
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous improvement of the thermostability and activity of lactic dehydrogenase from Lactobacillus rossiae through rational design
Xi Luoa,
Yifeng Wangab,
Weilong Zhenga,
Xiaolong Suna,
Gaowei Hua,
Longfei Yina,
Yingying Zhanga,
Fengwei Yina and
Yongqian Fu *a
*a
aInstitute of Biomass Resources, Taizhou University, Taizhou 318000, Zhejiang, People's Republic of China. E-mail: bioengineer@163.com; Fax: +86-576-88660338; Tel: +86-576-88660338
bSchool of Food Science and Pharmaceutical Engineering, Nanjing Normal University, Nanjing, Jiangsu 210023, China
First published on 21st November 2022
Abstract
D-Phenyllactic acid, is a versatile organic acid with wide application prospects in the food, pharmaceutical and material industries. Wild-type lactate dehydrogenase LrLDH from Lactobacillus rossiae exhibits a high catalytic performance in the production of D-phenyllactic acid from phenylpyruvic acid or sodium phenylpyruvate, but its industrial application is hampered by poor thermostability. Here, computer aided rational design was applied to improve the thermostability of LrLDH. By using HotSpot Wizard 3.0, five hotspot residues (N218, L237, T247, D249 and S301) were identified, after which site-saturation mutagenesis and combined mutagenesis were performed. The double mutant D249A/T247I was screen out as the best variant, with optimum temperature, t1/2, and T1050 that were 12 °C, 17.96 min and 19 °C higher than that of wild-type LrLDH, respectively. At the same time, the kcat/Km of D249A/T247I was 1.47 s−1 mM−1, which was 3.4 times higher than that of the wild-type enzyme. Thus rational design was successfully applied to simultaneously improve the thermostability and catalytic activity of LrLDH to a significant extent. The results of molecular dynamics simulations and molecular structure analysis could explain the mechanisms for the improved performance of the double mutant. This study shows that computer-aided rational design can greatly improve the thermostability of D-lactate dehydrogenase, offering a reference for the modification of other enzymes.
Introduction
Currently, chemical preservatives like sodium benzoate and potassium sorbate are widely used in the food industry. They play important roles in preventing the proliferation of spoilage microorganisms and food-borne pathogens, as well as reducing the generation of harmful microbial secondary metabolites. However, excessive or improper use of these preservatives may cause metabolic imbalances and lead to certain diseases.1 With increasing awareness of food safety issues, there is an urgent need to find safer alternatives. Phenyllactic acid (PLA), a natural organic acid with remarkable antimicrobial properties, is found in honey and some foods fermented by lactic acid bacteria (LAB).2,3 Due to the well-known safety of LAB in the food industry, the use of their metabolites, including PLA, is generally regarded as safe. By decreasing the rigidity of the cell wall and damaging the membrane integrity, PLA has antibacterial effects against a variety of bacteria and fungi, resulting in a broad antimicrobial spectrum.4–6 Moreover, PLA has prominent water solubility and excellent thermal stability while having no objectionable odor.7,8 Due to these advantages, PLA has great potential for use as a preservative in the food industry. Due to the presence of a chiral carbon atom, chemical synthesis results in the production of a mixture of D-PLA and L-PLA, whereby the former has a better antibacterial effect.9 In addition, D-PLA can be used as a building block in the synthesis of pharmaceuticals, including the hypoglycemic agent englitazone, the coronary heart disease treatment drug danshensu and the anthelmintic agent PF1022A.10–12 In addition, D-PLA can act as a monomer to produce poly-PLA, a biodegradable material with excellent thermostability and ultraviolet absorption properties.13 Hence, D-PLA has broad application potential in the food, pharmaceutical and material industries, which has attracted considerable interest in its manufacturing.A variety of microorganisms can be used for PLA fermentation, especially LAB.14 However, although many effort including metabolic engineering and process optimization have been made, both the output and optical purity were unsatisfactory.15,16 Moreover, the excessive by-products drive up the cost, due to the complex process of product separation. PLA is the by-product of phenylalanine metabolism in the cells of LABs, and phenylpyruvate (PPA) is the direct precursor of PLA. D-PLA is generated by PPA reduction catalyzed by D-lactate dehydrogenase (D-LDH).17 Owing to its excellent catalytic activity and stereoselectivity, fewer by-products and eco-friendly reaction conditions, biocatalytic conversion is considered a more efficient and straightforward strategy for D-PLA production than fermentation. Zhou et al.,18 cloned a D-LDH gene from Lactobacillus sp. ZX1 and expressed it in Escherichia coli. Subsequently, a fed-batch conversion using whole cells was conducted, and 18.21 g l−1 D-PLA was generated from PPA within 8 h. To improve NADH regeneration and the yield of D-PLA, Qin et al. fusion expressed D-LDH and glycerol dehydrogenase in E. coli. Catalyzed by this strain, a productivity of 5.83 g l−1 h−1 was achieved in a 5 l reactor.19 In our previous work, a gene encodes D-LDH was cloned and heterologous expressed in E. coli. The obtained recombinant protein LrLDH has the ability to asymmetrically reduce PPA to D-PLA with high activity, but the half-life of LrLDH at 50 °C is only 3.70 min, indicating poor thermostability.20 Thermostability is an important process parameter, which reflects the ability of enzyme maintain the integrity of its structure and function at a certain temperature. Moreover, high temperature can improve the reaction rate and reduce the viscosity of the feed liquid.21 Therefore, the poor thermostability of D-LDH is a serious obstacle for its application in the production of D-PLA.
Protein engineering approaches, including directed evolution and rational design, are powerful tools for overcoming the shortcomings of natural enzymes such as unsatisfactory thermostability.22 Compared with directed evolution, rational design based on computer-assisted techniques is more rapid and targeted.23 This strategy is usually employed to enhance enzyme thermostability and numerous successful examples have been reported. Zhao et al. predicted candidate residue pairs suitable for substitution by cysteine using the DbD computational tool, and the additional disulfide bridges were introduced into D-psicose-3-epimerase. After B-factor value analysis and screening, three mutant enzymes were obtained with varying degrees of improvement in optimum catalytic temperature and half-life (t1/2).24 Based on protein surface topography, multiple sequence alignment and calculations of the changes in free energy of folding (ΔΔG), hot spots related to thermal stability of endoglucanase II from Penicillium verrucosum were predicted and site-specific mutagenesis were conducted. The mutants showed a remarkable improvement in thermostability without changes of specific activity.25
Under extreme conditions such as high temperature or extreme pH, enzymatic denaturation begins with partial unfolding of the peripheral protein chains, followed by exposure of the protein interior and aggregation of the hydrophobic regions.26 Therefore, the rigidity of the structure outside the active center plays an important role in determining the thermostability of enzymes. To improve the thermostability of LrLDH, we engineered the enzyme using computationally assisted rational design. The appropriate substitutions of amino acid residues were selected with the aid of HotSpot Wizard 3.0 (https://loschmidt.chemi.muni.cz/hotspotwizard/), followed by site-saturation mutagenesis and combinatorial mutations. The variant D249A/T247I with both enhanced thermostability and improved catalytic activity with PPA was obtained, and the underlying molecular mechanisms were analyzed.
Experimental
Reagents
Sodium phenylpyruvate (PPA), β-nicotinamide adenine dinucleotide disodium salt (NADH), kanamycin and Ni-TED pre-packed gravity column (5 ml) were purchased from Sangon Biotech (Shanghai, China) Co., Ltd. Tryptone and yeast extract were obtained from Oxoid Co., Ltd (United Kingdom). PrimeSTAR® HS DNA polymerase, QuickCut™ restriction enzymes, PCR reagents, loading buffers for nucleic acid electrophoresis and SDS-PAGE, DNA and protein markers, as well as the Bradford protein assay kit were purchased from Takara Biotechnology (Dalian, China) Co., Ltd. The plasmid miniprep kits were purchased from Axygen Biotechnology (Hangzhou, China) Co., Ltd. Oligonucleotide synthesis and DNA sequencing were conducted by Sangon Biotech Co., Ltd (Shanghai, China). Unless specified otherwise, all other reagents and chemicals used in this study were obtained from general commercial suppliers and used without further purification.E. coli BL21 (DE3) and pET-28a (+) were used as the host strain and vector for protein expression. The plasmid pET28a-lrldh with the lactic dehydrogenase gene from Lactobacillus rossiae was used as the template for the site-saturation mutagenesis.
The engineered E. coli strains were grown in Luria-Bertani (LB) medium (0.5% yeast extract, 1.0% tryptone and 1.0% NaCl), supplemented with 50 μg ml−1 kanamycin.
Molecular modelling and docking
The three-dimensional (3D) homology models of LrLDH and its mutants were generated using Rosetta CM based on the crystal structure of lactic dehydrogenase from Lactobacillus helveticus in complex with NADH (PDB ID: 2DLD) as the template. A total of 1000 models were generated and the model with the lowest score was used as the input structure for further calculations. Molecular docking was then performed between LrLDH or its variants and PPA according to the program Autodock 4.2. The detailed parameters were set refer to the previous article.27 And the images were created with PyMOL.Construction of the mutant library
The 3D homology model of wild-type LrLDH was submitted to the HotSpot Wizard 3.0 webserver, and the potential thermostability-related residues were predicted as described before.28 Site-directed mutations were performed via polymerase chain reactions (PCR) using pET28a (+) recombinant plasmid with wild-type or mutant lrldh genes as the template. The primers used for the saturation mutagenesis at each site are listed in Table 1. The PCR was performed as described previously.29 To degrade the template, the QuickCut™ restriction enzyme DpnI was added to the PCR reaction mixture and incubated at 37 °C for 10 min. The products were used to transform E. coli BL21 (DE3) competent cells and cultivated on LB agar plates containing 50 μg ml−1 kanamycin. The transformants were verified by DNA sequencing.| Primer | Oligonucleotide sequences (5′–3′) |
|---|---|
| a N stands for A, T, G or C. The mutated nucleotides are underlined. | |
| S301-F | GTGTTAGTGNNNCCGCACATCGCGTTCTAC |
| S301-R | GATGTGCGGNNNCACTAACACGTTTTCACGTT |
| T247-F | CTGGTTGATNNNGATGATCTGATCAAAGCGC |
| T247-R | CAGATCATCNNNATCAACCAGATCCCCACG |
| D249-F | ATACCGATNNNCTGATCAAAGCGCTGGATT |
| D249-R | TTTGATCAGNNNATCGGTATCAACCAGATC |
| N218-F | ACCAAAGAANNNATGCACATGCTGAACGCTG |
| N218-R | CATGTGCATNNNTTCTTTGGTTGCCGGAGC |
| L237-F | GTTTATATANNNAACCCGGCGCGTGGGG |
| L237-R | CGCCGGGTTNNNTATATAAACACCATCTTTCATTTTG |
| S301M-F | TGTTAGTGATGCCGCACATCGCGTTCTAC |
| S301M-R | GATGTGCGGCATCACTAACACGTTTTCACGT |
| S301R-F | GTGTTAGTGCGCCCGCACATCGCGTTC |
| S301R-R | TGTGCGGGCGCACTAACACGTTTTCACGTT |
| N218V-F | ACCAAAGAAGTTATGCACATGCTGAACGCTG |
| N218V-R | CATGTGCATAACTTCTTTGGTTGCCGGAGC |
| N218T-F | CCAAAGAAACCATGCACATGCTGAACGCT |
| N218T-R | ATGTGCATGGTTTCTTTGGTTGCCGGAGC |
| L237K-F | GTTTATATAAAAAACCCGGCGCGTGGGG |
| L237K-R | CGCCGGGTTTTTTATATAAACACCATCTTTCATTTTG |
| L237C-F | GTTTATATATGTAACCCGGCGCGTGGGG |
| L237C-R | CGCCGGGTTACATATATAAACACCATCTTTCATTTTG |
| D249A-F | ATATTGATGCGCTGATCAAAGCGCTGGATT |
| D249A-R | TTTGATCAGCGCATCAATATCAACCAGATC |
Protein expression and purification
E. coli cells harboring recombinant plasmids encoding lrldh or its variants were cultured in 50 ml LB medium containing 50 μg ml−1 kanamycin at 37 °C and 180 rpm until the optical density at 600 nm (OD600) reached approximately 0.8. Subsequently, lactose was added as an inducer to a final concentration of 10 g l−1, and the cultures were cultured at 28 °C and 150 rpm for another 12 h. The cells were harvested by centrifuged at 4 °C and 8000×g for 10 min, washed twice with potassium phosphate buffer (20 mM, pH 8.0), resuspended in the same buffer with a wet cell concentration of 50 g l−1, and disrupted by ultrasonication (300 W, 10 min) in an ice bath followed by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g, 4 °C, 20 min) to remove the cell debris. The supernatants were collected and loaded onto a 5 ml Ni-TED pre-packed gravity column pre-equilibrated with buffer A (20 mM imidazole and 500 mM NaCl in 20 mM potassium phosphate buffer, pH 8.0), and washed with 25 ml buffer A to remove the unbound proteins. The fractions were collected by elution with buffer B (500 mM imidazole and 500 mM NaCl in 20 mM potassium phosphate buffer, pH 8.0) and dialyzed overnight against potassium phosphate buffer (20 mM, pH 8.0). All purification steps were conducted at 0–4 °C. The purity of the target protein was checked by SDS-PAGE with a 12% acrylamide separating gel under denaturing conditions. The concentration of purified enzymes was measured using the Bradford protein assay kit with bovine serum albumin (BSA) as the protein standard.
000×g, 4 °C, 20 min) to remove the cell debris. The supernatants were collected and loaded onto a 5 ml Ni-TED pre-packed gravity column pre-equilibrated with buffer A (20 mM imidazole and 500 mM NaCl in 20 mM potassium phosphate buffer, pH 8.0), and washed with 25 ml buffer A to remove the unbound proteins. The fractions were collected by elution with buffer B (500 mM imidazole and 500 mM NaCl in 20 mM potassium phosphate buffer, pH 8.0) and dialyzed overnight against potassium phosphate buffer (20 mM, pH 8.0). All purification steps were conducted at 0–4 °C. The purity of the target protein was checked by SDS-PAGE with a 12% acrylamide separating gel under denaturing conditions. The concentration of purified enzymes was measured using the Bradford protein assay kit with bovine serum albumin (BSA) as the protein standard.
Enzyme activity assay
The enzymatic activity of wild-type LrLDH and its mutants was measured at 30 °C by monitoring the decrease in the absorbance of NADH at 340 nm using a microplate reader. The standard assay mixture was composed of potassium phosphate buffer (100 mM, pH 6.5), 0.5 mM NADH, 1 mM PPA, and the appropriate diluted enzyme in a total volume of 200 μl. All reactions were pre-mixed at 30 °C and initiated by adding NADH. One unit of enzyme activity was defined as the amount of enzyme that catalyzes the oxidation of 1 μmol of NADH per minute.Optimal temperature and thermostability analysis
The optimal reaction temperature of wild-type LrLDH and its mutants was determined by measuring the enzyme activity of each sample at temperatures ranging from 30 and 60 °C using the standard assay conditions.To assess the thermostability of wild-type LrLDH and its mutants, the residual activity of the purified enzyme was measured after 30 min at 50 °C, with non-incubated enzyme as the control. The half-lives (t1/2) were calculated according to the first-order deactivation function: ln(residual activity) = −kDt, t1/2 = ln2/kD, where kD represents the deactivation rate constant.30
For measuring the half inactivation temperature (T1050) of wild-type LrLDH and its mutants, defined as the temperature at which enzymes lose 50% of the initial activity after incubation for 10 min, the purified enzymes were incubated at temperatures ranging from 20 to 60 °C for 10 min, and the residual activity was determined using the standard assay conditions with non-incubated enzyme as the control.
Determination of kinetic parameters
The kinetic parameters of wild-type LrLDH and its mutants were determined by measuring the enzyme activity at varying PPA concentrations (1–5 mM) and a fixed NADH concentration (0.5 mM). The data were fitted to the Michaelis–Menten model using the software Origin 9.0.Molecular dynamics simulations
The molecular dynamics simulations were performed with Gromacs 2018, using “chain A” of wild-type LrLDH and its variants. The force field was selected with GROMOS 53a6. The protein was solvated in water. spc216.gro was used as the solvent configuration for simple point charge (SPC) water. To balance the system, Na+ or Cl− ions were added to the simulation box to replace the solvent. Then, the structure was relaxed through energy minimization. Furthermore, the NVT equilibration was performed at 343 K for 100 ps and 1 bar in the NPT equilibration for 100 ps. Finally, the MD simulations were carried out for 100 ns. The root mean square deviation (RMSD) and radius of gyration (Rg) of wild-type LrLDH and its variants were calculated.31Results and discussion
Identification of hotspot residues
Our previous study suggested that LrLDH from Lactobacillus rossiae is a promising biocatalyst for the efficient synthesis of D-phenyllactic acid and other fine chemicals. However, the poor thermostability (t1/2 at 50 °C of only 3.7 min) hampered its practical application.20 To better meet the requirements of industrial application, we attempted to improve the thermostability of LrLDH. HotSpot Wizard is a popular tool for the automated prediction of hotspots for rational enzyme improvement based on structural, functional and evolutionary information obtained from different databases.28 Many vivid examples have demonstrated its effectiveness not only in tailoring the catalytic activity, but also in improving the thermostability of enzymes.32–35 To predict residues that might affect the enzyme's thermostability, a 3D homology models of LrLDH was constructed (Fig. 1a) and uploaded to the HotSpot Wizard web server. After calculating, 8 stability hot spots (Pro 239, Phe 306, Ser 301, Phe 274, Leu237, Asn 218, Asp 249, T247) were shown in the website. The amino acid residues around the catalytic site, including the residues in catalytic pocket and the tunnels, may appreciable impact the catalytic activity of enzyme.36 Excluding these residues, 5 amino acid residues located far from the active center were selected (N218, L237, T247, D249 and S301) and chosen as engineering targets for the thermostability mutagenesis (Fig. 1b).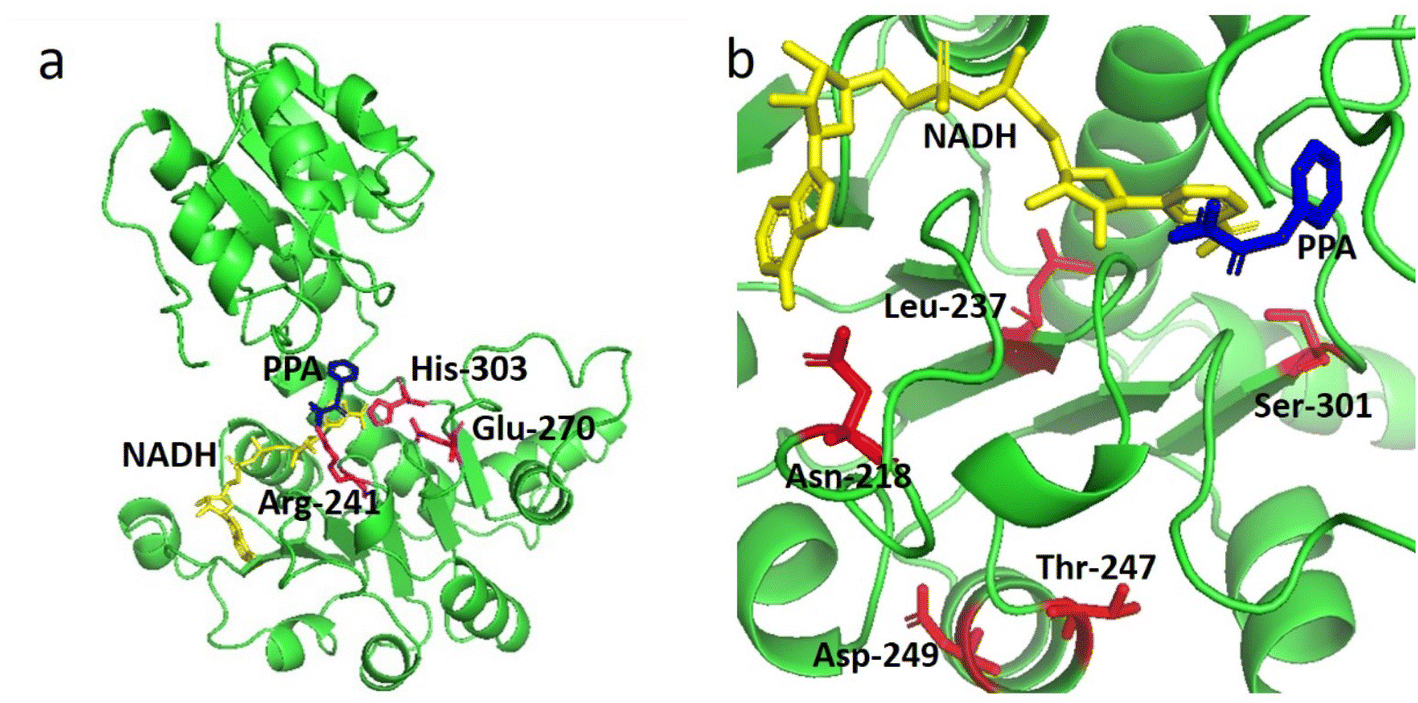 | ||
| Fig. 1 (a) Three-dimensional structure of LrLDH (green cartoon representations). (b) Locations of mutations (red sticks representations) in the structure of LrLDH. The catalytic triad (Arg241, Glu270, and His303), PPA and NADH are indicated by magentas, blue and yellow sticks, respectively. | ||
Construction and screening of mutant libraries
Subsequently, site-saturation mutagenesis was conducted by PCR with the plasmid pET28a-lrldh as the template. The generated mutant plasmids were introduced into E. coli for expression. According to the gene sequence analysis, all 95 mutants covering each site were obtained, and a single site mutant library was generated.The strains harboring the wild-type lrldh or its mutants were cultivated and induced with lactose. The protein-expressing cells were disrupted and the enzymes were purified by Ni2+-chelating affinity chromatography. We subsequently performed thermostability screening by incubated the purified enzymes at 50 °C for 5 min, followed by immediate cooling in an ice-bath and measurement of the residual activity. An enzyme sample that was not heated was included as a control. As shown in Table 2, 10 single mutants (S301M, S301R, T247I, T247C, T247W, D249A, N218V, N218T, L237K and L237C) presented better thermostability than the wild-type LrLDH (with a residual activity of 37.7%). The T247I variant was the best single mutant, and it retained 93.31% residual activity after heat treatment. Almost all of the mutants (including T247I) suffered a decline of specific activity relative to wild-type LrLDH, as there is a general trade-off between catalytic activity and stability.30 To our surprise, the variant D249A exhibited a simultaneous improvement of catalytic activity and thermostability, with approximately 6-fold higher specific activity (3.10 U mg−1) and nearly 60% higher residual activity (62.03%) than the wild-type LrLDH. To combine the advantages, double and triple mutants were also constructed. After screening, the double-mutant variants T247I/N218V, D249A/T247W, and L237K/T247C, as well as the triple-mutant variants D249A/T247I/S301M, D249A/T247I/S301R, D249A/T247I/N218T, D249A/T247I/L237K, and D249A/T247I/L237C displayed enhanced thermostability but a lower specific activity than the wild-type enzyme. Conversely, the variants D249A/N218T and D249A/T247I/N218V showed a better specific activity but a poor thermal stability. However, we fortunately obtained the double-mutant variant D249A/T247I with a specific activity of 1.61 U mg−1 and a residual activity of 89.07%. In comparison with the wild-type enzyme, that was an approximately 2-fold improvement of catalytic activity and nearly 40% higher residual activity. Although the mutant D249A/T247I sacrificed a small amount of thermostability, the catalytic activity was significantly improved compared with the mutant T247I (Table 2). This variant therefore strikes a good balance of improved thermostability and catalytic activity. Additionally, The SDS-PAGE results indicated that both the mutant D249A and D249A/T247I exhibited better soluble expression in E. coli (Fig. 2), which is consistent with their improved stability.
| Enzyme | Specific activitya (U mg−1) | Residual activityb (%) | Enzyme | Specific activity (U mg−1) | Residual activity (%) |
|---|---|---|---|---|---|
| a The enzyme activity was measured under the standard assay conditions.b LrLDH and the mutants were incubated in 100 mM, pH 6.5 potassium phosphate at 50 °C for 5 min, the residual activity was detected under the standard assay conditions, untreated enzyme was taken as the control.c All reactions were performed in triplicate, and activities expressed as means ± standard deviations. | |||||
| WT | 0.51 ± 0.08 | 37.70 ± 2.20 | D249A/T247I | 1.61 ± 0.08 | 89.07 ± 3.55 |
| S301M | 0.05 ± 0.00 | 66.85 ± 5.75 | T247I/N218V | 0.02 ± 0.00 | 70.00 ± 2.89 |
| S301R | 0.01 ± 0.00 | 73.50 ± 4.83 | L237K/T247W | 0.15 ± 0.02 | 62.30 ± 3.68 |
| T247I | 0.38 ± 0.06 | 93.31 ± 1.57 | L237K/T247C | 0.09 ± 0.00 | 58.30 ± 1.37 |
| T247C | 0.03 ± 0.00 | 84.57 ± 3.25 | D249A/N218T | 0.90 ± 0.10 | 24.70 ± 2.74 |
| T247W | 0.01 ± 0.00 | 84.14 ± 3.86 | D249A/T247I/S301M | 0.09 ± 0.01 | 55.36 ± 3.33 |
| D249A | 3.10 ± 0.17 | 62.03 ± 2.75 | D249A/T247I/S301R | 0.13 ± 0.01 | 51.65 ± 2.79 |
| N218V | 0.21 ± 0.03 | 56.75 ± 2.59 | D249A/T247I/N218V | 0.59 ± 0.06 | 35.38 ± 3.45 |
| N218T | 0.35 ± 0.03 | 56.60 ± 4.69 | D249A/T247I/N218T | 0.36 ± 0.05 | 41.27 ± 2.65 |
| L237K | 0.25 ± 0.07 | 52.94 ± 2.92 | D249A/T247I/L237K | 0.38 ± 0.03 | 52.58 ± 3.58 |
| L237C | 0.13 ± 0.02 | 44.96 ± 3.20 | D249A/T247I/L237C | 0.44 ± 0.04 | 46.95 ± 4.42 |
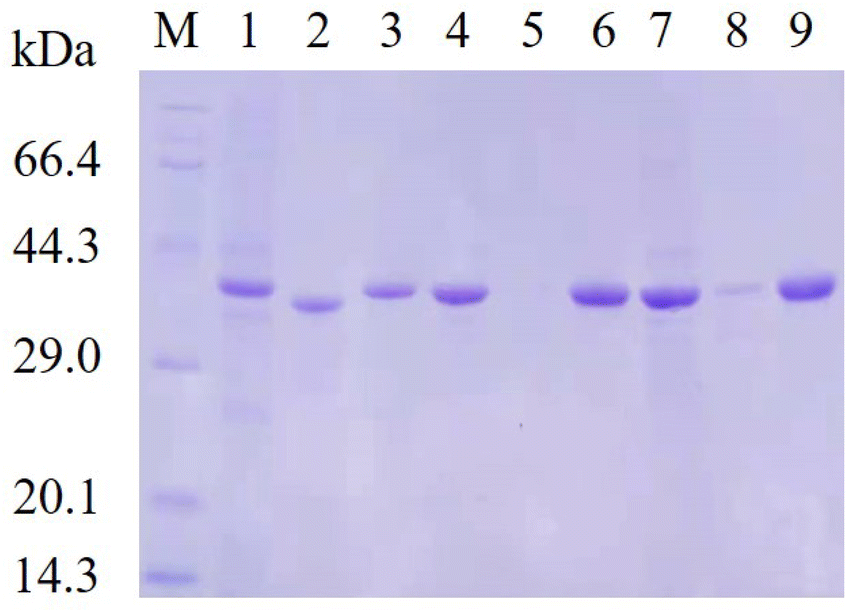 | ||
| Fig. 2 SDS-PAGE analysis. M: protein molecular weight marker; lane 1, 4 and 7: soluble fractions from lactose induced E. coli/pET28a-lrldhWT, E. coli/pET28a-lrldhD249A and E. coli/pET28a-lrldhD249A/T247I, respectively; lane 2, 5 and 8: precipitates from lactose induced E. coli/pET28a-lrldhWT, E. coli/pET28a-lrldhD249A and E. coli/pET28a-lrldhD249A/T247I, respectively; lane 3, 6 and 9: the purified WT, mutant D249A and mutant D249A/T247I, respectively. | ||
Effects of temperature
The T1050 values were estimated by measuring the residual activity after incubation for 10 min at different temperatures in the range of 20–60 °C (Fig. 3a).The thermal inactivation curves were recorded by measuring the time-variance of residual enzyme activity at 50 °C. As illustrated in Fig. 3b, wild-type LrLDH, as well as mutant D249A and D249A/T247I, all suffered a rapid decline of activity. The data were fitted nonlinearly using the software Origin 9.0 to calculate the t1/2. As shown in Table 3, the t1/2 values of wild-type LrLDH, D249A and D249A/T247I were 3.70, 7.88 and 21.66 min, respectively, while the T1050 values were 38, 44 and 57 °C. This result indicated that the thermostability of LrLDH was significantly enhanced by this rational mutagenesis strategy.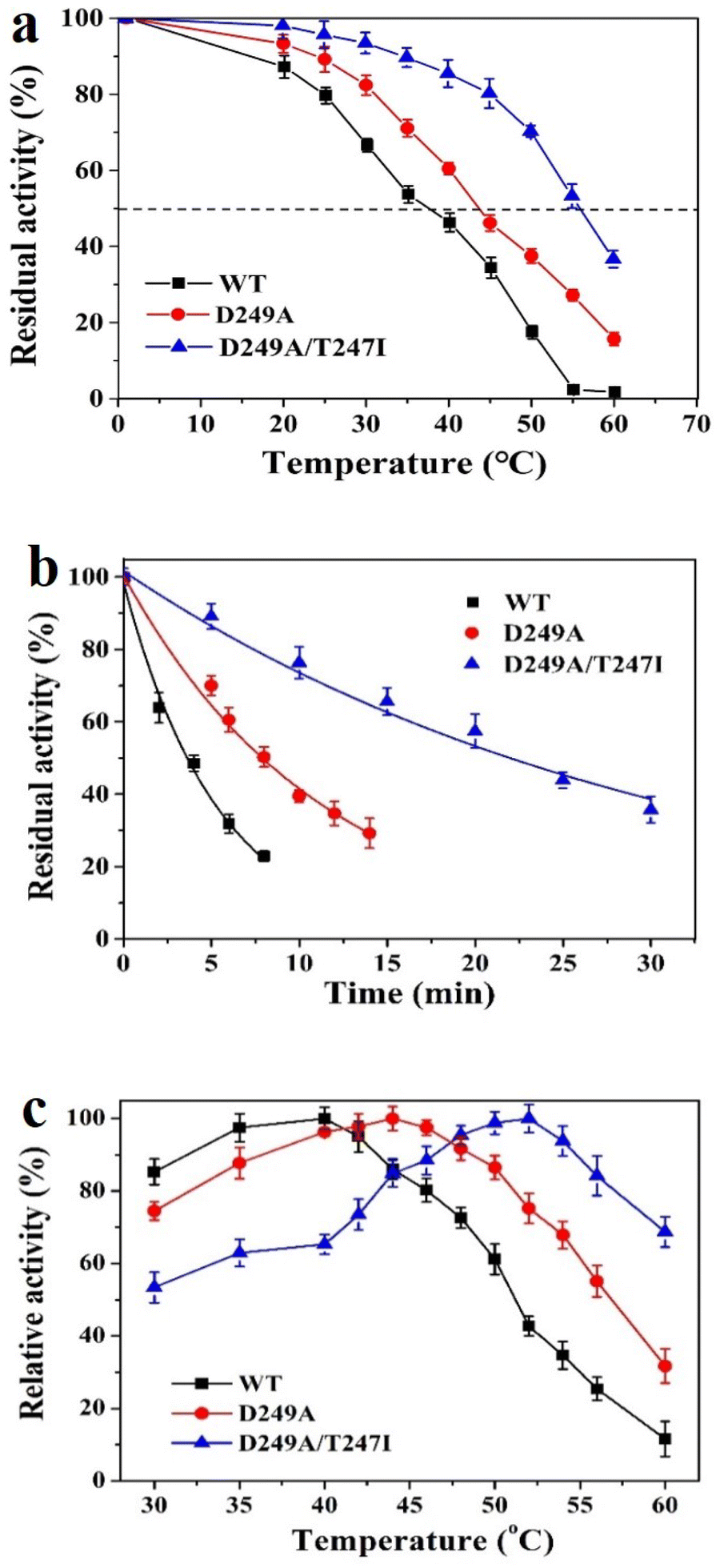 | ||
| Fig. 3 Influence of temperature on the activity of wild-type LrLDH and the variants. (a) T1050, the WT and engineered mutants were incubated in 100 mM, pH 6.5 potassium phosphate at 20–60 °C for up to 10 min, the residual activity was detected under the standard assay conditions. (b) Thermostability, WT and the mutants were incubated in 100 mM, pH 6.5 potassium phosphate at 50 °C for up to 30 min, the residual activity was detected under the standard assay conditions, untreated LrLDH or the mutant was taken as the control. (c) Optimal temperature, assays were carried in 100 mM potassium phosphate buffer, pH 6.5, at 30–60 °C for 1 min. All reactions were performed in triplicate, and error bars represent the standard error of mean. | ||
| Enzyme | Km (mM) | Vmax (U mg−1) | kcat (s−1) | kcat/Km (s−1 mM−1) | t1/2 (min) | T1050 (°C) |
|---|---|---|---|---|---|---|
| WT | 2.83 | 1.95 | 1.23 | 0.43 | 3.70 | 38 |
| D249A | 1.67 | 8.01 | 5.17 | 3.10 | 7.88 | 44 |
| D249A/T247I | 2.32 | 5.28 | 3.41 | 1.47 | 21.66 | 57 |
To investigate the optimal temperature of wild-type LrLDH and the mutants, the catalytic activity towards PPA was determined at different temperatures in the range of 30–60 °C. As shown in Fig. 3c, an Asp to Ala substitution at position 249 resulted in a small increase of the optimum temperature from 40 °C (wild-type) to 44 °C. After combination with the T247I mutation, the double-mutant variant D249A/T247I exhibited a further increase of the optimal temperature to 52 °C.
Determination of kinetic parameters
The specific enzyme activity of wild-type LrLDH, the single mutant D249A and the double mutant D249A/T247I at different substrate concentrations (1–5 mM PPA) was determined at a fixed NADH concentration. The data were subjected to nonlinear fitting to the Michaelis–Menten equation (Fig. 4). As illustrated in Table 3, the maximum reaction velocities (Vmax) of wild-type LrLDH, single mutant D249A and double mutant D249A/T247I were 1.95, 8.01 and 5.28 U mg−1, respectively. The corresponding Michaelis constants (Km) were 2.83, 1.67 and 2.32 mM. The mutation of aspartate 249 to alanine resulted in a more than 3-fold improvement of Vmax and an approximately 41% decrease in Km. As a result, the D249A mutant exhibited 6.21-fold increase of catalytic efficiency (kcat/Km) toward PPA. The addition of the T247I mutation resulted in a significant reduction in kcat/Km, but the value (1.47 s−1 mM−1) was still much higher than that of wild-type LrLDH (0.43 s−1 mM−1). Nakano et al.,37 obtained a double mutant D-LDH T75L/A234S by site-directed mutagenesis. Using pyruvate as the substrate, the mutation give a 6.8-fold improvement in kcat/Km, which was better than our results. Nevertheless, we got robust enzymes with both enhanced thermostability and higher specific activity, which has a good application value in industry.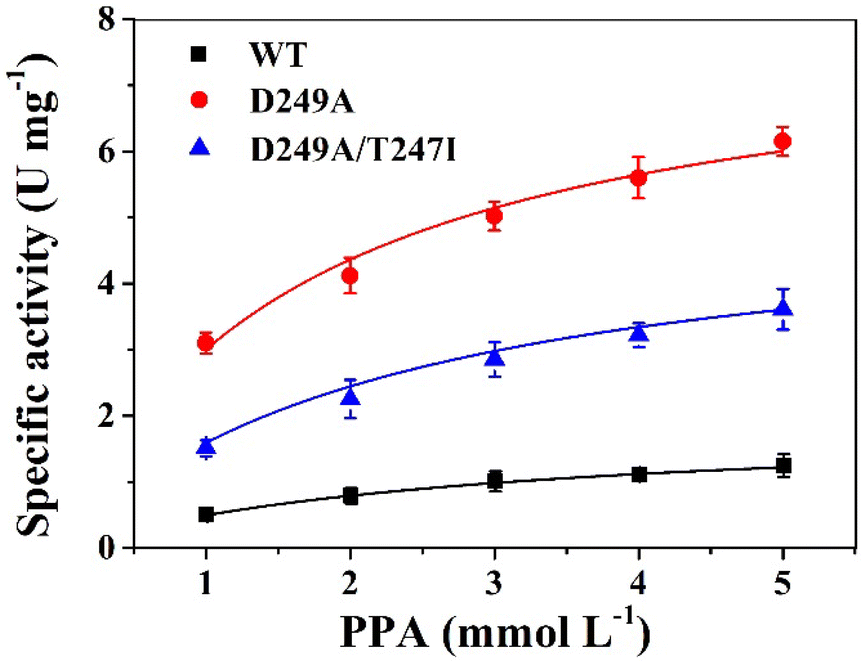 | ||
| Fig. 4 The kinetic curve of wild-type LrLDH (WT), mutant D249A and mutant D249A/T247I, respectively. | ||
Molecular dynamics simulations
To elucidate the mechanisms responsible for the enhanced thermostability of the best variant, molecular dynamics (MD) simulations were performed at 343 K (70 °C), in which root mean square deviation (RMSD) and radius of loop (Rg) values were recorded over 100 ns.RMSD represents the mean deviation of position between the protein structure and original conformation in 3D space at a specific time, which is an important indicator for evaluating the stability of enzymes. As shown in Fig. 5a, the RMSD value of wild-type LrLDH at 343 K was gradually increased and greatly fluctuated, indicating that the structure of the enzyme was unstable and could be easily inactivated at high temperature. By contrast, the RMSD values of the single mutant D249A and double mutant D249A/T247I were both basically stable at about 30 ns, and much lower than that of wild-type LrLDH. These two mutant enzymes therefore showed good structural stability at high temperature. Furthermore, compared with the single mutant enzyme D249A, the fluctuation of RMSD value of the double mutant D249A/T247I was smaller after 60 ns, indicating that the thermostability was further improved by combined mutation.
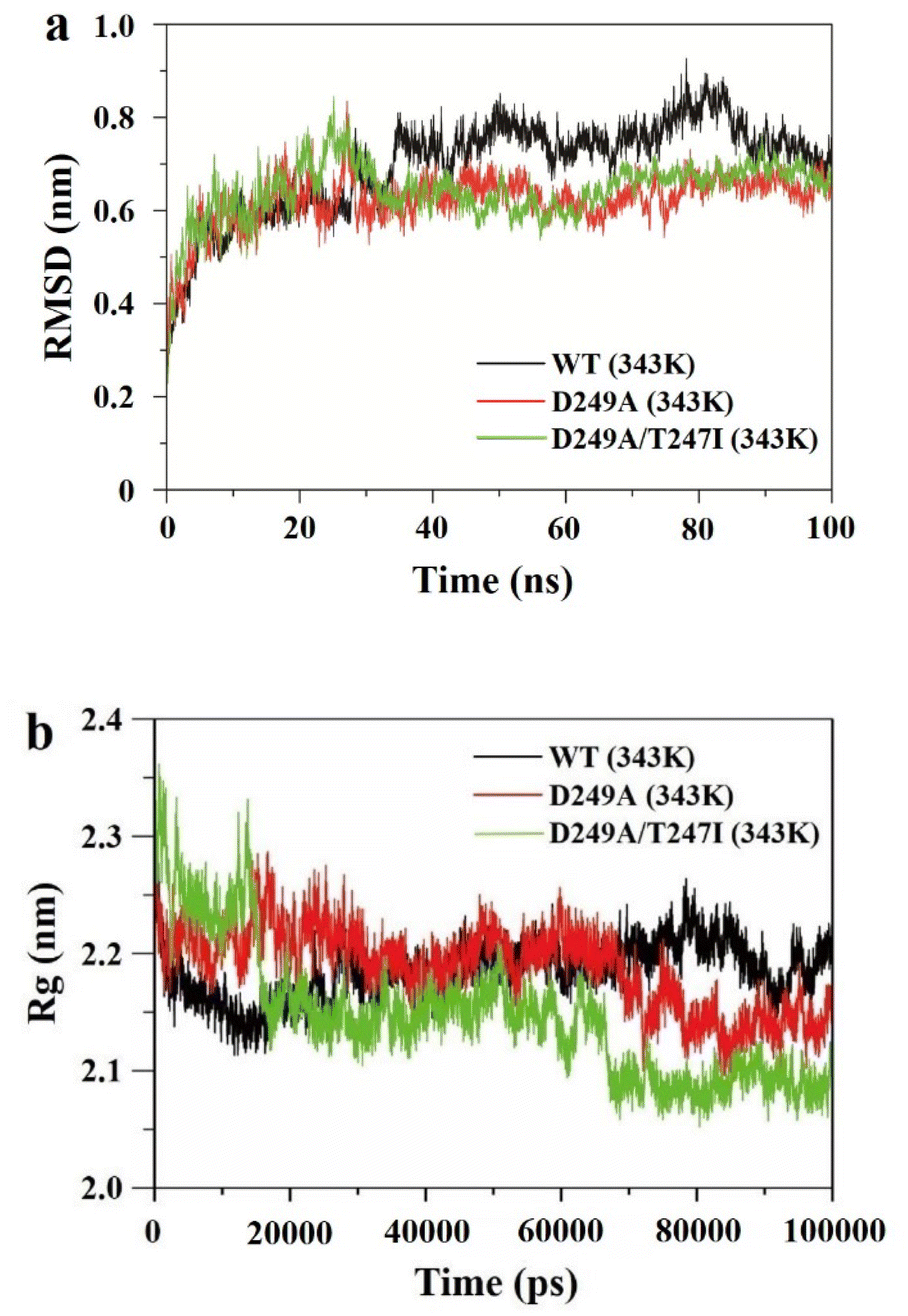 | ||
| Fig. 5 Molecular dynamics simulations of wild-type LrLDH (WT), mutant D249A and mutant D249A/T247I. (a) RMSD values at 343 K, (b) Rg values at 343 K. | ||
The Rg value reflects the compactness of the enzyme structure. As shown in Fig. 5b, The Rg value of wild-type LrLDH displayed an increasing trend over 100 ns, indicating that the enzyme expanded at high temperature due to poor thermostability. The Rg value of mutant D249A gradually decreased and stabilized at about 70 ns, indicating that the structure of the enzyme tends to contract. The average value was about 2.15, much lower than that of wild-type LrLDH, indicating better thermostability. In the double mutant D249A/T247I, the stabilization time of the Rg value shortened to 67 ns, and the average value was reduced to about 2.15 (Fig. 5b). These results indicated that the protein structure was packed tighter than in wild-type LrLDH and the single mutant D249A, which slowed down the enzyme's inactivation at high temperatures.
Consequently, the MD simulations suggested that both the single mutant D249A and double mutant D249A/T247I had increased thermostability, which may be due to the enhancement of structural rigidity of the enzyme molecule caused by the change of intramolecular forces in the peripheral structure.38 The calculated thermostability of mutant D249A/T247I was better than that of mutant D249A, which was consistent with the experimental results.
Elucidating the mechanism of the enhanced activity
To elucidate the molecular mechanism of the enhanced catalytic activity of the best variant, structural models of wild-type LrLDH, the single mutant D249A and the double mutant D249A/T247I were constructed based on the crystal structure of LDH from Lactobacillus helveticus (PDB ID: 2DLD). Subsequently, PPA and NADH were docked into the active site (Fig. 6). When the substrate approached, the active center of the enzyme underwent a conformational change and the substrate was bound mainly through hydrophobic interactions, after which the catalytic reaction commenced. As it’s stated in the literature of Wang et al.,39 the hydrophobic interactions between the active center and the substrate, as well as the conformation of the active center, especially in terms of steric hindrance, have significant influence on the catalytic activity of the enzyme. However, amino acid residues 247 and 249 are both far from the active center of the enzyme (Fig. 6a), and mutations at these two sites have no significant effect on steric hindrance or hydrophobicity at the active site. However, NADH is the hydride donor in the redox reactions catalyzed by LDHs, whereby the hydrogen on the fourth carbon atom of the nicotinamide ring is transferred to the carbonyl carbon of the substrate, whereby the carbonyl is reduced to a hydroxyl.27 Therefore, a closer distance between carbon 4 of the NADH nicotinamide ring and the carbonyl carbon of the substrate leads to higher hydride transfer efficiency, resulting in higher overall LDH catalytic activity. As shown in Fig. 6b–d, in the structures of wild-type LrLDH, single mutant D249A and double mutant D249A/T247I, the distances between carbon 4 of the NADH nicotinamide ring and the carbonyl carbon were 3.4, 2.5 and 2.9 Å, respectively. As a result, the catalytic activity of mutant D249A toward PPA was much higher than that of D249A/T247I and wild-type LrLDH. In addition, the Km values were in the order: wild-type LrLDH > D249A/T247I > D249A, which can also help explain the observed changes of catalytic activity.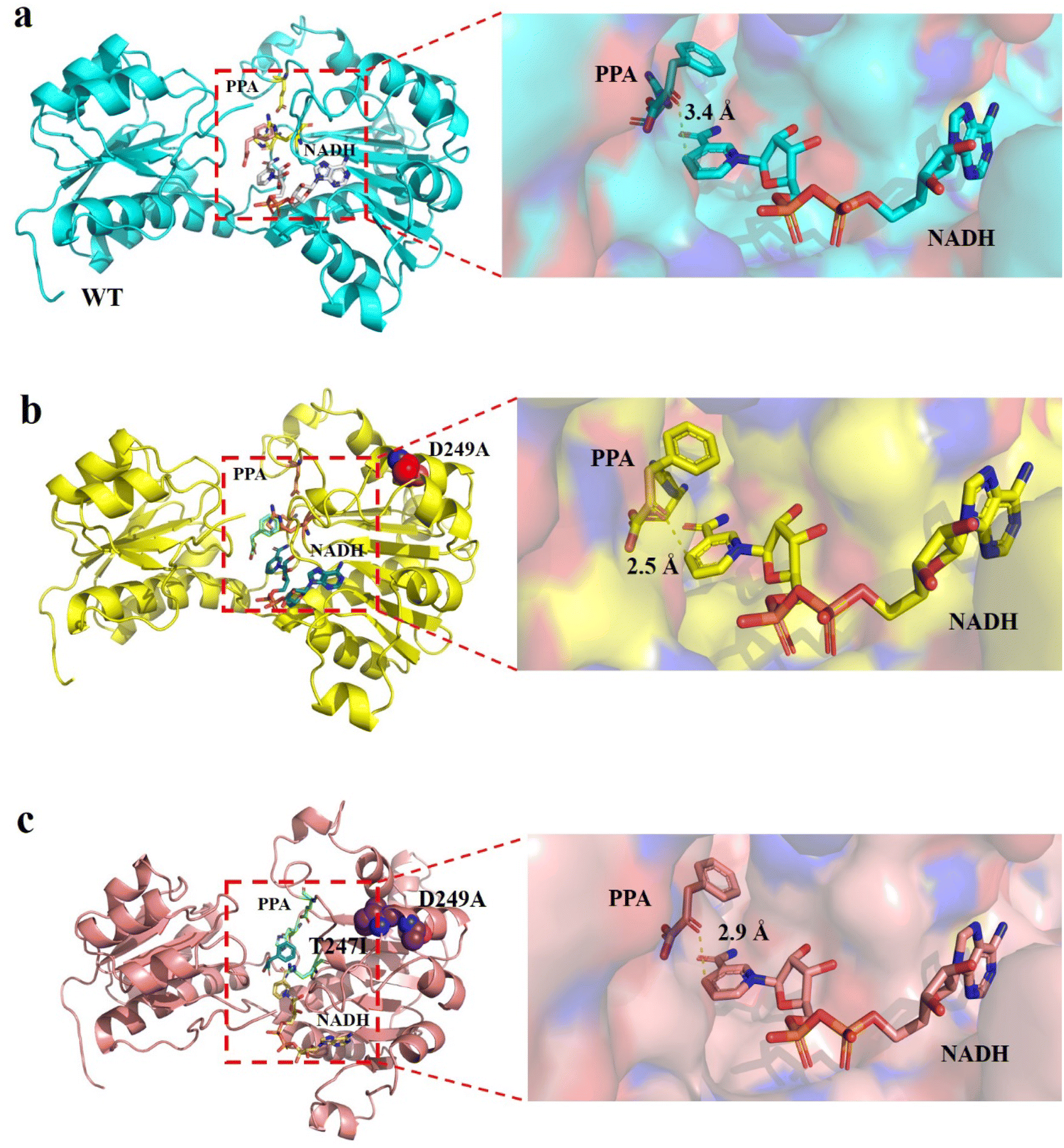 | ||
| Fig. 6 Structural analysis of WT (a), mutant D249A (b) and mutant D249A/T247I (c). PPA and NADH are shown as sticks, mutated residues are shown as spheres, and dotted lines depict the distances between 4-position carbon atom of NADH nicotinamide ring and the carbonyl carbon of PPA. | ||
Conclusions
In this study, the D-LDH LrLDH was engineered using computationally-assisted rational mutagenesis. The two mutants D249A and D249A/T247I showed significantly enhanced thermostability and catalytic activity compared with the wild-type enzyme. Furthermore, the double mutant D249A/T247I exhibited a favorable balance between the two catalytic properties. MD simulations indicated that both D249A and D249A/T247I enzyme molecules tend to contract at high temperatures rather than expanding and unfolding, leading to the enhancement of thermostability. According to the kinetic analysis and molecular docking, the mutants exhibited increased affinity for PPA, with a shortened distance between the C4 atom of the nicotinamide ring of NADH and the carbonyl carbon atom of PPA, which also helps explain the observed improvement of catalytic activity toward PPA.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Taizhou Scientific and Technological Project (2002gy11), the Natural Science Foundation for Young Scholars of Zhejiang Province (LQ19B060003) and the Key Research and Development Plan of Zhejiang Province (2020C02049).References
- H. Wu, C. E. Guang and W. L. Zhang, et al., Recent development of phenyllactic acid: physicochemical properties, biotechnological production strategies and applications, Crit. Rev. Biotechnol., 2021 DOI:10.1080/07388551.2021.2010645.
- S. Siedler, R. Baltiand and A. R. Neves, Bioprotective mechanisms of lactic acid bacteria against fungal spoilage of food, Curr. Opin. Biotechnol., 2019, 56, 138–146 CrossRef PubMed.
- F. Valerio, P. Lavermicocca and M. Pascale, et al., Production of phenyllactic acid by lactic acid bacteria: an approach to the selection of strains contributing to food quality and preservation, FEMS Microbiol. Lett., 2004, 233(2), 289–295 CrossRef PubMed.
- V. Dieuleveux, S. Lemarinierand and M. Gueguen, Antimicrobial spectrum and target site of D-3-phenyllactic acid, Int. J. Food Microbiol., 1998, 40(3), 177–183 CrossRef PubMed.
- Y. W. Ning, A. H. Yan and K. Yang, et al., Antibacterial activity of phenyllactic acid against Listeria monocytogenes and Escherichia coli by dual mechanisms, Food Chem., 2017, 228, 533–540 CrossRef PubMed.
- E. Debonne, A. Vermeulen and N. Bouboutiefski, et al., Modelling and validation of the antifungal activity of DL-3-phenyllactic acid and acetic acid on bread spoilage moulds, Food Microbiol., 2020, 88, 103407 CrossRef CAS PubMed.
- O. Cortes-Zavaleta, A. Lopez-Malo and A. Hernandez-Mendoza, et al., Antifungal activity of lactobacilli and its relationship with 3-phenyllactic acid production, Int. J. Food Microbiol., 2014, 173, 30–35 CAS.
- F. Valerio, P. Lavermicocca and M. Pascale, et al., Production of phenyllactic acid by lactic acid bacteria: an approach to the selection of strains contributing to food quality and preservation, FEMS Microbiol. Lett., 2004, 233(2), 289–295 CrossRef CAS.
- W. M. Mu, S. H. Yu and L. J. Zhu, et al., Recent research on 3-phenyllactic acid, a broad-spectrum antimicrobial compound, Appl. Microbiol. Biotechnol., 2012, 95(5), 1155–1163 Search PubMed.
- F. J. Urban and B. S. Moore, Synthesis of optically-active 2-benzyldihydrobenzopyrans for the hypoglycemic agent englitazone, J. Heterocycl. Chem., 1992, 29(2), 431–438 CrossRef CAS.
- T. Z. Xiong, P. Jia and J. Jiang, et al., One-pot, three-step cascade synthesis of D-danshensu using engineered Escherichia coli whole cells, J. Biotechnol., 2019, 300, 48–54 CrossRef CAS PubMed.
- W. Weckwerth, K. Miyamoto and K. Iinuma, et al., Biosynthesis of PF1022A and related cyclooctadepsipeptides, J. Biol. Chem., 2000, 275(23), 17909–17915 CrossRef CAS PubMed.
- H. Kawaguchi, C. Ogino and A. Kondo, Microbial conversion of biomass into bio-based polymers, Bioresour. Technol., 2017, 245, 1664–1673 CrossRef CAS PubMed.
- R. V. Rajanikar, B. H. Nataraj and H. Naithani, et al., Phenyllactic acid: a green compound for food biopreservation, Food Control, 2021, 128, 108184 CrossRef.
- H. Kawaguchi, H. Miyagawa and S. Nakamura-Tsuruta, et al., Enhanced phenyllactic acid production in Escherichia coli via oxygen limitation and shikimate pathway gene expression, Biotechnol. J., 2019, 14(6), 1800478 CrossRef PubMed.
- J. J. Xu, L. J. Fu and K. L. Si, et al., 3-phenyllactic acid production by free-whole-cells of Lactobacillus crustorum in batch and continuous fermentation systems, J. Appl. Microbiol., 2020, 129(2), 335–344 CrossRef PubMed.
- Z. Wu, S. Xu and Y. Yun, et al., Effect of 3-phenyllactic acid and 3-phenyllactic acid-producing lactic acid bacteria on the characteristics of alfalfa silage, Agriculture, 2019, 10(1), 10 CrossRef.
- X. H. Zhou, J. Zhou and F. X. Xin, et al., Heterologous expression of a novel D-lactate dehydrogenase from Lactobacillus sp ZX1 and its application for D-phenyllactic acid production, Int. J. Biol. Macromol., 2018, 119, 1171–1178 CrossRef CAS PubMed.
- Z. Qin, D. Wang and R. S. Luo, et al., Using unnatural protein fusions to engineer a coenzyme self-sufficiency system for D-phenyllactic acid biosynthesis in Escherichia coli, Frontiers in Bioengineering and Biotechnology, 2021, 9, 795885 CrossRef PubMed.
- X. Luo, Y. Y. Zhang and F. W. Yin, et al., Enzymological characterization of a novel D-lactate dehydrogenase from Lactobacillus rossiae and its application in D-phenyllactic acid synthesis, 3 Biotech, 2020, 10(3), 101 CrossRef PubMed.
- F. Hasan, A. A. Shahand and A. Hameed, Industrial applications of microbial lipases, Enzyme Microb. Technol., 2006, 39(2), 235–251 CrossRef CAS.
- H. P. Modarres, M. R. Mofrad and A. Sanati-Nezhad, Protein thermostability engineering, RSC Adv., 2016, 6(116), 115252–115270 RSC.
- J. H. Bi, S. H. Chen and X. H. Zhao, et al., Computation-aided engineering of starch-debranching pullulanase from Bacillus thermoleovorans for enhanced thermostability, Appl. Microbiol. Biotechnol., 2020, 104(17), 7551–7562 CrossRef CAS PubMed.
- J. Y. Zhao, J. Chen and H. Y. Wang, et al., Enhanced thermostability of D-psicose 3-epimerase from Clostridium bolteae through rational design and engineering of new disulfide bridges, Int. J. Mol. Sci., 2021, 22(18), 10007 CrossRef CAS PubMed.
- A. S. Dotsenko, A. M. Rozhkova and I. N. Zorov, et al., Protein surface engineering of endoglucanase Penicillium verruculosum for improvement in thermostability and stability in the presence of 1-butyl-3-methylimidazolium chloride ionic liquid, Bioresour. Technol., 2020, 296, 122370 CrossRef CAS PubMed.
- M. Ladero, G. Ruiz and B. C. C. Pessela, et al., Thermal and pH inactivation of an immobilized thermostable β-galactosidase from Thermus sp. strain T2: comparison to the free enzyme, Biochem. Eng. J., 2006, 31(1), 14–24 CrossRef CAS.
- J. Deng, Z. Q. Yao and K. L. Chen, et al., Towards the computational design and engineering of enzyme enantioselectivity: a case study by a carbonyl reductase from Gluconobacter oxydans, J. Biotechnol., 2016, 217, 31–40 CrossRef CAS.
- L. Sumbalova, J. Stourac and T. Martinek, et al., HotSpot Wizard 3.0: web server for automated design of mutations and smart libraries based on sequence input information, Nucleic Acids Res., 2018, 46, 356–362 CrossRef PubMed.
- X. Luo, Y. J. Wang and W. Shen, et al., Activity improvement of a Kluyveromyces lactis aldo-keto reductase KlAKR via rational design, J. Biotechnol., 2016, 224, 20–26 CrossRef CAS PubMed.
- X. M. Gong, Z. Qin and F. L. Li, et al., Development of an engineered ketoreductase with simultaneously improved thermostability and activity for making a bulky atorvastatin precursor, ACS Catal., 2019, 9(1), 147–153 CrossRef CAS.
- Y. Chen, D. Ming and L. Zhu, et al., Tailoring the Tag/Catcher System by integrating covalent bonds and noncovalent interactions for highly efficient protein self-assembly, Biomacromolecules, 2022, 23, 3936–3947 CrossRef CAS PubMed.
- L. Kiermund, A. Riederer and A. Hunger, et al., Protein engineering of a bacterial N-acyl-D-glucosamine 2-epimerase for improved stability under process conditions, Enzyme Microb. Technol., 2016, 87–88, 70–78 CrossRef.
- X. Y. Wang, H. Y. Luo and W. N. Yu, et al., A thermostable Gloeophyllum trabeum xylanase with potential for the brewing industry, Food Chem., 2016, 199, 516–523 CrossRef PubMed.
- F. Xue, Z. Q. Liu and N. W. Wan, et al., Engineering the epoxide hydrolase from Agromyces mediolanus for enhanced enantioselectivity and activity in the kinetic resolution of racemic epichlorohydrin, RSC Adv., 2015, 5(40), 31525–31532 RSC.
- S. F. Li, J. Y. Xie and S. Qiu, et al., Semirational engineering of an aldo-keto reductase KmAKR for overcoming trade-offs between catalytic activity and thermostability, Biotechnol. Bioeng., 2021, 118(11), 4441–4452 CrossRef PubMed.
- K. S. Siddiqui, D. M. Parkin and P. M. G. Curmi, et al., A novel approach for enhancing the catalytic efficiency of a protease at low temperature: reduction in substrate inhibition by chemical modification, Biotechnol. Bioeng., 2009, 103(4), 676–686 CrossRef PubMed.
- K. Nakano, S. Sawada and R. Yamada, et al., Enhancement of the catalytic activity of D-lactate dehydrogenase from Sporolactobacillus laevolacticus by site-directed mutagenesis, Biochem. Eng. J., 2018, 133, 214–218 CrossRef CAS.
- Z. P. Zhou and X. Wang, Rational design and structure-based engineering of alkaline pectate lyase from Paenibacillus sp. 0602 to improve thermostability, BMC Biotechnol., 2021, 21, 32 CrossRef CAS PubMed.
- F. H. Wang, M. L. Zhu and Z. Song, et al., Reshaping the binding pocket of lysine hydroxylase for enhanced activity, ACS Catal., 2020, 10(23), 13946–13956 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |