
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular redox cyclization reaction access to cinnolines from 2-nitrobenzyl alcohol and benzylamine via intermediate 2-nitrosobenzaldehyde†
Yun Saa,
Mao-Xin Caia,
Xin Lva,
Ai-Bin Wua,
Wen-Ming Shu *ab and
Wei-Chu Yu*a
*ab and
Wei-Chu Yu*a
aHubei Engineering Research Centers for Clean Production and Pollution Control of Oil and Gas Fields, College of Chemistry and Environmental Engineering, Yangtze University, Jingzhou 434023, P. R. China. E-mail: shuwm@yangtzeu.edu.cn; yuweichu@126.com
bHubei Key Laboratory of Pollutant Analysis & Reuse Technology, Hubei Normal University, Huangshi 435002, P. R. China
First published on 21st November 2022
Abstract
A transition-metal-free intramolecular redox cyclization reaction for the synthesis of cinnolines has been developed from 2-nitrobenzyl alcohol and benzylamine. Mechanistic investigations disclosed the involvement of a key intramolecular redox reaction, followed by condensation, azo isomerization to hydrazone, cyclization, and aromatization to form the desired products. Notably, the formation of intermediate 2-nitrosobenzaldehyde and (E)-2-(2-benzylidenehydrazineyl) benzaldehyde plays an important role in this transformation.
Cinnolines are important nitrogen-containing heterocyclic organic compounds which have attracted widespread attention in the field of organic, materials and pharmaceutical chemistry.1 They are known to display interesting biological activities such as inhibitory activity toward CSF-1R,2 antitumor,3 antiviral,4 anti-inflammatory5 and sedative effects6 (Fig. 1).
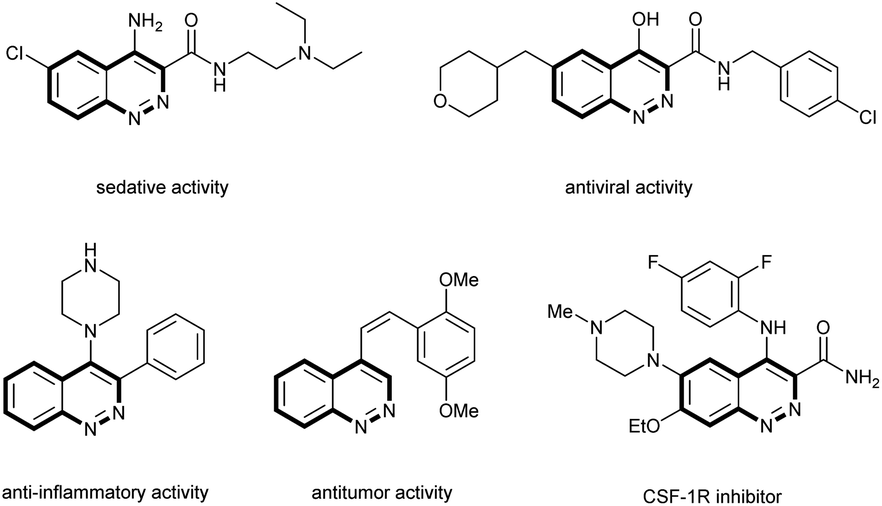 | ||
| Fig. 1 Bioactive cinnoline derivatives. | ||
The classical synthetic approaches to cinnolines start from N-phenylhydrazones or N-phenylazo compounds via Rh-catalyzed C–H activation,7 Pd-catalyzed annulation,8 or Cu-catalyzed C–H functionalization/cyclization strategies.9 For example, in 2012, Ge's group reported a Cu-catalyzed aerobic intramolecular dehydrogenative cyclization reaction of N-methyl-N-phenylhydrazones for the formation of cinnolines (Scheme 1a).10 Subsequently, Lin and coworkers described a Rh-catalyzed redox-neutral annulation of azo and diazo compounds through a tandem C–H activation and C–N bond formation strategy (Scheme 1b).11 More recently, Xu et al. developed a copper-catalyzed aerobic annulation involving a dehydrogenative amination to afford cinnolines (Scheme 1c).12 Interestingly, Wang's group reported a Cu-catalyzed cascade cyclization to achieve regioselective synthesis of functionalized cinnolines (Scheme 1d).13 Although great achievements have been made,14 the development of a novel transition-metal-free mediated reaction from simple substrates to obtain the cinnoline framework is highly desirable. For example, in 2016, Wang and coworkers developed a one-pot cascade reaction for the construction of 4-amido-cinnoline derivatives (Scheme 1e).15 In our previous works, we developed a novel transition-metal-free multicomponent coupling cyclization reaction for synthesis of cinnoline derivatives.16 Inspired by these works, herein, we reported a transition-metal-free intramolecular redox cyclization reaction for straightforward access to cinnolines from 2-nitrobenzyl alcohol and benzylamine (Scheme 1f).
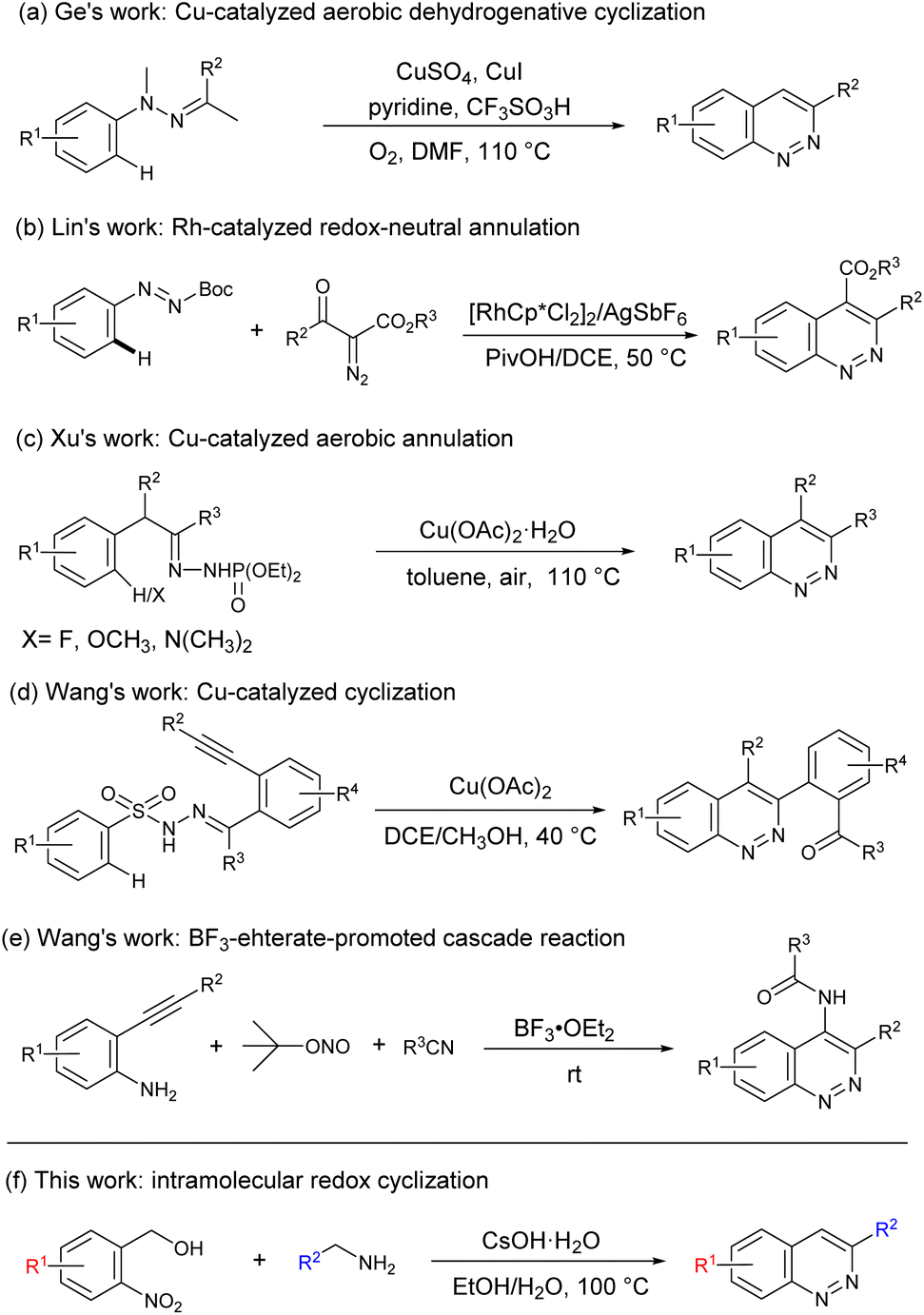 | ||
| Scheme 1 Strategic access to cinnolines. | ||
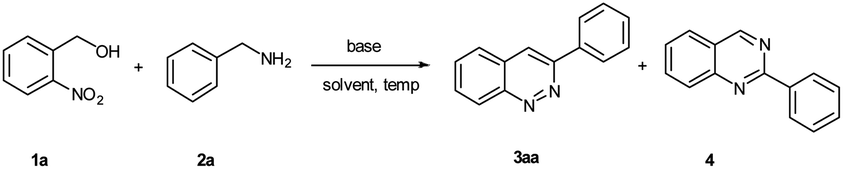
Initially, the reaction was investigated using 2-nitrobenzyl alcohol (1a) and benzylamine (2a) as model substrates (Table 1). The desired product, 3-phenylcinnoline (3aa), was furnished with 15% yield when the reaction proceeded in the presence of 20.0 equiv. of KOH at 100 °C for 3 h in iPrOH (entry 1). At the same time, 2-phenylquinazoline (4) was also obtained in 12% yield. Unfortunately, the target product 3aa was not obtained when the reaction proceeded in H2O (entry 2). Intriguingly, the yield of 3-phenylcinnoline (3aa) was increased to 36% when the reaction was carried out in the mixed solvent iPrOH/H2O (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 100 °C for 3 h (entry 3). Encouraged by this result, we continued to optimize the reaction conditions. Several bases, solvents, and temperatures were examined, and the results were summarized in Table 1. Remarkably, when the reaction was conducted in the presence of CSOH·H2O, the yield of 3aa was given to 56% (entry 4). Other bases (NaOH and LiOH) were found to give lower yields (entries 5 and 6). However, we did not detect the desired product when using some inorganic bases (K2CO3, Cs2CO3 and Na2CO3) and organic bases (DABCO, t-BuOK and DBU) (entries 7–12). Next, the amount of CSOH·H2O was examined (entries 13–15). The reaction could proceed smoothly with 59% yield, when only 3.0 equiv. of CSOH·H2O was used in the reaction (entry 15). Subsequently, examination of different mixed solvents (MeOH/H2O, DMSO/H2O, dioxane/H2O, EtOAc/H2O, THF/H2O and EtOH/H2O) revealed that EtOH/H2O was the best choice for the reaction, and the corresponding yield increased to 63% (entries 16–21). Following the above investigations, the ratio of EtOH/H2O was evaluated (entries 22–26), and a higher yield (81%) was observed with the ratio 2:1 (entry 23). Temperature screening suggested that neither a rise nor a drop of the temperature promoted the reaction (entries 27 and 28).
:1) at 100 °C for 3 h (entry 3). Encouraged by this result, we continued to optimize the reaction conditions. Several bases, solvents, and temperatures were examined, and the results were summarized in Table 1. Remarkably, when the reaction was conducted in the presence of CSOH·H2O, the yield of 3aa was given to 56% (entry 4). Other bases (NaOH and LiOH) were found to give lower yields (entries 5 and 6). However, we did not detect the desired product when using some inorganic bases (K2CO3, Cs2CO3 and Na2CO3) and organic bases (DABCO, t-BuOK and DBU) (entries 7–12). Next, the amount of CSOH·H2O was examined (entries 13–15). The reaction could proceed smoothly with 59% yield, when only 3.0 equiv. of CSOH·H2O was used in the reaction (entry 15). Subsequently, examination of different mixed solvents (MeOH/H2O, DMSO/H2O, dioxane/H2O, EtOAc/H2O, THF/H2O and EtOH/H2O) revealed that EtOH/H2O was the best choice for the reaction, and the corresponding yield increased to 63% (entries 16–21). Following the above investigations, the ratio of EtOH/H2O was evaluated (entries 22–26), and a higher yield (81%) was observed with the ratio 2:1 (entry 23). Temperature screening suggested that neither a rise nor a drop of the temperature promoted the reaction (entries 27 and 28).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent (ratio) | Base (equiv.) | T (°C) | Yieldb (%) | |
| 3aa | 4 | ||||
| a Reaction conditions: 1a (0.5 mmol, 1.0 equiv.), 2a (1.5 mmol, 3.0 equiv.), base (1.5 mmol, 3.0 equiv.) and solvent (6 ml) heated in oil bath for 3 h in sealed tube.b Isolated yields. | |||||
| 1 | iPrOH | KOH (20) | 100 | 15 | 12 |
| 2 | H2O | KOH (20) | 100 | 0 | 0 |
| 3 | iPrOH/H2O (5/1) | KOH (20) | 100 | 36 | 20 |
| 4 | iPrOH/H2O (5/1) | CSOH·H2O (20) | 100 | 56 | 19 |
| 5 | iPrOH/H2O (5/1) | NaOH (20) | 100 | 35 | 21 |
| 6 | iPrOH/H2O (5/1) | LiOH (20) | 100 | 42 | 10 |
| 7 | iPrOH/H2O (5/1) | K2CO3 (20) | 100 | 0 | 0 |
| 8 | iPrOH/H2O (5/1) | Cs2CO3 (20) | 100 | 0 | 0 |
| 9 | iPrOH/H2O (5/1) | Na2CO3 (20) | 100 | 0 | 0 |
| 10 | iPrOH/H2O (5/1) | DABCO (20) | 100 | 0 | 0 |
| 11 | iPrOH/H2O (5/1) | t-BuOK (20) | 100 | 0 | 0 |
| 12 | iPrOH/H2O (5/1) | DBU (20) | 100 | 0 | 0 |
| 13 | iPrOH/H2O (5/1) | CSOH·H2O (10) | 100 | 57 | 25 |
| 14 | iPrOH/H2O (5/1) | CSOH·H2O (5) | 100 | 55 | 27 |
| 15 | iPrOH/H2O (5/1) | CSOH·H2O (3) | 100 | 59 | 22 |
| 16 | MeOH/H2O (5/1) | CSOH·H2O (3) | 100 | 17 | <5 |
| 17 | DMSO/H2O (5/1) | CSOH·H2O (3) | 100 | <5 | 51 |
| 18 | Dioxane/H2O (5/1) | CSOH·H2O (3) | 100 | 41 | <5 |
| 19 | EtOAc/H2O (5/1) | CSOH·H2O (3) | 100 | 0 | 0 |
| 20 | THF/H2O (5/1) | CSOH·H2O (3) | 100 | 0 | 0 |
| 21 | EtOH/H2O (5/1) | CSOH·H2O (3) | 100 | 63 | 31 |
| 22 | EtOH/H2O (3/1) | CSOH·H2O (3) | 100 | 67 | 23 |
| 23 | EtOH/H2O (2/1) | CSOH·H2O (3) | 100 | 81 | <5 |
| 24 | EtOH/H2O (1/1) | CSOH·H2O (3) | 100 | 73 | <5 |
| 25 | EtOH/H2O (1/2) | CSOH·H2O (3) | 100 | <5 | <5 |
| 26 | EtOH/H2O (1/3) | CSOH·H2O (3) | 100 | <5 | <5 |
| 27 | EtOH/H2O (2/1) | CSOH·H2O (3) | 90 | 70 | 11 |
| 28 | EtOH/H2O 2/1) | CSOH·H2O (3) | 110 | 74 | <5 |
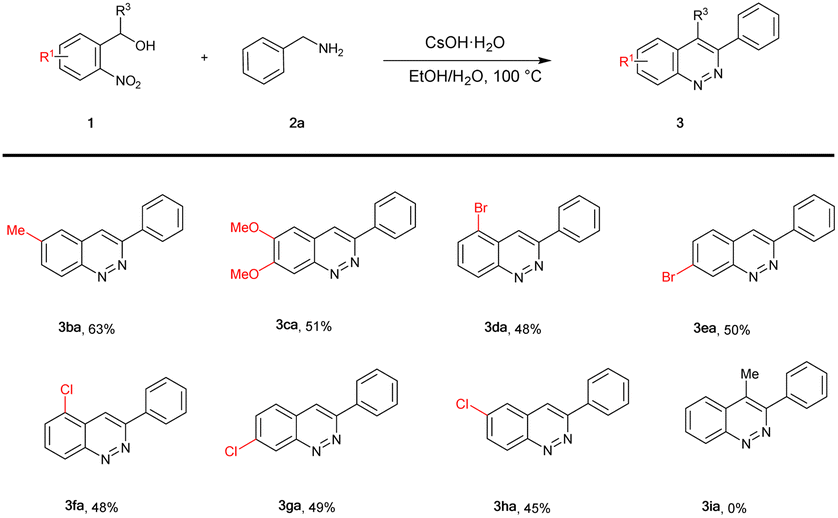
After optimizing the reaction conditions, we explored the substrate scope of benzylamine (2), as shown in Table 2. It is noteworthy that the electronic properties of the substituents on the aromatic ring system were shown to have a significant influence on the efficiency of this transformation. The benzylamine bearing electron-neutral (H) and electron-donating groups (2-Me, 3-Me, 4-Me, 3,4-2Me, 2-OMe, 4-OMe, –NH2, 3,4-(OCH2O)–) attached to the benzene ring, transformed smoothly into the corresponding products in moderate to good yields (53–81%; 3aa–3ai). Much to our satisfaction, halo-substituted (2-Br, 3-Br, 4-Br, 2-Cl, 4-Cl, 4-F, 4-I) substrates were also compatible, giving moderate yields (45–60%; 3aj–3ap). Fortunately, heterocyclic substituents (3-pyridyl, 4-pyridyl, 2-furyl, 2-thienyl) were also compatible, affording the expected products (3aq–3at) in 45–56% yields. In addition, naphthyl group substrates were also compatible, affording the expected products 3au and 3av in 46% and 50% yields, respectively. However, when the substrate was 2-aminoacetonitrile, the desired product 3ax was not afforded.
| a Reaction conditions: 1a (1.0 mmol, 1.0 equiv.), 2 (3.0 mmol, 3.0 equiv.) and CSOH·H2O (3.0 mmol, 3.0 equiv.) was stirred in EtOH/H2O (4 ml/2 ml) at 100 °C heated in oil bath for 3 h in sealed tube. Isolated yields. |
|---|
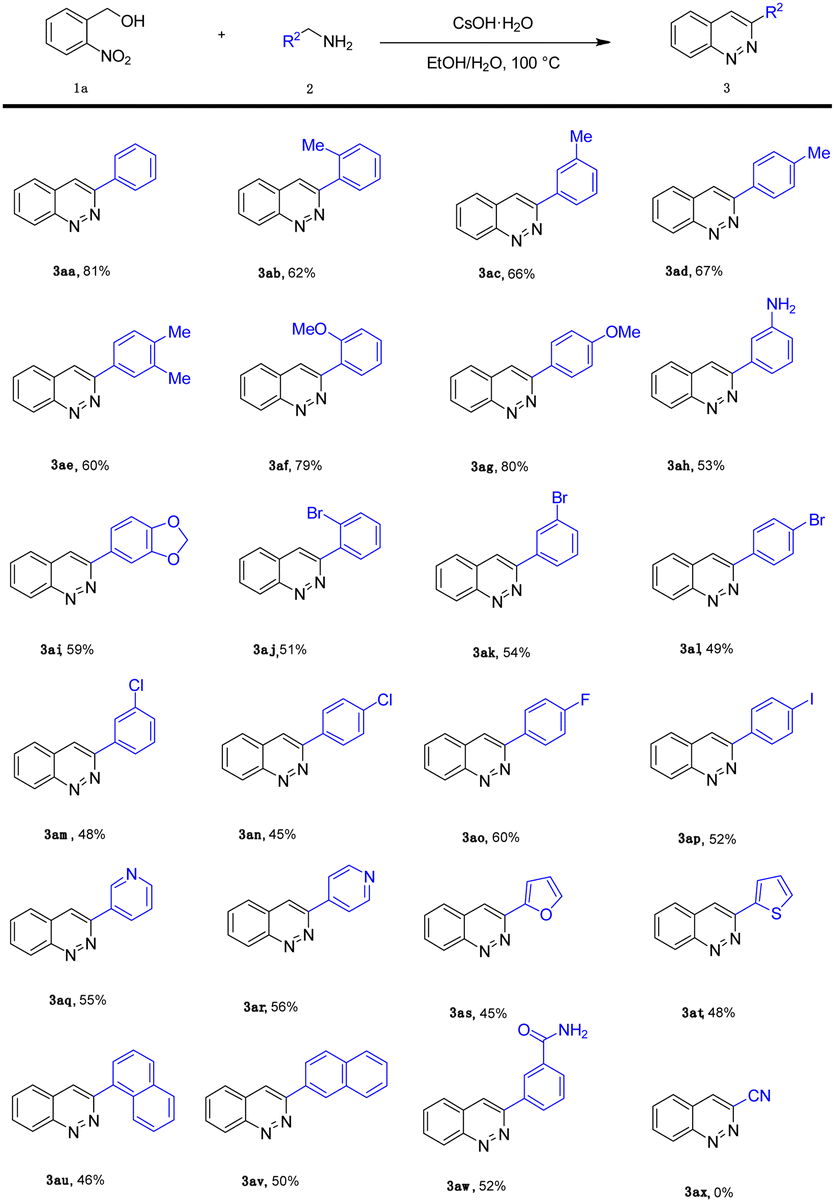 |
Encouraged by the results described above, the scope of 2-nitrobenzyl alcohol (1) was subsequently examined (Table 3). At first, electron-donating substituted 2-nitrobenzyl alcohols 1b and 1c were explored, and the corresponding products 3ba and 3ca were obtained in 63% and 51% yields, respectively. Halo-substituted (2-Br, 4-Br, 2-Cl, 4-Cl, 5-Cl) groups substrates were also compatible, affording the expected products (45–50%; 3da–3ha). In addition, when the substrate was secondary alcohol 1i, the desired product 3ia was not afforded.
| a Reaction conditions: 1 (0.5 mmol, 1.0 equiv.), 2a (1.5 mmol, 3.0 equiv.) and CSOH·H2O (1.5 mmol, 3.0 equiv.) was stirred in EtOH/H2O (4 ml/2 ml) at 100 °C heated in oil bath for 3 h in sealed tube. Isolated yields. |
|---|
 |
To gain insights into the reaction process, control experiments were performed (Scheme 2). The reaction of 2-nitrobenzyl alcohol 1a converted to 2-nitrosobenzaldehyde 5 in 33% yield under the standard conditions (eqn 1). Later, the reaction between compound 5 and 2a proceeded to give the target product 3aa in 39% yield (eqn 2). To our surprise, when the reaction was carried out in EtOH/H2O at 100 °C for 20 min in the presence of 3.0 equiv. CSOH·H2O, the desired product (3aa) was obtained in 20% yield together with unexpected (E)-2-(2-benzylidenehydrazineyl) benzaldehyde 6 (16%) (eqn 3). Moreover, compound 6 converted to the desired product 3aa in excellent yield (90%) under standard conditions (eqn 4). These results identified that compounds 5 and 6 were very probably intermediates for this reaction.
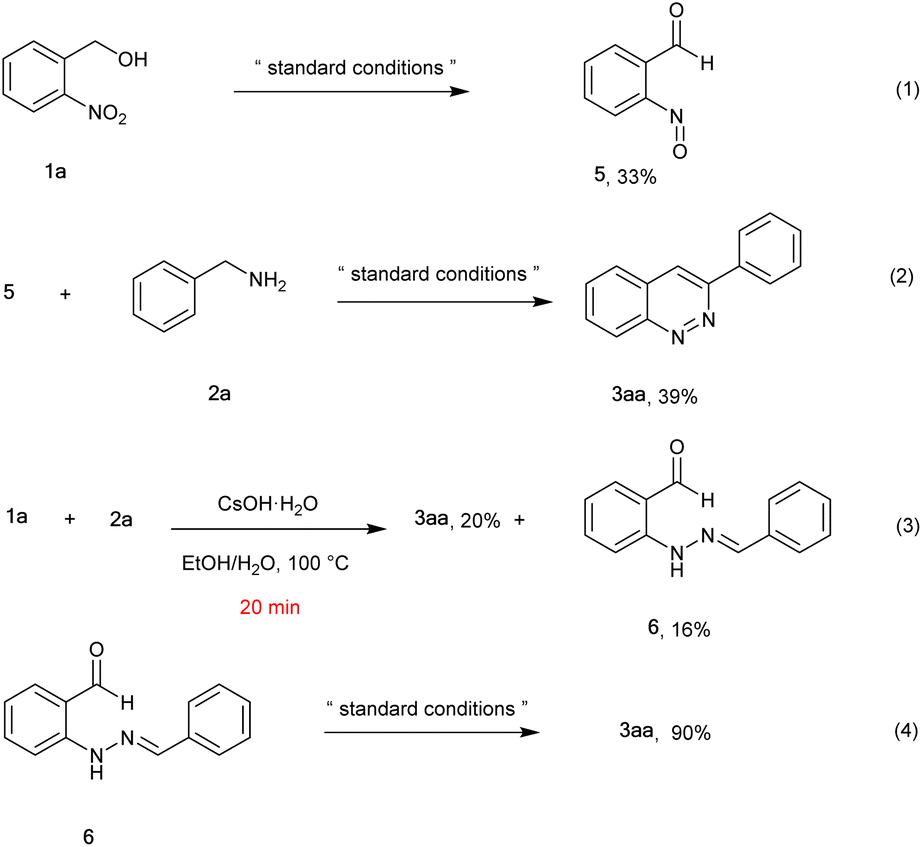 | ||
| Scheme 2 Control experiments. | ||
On the basis of the above-mentioned experimental results, a possible mechanism for this reaction is proposed as shown in Scheme 3 (using 3aa as an example). Initially, 2-nitrobenzyl alcohol 1a undergoes deprotonation in the presence of CSOH·H2O to form 2-nitrosobenzaldehyde 5,17 which is generated in situ via intramolecular redox reaction. Subsequently, intermediate 5 reacts with benzylamine 2a through a condensation reaction with elimination of H2O to afford (E)-2-(benzyldiazenyl)benzaldehyde C. Meanwhile, ortho-azo intermediate C is isomerized to form (E)-2-(2-benzylidenehydrazineyl) benzaldehyde 6. Subsequently, hydrazone 6 undergoes an intramolecular cyclization process to afford intermediate D. Finally, intermediate D is converted to the desired product 3aa through an aromatization reaction with elimination of H2O.
 | ||
| Scheme 3 A possible mechanism. | ||
In order to demonstrate the potential applications for organic synthesis, the reaction of 2-nitrobenzyl alcohol (1a) and benzylamine (2a) was performed on a gram scale (Scheme 4). To our satisfaction, the reaction was carried out very well, and the 3aa was furnished in 57% yield.
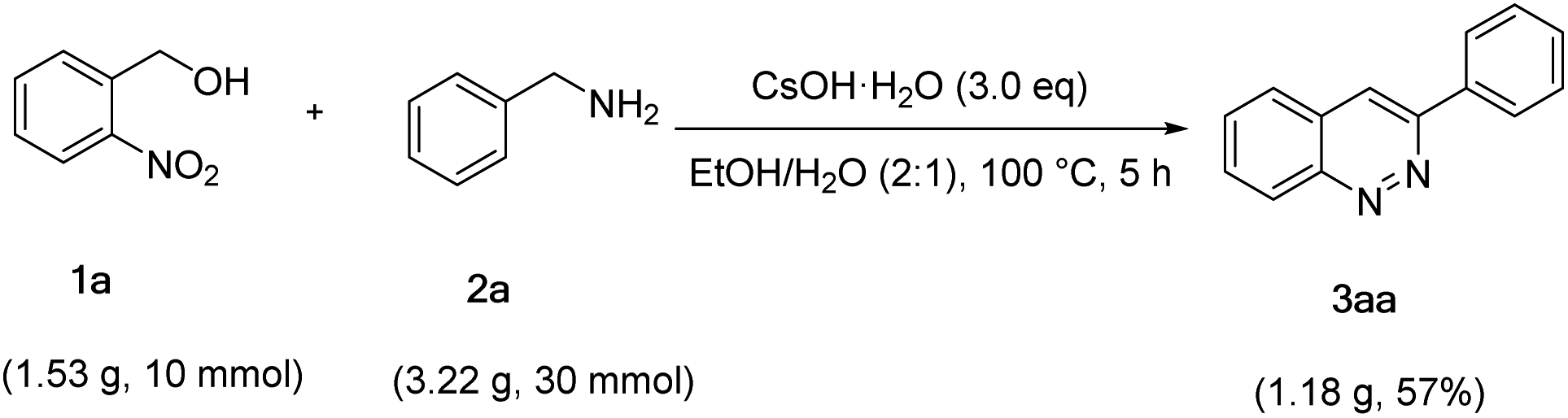 | ||
| Scheme 4 Gram scale experiment. | ||
In summary, we have developed a novel transition-metal-free intramolecular redox cyclization reaction for the efficient synthesis of cinnoline derivatives by simple treatment with CSOH·H2O in EtOH/H2O. This method obtained 2-nitrobenzaldehyde intermediate by intramolecular redox reaction of 2-nitrobenzyl alcohol, which then condenses with benzylamine, followed by azo is isomerized to form hydrazone, cyclization, and aromatization processes to form cinnoline compounds. Further studies on this method for the synthesis of other bioactive compounds and applications are in progress in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (Grant 21801022) for financial support. This work was also sponsored by CNPC Innovation Found (2021DQ02-0203).References
- W. Lewgowd and A. Stanczak, Arch. Pharm. Chemi, 2007, 340, 65–80 CrossRef CAS.
- D. A. Scott, L. A. Dakin, D. J. Del Valle, R. B. Diebold, L. Drew, T. W. Gero, C. A. Ogoe, C. A. Omer, G. Repik, K. Thakur, Q. Ye and X. L. Zheng, Bioorg. Med. Chem. Lett., 2011, 21, 1382–1384 CrossRef CAS.
- B. Parrino, A. Carbone, M. Muscarella, V. Spano, A. Montalbano, P. Barraja, A. Salvador, D. Vedaldi, G. Cirrincione and P. Diana, J. Med. Chem., 2014, 57, 9495–9511 CrossRef CAS PubMed.
- (a) A. L. Ruchelman, S. K. Singh, A. Ray, X. H. Wu, J.-M. Yang, T.-K. Li, A. Liu, L. F. Liu and E. J. LaVoie, Bioorg. Med. Chem., 2003, 11, 2061–2073 CrossRef CAS PubMed; (b) A. L. Ruchelman, S. K. Singh, A. Ray, X. Wu, J. M. Yang, N. Zhou, A. Liu, L. F. Liu and E. J. LaVoie, Bioorg. Med. Chem., 2004, 12, 795–806 CrossRef.
- C. Tian, C. Yang, T. Wu, M. Lu, Y. Chen, Y. Yang, X. Liu, Y. Ling, M. Deng, Y. Jia and Y. Zhou, Bioorg. Med. Chem. Lett., 2021, 48, 128271 CrossRef.
- X. Yao, X. Sun, S. Jin, L. Yang, H. Xu and Y. Rao, J. Med. Chem., 2019, 62, 6561–6574 CrossRef PubMed.
- (a) K. Muralirajan and C. H. Cheng, Chem.–Eur. J., 2013, 19, 6198–6202 CrossRef; (b) D. Zhao, Q. Wu, X. Huang, F. Song, T. Lv and J. You, Chem.–Eur. J., 2013, 19, 6239–6244 CrossRef CAS.
- (a) C. Zhu and M. Yamane, Tetrahedron, 2011, 67, 4933–4938 CrossRef CAS; (b) N. A. Danilkina, P. S. Vlasov, S. M. Vodianik, A. A. Kruchinin, Y. G. Vlasov and I. A. Balova, Beilstein J. Org. Chem., 2015, 11, 373–384 CrossRef CAS PubMed.
- C. J. Ball, J. Gilmore and M. C. Willis, Angew. Chem., Int. Ed., 2012, 51, 5718–5722 CrossRef CAS.
- G. Zhang, J. Miao, Y. Zhao and H. Ge, Angew. Chem., Int. Ed. Engl., 2012, 51, 8318–8321 CrossRef CAS PubMed.
- P. Sun, Y. Wu, Y. Huang, X. Wu, J. Xu, H. Yao and A. Lin, Org. Chem. Front., 2016, 3, 91–95 RSC.
- C. Lan, Z. Tian, X. Liang, M. Gao, W. Liu, Y. An, W. Fu, G. Jiao, J. Xiao and B. Xu, Adv. Synth. Catal., 2017, 359, 3735–3740 CrossRef.
- B. Yao, T. Miao, W. Wei, P. Li and L. Wang, Org. Lett., 2019, 21, 9291–9295 CrossRef.
- (a) A. M. Amer, Monatsh. Chem., 2001, 132, 859–870 CrossRef; (b) A. M. Amer, M. M. El-Mobayed and S. Asker, Monatsh. Chem., 2004, 135, 595–604 CrossRef; (c) L. Forlani, O. A. Attanasi, C. Boga, L. De Crescentini, E. Del Vecehio, G. Favi, F. Mantellini, S. Tozzi and N. Zanna, Eur. J. Org. Chem., 2008, 2008, 4357–4366 CrossRef; (d) S. Vikas and S. Darbhamulla, Afr. Health Sci., 2009, 9, 275–278 Search PubMed; (e) C. T. Bui and B. L. Flynn, Mol. Divers., 2011, 15, 83–89 CrossRef CAS; (f) P. Gogoi, S. R. Gogoi, N. Devi and P. Barman, Synth. Commun., 2014, 44, 1142–1148 CrossRef CAS; (g) A. S. Al-zagameem, M. M. El-Abadelah, M. A. Zihlif, R. G. Naffa, M. L. Al-Smadi and M. S. Mubarak, J. Heterocycl. Chem., 2016, 53, 1771–1777 CrossRef; (h) X. Pang, L. Zhao, D. Zhou, P. Y. He, Z. An, J. X. Ni and R. Yan, Org. Biomol. Chem., 2017, 15, 6318–6322 RSC; (i) N. G. Khaligh, T. Mihankhah, M. R. Johan and J. J. Ching, Monatsh. Chem., 2018, 149, 1083–1087 CrossRef.
- G. C. Senadi, B. S. Gore, W. P. Hu and J. J. Wang, Org. Lett., 2016, 18, 2890–2893 CrossRef PubMed.
- W. M. Shu, J. R. Ma, K. L. Zheng and A. X. Wu, Org. Lett., 2016, 18, 196–199 CrossRef CAS PubMed.
- (a) K. Inamoto, M. Katsuno, T. Yoshino, Y. Arai, K. Hiroya and T. Sakamoto, Tetrahedron, 2007, 63, 2695–2711 CrossRef CAS; (b) J. S. Zhu, N. Kraemer, M. E. Shatskikh, C. J. Li, J. H. Son, M. J. Haddadin, D. J. Tantillo and M. J. Kurth, Org. Lett., 2018, 20, 4736–4739 CrossRef CAS PubMed; (c) Y. Ou, T. Yang, N. Tang, S. F. Yin, N. Kambe and R. Qiu, Org. Lett., 2021, 23, 6417–6422 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06523a |
| This journal is © The Royal Society of Chemistry 2022 |