conductivity,7etc., was the outcome of strong light–matter interaction. Placing a molecular vibrational transition in an optical/plasmonic mode generates an exchange of photons when they come into resonance, creating two new eigenstates with equidistant energy from the fundamental transition called vibro-polaritonic states (VP+ and VP−; Fig. 1a).8 The separation between the polaritonic states is called Rabi splitting (ħΩVR), and the relation can be express as: | (1) |
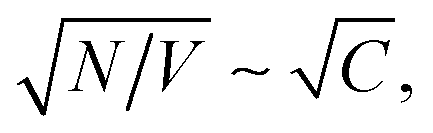 where N is the number and C the concentration of the coupled molecules.10 This collective coupling is critical in observing changes in the chemical and physical properties of the coupled system.
where N is the number and C the concentration of the coupled molecules.10 This collective coupling is critical in observing changes in the chemical and physical properties of the coupled system.
 | ||
| Fig. 1 (a) Schematic illustration of cooperative VSC of solute and solvent molecules in a FP cavity. (b) A tentative energy diagram of change in free-energy of a chemical reaction in a cavity catalysis process. | ||
Laser control of chemical reactions was a long-standing goal in coherent chemistry.11 The availability of 3N − 6 degrees of freedom makes an efficient distribution of energy within the system, decreases the quantum yield of a chemical conversion process.12 Here, light-matter strong coupling offers a unique way to control a chemical reaction by vacuum field dressing of reactant molecules.13,14 It also provides the same effect without an external source, makes them promising for next-generation chemical reactors.15 Ebbesen and co-workers proposed the first conceptual work in 2012 based on a photochemical reaction-switching of merocyanine to spiropyran in a Fabry–Perot (FP) cavity.16 The photochemical switching was slowed down due to the collective protection of the polaritonic states, thereby shifting the photostationary state. Here, the electronically excited state was coupled to the cavity mode. The above experiment seeded the idea of vibrational strong coupling (VSC) and helped to control chemical reactions at the molecular level. The first attempt in this direction was to couple C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band of a polymer, polyvinyl acetate, to an infrared FP cavity.8 Later, many solids and liquids are shown to couple to the FP cavity.17–20 The flow cell configuration of FP cavity is beneficial for homogeneous chemical reactions in the liquid state (Fig. 2).21 The first experimental observation under VSC was a desilylation reaction.22 Here, the reaction got decelerated at ON resonance coupling of C–Si band to an FP cavity mode. Selective coupling of C–Si band to the cavity mode decreased the reaction rate by more than four times at room temperature.23 Another attempt was to try tilting the energy landscape through selective VSC of different functional groups in a molecule.24
O stretching band of a polymer, polyvinyl acetate, to an infrared FP cavity.8 Later, many solids and liquids are shown to couple to the FP cavity.17–20 The flow cell configuration of FP cavity is beneficial for homogeneous chemical reactions in the liquid state (Fig. 2).21 The first experimental observation under VSC was a desilylation reaction.22 Here, the reaction got decelerated at ON resonance coupling of C–Si band to an FP cavity mode. Selective coupling of C–Si band to the cavity mode decreased the reaction rate by more than four times at room temperature.23 Another attempt was to try tilting the energy landscape through selective VSC of different functional groups in a molecule.24
 | ||
| Fig. 2 (a) Open sketch and (b) photograph of a FP cavity used for cavity catalysis experiment. | ||
This experimental observation generated a spur in the physical chemistry community as coherent chemical reaction control and improving the quantum yield was a dream for the researchers for decades. There were many theoretical approaches worldwide to understand the basic mechanism of cavity-mediated chemical reactions-called polariton chemistry.25 In a first attempt, Feist and co-workers proved the collective nature of vibro-polaritons in chemical reactions.26–28 A QE-DFT approach was also used to understand the potential energy surface and the reaction dynamics of small molecules like H32+ under VSC.3 More theoretical papers appeared with suggestions on the dissipative nature and equilibrium consideration of VSC and their influence on reaction control.29,30 Few spectroscopic experimental evidence suggests a better energy transfer mechanism in the coupled system.31,32 The vibro-polaritonic state can be used as a new decay channel in the reaction dynamics.33,34 Other models such as the multi-level quantum Rabi (MLQR) model were introduced to understand the intermolecular interaction of the coupled molecules.35 A very recent approach in this direction is the non-equilibrium behavior of VSC.30 Some recent reviews cover detailed discussion about the nature of vibro-polaritonic states and their applications.1–4 Even though there are significant attempts on the theoretical side, a general mechanism of polariton chemistry is not well understood.
There were very few attempts from the experimental side to understand the role of VSC in chemical reactions.36 Hirai et al., shown that VSC can control a cycloaddition reaction.37 Thermodynamic experiments suggest that the polarity of the CO group got affected under VSC that decelerated the reaction rate. Charge transfer complexation of trimethylated-benzene–I2 was studied, and the complexation equilibrium is shifting based on the symmetry of the vibrationally coupled state.38 Another interesting work is to see the breaking of the Woodward–Hoffmann rule and the modification of stereoselectivity under VSC.39 We have recently shown that cooperative VSC of solute to solvent molecules can drastically modify the chemical reaction rate.40 A similar approach was also used to control enzyme hydrolysis by coupling to water molecules.41 The main advantage of cooperative VSC is the bulk coupling of the solvent molecules in large numbers, thereby increasing the collective coupling strength and its effect on a chemical reaction.
Thermodynamic studies of all the chemical reactions under VSC show that the enthalpy and entropy of activation are drastically changing in the coupled system. Herein, we report cooperative VSC of a transesterification reaction decreases the free energy of activation by 2–5 kJ mol−1, thereby catalyzing the reaction (Fig. 1b). Thus, the origin of cavity catalysis can be due to transition state (TS) stabilization triggered through better solvent reorganization. In the current work, we looked into the effect of substituents and the structure–reactivity relationship on the transesterification process of p-nitrophenyl benzoate molecules under VSC. The overall idea is to study the linear free-energy relationship and other thermodynamic correlations to understand the mechanism of cavity catalysis.
Results and discussion
The idea of cavity catalysis was materialized by the ergonomic design of an infrared microfluidic flow cell.21 The cell is demountable, and a mylar spacer separates two substrates (windows) to complete the geometry of the flow cell-non-cavity. The spacer will define the pathlength, and it varies from 6–25 μm. The substrates mainly used for the experiments are BaF2, CaF2, ZnSe, etc. Here, BaF2 is selected for a specific reason to experiment in the UV-VIS to IR regime (BaF2 is transparent from 0.2 μm to 10 μm in the wavelength spectrum). The inner side of the BaF2 windows are sputtered with 10 nm Au mirrors to complete a Fabry–Perot design, as shown in Fig. 2. All the experiments in cavity and non-cavity are conducted with a mylar spacer thickness of 12 μm. The FP cavity is designed to manually move the upper mirror by tightening and loosening the four screws on the top of the cell. The mirrors' flatness is ensured by observing Newton's rings formed in the center of the cavity structure (see the interference pattern formed in Fig. 2b and the ESI†). Please note that the elasticity of the spacer will allow moving the mirror precisely to achieve 1 cm−1 free spectra range (FSR) resolution. FSR can be calculated for the cavity using the formula k (cm−1) = 104/2nL where n is the refractive index (RI) of medium and L is the length between the mirrors. FSR value pre-calculated using the refractive index of the medium will help to attain ON resonance condition after injection of the reaction mixture. The cell design given in Fig. 2 is having a pathlength of one order of magnitude smaller than a conventional microfluidic setup. The smaller pathlength of the cavity forces us to work in a static configuration. The above design is unique and will help us study in situ reaction kinetics while preserving the coupling condition.VSC of isopropyl acetate (IPAc) is achieved by coupling 7th mode of the cavity to the CO stretching band (1739 cm−1) of the solvent molecules. New vibro-polaritonic states (VP+ and VP−) are formed due to VSC, observed using an FTIR setup in the transmission mode configuration (Fig. 3a). Neat coupling of the solvent gives a Rabi splitting of 136 cm−1 with VP+ and VP− at 1814 cm−1 and 1678 cm−1, respectively. VSC of liquid esters shows a similar coupling strength as reported in the literature.40 There are five derivatives of p-nitrophenyl benzoate used for the experiment (Scheme 1). All the solute molecules selected have matching CO stretching bands with respect to the solvent molecules. This allows the system to enter into cooperative VSC. Here, the solute concentration is 100 times lower than the bulk solvent, and hence the coupling strength is preserved during the course of the reaction. Angle-dependent measurements suggest a dispersive nature of polaritonic states with minimum splitting energy at ON resonance condition (Fig. 3b). Vibro-polaritonic states and their spectroscopic studies are already known in the literature.8 Our primary focus in the current work is to understand the influence of vibro-polaritonic states on the reaction rate.
 | ||
| Fig. 3 (a) FTIR spectra of neat isopropyl acetate (green trace), p-CH3 in chloroform (red trace) and cavity at ON-resonance (blue trace) condition; (b) angle dependent dispersion measurement of COstr band of solute and solvent molecules after coupling to 7th mode of an IR cavity. | ||
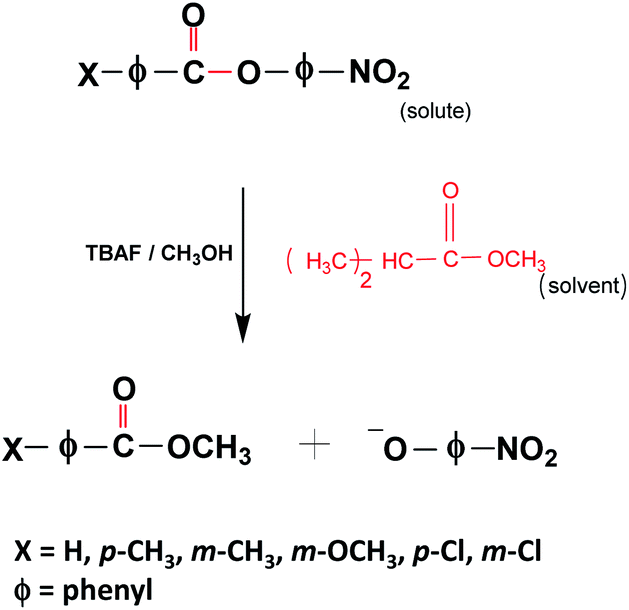 | ||
| Scheme 1 Scheme of PNPB transesterification reaction. | ||
Stock solutions of p-nitrophenyl benzoate (0.1 M; PNPB) and all other derivatives (p-toluate (p-CH3); m-toluate (m-CH3); m-anisate (m-OCH3); p-chlorobenzoate (p-Cl); m-chlorobenzoate (m-Cl)) were prepared in IPAc, and tetrabutylammonium fluoride (0.1 M; TBAF) was prepared in methanol. All the derivatives were synthesized by following a reported procedure using substituted benzoyl chloride and p-nitrophenol (ESI†).42 The final products were characterized by IR and 1H NMR spectroscopy. The solution containing solute molecules was prepared fresh and used for all the kinetic experiments. The final reaction mixture was prepared by mixing 190 μl of the solute and 10 μl of TBAF solution. The reaction mixture was immediately injected into an FP cavity (the FP cavity was kept at RT for about 30 minutes for stabilization before the injection) using a disposable syringe. The reaction rate was studied at 400 nm in a UV-visible spectrophotometer (Cary-5000) for 600 seconds. The reaction follows a pseudo-first-order, base-catalyzed (BAc2) SN2 mechanism.43 The nucleophilic attack on the CO carbon and formation of the negatively charged tetrahedral TS is the rate-determining step (RDS) of the reaction. The evolution of the spectra at 400 nm shows the formation of the product para-nitrophenoxide ion (PNP−). All the derivatives follow a similar reaction mechanism, whereas the substituents at m- and p-position control the reaction rate, either by inductive (±I) or/and by resonance (±R) effects.42
Kinetic traces obtained from the experiments conducted in the non-cavity condition for PNPB shows an apparent rate (kapp) of 1.05 × 10−3 s−1 (kH). The reaction rate changed approximately 1.5 times (1.55 × 10−3 s−1) in the cavity condition by coupling the CO stretching band of the solute and solvent molecules (see Section 4 of the ESI†). Similarly, we have conducted kinetic experiments for all the derivatives in non-cavity and cavity conditions. kapp is increased from 1.5 times to 6 times for different substituents, as shown in Table 1. Very interestingly, moving the mirror position (FSR variation) from ON resonance to OFF resonance condition brings down the rate enhancement to non-cavity rate (Fig. 4). The rate enhancement more or less follows the vibrational envelope of the solute/solvent molecules, indicating a cooperative VSC mechanism (see also Section 6 of the ESI†). However, we cannot extract the linewidth from the kinetic data due to limitations in obtaining kinetic traces within 1 cm−1 FSR precision. At least, the rate enhancement lineshape changes slightly from p-Cl to m-CH3 depending upon the position of the CO stretching band. The same effect was reported previously by a similar class of molecules.40
| Sl No. | Compound | COstr (cm−1) |
Non-cavity kapp (s−1) | Cavity kappa (s−1) |
|---|---|---|---|---|
| a Cavity experiments repeated two times at ON resonance condition, and the percentage error in kapp is ±16%. | ||||
| 1 | PNPB | 1744 | 1.05 × 10−3 | 1.55 × 10−3 |
| 2 | m-CH3 | 1742 | 7.73 × 10−4 | 2.58 × 10−3 |
| 3 | p-CH3 | 1741 | 5.54 × 10−4 | 3.50 × 10−3 |
| 4 | m-OCH3 | 1743 | 1.19 × 10−3 | 1.86 × 10−3 |
| 5 | m-Cl | 1748 | 1.63 × 10−3 | 7.26 × 10−3 |
| 6 | p-Cl | 1741 | 1.49 × 10−3 | 7.50 × 10−3 |
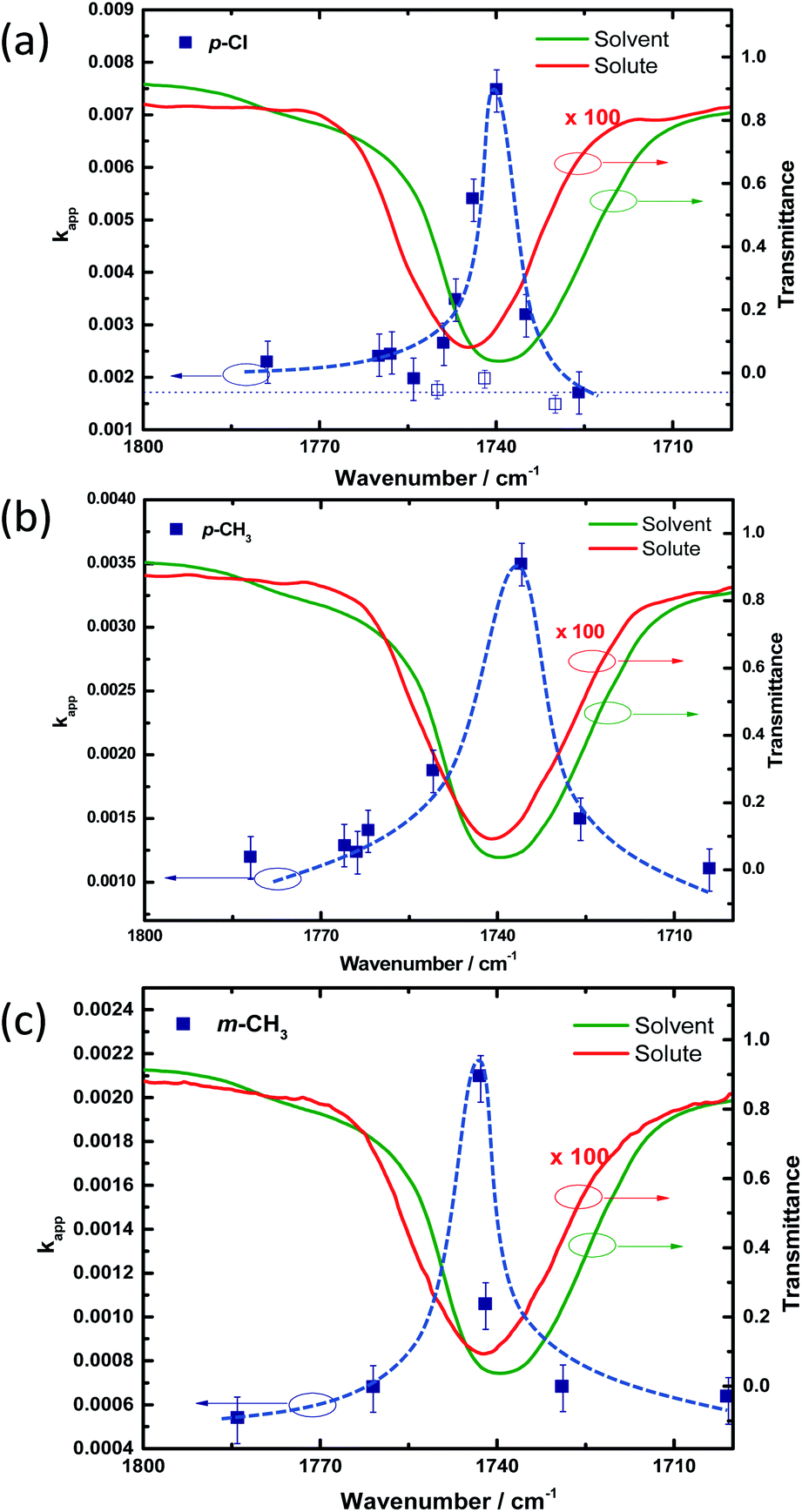 | ||
| Fig. 4 Kinetic action spectra for cavity catalysis measured at 298 K. Cavity mode position (7th) moved slowly from 1680 cm−1 to 1780 cm−1 in each experiment, and the corresponding kapp was plotted against the IR transmission spectra of COstr band of solute (red trace) and solvent (IPAc; green trace) molecules. A sharp increase in the kapp was observed (blue square) at the ON resonance condition for (a) p-Cl, (b) p-CH3, and (c) m-CH3 in similar conditions [dotted lines are guide to the eyes]. | ||
Next, we have studied the rate enhancement ratio (kX/kH) for the cavity and non-cavity condition and compared it with the sigma (σ) (otherwise called substituent constant) of the benzoic acids. σ value is the difference in pKa of unsubstituted to substituted benzoic acid.44 The σ value varies from negative to positive depending upon ±I and ±R effects of the substituents. Plotting log(kX/kH) against σ value gives a straight line for the non-cavity system as it follows linear free-energy relationship, otherwise called Hammett relationship.44 A linear regression fit will give the slope as reactivity constant (ρ). ρ value of PNPB based ester transesterification in IPAc is found to be +0.89 (Fig. 5). Please note that ρ value can vary depending upon the reaction condition and the solvent medium.45
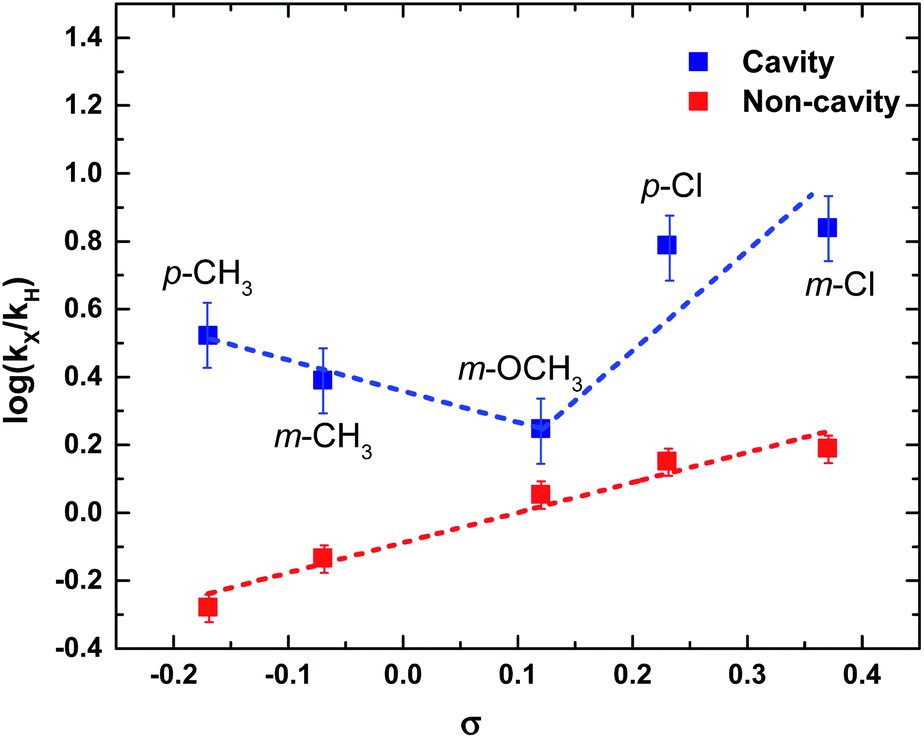 | ||
| Fig. 5 Hammett plot for a series of ester solvolysis (p-CH3, m-CH3, m-OCH3, p-Cl, and m-Cl) in non-cavity (red squares) and ON-resonance cavity conditions (blue squares) [blue dotted lines are guide to the eyes; red dotted line is a linear regression fit]. | ||
Interestingly, experiments conducted in the cavity condition show the breaking of the linear free-energy relationship (Fig. 5). Here, all the experimental parameters are the same except the VSC effect (kH is taken from the ON resonance rate of PNPB). Substituents with σ− show a negative ρ value, whereas σ+ show a higher positive ρ value at the ON resonance condition. Here, the effect of the cavity can be complex; it not only modifies the electron density around the CO group but also reshuffles the substituent effect at the reaction center by VSC. Please note that Hammett's empirical relation ignores the interaction of solvent molecules with different substituents (it only considers the dielectric constant of the solvent). Inductive and/or hyperconjugation effects are blocked in p-CH3 and m-CH3 under VSC, resulting in an increased reaction rate. PNPB and m-OCH3 show the minimal change in reaction rate, indicating that the energy distribution channel is not perturbed under VSC. At the same time, both p-Cl and m-Cl show a better stabilization of the TS by increasing its effective charge (ρ value is highly positive). Here, we assume that the σ value is not changing under VSC conditions; this approximation is essential to compare the structure–reactivity relationship in the cavity and non-cavity systems. Other than the VSC effect, electric field may affect the σ value, which is apparently difficult to evaluate and beyond the scope of the current studies. The non-linear behavior observed under VSC can be due to a change in the reaction mechanism or change in the RDS.46 These aspects are further checked by looking into the thermodynamic behavior of the system.
Further, activation parameters are extracted for four derivatives (including PNPB) by carrying out temperature dependant studies. In the first case, Eyring plot was obtained for PNPB in cavity and non-cavity conditions. Thermodynamics data suggest that the system follows a very similar trend of the previously reported ester solvolysis process (see Section 5 of the ESI†). Whereas electron-donating substitution (p-CH3) shows the enthalpy and entropy of activation of 79.15 kJ mol−1 and −44.30 J K−1 mol−1 for the non-cavity experiments. The corresponding cavity at ON resonance condition shows the enthalpy and entropy of activation of 139.27 kJ mol−1 and +175.07 J K−1 mol−1, respectively. The changes in the thermodynamic parameters between non-cavity and cavity are very significant. Please note that a temperature range of 19–35 °C is used for all the measurements, which gives a maximum error limit within ±16% for the cavity and non-cavity experiments (see Section 5 of the ESI†). The change in free-energy of activation (ΔΔG‡C–NC) from the cavity to non-cavity is found to be −5. 23 kJ mol−1. The fascinating observation here is that the entropy change is very high (219 J K−1 mol−1), which is relatively large for any chemical reaction at a given condition. A detailed analysis of the enthalpy and entropy of activation is shown in Table 2. In the second case, the σ+ (p-Cl) molecule shows a similar trend with a positive entropy of activation at the ON resonance condition. m-OCH3 molecules show the least variation expected from the kinetic data; here, the cavity coupling also reshuffles the thermodynamic parameters as in the previous cases. In the first two cases, the system enters into an entropy-driven process as TΔΔS‡C–NC > ΔΔH‡C–NC at RT (298 K). This observation is similar to solvent-triggered chemical reaction control in some of the reactions reported in the literature.46,47 Free energy of activation extracted for all the derivatives is shown in Fig. 6a. The non-cavity system clearly follows a linear relationship versus the substitution constant. In contrast, the activation parameters are non-linear under ON resonance coupling conditions.
| Sl no. | Name | σ | ΔH‡NC (kJ mol−1) | TΔS‡NC (kJ K−1 mol−1) | ΔH‡C (kJ mol−1) | TΔS‡C (kJ K−1 mol−1) | ΔΔG‡C–NC (kJ mol−1) |
|---|---|---|---|---|---|---|---|
| 1 | p-CH3 | −0.17 | 79.2 | −13.2 | 139.3 | 52.2 | −5.2 |
| 2 | p-Cl | 0.23 | 53.5 | −34.9 | 148.7 | 63.8 | −3.2 |
| 3 | m-OCH3 | 0.12 | 83.5 | −6.1 | 62.0 | −26.8 | −0.9 |
| 4 | PNPB | 0 | 71.3 | −19.2 | 28.7 | −59.0 | −2.8 |
 | ||
| Fig. 6 (a) Thermodynamic relation between free energy of activation versus σ for cavity (blue) and non-cavity (red) system. (b) Enthalpy–entropy compensation plot for cavity versus non-cavity experiments listed in Table 2. | ||
Now we try to compare the change in the thermodynamic parameters for the cavity and the non-cavity system. Plotting ΔΔH‡C–NC against ΔΔS‡C–NC gives a straight line indicating the reaction mechanism may not be affected as it follows an isokinetic relationship.48 The free energy of activation doesn't change much (ΔΔG‡C–NC = 2–5 kJ mol−1) as the entropy compensates for the enthalpy of activation (Fig. 6b and Table 2). Please note that the Helmholtz free energy is not affected much under VSC as the ground state energy shift is minimal. All these observations point to the fact that the vibrational control of the CO stretching band by VSC can reshuffle the electron density locally in the reaction center and generate a non-equilibrium energy distribution, as evident from the breaking of the linear free-energy relationship. This non-equilibrium behavior may be originating from the control of electron density flow through inductive and resonance effects.
As evident from the kinetic and thermodynamic experiments, VSC plays a crucial role in controlling the chemical reaction dynamics in the system. Many factors can control reaction dynamics, which include: (i) intramolecular vibrational relaxation process within the solute molecule, (ii) intermolecular vibration distribution channel (self-interaction), and finally (iii) solute–solvent vibrational energy transfer. Cavity tuning experiments in Fig. 4 is a clear indication of the cooperative VSC effect. The rate enhancement follows the vibrational envelope suggesting a strong energy redistribution process with the cavity and the molecular state. The first-order perturbation to molecular transition by cavity coupling (v1 state of Fig. 1a) may reshuffle its entire chemical reaction coordinate as suggested by many theoretical works.3,49,50 The cavity resonance effect observed in three of the substituted derivatives (Fig. 4) pointing to the impact of the energy level modification by cooperative VSC. At the same time, one can argue that a cavity can act as a thermal reservoir that can provide sufficient thermal energy to undergo multiple collision processes to overcome the barrier. This observation was already proved wrong by doing the experiments without strong coupling (in the absence of a strong oscillator), by only having the cavity mode as a reservoir.40
Now, let us look into the structure–reactivity relationship under VSC. The deviation from the linear free-energy relationship under VSC may have a different origin. Here, one can look into the electron density flow between the substituent and the reaction center through sigma and pi bonds. The electron-withdrawing and donating ability of the substituents plays a crucial role in the chemical reaction rate. Here, VSC of the CO stretching band of the solute may favor/disfavor the electron density flow within the system, either through sigma or pi bonds. Previous experiments show that the resonance coupling of vibrational states to specific modes of the substituents can accelerate the energy transfer rate up to 50 times by controlling the IVR of m- and p-substituted derivatives.51 VSC of the CO band may increase the coupling state density of the substituents to the reaction center and provide a strong gateway for IVR within the coupled system. Another possibility is the play of a spectator bond or direct coupling of the substituents in the far IR region. The current experimental setup won't allow us to probe below 1000 cm−1. p-CH3 shows +R and +I effect; reaction rate gets amplified compared to m-CH3 (+I effect), much faster than m-OCH3 under VSC condition. On the other side, p-Cl and m-Cl has dominating –I effect also get amplified under VSC. This experimental observation is a strong argument on the role of VSC to control the electron density flow across the aromatic ring.
Thermodynamic experiments suggest that VSC brings an overall increase in entropy of activation. Simultaneously, a doubling of enthalpy of activation in p-CH3, suggesting a change of mechanism. The linear correlation in ΔΔH‡C–NC against ΔΔS‡C–NC makes to believe the coupled system behaves similarly towards σ+ and σ− substituents. The isokinetic relationship observed here suggests a similar mechanism for cavity and non-cavity conditions. This happens if the potential energy surface changes under VSC (like early barrier versus late barrier). However, we do not have any evidence to substantiate this claim. Another possibility is the non-equilibrium behavior of the coupled molecules. Here, the Eyring equation may not be valid, which leads to a difference in frame of reference between cavity and non-cavity conditions. It is exciting to note that TΔΔS‡C–NC > ΔΔH‡C–NC (298 K) for p-Cl and p-CH3 indicates a pure entropy-driven process triggered through cooperative VSC. Along with that, the enthalpy–entropy compensation is a clear sign of a catalysis process. Solute–solvent interaction is a complex phenomenon and understanding the kinetics and thermodynamics under VSC requires high-level theoretical intervention.
Conclusion
Here, we observed the breaking of linear free-energy relationship in cavity catalysis by cooperative VSC of solute and solvent molecules. Thermodynamic correlations suggest that the cavity catalysis process did not affect the actual reaction mechanism. All the derivative shows cavity catalysis, whereas the rate enhancement varies from one substituent to another. The non-linear trend in the reaction rate can be due to IVR between the substituents and the reaction center under VSC conditions. The origin of this can also be due to intra/intermolecular interactions.52 In all the cases, the system is trying to compensate entropy versus enthalpy of activation. Here, cooperative VSC is used not only as a tool to understand the transesterification reaction of PNPB but also to boost its chemical conversion. The above results support the application of vibrational strong coupling to control chemical reactions at the molecular level. New experiments and theoretical studies are further required to understand the exact mechanism of cavity catalysis.Author contributions
J. G. conceived the idea and supervised the project. J. L., T. A. N. K. synthesized the compounds, conducted the experiments. J. L., J. S., and J. G. wrote the manuscript. All the authors contributed to the discussion and analysis of the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Srihari Keshavamurthy for his helpful discussions. DST-SERB, Core Research Grant (EMR/2017/003455) is acknowledged for funding. MoE-Scheme for Transformational and Advanced Research in Sciences (MoE-STARS/STARS-1/175) is also thanked for utilizing major instruments for the current work. J. L. thank IISER Mohali, T. A. N. K. thank DST inspire, and J. S. thank CSIR for the fellowships.Notes and references
- T. W. Ebbesen, Acc. Chem. Res., 2016, 49, 2403–2412 CrossRef CAS PubMed.
- M. Hertzog, M. Wang, J. Mony and K. Börjesson, Chem. Soc. Rev., 2019, 48, 937–961 RSC.
- J. Flick, M. Ruggenthaler, H. Appel and A. Rubio, Proc. Natl. Acad. Sci., 2017, 114, 3026–3034 CrossRef CAS.
- F. J. Garcia-Vidal, C. Ciuti and T. W. Ebbesen, Science, 2021, 373, eabd0336 CrossRef CAS PubMed.
- J. D. Plumhof, T. Stöferle, L. Mai, U. Scherf and R. F. Mahrt, Nat. Mater., 2014, 13, 247–252 CrossRef CAS PubMed.
- S. Kena-Cohen and S. R. Forrest, Nat. Photonics, 2010, 4, 371–375 CrossRef CAS.
- E. Orgiu, J. George, J. A. Hutchison, E. Devaux, J. F. Dayen, B. Doudin, F. Stellacci, C. Genet, J. Schachenmayer, C. Genes, G. Pupillo, P. Samorì and T. W. Ebbesen, Nat. Mater., 2015, 14, 1123–1129 CrossRef CAS.
- A. Shalabney, J. George, J. Hutchison, G. Pupillo, C. Genet and T. W. Ebbesen, Nat. Commun., 2015, 6, 5981 CrossRef CAS.
- S. Haroche and D. Kleppner, Phys. Today, 1989, 42, 24–30 CrossRef CAS.
- M. Brune, F. Schmidt-Kaler, A. Maali, J. Dreyer, E. Hagley, J. M. Raimond and S. Haroche, Phys. Rev. Lett., 1996, 76, 1800–1803 CrossRef CAS.
- P. Brumer and M. Shapiro, Acc. Chem. Res., 1989, 22, 407–413 CrossRef CAS.
- H. Frei and G. C. Pimentel, J. Chem. Phys., 1983, 78, 3698–3712 CrossRef CAS.
- C. A. DelPo, B. Kudisch, K. H. Park, S.-U.-Z. Khan, F. Fassioli, D. Fausti, B. P. Rand and G. D. Scholes, J. Phys. Chem. Lett., 2020, 11, 2667–2674 CrossRef CAS PubMed.
- G. D. Scholes, C. A. DelPo and B. Kudisch, J. Phys. Chem. Lett., 2020, 11, 6389–6395 CrossRef CAS.
- C. Genet, J. Faist and T. W. Ebbesen, Phys. Today, 2021, 74, 42–48 CrossRef.
- J. A. Hutchison, T. Schwartz, C. Genet, E. Devaux and T. W. Ebbesen, Angew. Chem., Int. Ed., 2012, 51, 1592–1596 CrossRef CAS PubMed.
- J. P. Long and B. S. Simpkins, ACS Photonics, 2015, 2, 130–136 CrossRef CAS.
- B. S. Simpkins, K. P. Fears, W. J. Dressick, B. T. Spann, A. D. Dunkelberger and J. C. Owrutsky, ACS Photonics, 2015, 2, 1460–1467 CrossRef CAS.
- M. Hertzog, P. Rudquist, J. A. Hutchison, J. George, T. W. Ebbesen and K. Börjesson, Chem.–Eur. J., 2017, 23, 18166–18170 CrossRef CAS.
- R. Damari, O. Weinberg, D. Krotkov, N. Demina, K. Akulov, A. Golombek, T. Schwartz and S. Fleischer, Nat. Commun., 2019, 10, 3248 CrossRef.
- J. George, A. Shalabney, J. A. Hutchison, C. Genet and T. W. Ebbesen, J. Phys. Chem. Lett., 2015, 6, 1027–1031 CrossRef CAS.
- A. Thomas, J. George, A. Shalabney, M. Dryzhakov, S. J. Varma, J. Moran, T. Chervy, X. Zhong, E. Devaux, C. Genet, J. A. Hutchison and T. W. Ebbesen, Angew. Chem., Int. Ed., 2016, 55, 11462–11466 CrossRef CAS.
- A. Thomas, A. Jayachandran, L. Lethuillier-Karl, R. M. A. Vergauwe, K. Nagarajan, E. Devaux, C. Genet, J. Moran and T. W. Ebbesen, Nanophotonics, 2020, 9, 249–255 CAS.
- A. Thomas, L. Lethuillier-Karl, K. Nagarajan, R. M. A. Vergauwe, J. George, T. Chervy, A. Shalabney, E. Devaux, C. Genet, J. Moran and T. W. Ebbesen, Science, 2019, 363, 615–619 CrossRef CAS.
- J. Yuen-Zhou and V. M. Menon, Proc. Natl. Acad. Sci., 2019, 201900795 Search PubMed.
- J.-D. Pino, J. Feist and F. J. Garcia-Vidal, New J. Phys., 2015, 17, 053040 CrossRef.
- J. Galego, F. J. Garcia-Vidal and J. Feist, Nat. Commun., 2016, 7, 13841 CrossRef CAS PubMed.
- J. Galego, F. J. Garcia-Vidal and J. Feist, Phys. Rev. Lett., 2017, 119, 136001 CrossRef PubMed.
- T. Li, A. Nitzan and J. E. Subotnik, J. Chem. Phys., 2020, 152 23, 234107 CrossRef.
- T. E. Li, A. Nitzan and J. E. Subotnik, Angew. Chem., Int. Ed., 2021, 60, 15533–15540 CrossRef CAS PubMed.
- B. Xiang, R. F. Ribeiro, M. Du, L. Chen, Z. Yang, J. Wang, J. Yuen-Zhou and W. Xiong, Science, 2020, 368, 665 CrossRef CAS.
- B. Xiang, J. Wang, Z. Yang and W. Xiong, Sci. Adv., 2021, 7, eabf6397 CrossRef CAS PubMed.
- J. A. Campos-Gonzalez-Angulo, R. F. Ribeiro and J. Yuen-Zhou, Nat. Commun., 2019, 10, 4685 CrossRef PubMed.
- F. Herrera and F. C. Spano, Phys. Rev. Lett., 2016, 116, 238301 CrossRef PubMed.
- F. J. Hernández and F. Herrera, J. Chem. Phys., 2019, 151, 144116 CrossRef.
- K. Hirai, J. A. Hutchison and H. Uji-i, ChemPlusChem, 2020, 85, 1981–1988 CrossRef CAS.
- K. Hirai, R. Takeda, J. A. Hutchison and H. Uji-i, Angew. Chem., Int. Ed., 2020, 59, 5332–5335 CrossRef CAS PubMed.
- Y. Pang, A. Thomas, K. Nagarajan, R. M. A. Vergauwe, K. Joseph, B. Patrahau, K. Wang, C. Genet and T. W. Ebbesen, Angew. Chem., Int. Ed., 2020, 59, 10436–10440 CrossRef CAS.
- A. Sau, K. Nagarajan, B. Patrahau, L. Lethuillier-Karl, R. M. A. Vergauwe, A. Thomas, J. Moran, C. Genet and T. W. Ebbesen, Angew. Chem., Int. Ed., 2021, 60, 5712–5717 CrossRef CAS PubMed.
- J. Lather, P. Bhatt, A. Thomas, T. W. Ebbesen and J. George, Angew. Chem., Int. Ed., 2019, 58, 10635–10638 CrossRef CAS.
- J. Lather and J. George, J. Phys. Chem. Lett., 2021, 12, 379–384 CrossRef CAS.
- S. L. Keenan, K. P. Peterson, K. Peterson and K. Jacobson, J. Chem. Educ., 2008, 85, 558 CrossRef CAS.
- C. G. Mitton, R. L. Schowen, M. Gresser and J. Shapley, J. Am. Chem. Soc., 1969, 91, 2036–2044 CrossRef CAS.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- G. Cainelli, P. Galletti and D. Giacomini, Chem. Soc. Rev., 2009, 38, 990–1001 RSC.
- Y. Inoue, H. Ikeda, M. Kaneda, T. Sumimura, S. R. L. Everitt and T. Wada, J. Am. Chem. Soc., 2000, 122, 406–407 CrossRef CAS.
- W. Linert and R. F. Jameson, Chem. Soc. Rev., 1989, 18, 477–505 RSC.
- M. Ruggenthaler, N. Tancogne-Dejean, J. Flick, H. Appel and A. Rubio, Nat. Rev. Chem., 2018, 2, 0118 CrossRef CAS.
- X. Li, A. Mandal and P. Huo, Nat. Commun., 2021, 12, 1315 CrossRef CAS PubMed.
- P. J. Timbers, C. S. Parmenter and D. B. Moss, J. Chem. Phys., 1994, 100, 1028–1034 CrossRef CAS.
- A. Kadyan, A. Shaji and J. George, J. Phys. Chem. Lett., 2021, 4313–4318 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04707h |
| ‡ Jyoti Lather and Thabassum Ahammad N. K. contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2022 |