
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning dibenzosilole core based, A2–π–A1–π–D–π–A1–π–A2 type donor molecules for promising photovoltaic parameters in organic photovoltaic cells
Saima Rania,
Nabil Al-Zaqri *b,
Javed Iqbal*af,
Sahar Javaid Akrama,
Ahmed Boshaalacd,
Rana Farhat Mehmoode,
Muhammad Umar Saeeda,
Ehsan Ullah Rashida and
Rasheed Ahmad Khera*a
*b,
Javed Iqbal*af,
Sahar Javaid Akrama,
Ahmed Boshaalacd,
Rana Farhat Mehmoode,
Muhammad Umar Saeeda,
Ehsan Ullah Rashida and
Rasheed Ahmad Khera*a
aDepartment of Chemistry, University of Agriculture, Faisalabad 38000, Pakistan. E-mail: rasheedahmadkhera@yahoo.com; rasheed.ahmad.khera@uaf.edu.pk; javedkhattak79@gmail.com; Javed.iqbal@uaf.edu.pk
bDepartment of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia. E-mail: nalzaqri@ksu.edu.sa
cResearch Centre, Manchester Salt & Catalysis, Unit C, 88-90 Chorlton Rd, M15 4AN Manchester, UK
dLibyan Authority for Scientific Research, P. O. Box 80045, Tripoli, Libya
eDepartment of Chemistry, Division of Science and Technology, University of Education, Township, Lahore 54770, Pakistan
fDepartment of Chemistry, College of Science, University of Bahrain, Zallaq, Bahrain
First published on 13th October 2022
Abstract
In this research work, four new molecules from the π–A–π–D–π–A–π type reference molecule “DBS-2PP”, were designed for their potential application in organic solar cells by adding peripheral A2 acceptors to the reference. Under density functional theory, a comprehensive theoretical investigation was conducted to examine the structural geometries, along with the optical and photovoltaic parameters; comprising frontier molecular orbitals, density of states, light-harvesting effectiveness, excitation, binding, and reorganizational energies, molar absorption coefficient, dipole moment, as well as transition density matrix of all the molecules under study. In addition, some photo-voltaic characteristics (open circuit photo-voltage and fill factor) were also studied for these molecules. Although all the developed compounds (D1–D4) surpassed the reference molecule in the attributes mentioned above, D4 proved to be the best. D4 possessed the narrowest band-gap, as well as the highest absorption maxima and dipole moment of all the molecules in both the evaluated phases. Moreover, with PC61BM as the acceptor, D4 showed the maximum VOC and FF values. Furthermore, while D3 had the greatest hole mobility owing to its lowest value of hole reorganization energy, D4 exhibited the maximum electron mobility due to its lowermost value of electron reorganization energy. Overall, all the chromophores proposed in this study showed outstanding structural, optical, and photovoltaic features. Considering this, organic solar cell fabrication can be improved by using these newly derived donors at the donor–acceptor interfaces.
1. Introduction
Despite generating more than 80% of the world's energy, non-renewable resources produce large amounts of CO2 and have antagonistic impacts on the environment.1 In addition, these resources are rapidly depleting over time, so the world is turning towards clean and renewable sources of energy.2 In this regard, solar energy seems to be quite abundant, cheap, carbon-free, and safe for the environment. Furthermore, solar panels are easy to install and can function in remote areas where it is very challenging to provide electricity using other approaches. With a lifespan of more than 20 years, they demand little maintenance and require no additional land space, because of their effective installation on the roofs of homes, schools, or any other building. Consequently, they can be a good substitute for conventional electricity sources, which are overpriced and difficult to install in distant areas.3,4 The fundamental component of a solar panel is the solar cell, which works on the idea of the photovoltaic effect. So, when photons with specific energy are absorbed by a semiconducting material, holes–electrons form and flow toward opposing electrodes to produce an electric current.5–7 In the solar cells of the early stages, these semiconducting materials were limited to inorganic silicon ones, which despite giving good efficiencies, had constrained applications owing to their inflexible design, low competence as well as high fabricating cost.8,9 Thus, researchers have focused their efforts on developing solar materials that are more efficient and reliable than silicon ones.10Recently organic photovoltaics (OPVs) have gained considerable interest as a possible alternative to silicon-based materials. These OPVs are considered more attractive because of their varied and tunable energy levels, adjustable morphology, as well as excellent purity.11 The active layer of OPVs, responsible for the current generation by light absorption, comprises donor and acceptor materials either separated or in a bulk-heterojunction (BHJ). Between these two, the bulk-heterojunction (active layer made up by blending donor and acceptor materials) proves to be exceptional in terms of boosting the efficiencies of OPVs. Additionally, due to its tunable organic materials, the efficiencies of BHJ-based OPVs can be further enhanced by experimenting with different blends of donors and acceptors.
Between the small molecule and polymer-based donor materials, the former are preferred for bulk-heterojunctions in the fabrication of OPVs. This could be because they have a well-defined structure, are easier to purify, and also exhibit improved batch-to-batch reproducibility.12,13 Effective device manufacturing, as well as the utilization of novel small molecule donor (SMD) materials, is credited to have achieved PCEs of up to 11 percent when fullerene acceptor molecules such as PC61BM and PC71BM are blended with them.14 In addition, a variety of remodeling techniques, including end-capped engineering, central core remodeling, and pi-bridge placement or substitution, can be used on SMDs to improve their various photovoltaic properties. End-capped engineering, when compared to other approaches, has several advantages. For instance, by only adding small end-capped acceptors to a molecule, the consequent morphology of the whole blend can be impacted, which as a result affects the light absorption ranges, energy levels of frontier molecular orbitals, pi-bond conjugation, and the mobilities of charge carriers, all of which have a major influence on the overall performance of OPVs. So in this research work, with the aforementioned capabilities of end-capped modification in mind, DBS-2DPP is taken as the reference molecule (R) and different side-chained acceptors are attached to its peripheral thiophene moieties.
DBS-2DPP is a dibenzosilole (DBS) core-based acceptor molecule with diketopyrrolopyrrole (DPP) arms attached to the DBS core via thiophene bridges, which are further attached to a second set of thiophene rings on each peripheral end, as shown in Fig. 1. The DBS core contains a silole moiety fused attached to two six-membered rings, and demonstrates considerable planarity and proficient charge transfer properties, whereas the DPP fragments are known to exhibit remarkable electron withdrawing properties. Collectively, the DBS-2DPP molecule shows good thermal stability and open circuit photo-voltage, voltage, along with an optical band-gap of 1.83 eV in thin film. But, when blended with a P3HT donor molecule to investigate its capabilities in OPVs, this molecule showed a photo-conversion efficiency (PCE) of only 2.05%, which might be due to its lower charge mobility, larger band-gap, and smaller absorption range.15–17 As a result, by modifying its morphology through the addition of strongly electron-withdrawing acceptor groups at the thiophene fringes of this molecule, four new molecules (D1–D4) have been reported, which due to their better opto-electronic properties and smaller band-gaps prove themselves to be better than the DBS-2PP (R) molecule, and thus could exhibit increased PCE if taken as donors and blended with some prominent electron acceptor molecule, such as PC61BM.
 | ||
| Fig. 1 (a) ChemDraw sketches of R and D1–D4 molecules, (b) molecular map of the designing strategy. | ||
Precisely, in the formulation of the newly reported molecules, the hydrogen of the carbon closest to the sulphur of the terminal thiophene rings of the R molecule has been substituted with a strong electron accepting group. The reason behind the selection of this position is its reported strong electron transferring capabilities due to its seemingly planar orientation and low steric hindrance with the rest of the molecule.18 In D1, the newly attached acceptor moiety was; 2-(2-ethylidene-5,6-difluoro-3-oxo-indan-1-ylidene)-malononitrile, in D2 it was; 5-ethylidene-3-methyl-2-thioxo-thiazolidin-4-one, in D3; 2-(1-chloro-5-ethylidene-6-oxo-5,6-dihydro-cyclopenta [c]thiophen-4-ylidene)-malononitrile, while in D4 this moiety was 6-cyano-1-dicyanomethylene-2-ethylidene-3-oxo-indan-5-carboxylic acid methyl ester. Chemdraw sketches of reference R and D1–D4 chromophores are given in Fig. 1(a).
2. Computational details
To explore various optoelectronic properties of the studied structures, a quantum mechanical technique based on density functional theory (DFT) was employed with the help of Gaussian 09 software.19 In order to avoid excessive computational cost, the long alkyl side chains of DBS-2DPP (taken as R in this work) were replaced with methyl groups based on their general inconsequential influence on the optical and electronic attributes of the overall molecule.20 GaussView 6.0 (ref. 21) was employed to draw the structures of R and D1–D4 molecules and to visualize various outputs as well.Firstly the R molecule was subjected to four different exchange-correlation functionals of DFT, i.e., B3LYP,22 CAM-B3LYP,23 MPW1PW91,24 and ωB97XD25 at 6-31G (d, p) basis set to attain any one functional with the best imitation of the experimental work.26 So, the maximum absorption wavelengths (λmax) of all the afore-stated functionals were compared to the experimental value of R. And for this reason, time dependent DFT was exercised to obtain the UV-Visible profile of R under solvent phase with these functionals. It should be noted that in the cited literature the absorption spectra of R were calculated using dichloromethane solvent, thus the very same environment of solvent was applied to the molecules of this study using the integral equation formalism variant of the polarizable continuum model (IEFPCM).27 As shown in the graph of absorption attained with the aid of Origin 6.0,28 the maximum absorption wavelength (592 nm) produced by the B3LYP functional is consistent with the published value (596 nm) of R (Fig. 2), while that of the other three functionals were too far off to be of consequence, as CAMB3LYP showed a λmax of 4845 nm, MPW1PW91 565 nm, and ωB97XD had its value of 471 nm. Subsequently, B3LYP functional with 6-31G (d, p) basis set was selected for further calculations of R and devised compounds.
 | ||
| Fig. 2 Absorption profile of R (DBS-2DPP) calculated by using four hybrid functionals in dichloromethane solvent. | ||
After the selection of the desired functional, the molecules were optimized at their ground state to understand their geometry. For this purpose, the bond length and dihedral angle of the carbon–carbon bond between the thiophene ring and the newly added acceptor group at their site of attachment were studied. To validate the geometry of the molecules, span of deviation from plane (SDP) and molecule planarity parameter (MPP) were also explored. These two parameters, in addition to, the transition density matrix (TDM) of all the examined molecules were created using the Multiwfn program,29,30 while VMD 1.9.3 (ref. 31) was utilized to visualize MPP and SDP. Furthermore, the frontier molecular orbitals (FMOs) were also generated at the ground state to visualize the wave-function of the highest occupied (HOMO) and the lowest unoccupied (LUMO) molecular orbitals, and from these FMOs, the band-gap was also calculated. In addition, the graphs of density of states (DOS) were plotted with the help of PyMOlyze 1.1 (ref. 32) to investigate the electronic densities at various energy levels and also to authenticate the FMOs' findings.33
At the excited state, the excitation energies, absorption maxima, dipole moment, oscillator strength, transition density matrix, binding energies, etc. were investigated to attain the optoelectronic attributes of the molecules after absorption of light. Similarly, the fill factor, light harvesting efficiencies, and open circuit photo-voltage were computed to scrutinize the photovoltaic properties of the molecules and to give a direction about their photovoltaic efficiencies.
Lastly, the internal reorganization energies of the explored molecules were also computed with the help of their optimized neutral, anionic, and cationic states. Thus, the following eqn (1) and (2), taken from Marcus theory, were used to compute the two components of internal reorganization energies, i.e., for electrons and holes, respectively.34
| λe = [E0− − E−] + [E0− − E0] | (1) |
| λh = [E0+ − E+] + [E0+ − E0] | (2) |
Optimized anionic and cationic geometries were used to compute molecular neutral energies E0− and E0+ respectively. E0+ and E0− are the single point energies of cations and anions at ground level, estimated by employing optimized neutral molecule geometries. E− along with E+ are the energies of anion and cation calculated using neutral molecule's anion and cation structural optimization. Finally, E0 demonstrates the neutral molecule's ground state energy.
3. Results and discussion
3.1. Ground state geometries and planarity measurements
Optimized structures of R and modified chromophores (D1–D4) at their ground state, computed through B3LYP functional, are presented in Fig. 3. From these optimized geometries, the planarity in the molecules was estimated to be quite substantial, which could be attributed to the two smaller thiophene entities present between the large acceptor and donor groups of the molecules. These thiophene rings, by acting as bridge between the acceptor and donor entities, reduced the overall steric hindrance within the molecule and increased the planarity, which as result, enhanced the conjugation in the molecules under scrutiny. To quantitatively understand the planarity and conjugation of the newly added acceptors with respect to the rest of the molecule, the dihedral angles and bond length values were obtained at their point of attachment with the thiophene ring of the molecule.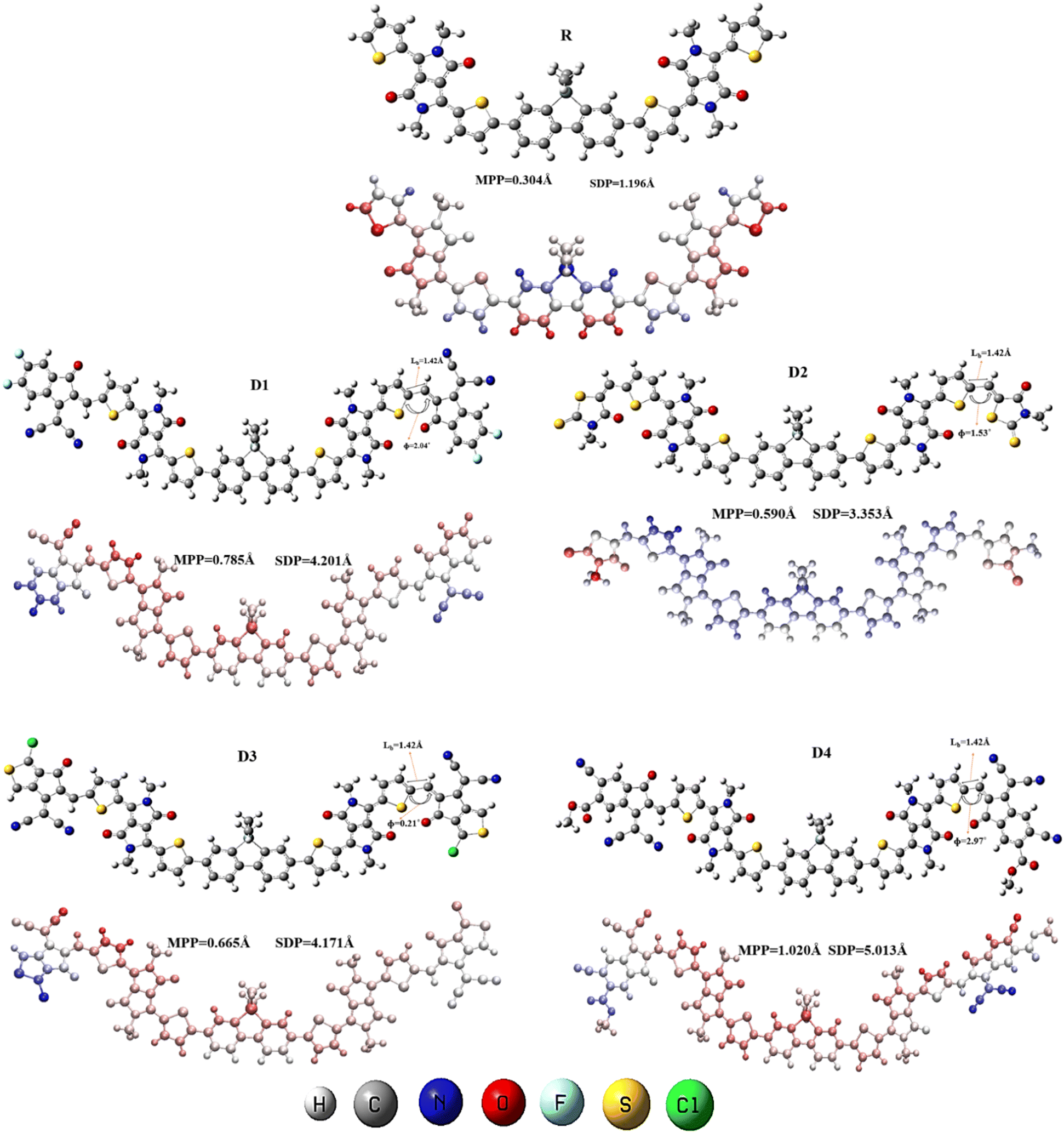 | ||
| Fig. 3 Optimized structures (along with the studied bond parameters of the designed molecules), the molecular planarity parameter (MPP), the span of deviation from the plane (SDP) of R and D1–D4. | ||
It was discovered that the studied bond length of all the newly reported compounds fell at 1.42 Å, indicating significant conjugation and charge transfer between the molecule and the newly added acceptors. This conjugation was evaluated with the fact that the range of a carbon–carbon bond to have conjugation in it is between the single bond (C–C; 1.54 Å) and double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C; 1.34 Å) lengths. The closer a C–C bond is to the double bond length, the greater could be its conjugation. Furthermore, in D1–D4, the measured dihedral angles ranged from 0.21° to 2.97°,35 as shown in Table 1. These low values of dihedral angles illustrated the planarity of the molecule at this bond.
C; 1.34 Å) lengths. The closer a C–C bond is to the double bond length, the greater could be its conjugation. Furthermore, in D1–D4, the measured dihedral angles ranged from 0.21° to 2.97°,35 as shown in Table 1. These low values of dihedral angles illustrated the planarity of the molecule at this bond.
| Molecules | Bond length (Lb) (Å) | Dihedral angle (Φ) (θ°) | Molecular planarity parameter (MPP) (Å) | Span of deviation from plane (SDP) (Å) |
|---|---|---|---|---|
| R | — | — | 0.304 | 1.196 |
| D1 | 1.42 | 2.04 | 0.785 | 4.201 |
| D2 | 1.42 | 1.53 | 0.590 | 3.353 |
| D3 | 1.42 | 0.21 | 0.665 | 4.171 |
| D4 | 1.42 | 2.97 | 1.020 | 5.013 |
But to evaluate the overall planarity of these compounds and to compare it to that of the reference molecule, the molecular planarity parameter (MPP) and the span of deviation from the plane (SDP) values were computed (Fig. 3). Here, it is known that the structure will be more planer if it has smaller values of both MPP and SDP. So, the lower MPP (0.304 Å) value indicates that the reference (R), i.e., the central region, except for the added acceptors, has quite a planer structure, which is also indicated by its SDP (1.196 Å) results. Whereas, amongst the designed compounds the maximum deviation from planarity has been observed by D4, which may be due to the steric hindrance caused by its bulky end-groups, which rendered the molecule slightly non-planer as compared to other structures. Correspondingly, D1, D2, and D3 also showed slight deviation from the plane, again attributed to their added end-groups. In Fig. 3, the deviations of these structures from the plane are shown in pictorial forms. Where the red color shows those areas, which are below the plane of paper, while the blue color areas are those which are above the plane of paper. This coloring scheme gives a very good idea about the different parts of these molecules, which deviate from the fitting plane. In R molecule, it is observable that there are a few blue and red parts at the DBS core and a few of them are also present at DPP acceptors, but the overall structure is quite planer.
But, in D1 molecule, there are very light red and blue hues on the central and right part of this molecule, which indicate planarity, but a little deviation occurs at the end-group on the left side. Similar deviation from the plane has been observed in the case of other the three derived molecules, in which the whole structure is quite planer, but deviations are observed in right end group of D2 and D4, as well as left end group of D3, resulting in the slight deviation of their structures from the plane.
Though the values of these two parameters, i.e., MPP and SDP of the four modified molecules are comparatively higher than the reference molecule, still their rather low values certify their planarity. Also, it should be noted that this slight deviation of the planarity of the reported molecules, could be supportive in case of their charge density dispersal. As molecules with high values of planarity tend to have a rather dispersed charge density, like the FMOs of the reference molecule (Fig. 6). While the designed molecules due to their deviation from planarity have retained the charge densities of their FMOs in such a way that it could be helpful in charge transfer from the HOMO towards the LUMO.
3.2. Optical parameters
The optical properties of studied molecular structures were investigated in order to evaluate the light harvesting efficiency (LHE) and performance of the organic solar systems of the corresponding molecule.36,37 A combination of the TD-DFT and the selected functional was used to determine the spectral and photo-physical properties of the newly constructed structures (D1–D4) along with R (reference molecule) in both the solvent (dichloromethane-DCM) and gaseous medium.38 The values of the computationally determined optical parameters, i.e., the absorption range, absorption maxima (λmax), excitation energy, oscillating power, and transition coefficient for each of the four designed molecules D1–D4 and R (reference), are provided in Tables 2 and 3.| Molecules | Calculated λmax (nm) | Experimental λmax (nm) | Ex (eV) | fos | LHE | Molecular states | Transition character |
|---|---|---|---|---|---|---|---|
| R | 592 | 596 | 1.98 | 1.23 | 0.940 | S0 → S1 | 70% |
| D1 | 840 | — | 1.42 | 1.97 | 0.989 | S0 → S1 | 67% |
| D2 | 744 | — | 1.62 | 1.95 | 0.988 | S0 → S1 | 67% |
| D3 | 858 | — | 1.40 | 2.02 | 0.990 | S0 → S1 | 66% |
| D4 | 876 | — | 1.36 | 1.84 | 0.985 | S0 → S1 | 68% |
| Molecules | Calculated λmax (nm) | Ex (eV) | fos | LHE | Molecular states | Transition character |
|---|---|---|---|---|---|---|
| R | 578 | 2.02 | 1.00 | 0.901 | S0 → S1 | 70% |
| D1 | 781 | 1.51 | 1.63 | 0.976 | S0 → S1 | 68% |
| D2 | 702 | 1.69 | 1.60 | 0.974 | S0 → S1 | 68% |
| D3 | 794 | 1.48 | 1.65 | 0.977 | S0 → S1 | 68% |
| D4 | 802 | 1.47 | 1.62 | 0.976 | S0 → S1 | 68% |
Interestingly, all of the newly formulated compounds exhibited a bathochromic shift in their absorbance maxima, supporting our claim that the added peripheral acceptor groups have increased the conjugation and, by association, the electron transfer within the new molecules. In addition, the presence of the DCM solvent has enhanced the absorption profile of the molecules formulated in this study. Consequently, the absorption spectra (Fig. 4) indicates that dichloromethane shifted the absorption peaks of the electromagnetic spectrum in the higher visible region for D2 and near infrared regions for other newly created molecules. Precisely in the studied solvent, R has a maximum wavelength of 592 nm, while the maximum absorption wavelengths of the newly reported compounds (D1–D4) are 840, 744, 858, and 876 nm, respectively. Similarly, in the gas medium, the order of absorption maxima followed by the studied molecules is the same as of DCM solvent; D4 > D3 > D1 > D2 > R. Furthermore, the absorption range of R in both the phases is narrower as compared to the broad ranges of all the newly formulated D1–D4. These results imply that the newly created compounds under research have a better absorption profile as compared to R molecule. Individually, because of the presence of the unsaturated acetyle and cyano groups in the acceptor unit in D4, it has the maximum absorption value of 876 nm and 802 nm in the solvent and gaseous medium, respectively. This implied that D4 might also have the lowest band-gap energy among all, solidifying its candidature in efficient photovoltaic devices. The reason behind this assumption is that generally, molecules with high absorption peaks tend to have lower band-gap, for easier transfer of electrons between the FMOs upon absorption of photons.39
 | ||
| Fig. 4 UV-Vis spectra of scrutinized D1–D4 along with R in the (1) solvent phase (dichloromethane) and (2) in gaseous medium. | ||
One of the properties related to photo-absorption is excitation, which occurs when a low-energy state electron is stimulated electrically to a high-energy state. Effective electron transport, along with greater spectral absorption at a longer wavelength may be accomplished, as a consequence of a smaller energy difference between the two states in which the excitation is to occur.40 This energy difference could be attributed to the energy needed for excitation, i.e., excitation energy, a significant optical feature.41 According to the results tabulated in Tables 2 and 3 for the first excitation energy values, i.e., between the FMOs, all of the newly developed molecules had lower excitation energies than reference, suggesting that the electron transport process between their molecular orbitals (HOMO and LUMO) might be more accelerated than reference. Actually, the effective electron-withdrawing newly added acceptors of the designed molecules increase the possibility of electronic excitation and thus lowers the required excitation energy.
The oscillation strength is generally used to compute the statistical probability of an electronic transition from the initial ground level to an excited level (first in our case) after light absorption by the molecule. It has been shown in literature that strong acceptor moieties are responsible for the higher oscillation strength of any molecule.42 The oscillator strength values in Tables 2 and 3 of optical properties, illustrates the higher oscillation strengths of the derived molecules as opposed to the reference. Here, the studied molecules followed the increasing order of R < D4 < D2 < D1 < D3 in the solvent medium and of R < D2 < D4 < D1 < D3 in the gas medium. The highest oscillation strength of D3 in both mediums demonstrates its high aptitude for utilization in organic photovoltaics. In addition, to determine the probability of electronic transition of an electron between orbitals of study, oscillation strength plays a momentous role in calculating the light harvesting efficiencies of a molecule, the values for which are also tabulated in Tables 2 and 3
3.3. Light harvesting efficiency (LHE)
LHE of solar materials is used to assess the optical qualities, JSC, and efficacy of the cell, as it estimates the production of the charges produced after the absorption of incoming photons.43 It is directly dependent on oscillation strength and is determined by given eqn (3).| LHE = 1 − 10−f | (3) |
Among all the newly tailored structures, D3 had the highest LHE because of the highest overlapping of pi-orbitals in combination with the lowest dihedral angle between its added peripheral and core of the molecule, which permits a more efficient electronic transition.38 It is clear from the values of light harvesting efficiency that the newly added acceptors at the peripheries of the molecules have directed the transfer of charges towards themselves and enhanced the conjugation, which benefited the molecule in harvesting light effectively and transporting the thus generated charges within itself. Thus, as opposed to the reference molecule, all newly created compounds could be taken into consideration as conspicuous small molecules donors in organic photo-active layers of photovoltaic cells. Till now, amongst all the newly developed molecular structures, D4 with its lowest excitation energy and highest absorption maxima, along with D3 with its second highest absorption maxima, and highest oscillator strength, and by association, highest LHE seems to be the most effective organic chromophores for their exploitation in OPVs.
3.4. Quantum mechanical descriptors [HOMO and LUMO]
FMOs (Frontier molecular orbitals) of a molecule can be described as; LUMO-the lowest unoccupied molecular orbital and HOMO-the highest occupied molecular orbital. When characterizing the optical characteristics of a molecule, FMOs' quantum mechanical analysis reveals the presence of HOMO–LUMO's electronic distribution patterns in the molecular electronic structures.44,45 The electronic distribution of FMOs has been utilized to anticipate the intramolecular charge transfer (ICT), during which the electron density moves from the ground state's valence band (HOMO) to the excited state's conduction band (LUMO). From Fig. 6, it is distinctive that in the reference molecule, electron density is uniformly distributed throughout the structure in both the studied FMOs. This diffusion of electronic density over the whole molecule could be due to the highly planar geometry of the molecule, as evaluated by the study of its MPP and SDP.But for the efficient transfer of an electron from the valence-HOMO to the conduction-LUMO band, the most proficient approach is for the HOMO to be present around the donor region and the LUMO to be over the acceptor region. In other words, the localization of both FMOs should be at different sites. Similar is the case in the D1–D4 structures, where the electron density of HOMO is dispersed uniformly on the DBS core, benzothiazole acceptors, and thiophene bridges, while in the LUMO state this electron density has moved towards the newly added peripheral acceptors. This peculiarity of charge density is evaluated to be because of the added dimensionality in the molecules created in this study. Also, it was noted that the charge density is present around the thiophene rings in both the FMOs, which exhibits their proficient contribution to the transfer of electrons (Fig. 6).
Overall, in the considered molecules, the order of stabilization for HOMOs is D4 > D1 > D3 > D2 > R, while the order in which LUMO energies are most stable is as follows: D4 > D3 > D1 > D2 > R, as seen in the graph of band-gap in Fig. 5. Values computed for FMOs in Fig. 5 indicate that the HOMO of D1–D4 exhibits greater stability and contains lower energy as compared to the R's HOMO. This greater stability is attributed to the added electron withdrawing acceptor groups in the derived molecules. Amongst all, the largest electron withdrawing acceptor group of D4 molecule with its prolonged conjugation has triggered this molecule to be the one with the most stability, which might also help it in being a proficient donor at the photo-active layer. In addition, the most significant charge separation is also present in the LUMO of this molecule, which could be attributed to its highest values of the evaluated planarity parameters.
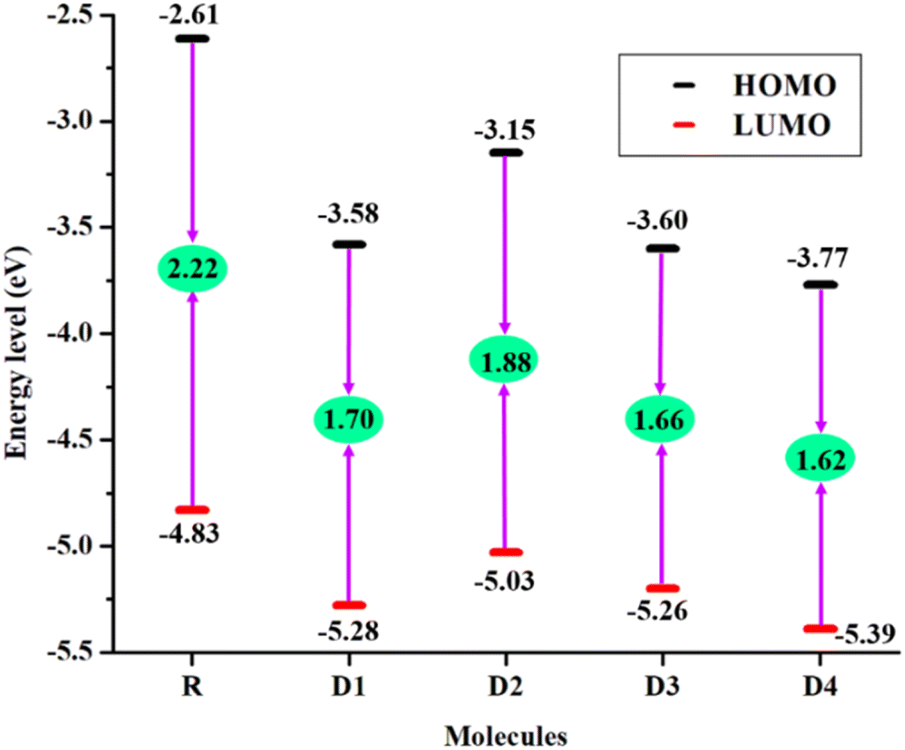 | ||
| Fig. 5 Band-gap plot of R along with D1–D4 designed chromophores. | ||
 | ||
| Fig. 6 HOMO–LUMO charge density dispersal of R and newly formulated small molecules D1–D4 at their optimized geometries. | ||
Usually, the greater the capability of an electron withdrawing group to extract the electrons from the donor region, the decreased will be value of the HOMO and LUMO energies.46 The band-gap, between the energy values of the FMOs, is closely related to the absorption maxima, as molecules with a lower band-gap tend to have a higher value of this important parameter. In addition, a lower band-gap increases the reactivity and also the charge transfer within the molecule. This band-gap is the energy difference between the evaluated valence and conduction bands of a molecule. From Fig. 5 it's perceived that the reference molecule possessed an Eg of 2.22 eV, while the Eg of D1–D4 molecules were much smaller when compared with R molecule. The order of band gap of the molecules of this study is R (DBS-2DPP) > D2 > D1 > D3 > D4. The reason behind these lowered values of band-gap of the newly modified molecules is the added peripheral acceptor groups, which by enhancing the conjugation have reduced the values of the band-gap, and thus made the transfer of electrons between the FMOs much easier. Though all the newly created molecules have a low band-gap, D4 has the lowest band-gap of all because of its most stable and differentiated FMOs and strongly conjugated end-capped electron acceptors containing cyano and acetyl groups. So, we could assume that amongst all the molecules under study, D4 might be the best possible candidate for possible future applications in organic photovoltaics, with D3 being a close second.
3.5. Density of state (DOS)
DOS offers specific information about the availability of charges in different energy levels along with their possible electronic excitation.45 In the plots of DOS, both the partial (PDOS) and total (TDOS) graphs are provided for a proper understanding of each fragment's contribution to total DOS (Fig. 7). Through the plots of density of states at the selected functional, plotted through PyMOlyze 1.1, further investigation to infer the validity of results drawn from FMOs was made possible. The relative intensity in the DOS graphs is represented at the vertical axis, whereas energy, (measured in electron volts) is represented at the horizontal axis. The graphs essentially depict the number of electronic states present, at a specific energy level and the contribution of a specific fragment in the elevation of that energy level.47 In regards to the FMOs, the planar zone in these plots, having no peak, depicts the band-gap, while the first peak towards its left represents the HOMO and that towards the right; the LUMO, and the values for which are in agreement with the values attained from the FMOs analysis above.48,49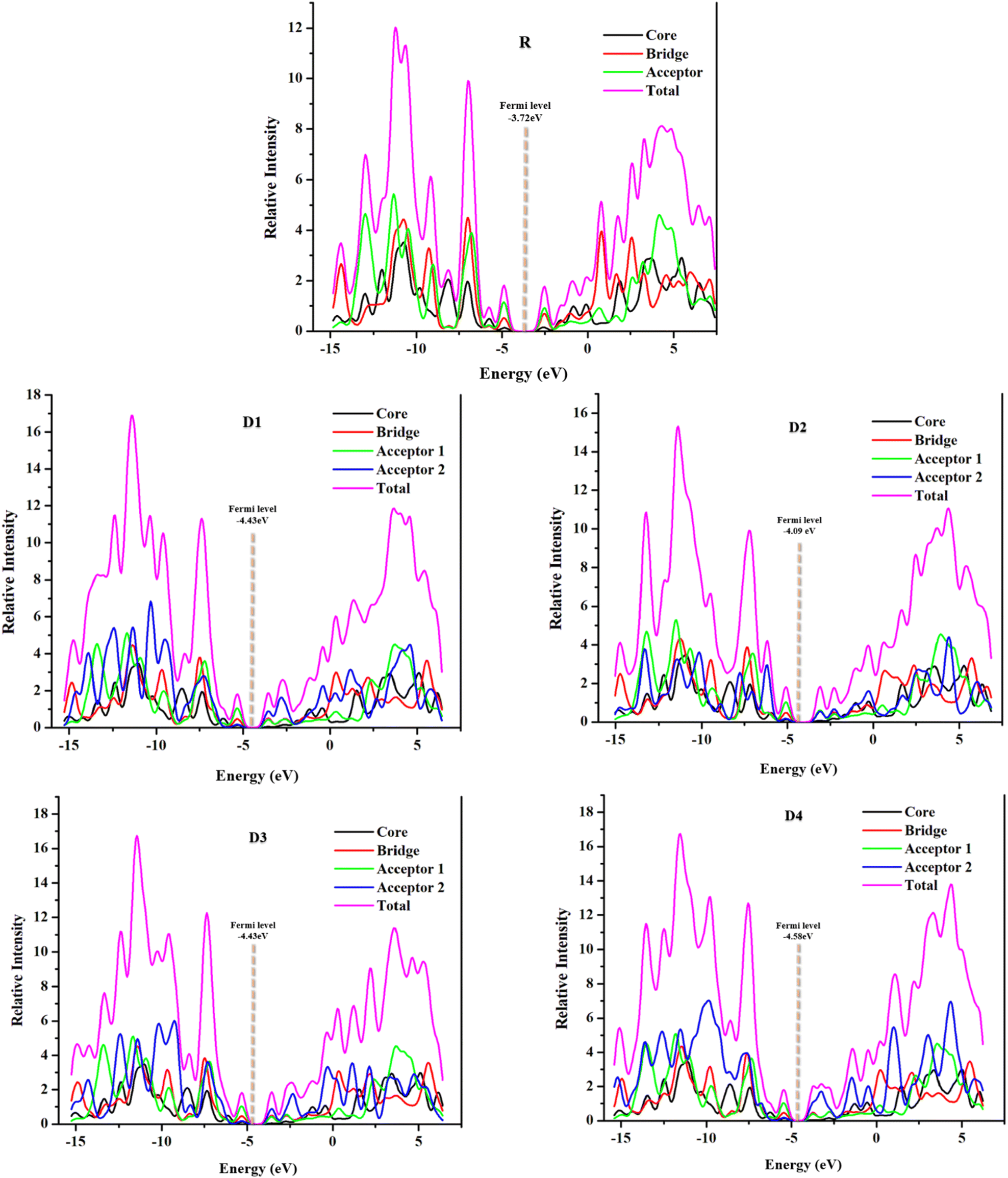 | ||
| Fig. 7 DOS curve plots of D1–D4 and R. | ||
Precisely in the DOS plots, the molecules were subdivided into four portions to comprehend their probable discrete contributions to the total electron density states, these parts are referred to as the bridge, the core (donor), the acceptor 1 (or acceptor in case of reference), and the acceptor 2 (the newly added one). These plots depict the overall molecular absorption (total DOS) by magenta line, while the individual contributions (PDOS) of the fragments are shown by red, black, green, and blue lines, respectively. Because of the added acceptor groups, the newly formulated molecules have high-intensity peaks of TDOS in their conduction band (towards the right of LUMO), illustrating the enhanced probability of residence of electron in this band, i.e., better electron transfer than reference. Individually, the peaks of acceptor 2 (blue) are the highest in case of D4 molecule, attributed to its most proficient abilities till now. Similarly, the acceptor groups in all the newly formulated compounds have varying strengths and conjugation levels, making the peaks of graphs differ from each other.
The Mulliken data of all the evaluated molecules, illustrating the percentage contribution of each fragment in raising the FMOs are presented in Table 4. In case of the reference molecule, all three fragments showed somewhat equal contribution in elevating its FMOs, which could retard the probable charge transfer from the HOMO towards the LUMO in the molecule. On the other hand, upon the addition of peripheral acceptors (acceptor 2), the percentage contribution of each fragment has improved drastically. The contribution of the donor in the LUMO has reduced from a 13% (R) to just 2.6–5.3% in the derived compounds, which could mean that the added acceptor fragments have shifted the LUMO electronic density towards themselves. Thus, the percentage contribution of both the acceptor fragments (1 and 2) in LUMO charge density have also increased in the designed molecules as compared to the acceptor 1 of the reference. These results signify the aptitude of the newly added fragments in differentiating the HOMO and LUMO in donor and acceptor, respectively, which could enhance the transfer of charges in the molecules. On a side note, the relatively unchanged contribution of the thiophene (bridge) fragments in the FMOs of the derived molecules validates their prominent charge transfer between the core and the peripherally added acceptors.
| Molecules | FMOs | Acceptor 1 (%) | Acceptor 2 (%) | Bridge (%) | Donor (%) |
|---|---|---|---|---|---|
| R | HOMO | 58.5 | — | 30.2 | 11.2 |
| LUMO | 48.5 | — | 38.6 | 13.0 | |
| D1 | HOMO | 52.8 | 8.6 | 26.9 | 11.6 |
| LUMO | 24.5 | 44.7 | 27.8 | 3.0 | |
| D2 | HOMO | 53.0 | 8.9 | 27.7 | 10.4 |
| LUMO | 34.3 | 27.7 | 32.7 | 5.3 | |
| D3 | HOMO | 52.7 | 9.3 | 26.7 | 11.4 |
| LUMO | 23.9 | 45.7 | 27.5 | 2.9 | |
| D4 | HOMO | 51.9 | 9.4 | 26.7 | 11.9 |
| LUMO | 21.7 | 50.1 | 25.6 | 2.6 |
Furthermore, the Fermi levels of the studied molecules were also calculated and are represented in Fig. 7 of the DOS spectra for each molecule. These values are the average of the FMO energy levels and represent the tendency of an electron to reside, either in the conduction band or the valence band. Here, to the left of the central planar region is the valence band and towards its right is the conduction band.50 The more the Fermi level is towards the conduction band the greater is its tendency to be an acceptor, while the opposite is true for a molecule to show donating properties.41
3.6. Transition density matrix (TDM)
By using the transition density matrix; in conjunction with electron excitation predictions, delocalization and coherence length of electron and hole pairs can be predicted. Furthermore, the relationship among the core (donor), bridges, and acceptors can be determined by using the transition density matrix.44,51 Precisely, during excitation, interaction among donating and receiving moieties, in addition to the quantum geometry of the overall molecule, is determined with the help of TDM.52,53 The TDM plots are the spatial maps used to describe electronic excitation procedures in light harvesting devices like OPVs, in which electron density travels from the core to the acceptor moiety via pi-conjugation inside the molecules.54 In TDM graphs of Fig. 8, hydrogen atoms were excluded and disregarded owing to their negligible share in the overall excitation. So, at B3LYP/6-31G (d,p), every molecule, except for reference, has been distributed into four moieties; donor (D), mid acceptors (A1), peripheral acceptors (A2), and bridge (B), while the reference constitutes three fragments, by excluding the A2 fragments (Fig. 8). This differentiation of fragments was done to better describe how the electrical charge density is localized or delocalized at these moieties. Varying fragments (ranging from one to the total no. of atoms in the molecule, except hydrogen) are labelled on the bottom horizontal and left vertical axis, while the electron density of the molecule is indicated through a colored bar on the right y-axis of the TDM maps. Uniform charge distribution, as well as coherency, is seen in the designed molecules, attributed to their added A2 component. Furthermore, the charge transfer is present discreetly in the diagonal and off-diagonal directions of these molecules. Overall, the donor region of all the molecules, including reference, shows a rather low charge density, while brightly colored zones in the acceptor regions of the molecules depict the high charge density in them, meaning effective charge distribution towards acceptors. | ||
| Fig. 8 2-D TDM plots for R (DBS-2DPP) and all (D1–D4) newly developed molecules. | ||
3.7. Exciton binding energy
A constructive aspect of OPVs, the binding energy (Eb), is generally exploited to make predictions regarding electrical and optical characteristics, or the overall performance of an OSC.55 Bound charge carriers (excitons) are produced at the photo-active layer of an OPV after photon absorption by the organic chromophores. It is necessary to disintegrate the exciton into opposite charge carriers of holes and electrons, in order to make their migration towards their respective electrodes possible. As a result of the strong coulombic attractive interactions between the generated charge carriers in the organic conjugated chromophores, their dissociation into separate charges becomes difficult. In order to elevate this phenomenon of dissociation, a convenient alternative method is to lower the binding energy of the respective molecule. This binding energy could be lowered by separating the acceptor and donor components within a molecule so that the respective holes and electrons could easily travel towards either of the two portions of the molecule. In the designed chromophores, the existence of the newly added acceptor moieties has distinguished the acceptor and donor regions within the molecules effectively, making it feasible for the molecule to have lowered binding energies between the charge carriers of the excitons due to the lowered coulombic attractive forces between them. Individually, the greater the electron withdrawing capability of the added acceptor fragment, the lowered the binding energy. For this reason, the binding energy of the D4 molecule is the lowest one of only 0.15 eV in the gas phase. Similarly, the designed chromophores in the gas medium, all have lowered binding energies than the reference, due to their prominently electron withdrawing added terminal acceptors. Overall, the declining order of exciton binding energy in the gas phase followed by all the molecules of this study is; R > D1 = D2 > D3 > D4. But in the dichloromethane solvent, the trend is as; D1 > D2 = D3 = D4 > R. Although in the solvent medium the designed chromophores have comparatively higher binding energies than reference, these values illustrate the enhanced capability and aptitude of the derived molecules with the polar solvent studied.33 Though the calculation of binding energy through theoretical evaluation is rather difficult, but the estimated guess is presented in this research work with the help of eqn (4). The values of the binding energies of all the molecules in both the gas medium and DCM solvent were calculated from eqn (4) and are demonstrated in Table 5.56| Eb = Eg – Ex | (4) |
| Molecule | Eb gas (eV) | Eb DCM (eV) |
|---|---|---|
| R | 0.20 | 0.24 |
| D1 | 0.19 | 0.28 |
| D2 | 0.19 | 0.26 |
| D3 | 0.18 | 0.26 |
| D4 | 0.15 | 0.26 |
3.8. Reorganizational energy (RE)
Extent of charge carrier transit characteristics is most likely to be done using reorganization energy (RE), measured in eV.57,58 The internal and the external RE are the two forms of reorganization energies required by a molecule. Internal reorganization energy informs about the variation in internal geometry of a molecule upon charge dispersal. While “external reorganization energy” is a term used to describe the effects of the environment outside the optical system. Because external reorganization energy has less impact on our present research (due to the unchanging external environment) and also is difficult to assess, it has been disregarded here.37The mobility of charges between donor, as well as acceptor components, in each molecule, is assessed using internal RE. Charge mobility rate has an inverse relation with RE values. The strength of the charge transfer between donor and acceptor units is estimated using the internal RE.59 Specifically, when the internal reorganization energy is decreased, the value of the rate of charge mobility is projected to be increased accordingly. The likelihood of electron and hole transfer in donors and acceptors is highly dependent on the structural characteristics of the moieties involved. The studied reorganization energy values were characterized statistically by the use of eqn (1) and (2) of Marcus theory, which emphasizes the structure of molecules and their stacking arrangement.60
Reorganization energy values for electrons (λe) and for holes (λh) for reference and the architectured molecules (D1–D4) are tabulated in Table 6. Attributed to the presence of a powerful electron-pulling moiety, all architectured molecules could have the higher electron and hole mobilities in comparison to the reference. D1–D4 have computed values of λe of 0.12354, 0.14450, 0.11266, and 0.10368 eV, respectively, while reference has a value of 0.15429 eV. Thus, the electron mobility of D4 seems to be the highest one amongst all, because of the lowest value of λe in charge transfer between the donating and accepting components shown by this molecule.
| Molecule | λe (electron) | λh (hole) |
|---|---|---|
| R | 0.15429 | 0.17633 |
| D1 | 0.12354 | 0.14531 |
| D2 | 0.14450 | 0.15266 |
| D3 | 0.11266 | 0.14123 |
| D4 | 0.10368 | 0.14395 |
The hole reorganization energy for reference R is 0.17933 eV, while other molecules (D1–D4) have λh values of 0.14531, 0.15266, 0.14123, and 0.14395 eV, correspondingly as provided in (Table 6). Here, the trend followed by the molecules under study for λh is R > D2 > D1 > D4 > D3. So, the highest hole mobility could be D3 molecule, attributed to its lowest λh of all. While, the hole mobility of D4 might also be a close second to D3 molecule due to the slight difference in their λh.
Concisely, the D4 molecule might have the highest electron mobility, while the D3 molecule could have the highest hole mobility among other developed molecules. Thus, both of these developed molecules are thought to be the most effective charge-transporting materials for future applications in solar cells.
3.9. Global reactivity descriptors
To analyze global reactivity indices, some useful measurements for characterizing chemical reactivity and kinetic stability, HOMO–LUMO energy levels, and their difference, i.e., the band-gap, can be utilized. The molecules having narrow band-gaps are more reactive when compared to those which have broader band-gaps and higher stability.61 Ionization potential and electron affinity are rather important global reactivity indices when evaluating charge carriers' transfer properties in molecules. The higher molecular reactivity of the molecules is associated with high EA values, and their higher stability is related to high IP values. The values of both the studied reactivity indices were calculated using eqn (5) and (6), shown below, and are presented in Table 7.| IP = (E+ − E0) | (5) |
| EA = (E0 − E−) | (6) |
| Molecule | IP (eV) | EA (eV) |
|---|---|---|
| R | 5.56 | 1.87 |
| D1 | 5.89 | 3.00 |
| D2 | 5.67 | 2.53 |
| D3 | 5.87 | 3.02 |
| D4 | 5.99 | 3.20 |
A significant connection is seen between ionization potentials and HOMO energies, while electron affinity is shown to be related to LUMO energies. Thus, as a result of the fact that R has the greatest HOMO energy of all the molecules investigated, it possesses the lowest IP value and is the least stable molecule. While D4 has the highest IP value due to its lowest HOMO energy, and thus is the most stable molecule and due to this stability could act proficiently as a donor in the photo-active layer.62 Similarly, higher values of EA were demonstrated by all newly developed molecules because of their lower LUMO energy levels. EA values for reference and entire designed chromophores are grouped in a descending order as D4 (3.20) > D3 (3.02) > D1 (3.00) > D2 (2.53) > R (1.87), showing their enhanced reactivity than the reference. Even here, D4 molecule seems to be an effective chromophore, attributed to its highest reactivity, while retaining its stability as well (Fig. 9).
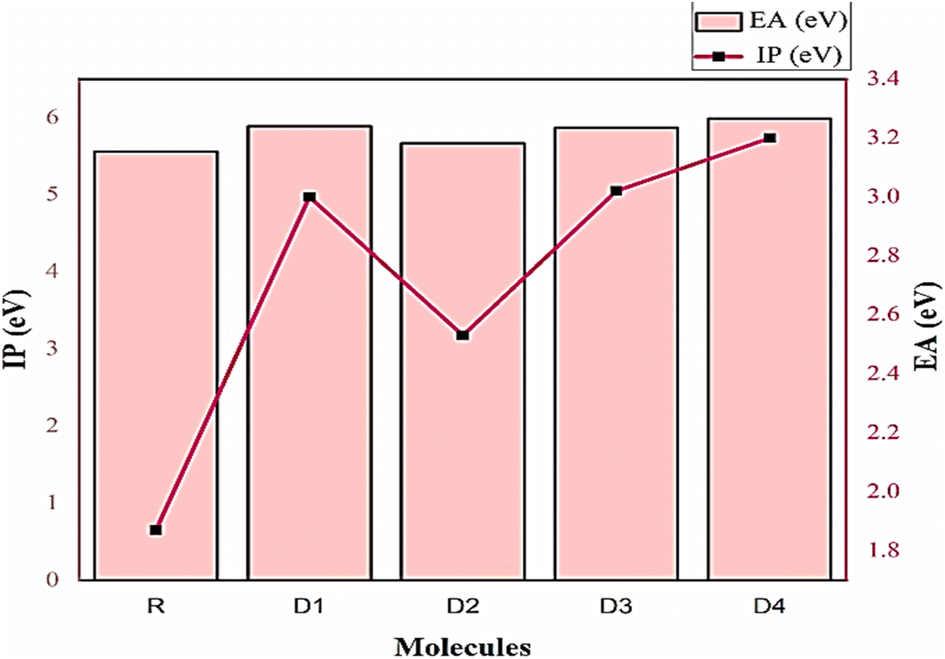 | ||
| Fig. 9 Global reactivity descriptors (IP and EA) plot for R and all newly developed molecules. | ||
3.10. Dipole moments
The solubility, dipole moment, and charge mobility of organic molecules are all interrelated to one another. A molecule with a high value of dipole moment tends to be highly soluble in a polar organic solvent due to the high degree of charge mobility in it.63 The increased dipole moments cause the respective molecules to easily self-assemble in a packed and antiparallel orientation, which results in an increased charge transfer rate at the active-layer.64 A smooth surface of the donor–acceptor interface of the active layer enables the molecule to be readily dissolved, resulting in a more efficient solar device assembly.65 With the help of the information attained about the solubility, polarization, and crystallinity of any molecule, attained from its dipole moment value, its utilization in the solution processing techniques for the construction of OPVs can be made possible. Planarity plays a contributing role in enhancing or diminishing the dipole moment of a molecule. Generally, the planar and organized geometries of molecules with increased dipole moments result in a tighter molecular assembly, improved crystallinity, and higher solubility in polar solvent. But, in a highly planar molecule due to the cancellation of opposite charges within its structure, the dipole moment is generally quite lowered.66 The values of dipole moments in the gas phase, as well as DCM solvent, are summarized in Table 8. In the gas medium, the dipole moment is following the declining order of D4 (11.73 D) > D1 (7.16 D) > D3 (6.51 D) > D2 (2.36 D) > R (0.83 D), while D4 (14.75 D) > D1 (8.80 D) > D3 (8.13 D) > D2 (3.45 D) > R (1.09 D) is the observed trend of molecules in DCM, as seen in Fig. 10. As a result of these findings, dipole moments in the gaseous and solvent states are found to be significantly higher than that of the reference's value. In dichloromethane, all proposed SMs were shown to have a superior solubility effect than R, since their π-interactions in the polar DCM solvent are stronger.| Molecules | μg (Debye) | μe (Debye) |
|---|---|---|
| R | 0.8311 | 1.0911 |
| D1 | 7.1599 | 8.8045 |
| D2 | 2.3572 | 3.4519 |
| D3 | 6.5144 | 8.1342 |
| D4 | 11.7339 | 14.7530 |
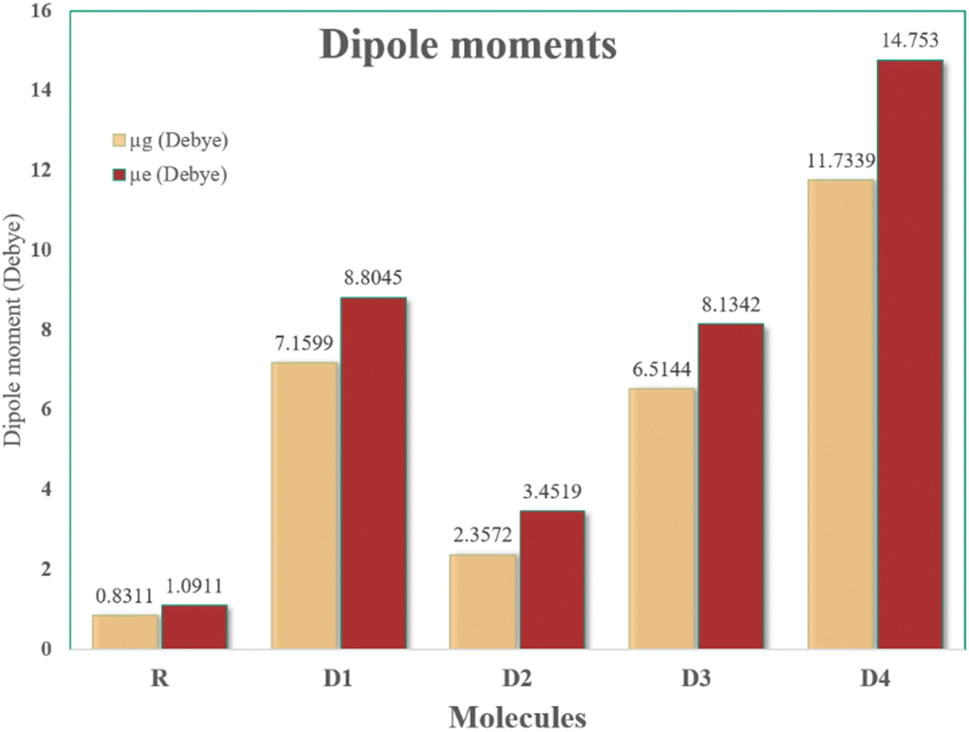 | ||
| Fig. 10 Colored plot of dipole moments (μg along with μe) of newly devised D1–D4 as well as R molecules. | ||
3.11. Molecular electrostatic potential surface (MEPS)
The probable charge transfer passage among donor, bridges, and acceptor moieties at the ground state was measured using the molecular electrostatic potential surface (MEPS) maps.63 The electrophilic and nucleophilic regions of the molecules under investigation were found via the use of MEPS simulations run at the designated DFT functional.67 In the red zones of the MEPS plots, a negative electrostatic potential, because of the excess of electrons is predicted. While in the blue zones, the opposite seems to be true, due to the deficiency of electrons and thus positive electrostatic potential of those areas. In addition, the green colored regions indicate the presence of the neutral sites. As the red regions have high negative surface potential, they are predicted to be zones for any electrophilic attack, while the blues zone, due to their high positive potential, are susceptible to attack by an incoming nucleophile. For easier understanding, the observed order of electrostatic potential from colored bar of Fig. 11 is written as; red > green > yellow > blue, shifting from negative to positive maxima. | ||
| Fig. 11 Molecular electrostatic potential surface (MEPS) colored maps of newly devised D1–D4 molecules, along with R. | ||
There are quite plenty of red and yellow patches dispersed within the blue donor region on the MEPS of the reference molecule, suggesting the relatively indistinguishable electrophilic and nucleophilic attacking sites in it. Although, the terminal unsaturated oxygen atoms of the reference molecule are bright red zones of nucleophilic attack, just like the unsaturated nitrogen and oxygen atoms of the derived molecules. But in the derived molecules, there are distinct red zones at the peripheral acceptor regions, and blue zones at the rest of the molecule. The majority of blue regions in the derived chromophores point towards their better aptitude as donors with their positive potential in the active layer. The majority of blue regions in the derived chromophores point towards their better aptitude as donors with their positive potential in the active layer. The maximum condensed red patches at the peripheries of the D4 molecule could be due to its significantly conjugated and electron withdrawing added acceptor group.39
3.12. Device efficiency
The open circuit photo-voltage, fill factor, and short circuit current density of any chromophore, are all important factors in determining the overall performance of the corresponding solar cell.The maximum open circuit photo-voltage that may be generated by a SC (due to the photoelectric effect) is governed by intensity of incident light, charge mobility, temperature of device, and the FMOs of the donor and acceptor molecules at the photo-active layer. Here, the VOC of the considered donors was calculated by taking a theoretically proficient and effective acceptor; PC61BM.69 The selected acceptor, with a HOMO of −6.3 eV and LUMO of −3.7 eV, has been known to be a powerful fullerene acceptor and provides acceptable efficiency for the respective solar cells. The reason behind the selection of our proposed molecules to be donors is the fact that a high value of VOC is achievable by lowering the energy level of HOMO in donor material and increasing the energy levels of LUMO in acceptor material of the photo-active layer.70 So, because of their low HOMO level than reference, all of the derived chromophores are predicted to have improved charge conduction and function as donor materials.
Actually, the open circuit photo-voltage (VOC) was obtained by subtracting the studied donors' HOMO from the acceptor's LUMO and dividing it with the charge of the molecule (taken as 1). In addition, a value of 0.3 was also subtracted as a common empirical value in fullerene acceptor based solar cells.71 Eqn (7) mathematically represents the relation stated above ,66 and the thus attained values are represented pictorially in Fig. 12.
 | (7) |
 | ||
| Fig. 12 VOC of proposed compounds, as well as the reference molecule, calculated using PC61BM as the acceptor. | ||
The voltaic strength of reference and designed compounds studied in terms of VOC rises in the trend as; R (0.83 V) < D2 (1.03 V) < D3 (1.26 V) < D1 (1.28 V) < D4 (1.39 V). From these values, it seems that all the newly proposed compounds could act as better donor materials in the photo-active layer of SC than reference. Among the proposed molecules, D4 retained its position as the best amongst all due to its relatively higher VOC than all others in the group. Specifically, with the highly electron-withdrawing cyano and acetyl groups of the added end-capped acceptor stabilizing its HOMO value, D4 has the greatest VOC value of any molecule.
 | (8) |
In this equation, KB represents the Boltzmann constant and T represents the temperature in Kelvin of 8.61733034 × 10−5 eV and 300 K, respectively. While the whole term eVOC/KBT is collectively represented as the normalized VOC.75 Table 9 presents a summary of the theoretical findings of the normalized VOC and FF. FF of all developed molecules is in the same order as VOC. So, upon comparison, it was evaluated that all the newly synthesized D1–D4 chromophores have higher FF than reference, with D4 having the highest one amongst all. The increment in the FF of newly constructed molecules proves their better efficacy than the reference, and thus they could be more effective in the photo-active layer of OPVs.
| Molecule | Normalized VOC
|
Fill factor |
|---|---|---|
| R | 32.11 | 0.864 |
| D1 | 49.51 | 0.903 |
| D2 | 39.84 | 0.885 |
| D3 | 48.74 | 0.901 |
| D4 | 53.77 | 0.909 |
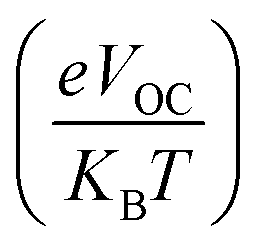
 | (9) |
 | (10) |
4. Conclusion
In this investigation, four A2–π–A1–π–D–π–A1–π–A2 type small-molecule donors were theoretically designed and explored under DFT, in order to possibly improve the photovoltaic properties and efficiency of OSCs. Here, a dibenzosilole-based DBS-2DPP molecule was taken as the reference and four new molecules were developed from it, with the addition of terminal acceptors at its peripheries. These added acceptor groups by shifting the electron density towards themselves, better differentiated the donor and acceptor components of the molecules, helping them in effective charge transport within their structures. Attributed to the A2 acceptors, the performed quantum chemical computations of the newly proposed molecules were seen to be rather substantially improved when compared to the reference. All of the proposed structures demonstrated extensive absorption in the near IR region in both the studied phases. Their band-gaps, excitation and exciton binding energies, reorganization energies, oscillation strengths and dipole moments all pointed towards their better performance than the reference. The studied FMOs, MEPS, TDM, and DOS analysis also showed the effective charge density distribution in the derived molecules. In addition, their calculated PCE factors with PC61BM acceptor, i.e., open circuit photo-voltage, light harvesting efficiencies, and fill factor, were also transparently boosted. Although all the newly derived chromophores were better than reference in all the studied aspects of solar cells, D4 seemed to be the best of all. With its highest dihedral angle, SDP, and MPP, this molecule while retaining its planar identity maintained discrete acceptor and donor regions in its structure, which helped it in lowering its exciton binding energy in gas phase (0.15 eV), increasing its dipole moment in both DCM (14.7530 D) and gas (11.7339 D) phase, as well as reducing its required λe (0.10368 eV). In addition, attributed to the strongly electron-withdrawing peripheral (A2) acceptors of D4, this molecule had the most highly stabilized FMOs amongst all, due to which it showed the lowest band-gap (1.62 eV), the highest VOC (1.39 V), and the maximum IP and EA of 5.99 and 320 eV, respectively. Most importantly, due to the highly conjugated cyano and acetyl groups in its added acceptor, D4 demonstrated the highest λmax of 876 nm in DCM and 802 nm in gas phase, and the lowest excitation energies of 1.47 eV in gas phase and 1.36 nm in the DCM solvent. Amongst all the studied parameters, only its binding energy in the solvent phase (0.26 eV), just like other proposed molecules was somewhat inferior to the reference molecule (0.24 eV), but this could be attributed to their enhanced compatibility with the studied solvent. Although D3 molecule seemed to be a close second to D4 molecule, due to its studied values being quite adjacent to D4 with some of them even exceeding it, such as its lowest λh, highest oscillator strength, and the highest light harvesting efficiencies. But, it's relatively lowered open circuit photo-voltage and fill factor reduced its aptitude in comparison to D4. In short, the strategy of adding new acceptor groups in the reference molecule has provided us with quite effective molecules, with D4 being the most effective one of all. Thus, all of them could be applied for their possible practical applications in future organic photovoltaic cells.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the technical provided by Department of Chemistry, University of Agriculture (UAF), Faisalabad 38000, Pakistan. The authors extend their appreciation to the Researchers Supporting Project number (RSP-2021/396), King Saud University, Riyadh, Saudi Arabia. The authors also thankful to Dr Khurshid Ayub, COMSATS University, Islamabad, Abbottabad Campus, Pakistan for additional resources.References
- W. Fulkerson, R. R. Judkins and M. K. Sanghvi, Sci. Am., 1990, 263, 128–135 CrossRef.
- A. M. Omer, Renew. Sustain. Energy Rev., 2008, 12, 2265–2300 CrossRef CAS.
- K. Tromly, Renewable Energy: An Overview, United States, N. p., 2001 Search PubMed.
- M. Ans, J. Iqbal, I. A. Bhatti and K. Ayub, RSC Adv., 2019, 9, 34496–34505 RSC.
- A. K. Abdelsalam, H. T. Shahin and I. Morsi, IEEE Second International Conference on DC Microgrids (ICDCM), 2017, pp. 406–414 Search PubMed.
- R. Ma, T. Yang, Y. Xiao, T. Liu, T. Yang, R. Ma, L. Zhan, Z. Luo, G. Zhang and H. Yan, Joule, 2021, 5(4), 914–930 CrossRef.
- P. Würfel, Phys. E, 2002, 14, 18–26 CrossRef.
- M. Mrinalini, N. Islavath, S. Prasanthkumar and L. Giribabu, Chem. Rec., 2019, 19, 661–674 CrossRef CAS PubMed.
- K. J. Yu, Z. Yan, M. Han and J. A. Rogers, npj Flexible Electron., 2017, 1(1), 1–14 CrossRef CAS.
- Q. Tan, X. Yang, M. Cheng, H. Wang, X. Wang and L. Sun, J. Phys. Chem. C, 2014, 118, 16851–16855 CrossRef CAS.
- Y. Xu, F. Zhang and X. Feng, Small, 2011, 7, 1338–1360 CrossRef CAS PubMed.
- R. Saleem, A. Farhat, R. A. Khera, P. Langer and J. Iqbal, Comput. Theor. Chem., 2021, 1197, 113154 CrossRef CAS.
- M. Ans, K. Ayub, I. A. Bhatti and J. Iqbal, RSC Adv., 2019, 9, 3605–3617 RSC.
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470–488 RSC.
- B. Walker, J. Liu, C. Kim, G. C. Welch, J. K. Park, J. Lin, P. Zalar, C. M. Proctor, J. H. Seo and G. C. Bazan, Energy Environ. Sci., 2013, 6, 952–962 RSC.
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. Janssen, Acc. Chem. Res., 2016, 49, 78–85 CrossRef CAS PubMed.
- Y. Lin, Y. Li and X. Zhan, Adv. Energy Mater., 2013, 3, 724–728 CrossRef CAS.
- S. J. Akram, N. Hadia, J. Iqbal, R. F. Mehmood, S. Iqbal, A. M. Shawky, A. Asif, H. Somaily, M. Raheel and R. A. Khera, RSC Adv., 2022, 12, 20792–20806 RSC.
- F. Ogliaro, M. Bearpark, J. Heyd, E. Brothers, K. Kudin, V. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell and D. J. Fox, Gaussian 09, revision a. 02., Gaussian., Inc., Wallingford, CT, 2009 Search PubMed.
- S. J. Akram, J. Iqbal, M. Ans, Y. A. El-Badry, R. F. Mehmood and R. A. Khera, Sol. Energy, 2022, 237, 108–121 CrossRef CAS.
- R. D. I. I. Dennington, T. A. Keith and J. M. Millam, GaussView, version 6.0. 16., Semichem Inc Shawnee Mission KS., 2016 Search PubMed.
- J. P. Finley, Mol. Phys., 2004, 102, 627–639 CrossRef CAS.
- A. Alparone, Chem. Phys. Lett., 2013, 563, 88–92 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J. Zhang, H.-B. Li, S.-L. Sun, Y. Geng, Y. Wu and Z.-M. Su, J. Mater. Chem., 2012, 22, 568–576 RSC.
- M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Chem. Phys. Lett., 1998, 286, 253–260 CrossRef CAS.
- L. A. Deschenes and A. David Vanden Bout, University of Texas, 2000.
- T. Lu, J. Mol. Model., 2021, 27, 1–6 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- A. Tenderholt, in Stanford University, CA Stanford, 2006.
- S. J. Akram, J. Iqbal, R. F. Mehmood, S. Iqbal, Y. A. El-Badry, M. I. Khan, M. Ans and R. A. Khera, Sol. Energy, 2022, 240, 38–56 CrossRef CAS.
- T. Tani, T. Suzumoto and K. Ohzeki, J. Phys. Chem., 1990, 94, 1298–1301 CrossRef CAS.
- I. Bahar, M. Kaplan and R. Jernigan, Proteins: Struct., Funct., Bioinf., 1997, 29, 292–308 CrossRef CAS.
- M. Khalid, A. Ali, R. Jawaria, M. A. Asghar, S. Asim, M. U. Khan, R. Hussain, M. F. ur Rehman, C. J. Ennis and M. S. Akram, RSC Adv., 2020, 10, 22273–22283 RSC.
- S. Zahid, A. Rasool, M. Ans, M. Yaseen and J. Iqbal, Energy Fuels, 2021, 35, 15018–15032 CrossRef CAS.
- M. Rafiq, M. Salim, S. Noreen, R. A. Khera, S. Noor, U. Yaqoob and J. Iqbal, J. Mol. Liq., 2022, 345, 118138 CrossRef CAS.
- M. I. Khan, J. Iqbal, S. J. Akram, Y. A. El-Badry, M. Yaseen and R. A. Khera, J. Mol. Graph. Model., 2022, 113, 108162 CrossRef CAS PubMed.
- A. Naveed, R. A. Khera, U. Azeem, I. Zubair, A. Farhat, A. R. Ayub and J. Iqbal, Mater. Sci. Semicond. Process., 2022, 137, 106150 CrossRef CAS.
- M. Salim, M. Rafiq, R. A. Khera, M. Arshad and J. Iqbal, Sol. Energy, 2022, 233, 31–45 CrossRef CAS.
- M. U. Saeed, J. Iqbal, R. F. Mehmood, S. J. Akram, Y. A. El-Badry, S. Noor and R. A. Khera, Surface. Interfac., 2022, 30, 101875 CrossRef CAS.
- U. Mubashar, A. Farhat, R. A. Khera, N. Iqbal, R. Saleem and J. Iqbal, J. Mol. Model., 2021, 27, 1–13 CrossRef PubMed.
- R. A. Shehzad, J. Iqbal, M. U. Khan, R. Hussain, H. M. A. Javed, A. ur Rehman, M. U. Alvi and M. Khalid, Comput. Theor. Chem., 2020, 1181, 112833 CrossRef CAS.
- A. Rasool, S. Zahid, R. A. Shehzad, M. S. Akhter and J. Iqbal, Comput. Theor. Chem., 2021, 1203, 113359 CrossRef CAS.
- M. Miar, A. Shiroudi, K. Pourshamsian, A. R. Oliaey and F. Hatamjafari, J. Chem. Res., 2021, 45, 147–158 CrossRef CAS.
- T. Liu, Y. Zhang, Y. Shao, R. Ma, Z. Luo, Y. Xiao, T. Yang, X. Lu, Z. Yuan and H. Yan, Adv. Funct. Mater., 2020, 30, 2000456 CrossRef CAS.
- Z. Luo, R. Ma, T. Liu, J. Yu, Y. Xiao, R. Sun, G. Xie, J. Yuan, Y. Chen and K. Chen, Joule, 2020, 4, 1236–1247 CrossRef CAS.
- M. Ans, J. Iqbal, B. Eliasson and K. Ayub, Comput. Mater. Sci., 2019, 159, 150–159 CrossRef CAS.
- I. Zubair, R. A. Kher, S. J. Akram, Y. A. El-Badry, M. U. Saeed and J. Iqbal, Chem. Phys. Lett., 2022, 793, 139459 CrossRef CAS.
- S. Tretiak and S. Mukamel, Chem. Rev., 2002, 102, 3171–3212 CrossRef CAS PubMed.
- S. Anwar, N. Naeem, Z. M. Elqahtani, S. Siddique, J. Iqbal, M. Al-Buriahi and S. Alomairy, Chem. Phys. Lett., 2022, 139726 CrossRef CAS.
- M. Ans, J. Iqbal, B. Eliasson, M. J. Saif, H. M. A. Javed and K. Ayub, J. Mol. Model., 2019, 25, 1–12 CrossRef CAS PubMed.
- L. Bhattacharya, S. Sharma and S. Sahu, Int. J. Quantum Chem., 2021, 121, e26524 CrossRef CAS.
- K. Q. Kayani, U. Yaqoob, S. Jabeen, S. Iqbal, M. Yaseen, M. Khalid, M. S. Akhter and J. Iqbal, Comput. Theor. Chem., 2021, 1202, 113305 CrossRef.
- M. Schapira, M. Totrov and R. Abagyan, J. Mol. Recognit., 1999, 12, 177–190 CrossRef CAS PubMed.
- A. Rasool, S. Zahid, M. Ans, S. Muhammad, K. Ayub and J. Iqbal, ACS Omega, 2021, 7(1), 844–862 CrossRef PubMed.
- M. Ans, K. Ayub, X. Xiao and J. Iqbal, J. Mol. Liq., 2020, 298, 111963 CrossRef CAS.
- S. A. Siddique, M. B. A. Siddique, R. Hussain, X. Liu, M. Y. Mehboob, Z. Irshad and M. Adnan, Comput. Theor. Chem., 2020, 1191, 113045 CrossRef CAS.
- I. Yousaf, R. A. Khera, J. Iqbal, S. Gul, S. Jabeen, A. Ihsan and K. Ayub, Mater. Sci. Semicond. Process., 2021, 121, 105345 CrossRef CAS.
- A. Musawwir, A. Farhat, R. A. Khera, A. R. Ayub and J. Iqbal, Comput. Theor. Chem., 2021, 1201, 113271 CrossRef CAS.
- A. Naveed, S. J. Akram, M. Ans, J. Iqbal, I. Batool, R. F. Mehmood and R. A. Khera, J. Mol. Model., 2022, 28, 1–16 CrossRef PubMed.
- U. Azeem, R. A. Khera, A. Naveed, M. Imran, M. A. Assiri, M. Khalid and J. Iqbal, ACS Omega, 2021, 6, 28923–28935 CrossRef CAS PubMed.
- U. Yaqoob, S. Rafiq, S. U. Rehman, S. Bibi and J. Iqbal, Optik, 2021, 246, 167845 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- M. U. Saeed, J. Iqbal, R. F. Mehmood, M. Riaz, S. J. Akram, H. Somaily, A. M. Shawky, M. Raheel, M. I. Khan and E. U. Rashid, J. Phys. Chem. Solids, 2022, 170, 110906 CrossRef CAS.
- J. S. Puranen, M. J. Vainio and M. S. Johnson, J. Comput. Chem., 2010, 31, 1722–1732 CAS.
- D. Credgington and J. R. Durrant, J. Phys. Chem. Lett., 2012, 3, 1465–1478 CrossRef CAS PubMed.
- D. Chirvase, Z. Chiguvare, M. Knipper, J. Parisi, V. Dyakonov and J. C. Hummelen, J. Appl. Phys., 2003, 93, 3376–3383 CrossRef CAS.
- M. Ans, J. Iqbal, Z. Ahmad, S. Muhammad, R. Hussain, B. Eliasson and K. Ayub, ChemistrySelect, 2018, 3, 12797–12804 CrossRef CAS.
- M.-H. Lee, Sol. Energy, 2022, 234, 360–367 CrossRef CAS.
- A. Farhat, R. A. Khera, S. Iqbal and J. Iqbal, Opt. Mater., 2020, 107, 110154 CrossRef CAS.
- U. Yaqoob, A. R. Ayub, S. Rafiq, M. Khalid, Y. A. El-Badry, Z. M. El-Bahy and J. Iqbal, J. Mol. Liq., 2021, 341, 117428 CrossRef CAS.
- L. Zhang, W. Shen, R. He, X. Liu, X. Tang, Y. Yang and M. Li, Org. Electron., 2016, 32, 134–144 CrossRef CAS.
- M. Rafiq, R. A. Khera, M. Salim, M. Khalid, K. Ayub and J. Iqbal, Chem. Phys. Lett., 2021, 782, 139018 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |