
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene oxide reinforced silk fibroin nanocomposite as an electroactive interface for the estimation of dopamine†
Afifa Noorab,
Hamad Khalid*a,
Muhammad Aslamb,
Akhtar Hayata,
Ather Farooq Khana,
Muhammad Nasira,
Aqif Anwar Chaudhrya and
Mian Hasnain Nawaz *a
*a
aInterdisciplinary Research Centre in Biomedical Materials (IRCBM), COMSATS University Islamabad, Lahore Campus, 54000, Pakistan. E-mail: hamadkhalid@cuilahore.edu.pk; mhnawaz@cuilahore.edu.pk
bDepartment of Chemistry, Division of Science and Technology, University of Education, Lahore, 54000, Pakistan
First published on 13th October 2022
Abstract
The fabrication of 2D materials and polymer-based nanocomposites deposited on flexible conductive interfaces has unblocked new horizons to expedite reaction kinetics for developing highly selective and sensitive electrochemical biosensors. Herein, we developed a novel biosensing platform, comprising graphene oxide and a silk fibroin-based nanocomposite, drop-cast on a carbon cloth electrode. The fabricated interface was expected to be a robust and miniaturized sensing platform for precise detection of dopamine (DA). Characterization was performed by SEM, EDX, FTIR, XRD, UV-visible spectroscopy, contact angle measurement, fluorescence spectroscopy, particle size, and zeta potential analysis. CV, EIS, DPV, and chronoamperometry demonstrated the superior electrochemical properties of the working interface and revealed its enhanced active surface area, increased conductivity, and accelerated electron transfer rate. The designed interface exhibited low LoD (0.41 μM), admirable stability, good sensitivity (2.46 μA μM−1 cm−2), wide linearity ranging from 100–900 μM, excellent reproducibility, and superb selectivity against dopamine even in the presence of possible interfering analytes. These findings endorse the feasibility of the practical execution of such an integrated system in real sample analysis.
1 Introduction
Biomolecules are very important for the functioning of living organisms. Irregularities in biomolecules' concentrations can cause serious issues in any living organism. Therefore, it is highly required to design efficient processes for qualitative and quantitative detection of specific biological molecules.1Dopamine (DA), a neurotransmitter, performs major functions in human body metabolism, in particular in the central nervous, cardiovascular, renal, and hormonal systems.2 The imbalance in the concentration of DA is linked with various psychiatric and neurological disorders including Parkinson's disease,3 senile dementia, schizophrenia,4 HIV infection, hyperactivity, and restless syndrome.5 Society is paying a huge price for the treatment of mental disorders caused by DA dysfunctions.6 Hence, a precise and highly sensitive system is required for the signaling of low concentrations of DA for early diagnosis of such diseases. So far, different analytical techniques, including capillary electrophoresis,7 fluorescence,8 spectroscopy,9 calorimetry,10 and chromatography11 have been employed for the detection of DA. However, most of these techniques are either costly or require complicated sample pretreatment stages which are time-consuming and arduous. Among different methods of detection, the electrochemical method has acquired special attention for rapid and cost-effective monitoring of electroactive species.12
Although electrochemical methods have been exhibited to be the most effective and efficient methods for both quantitative and qualitative measurement of DA,13 however, there are still considerable challenges conjoined with the sensitivity and selectivity of DA detection. The interfering analytes including uric acid (UA) and ascorbic acid (AA), overlap the voltammetric response of DA, as their oxidation potentials are proximate to that of DA.14 Thus, a highly selective and sensitive interface is required to precisely discriminate and detect DA signals in complex biological systems.15
In these lines, numerous nanomaterials owing to their outstanding physical and chemical characteristics16 have been used for sensing and biosensing applications. Among them, Graphene-Based Nanomaterials (GBNs) have acquired substantial regard in the recent years due to their outstanding mechanical strength,17 large surface area, superb optical properties, high thermal and electrical conductivity, and good biocompatibility.18 GBNs comprise 2D sheets of carbon including pristine graphene, graphene oxide (GO), and reduced graphene oxide (rGO). Among them, GO is an outstanding filler due to its high binding potential, outstanding mechanical properties, high aspect ratio, chemical stability, superior processability, and high flexibility.19 Carbon nanotubes have also exhibited impressive potential in the recent years for detection of neurotransmitters.20,21
Similarly, towards biosensing applications, natural biomaterials have gained interest as an excellent candidate for nanocomposite matrices due to their enchanting morphologies, biodegradability, biocompatibility, and exceptional performances, particularly by silk materials.22 Silk fibroin (SF) is an exemplary biomaterial as reinforcing polymeric glue in biocomposites due to its repetitive multi-domain structure that could expedite the fabrication of extremely stable antiparallel β-sheets.23 It has played a dominant role in tissue engineering,24 drug delivery,25 regenerative medicines,26 soft electronics,27 and biosensors28 due to its long-term biodegradability, intrinsic mechanical properties, minimal flammability, impressive biocompatibility,29 low immunogenicity,30 and mechanical robustness.27 The flexibility and transparency of silk materials make it favorable to be used in wearable electrodes and sensors.31 However, despite its exceptional mechanical properties, native silk is an insulating material and shows poor electrical properties,32 so it is necessary to combine it with a conducting material to explore its conductive applications.33
Based on the remarkable mechanical strength of GO and SF, and their tendency to form a layered structure, GO-SF composites have commanded significant attention in various research fields.34 The synergistic combination of GO and SF results in fascinating properties and can be tailored for specific applications.30
In past years, flexible and stretchable sensors have acquired remarkable interest in modern electronics and interdisciplinary fields due to their fascinating attributes, including low cost, light modulus, high stretchability and flexibility, and light weight.35 Exploring adaptable conductive substrates for selective detection of various analytes36 has received much attention. Similarly, several types of carbon materials have been extensively explored as electrode materials.37 In this context, carbon cloth (CC) has arisen as an outstanding electrode substrate being inexpensive, having outstanding mechanical flexibility, chemical stability, high tensile strength,38 three-dimensional (3D) network, remarkable electrical conductivity, and large surface area.39
Herein, by utilizing the inherent properties of GO and SF, GO-SF nanocomposites can be synthesized and employed for modification of CC electrodes. This sensing platform is expected to behave as a robust, miniaturized and highly stable sensing interface for electrochemical signaling of DA and can exhibit superior electrochemical performance for selective and sensitive estimation of DA in the presence of possible interfering species and complex physiological environment.
2 Experimental section
2.1 Reagents and chemicals
Graphite powder particle size < 80 microns, sulfuric acid, ethanol, hydrogen peroxide, potassium permanganate and glucose (Sigma-Aldrich), potassium ferrocyanide and potassium ferricyanide (Unichem), phosphate buffer saline (PBS) (Bioworld), dopamine, lithium bromide, and uric acid (Alfa Aesar), silk cocoons (Changa Manga forest, District. Kasur, Punjab, Pakistan), ascorbic acid, phosphoric acid, sodium chloride, potassium chloride, sodium hydroxide, and magnesium chloride (Daejung), sodium carbonate (Scharlau), hydrochloric acid and acetone (Analar), and polyoxyethylene 20 (Biotech). All the chemicals were utilized as received.2.2 Instrumentation
All the electrochemical studies were carried out at Gamry Interface 1010 E potentiostat/galvanostat. A standard three-electrode system comprising carbon cloth as a working electrode (WE), Ag/AgCl electrode as a reference electrode (RE), and the counter-electrode (CE) was platinum wire. UV-visible (UV-vis) spectroscopy was performed on PerkinElmer Lambda 25 UV-Vis spectrophotometer. Fluorescence spectra were analyzed on Carry Eclipse Fluorescence spectrophotometer. Fourier transform infrared (FTIR) spectra were analyzed on Thermo Fisher Scientific Nicolet 6700 spectrometer in ATR mode. X-ray powder diffraction (XRD) spectra were recorded on XRD Rigaku Model mini flex 600C with HYPIX-400 MF. Contact angle was measured on Contact angle Biolin Scientific Attension Theta Flex Model TF300-Plus, Biolin Scientific, Sweeden. SEM images were taken from FEI Nova 450 NanoSEM equipped with Oxford energy dispersive X-ray (EDX) detector for elemental analysis. Particle size and zeta potential were analyzed on Malvern Zetasizer Nano-Z5 using dynamic light scattering (DLS).2.3 Synthesis of graphene oxide
Modified Hummers' method was used for synthesizing GO.40 In a typical procedure, 90 mL concentrated H2SO4 and 10 mL concentrated H3PO4 (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were taken and 2 g natural graphite powder was added to it. The resulting mixture was stirred for several minutes, subsequently cooled up to 5 °C in an ice bath, and was kept below 5 °C for 30 min. KMnO4 (8 g) was gradually added, to avoid rapid heat evolution, under constant stirring and cooling to maintain the temperature below 10 °C. The reaction mixture was stirred for 2 hours. 100 mL of DI H2O was added into it and stirring was done for 1 hour. Then 200 mL of DI H2O was added and stirring was done continuously for 24 hours at room temperature until a dark brown suspension was produced. Afterwards, 20 mL of 30% H2O2 was slowly added to the reaction mixture while keeping it in an ice bath, to reduce the residual KMnO4 and to terminate the reaction. The slurry was filtered and residues were washed 5 times with 5% HCl aqueous solution for removing metal ions and subsequently washed several times with DI H2O until pH reaches 6.
:1) were taken and 2 g natural graphite powder was added to it. The resulting mixture was stirred for several minutes, subsequently cooled up to 5 °C in an ice bath, and was kept below 5 °C for 30 min. KMnO4 (8 g) was gradually added, to avoid rapid heat evolution, under constant stirring and cooling to maintain the temperature below 10 °C. The reaction mixture was stirred for 2 hours. 100 mL of DI H2O was added into it and stirring was done for 1 hour. Then 200 mL of DI H2O was added and stirring was done continuously for 24 hours at room temperature until a dark brown suspension was produced. Afterwards, 20 mL of 30% H2O2 was slowly added to the reaction mixture while keeping it in an ice bath, to reduce the residual KMnO4 and to terminate the reaction. The slurry was filtered and residues were washed 5 times with 5% HCl aqueous solution for removing metal ions and subsequently washed several times with DI H2O until pH reaches 6.
GO was ground to a fine powder and was sieved through a 100 micron sieve plate. GO suspension was produced by dispersing GO in DI H2O and ultrasonicated for 10 min. Polyoxyethylene 20 was added to it and was again ultrasonicated for 20 min to produce a uniform suspension. Concentration of surfactant was 0.2% (v/v) w.r.t to suspension. Finally, the pH was adjusted to 10 by 1 M NaOH solution to functionalize GO just before its assembly with SF. The resulting suspension was 1 mg mL−1 GO.
2.4 Preparation of silk fibroin (SF) aqueous solution
Silkworm (Bombyx mori) cocoons were cleaned and were cut into small pieces. Degumming was done by boiling 2.5 g of cocoons for 30 min in 1000 mL aqueous solution of 0.02 M Na2CO3 at 100 °C. Degummed fibers were washed several times with DI H2O to remove sericin protein and were dried overnight at room temperature. The dried silk fibers were cut and then dissolved in 9.3 M LiBr aqueous solution (1:4 weight by weight) at 60 °C for 4 hours. The resultant solution was dialyzed in a cellulose tube against DI H2O for 4 days at 4 °C. The obtained silk solution was centrifuged at 1000 rpm for 15 min at 4 °C. The yield of SF aqueous solution was adjusted to 4% w/v and was kept at 4 °C till further use.41
2.5 Assembly of GO-SF nanocomposites and electrode modification
Three different concentrations of GO (1%, 2% and 5%) w.r.t to SF (w/w) nanocomposites were prepared. The resulting nanocomposites were labeled as C1 (1% GO w.r.t SF w/w), C2 (2% GO w.r.t SF w/w), and C3 (5% GO w.r.t SF w/w). Carbon cloth (CC) electrodes were cut, washed sequentially in acetone, ethanol, and DI H2O, and dried. 30 μL of GO, SF, C1, C2, and C3 were drop-casted on separate carbon cloth electrodes for electrochemical studies.3 Results and discussions
Surface morphology of the modified electrodes was analyzed by scanning electron microscopy and the results revealed successful modification of carbon cloth. Fig. 1 elucidated that graphene oxide was firmly adsorbed on the smooth surface of carbon cloth. This was due to GO′s sp2 graphite structure, which is favorable to carbon cloth.42 The results showed that GO, SF and GO-SF were chemically grafted onto the surface of the electrode and virtually enhanced the surface roughness of the carbon cloth. This strong bonding generated an electroactive surface of the interface exhibiting enormous active sites which eventually enhanced the adsorption of DA on the interface, thus improving its catalytic activity. The average diameter of fibers as calculated by Image J came out to be 9.62 μm, 9.53 μm, and 9.57 μm for GO, SF and GO-SF modified carbon cloths respectively. | ||
| Fig. 1 SEM images of GO/CC (A), SF/CC (B), and C2/CC (C) at 13k (A and C) and 7k (B) magnifications. | ||
Energy dispersive X-ray spectroscopy was utilized for determining elemental composition. Fig. S1† demonstrated the stoichiometric elemental composition of graphene oxide, silk fibroin and GO-SF (C2) modified carbon cloth. The peak of carbon can be evidenced dominantly in all the three samples along with oxygen.
FTIR spectra were recorded to demonstrate the functionalities of the pristine materials and nanocomposites. Absorbance spectra were measured in 4000–500 cm−1 range with 256 number of scans and a resolution of 8 cm−1, as can be envisioned in Fig. 2(A). The spectra showed O–H stretching vibrations around 3143.55 cm−1 due to hydroxyl group,43 and the peak at 1574.56 cm−1 due to carbonyl and carboxyl moieties.44 The peak at 1371.30 cm−1 indicated C–OH stretching vibrations while the peak at 803.70 cm−1 indicated epoxy group.45 Absorption peaks at 1056.45 cm−1 and 1016.13 cm−1 could be accredited to C–O stretching.46 The high intensity of major peaks proved the presence of active functional groups produced due to oxidation of graphite. The spectra of extracted SF indicate three major vibrational bands. These include C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of amide I (1621.94 cm−1), amide II (1512.89 cm−1) owing to C–N stretching and N–H bending within the plane of SF backbone, and C–N stretching due to amide III (1229.45 cm−1).47 Amide I (1700–1600 cm−1) and amide II (1600–1500 cm−1) vibrations gave information about the secondary structure (random coil conformation) of the silk protein backbone.48 A broad peak at 3270.32 cm−1 affirmed the β-sheet conformation.46
O stretching vibrations of amide I (1621.94 cm−1), amide II (1512.89 cm−1) owing to C–N stretching and N–H bending within the plane of SF backbone, and C–N stretching due to amide III (1229.45 cm−1).47 Amide I (1700–1600 cm−1) and amide II (1600–1500 cm−1) vibrations gave information about the secondary structure (random coil conformation) of the silk protein backbone.48 A broad peak at 3270.32 cm−1 affirmed the β-sheet conformation.46
 | ||
| Fig. 2 (A) FTIR spectra of GO (a), SF (b), C1 (c), C2 (d), and C3 (e) in the range of 4000–500 cm−1 (B) XRD patterns of GO (a), SF (b), C1 (c), C2 (d), and C3 (e) at 2θ 5–60°. | ||
XRD patterns of pure GO, SF, and GO-SF were used to analyze the crystalline phase and structure of compounds. The XRD pattern of pristine GO (Fig. 2(B)) corroborated its characteristic diffraction peak at 2θ = 11.67° with d-spacing of 7.57 Å.49 Large interlayer distances of GO could be assigned to intercalating oxygen-containing functional groups, and due to H2O molecules in layered carbon structure.50 In the case of pure SF, the peak at 21.82° with d-spacing 4.09 Å referred to the silk II crystalline structure of fibroin.51 Peaks at 24.98° and 25.97° could be attributed to silk I structure, which is non-crystalline.52 Thus, XRD spectra of SF manifested its both crystalline and amorphous structure. The three nanocomposites (C1, C2, and C3) exhibited an amorphous structure with some small crystalline regions, but the overall amorphous structure was dominant. A broad hump appeared around 21° in C1, C2, and C3, and its intensity increased by the increase in GO content. The main peak of graphite at 11.67° was not found in any nanocomposite, which provided strong evidence of exfoliation of the layers of GO and their dispersion in the matrix.53
UV-vis spectra of pristine GO, SF, and GO-SF were analyzed in the range of 200–800 nm as demonstrated in Fig. 3(A). Pure SF displayed a prominent absorption peak at λ = 275 nm which could be assigned to π → π* transitions in SF due to the existence of amino acid residues, including tyrosine, phenylalanine, and tryptophan molecules.54 In the case of pristine GO, two minor absorption peaks appeared, where the peak at λ = 230 nm could be ascribed to π → π* transitions in aromatic C–C bonds, and the peak at λ = 300 nm could be ascribed to n → π* transitions of the carbonyl groups.55 In nanocomposite, the peak appeared at λ = 275 nm, which could be due to amino acid residues of SF, but the intensity of this peak was lowered as compared to SF due to the presence of GO.
 | ||
| Fig. 3 (A) UV-vis absorption spectra of GO (a), SF (b), and GO-SF (c) in the range of 200–800 nm. (B) Fluorescence emission spectra of GO (a), SF (b), and GO-SF (c) in the range of 300–450 nm at λex of 300 nm. | ||
The fluorescence emission spectra of GO, SF and GO-SF excited at 300 nm can be envisioned in Fig. 3(B). Pristine GO displayed no fluorescence at λex = 300 nm while at this excitation wavelength, pure SF exhibited an emission spectrum at 355 nm. In SF, aromatic amino acids including tryptophan, phenylalanine, and tyrosine are responsible for intrinsic fluorescent probes of intermolecular interactions of protein, its conformation, and dynamics. Among these, tryptophan is the most important probe for the determination of structural changes in a protein and its interaction with other molecules.56 The emission spectrum at 355 nm was ascribed to tryptophan and was dependent on secondary structure confirmation.57 When GO-SF nanocomposite was excited at λex = 300 nm, it displayed emission at 355 nm due to tryptophan in SF, but the intensity of this peak was lowered due to the presence of GO which showed no fluorescence at this excitation wavelength.
Moreover, contact angle and surface energy provided information about hydrophilicity of the materials. Water was used as a liquid with drop size of 4 μL. Pure SF displayed a contact angle of 66.19° and surface energy of 44.38 mN m−158 as can be envisioned in Table 1. The hydrophilic surface of SF could be assigned to carboxyl, amine, and hydroxyl groups in its structure.59 However, silk also contains alanine-rich hydrophobic units.60 Thus, all the three nanocomposites exhibited lower contact angles as compared to SF when amphiphilic GO was added into them.61 C1 exhibited a contact angle of 23.94°, due to the addition of GO filler.60 As the content of GO was increased, the contact angle increased to 38.10° in C2 and 56.78° in case of C3 due to an increase in hydrophobic π–π interactions. Overall, hydrophilicity dominated in all the samples as their contact angle was < 90°.
| Samples | Contact angle (°) | Surface energy (mN m−1) |
|---|---|---|
| SF | 66.19 | 44.38 |
| C1 | 23.94 | 66.56 |
| C2 | 38.10 | 59.82 |
| C3 | 56.78 | 49.28 |
Particle size and zeta potential of GO particles were evaluated using dynamic light scattering (DLS). The dispersity of particles as calculated from their polydispersity index (PDI) which is 0.972 (Fig. S2†). The large value of PDI accounts for higher polydispersity, which will lead to higher particle size variation.62 The Z-Average of GO was determined to be 1.36 μm, which proved the submicron size of particles due to the existence of GO in sheet structure. Individual peaks appeared at 604.4, 8.169, and 108 nm, which proved nano as well as submicron size of particles.
GO exhibited a zeta potential of −8.42 mV (Fig. S3†) due to negatively charged functional groups including hydroxyl, carboxyl, and epoxy groups, occurring on the surface of GO. This value of zeta potential provided an evaluation of sign and charge density at the surface of colloid particle.63 It also suggested that the negative zeta potential of GO is due to be the ionization of the functional groups on the surface of GO.64
The sensing efficacy of the working electrodes was investigated by electroanalytical techniques including CV, EIS, DPV, and chronoamperometry.
Cyclic voltammetry (CV) was employed to probe the electrochemical response of bare and modified carbon cloth electrodes. CV was measured in 0.1 M PBS (pH = 7.4) at potential of −0.8 to +0.8 V and at a scan rate of 100 mV s−1. Fig. 4(A) demonstrated that the redox peak current increased after modification of CC with GO and SF. The change in peak current was greater in case of modification with all the nano composites. C1/CC exhibited peak current of 47.02 μA, C2/CC 78.38 μA, and C3/CC 86.18 μA at 0.55 V. This can be accredited to enhanced surface area and increased electrochemical activity due to an increase in GO content.
 | ||
| Fig. 4 Cyclic Voltammetric response of (a) bare CC (b) GO/CC, (c) SF/CC, (d) C1/CC, (e) C2/CC, and (f) C3/CC in PBS (pH = 7.4) (A) and in 5 mM [Fe (CN6)]4−/3− in 0.1 M KCl (B), at 100 mV s−1 scan rate. (C) EIS of (a) bare CC (b) GO/CC, (c) SF/CC, (d) C1/CC, (e) C2/CC, and (f) C3/CC in 5 mM [Fe (CN6)]4−/3− in 0.1 M KCl. | ||
Charge transfer kinetics of bare and modified CC electrodes were examined using ferro/ferri solution as a redox probe in 0.1 M KCl. CV was measured in ferro/ferri at a potential of −1.2 to +1.3 V and 100 mV s−1 scan rate. The redox peak current value of the bare CC increased radically as GO was deposited on it, as demonstrated in Fig. 4(B). This can be accredited to the active functional groups of GO. SF/CC also exhibited a higher peak current in comparison to bare CC. C1/CC exhibited a better response because of the GO content in it. C2/CC and C3/CC displayed even higher redox currents due to increased surface area and better conductivity of these nanocomposites. This higher peak value also suggested that GO-SF nanocomposites increased the effective surface area of WE and added active sites to it due to the mutual superior properties of the pristine materials.
EIS was used to probe the interfacial properties of the electrode and the electrolyte, providing a Nyquist plot. EIS response of bare and modified CC in ferro/ferri revealed the charge transfer resistance Rct, which is directly proportional to the diameter of the semicircle of Nyquist plot. A low Rct value indicated rapid transfer of electrons. Bare CC depicted the highest Rct, which considerably decreased when CC was modified with GO and SF and most prominently with nanocomposites as shown in Fig. 4(C). The decrease in the value of Rct can be accredited to the higher electron transfer rate of the materials and the findings were in compliance with results of CV.
The proficiency of bare and modified electrodes towards dopamine was evaluated by performing CV in 0.2 mM dopamine in 0.1 M PBS solution (pH = 7.4) at 100 mV s−1 scan rate (Fig. 5). Bare CC displayed a minor redox current, but on modification with GO, it exhibited prominent redox peaks in dopamine solution. Two oxidation peaks appeared; the peak at 0.34 V was due to the oxidation reaction of DA to dopamine-o-quinone (DAQ), by its interaction with GO. The redox peak at −0.47 V indicated reformation of DA by DAQ. Another oxidation peak was also observed at 0.64 V, possibly depicting the transformation of DAQ to leucodopaminochrome (LDAC) as a result of deprotonation of the amino group, which was accredited to intramolecular cyclization due to 1,4-Michael addition which particularly occurs at pH 7 and above in PBS (7.4 in the present case).65,66

 | ||
| Fig. 5 CV curves of (a) bare CC, (b) GO/CC, (c) SF/CC, (d) C1/CC, (e) C2/CC, and (f) C3/CC in 0.2 mM dopamine in PBS (pH = 7.4) and at a scan rate of 100 mV s−1. | ||
In the case of SF/CC, the redox peak currents were less prominent. The redox peak currents in the case of C1/CC, C2/CC, and C3/CC were prominent with a slight shifting in their peak current values. Among them, the highest value of oxidation peak current was indicated in C2/CC, and due to the better stability of C2 in comparison to C3, it was chosen as model composite concentration for further electrochemical studies.
CV of C2/CC was analyzed at different scan rates from 50 to 270 mV s−1 to study the reaction kinetics and electron transfer process along with the surface activity. Scan rate effect on faradaic current value was measured in 0.2 mM DA, as can be seen in Fig. 6, which depicts that the increase in scan rate led to an increase in redox current value. It could be reasoned that at higher scan rates, the surface of the electrode acquires more time and larger interactions with the analyte. It resulted in the higher value of faradaic current as well as enhancement in catalytic activity, so a single oxidation peak appeared at 50 mV s−1 scan rate, while on increasing the scan rate, another oxidation peak appeared due to transformation of DAQ to LDAC.66 Moreover, by the increase in scan rate, a slight shift in potential was observed along with the increase in redox peak currents, which was assigned to fast electron transfer kinetics, suggesting that our process is a surface-controlled procedure as shown in Fig. 6 (B).67
| Ip = 0.096 mV s−1 + 10.968 (R2 = 0.9946) |
 | ||
| Fig. 6 (A) CV response of C2/CC and (B) its linear graph, in the presence of 0.2 mM DA in PBS (pH = 7.4) at 50 to 270 mV s−1 scan rate. | ||
The square root of scan rate has a linear relationship with anodic peak current, as depicted in Fig. S4† which provided evidence of adsorption-controlled process68
| Ip = 2.04 mV s−1 + 1.04 (R2 = 0.968) |
Surface coverage (Γ) of DA on the working interface of C2/CC was evaluated by Lavirion equation.
 500 C mol−1), R represents general gas constant (8.314 J mol−1. K) and T refers to the temperature (298 K). The Γ of DA on the working interface came out to be 1.46 × 10−4 mol cm−2. This larger value of Γ elucidated fast electron transfer and increased electrochemical activity of the designed interface towards dopamine estimation.
500 C mol−1), R represents general gas constant (8.314 J mol−1. K) and T refers to the temperature (298 K). The Γ of DA on the working interface came out to be 1.46 × 10−4 mol cm−2. This larger value of Γ elucidated fast electron transfer and increased electrochemical activity of the designed interface towards dopamine estimation.
The effect on sensing efficacy of C2/CC by changing the concentration DA was analyzed by measuring its cyclic voltammetric response in 10 μM to 900 mM DA in PBS (pH = 7.4) at −0.8 to +0.8 V potential and 50 mV s−1 scan rate as evident in Fig. 7(A). On increasing DA concentration, peak current value increased linearly. This could be ascribed to the fact that at higher concentrations of DA, more active sites of the electrode interface were involved; as a result peak current increased.
 | ||
| Fig. 7 (A) CV curves of C2/CC at different concentrations of DA (10–900 μM) in PBS (pH = 7.4) and 50 mV s−1 scan rate. (B) DPV of C2/CC in 0.2 mM DA in PBS at pH = 3 (a), 4 (b), 5 (c), 6 (d), 7 (e), 7.4 (f), 8 (g). | ||
DPV was employed to investigate the electrochemical behavior of the developed C2/CC sensor in different pH ranges, due to its distinct current response, low background noise, magnified resolution, and its potential to reduce non-faradic response.69 The electrolyte's pH has a significant effect on electrochemical behavior because it alters the potential as well as the Faradaic current. The effect of change in pH on electrolytic oxidation of DA (0.2 mM) at the C2/CC interface was investigated by performing DPV in PBS from pH 3 to 8, as depicted in Fig. 7(B). The electrode revealed a better analytical response at pH 6, 7, and 7.4. Above and below these pH values, the peak current value decreased. It demonstrated that C2/CC can work well at the pH of the human body; hence pH 7.4 was selected for all the experiments.
Ascorbic acid (AA) and uric acid (UA) are the main interfering analytes in the signaling of DA, so the simultaneous detection of DA in the existence of these interfering analytes was evaluated through CV as shown in Fig. 8(A). CV was performed in 0.2 mM UA which showed very low peak current values. AA 0.2 mM also exhibited low peak current. In the case of DA 0.2 mM, ipc = 270 μA at 0.30 V, and ipa is −271 μA at −0.4 V were observed. When 0.2 mM of AA was added to 0.2 mM UA in PBS, an ipc of 171 μA was produced at 0.48 V whereas no reduction peak was produced. When 0.1 mM DA was added to the same solution, two oxidation peaks were observed, ipc of 272 μA at 0.29 V which was the characteristic peak of oxidation of DA to DAQ, and another peak of ipc = 265 μA at 0.44 V which depicted the oxidation of interfering species. The reduction peak of DA was also produced, which was slightly shifted due to the occurrence of UA and AA. This proved that the as-prepared electrode exhibited an efficient selectivity for low-level detection of DA in the occupancy of interfering analytes including UA and AA. On increasing the concentration of DA to 0.2 mM, there was a sharp increase in redox peak current values.
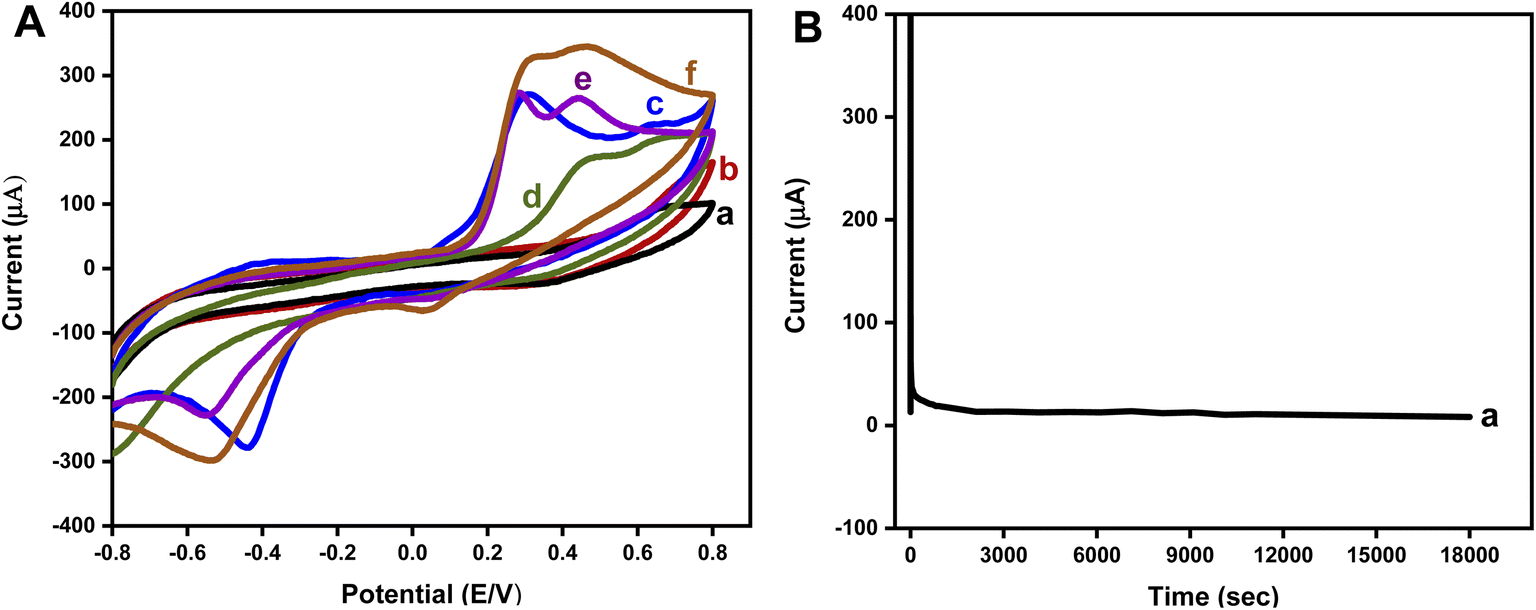 | ||
| Fig. 8 (A) Cyclic voltammetric response of C2/CC in (a) UA 0.2 mM, (b) AA 0.2 mM (c) DA 0.2 mM, (d) UA 0.2 mM + AA 0.2 mM, (e) UA 0.2 mM + AA 0.2 mM + DA 0.1 mM, (e) UA 0.2 mM + AA 0.2 mM + DA 0.2 mM, in PBS (pH = 7.4) at 50 mV s−1 scan rate. (B) Chronoamperometry of C2/CC in DA 0.2 mM in PBS (pH = 7.4) for 18000 s. | ||
Chronoamperometry was performed to evaluate the stability index and durability of the electrode. Response of C2/CC in 0.2 mM DA was analyzed for 5 hours as envisioned in Fig. 8(B). A potential of 0.34 V was applied, which was the oxidation potential of DA in C2/CC. The fabricated electrode exhibited admirable stability for up to 18000 seconds.
Similarly, selectivity of the C2/CC was investigated by CV measurements of the electrode in the presence of nine different analytes at a potential of −0.8 to +0.8 V and 50 mV s−1 scan rate. The prepared biosensor showed excellent selectivity towards DA as shown in Fig. 9(A). DA exhibited a peak current of 270 μA at 0.34 V, while the other analytes such as glucose, AA, MgCl2, UA, NaCl, urea, and KCl showed very small values of peak current at oxidation potential of DA. This proved the practical applicability of the developed electrode towards selective detection of DA even in complex environments.
 | ||
| Fig. 9 (A) Error bar diagram of C2/CC against 0.2 mM of DA, glucose, UA, AA, MgCl2, NaCl, H2O2, KCl, and Urea, in PBS (pH = 7.4) and 50 mV s−1 scan rate. (B) Response of C2/CC in DA 0.2 mM for 10 repeated cycles. | ||
The repeatability of developed biosensor was evaluated under analogous parameters by running 10 cycles of C2/CC in DA 0.2 mM at a scan rate of 100 mV s−1. The findings are shown in Fig. 9(B). The results proved that the fabricated electrode exhibited good precision and repeatability. It is even better than many of the reported materials in terms of LoD and linear range, as shown in Table 2.
| Sr. No | Electrode matrix | LoD (μM) | Sensitivity (μA μM−1 cm−2) | Linear range (μM) | Ref. |
|---|---|---|---|---|---|
| 1 | L-arginine/CPE | 0.5 | — | 50–100 | 70 |
| 2 | ITO/Au/GO | 1.28 | 0.53 | 0.1–30 | 71 |
| 3 | Graphene | 2.64 | — | 4–100 | 72 |
| 4 | GCE/N-rGO/MnO | 30 | 0.09 | 10–180 | 73 |
| 5 | GCE/N-rGO | 0.93 | — | 120–220 | 74 |
| 7 | Fe3O4-Gr/GCE | 0.7 | — | 5.0–160 | 74 |
| 8 | PEDOT-LSG | 0.33 | 0.220 ± 0.011 | 1–150 | 75 |
| 9 | Pd-NCd/rGo | 7.02 | 0.943 | 20–220 | 76 |
| 10 | GO-SF/CC | 0.41 | 2.46 | 100–900 | This work |
4 Conclusion
In this study, we fabricated a sensor based on GO and SF nanocomposite deposited on carbon cloth and was investigated for low-level detection of dopamine. The designed interface proved to be a highly proficient as well as miniaturized, robust, and simple sensing platform for estimation of DA. The prepared sensor exhibited enhanced selectivity and superior sensitivity in the occupancy of interfering analytes. The synergistic effect of GO and SF in GO-SF/CC led to enhanced active surface area and surface coverage of DA at sensing interface, consequently providing superior electrochemical performance. Low LoD and wide linear range offer the valuable potential of this fabricated sensor to be utilized for real samples and can have great practical applicability in the biomedical field.Conflicts of interest
Authors claim NO conflict of interest.Acknowledgements
MHN acknowledges the financial support provided by HEC (20–4993/R&D/HEC/14/614).References
- L. Barthelmebs, A. Hayat, A. W. Limiadi, J.-L. Marty and T. Noguer, Sens. Actuators, B, 2011, 156, 932–937 CrossRef CAS.
- D.-S. Kim, E.-S. Kang, S. Baek, S.-S. Choo, Y.-H. Chung, D. Lee, J. Min and T.-H. Kim, Sci. Rep., 2018, 8, 14049 CrossRef PubMed.
- W. Poewe, K. Seppi, C. M. Tanner, G. M. Halliday, P. Brundin, J. Volkmann, A.-E. Schrag and A. E. Lang, Nat. Rev. Dis. Primers, 2017, 3, 17013 CrossRef PubMed.
- A. F. Arnsten, R. R. Girgis, D. L. Gray and R. B. Mailman, Biol. Psychiatry, 2017, 81, 67–77 CrossRef CAS PubMed.
- M. Singh, I. Tiwari, C. W. Foster and C. E. Banks, Mater. Res. Bull., 2018, 101, 253–263 CrossRef CAS.
- M. G. Shapiro, G. G. Westmeyer, P. A. Romero, J. O. Szablowski, B. Küster, A. Shah, C. R. Otey, R. Langer, F. H. Arnold and A. Jasanoff, Nat. Biotechnol., 2010, 28, 264–270 CrossRef CAS PubMed.
- X. Wang, Y. Ma, X. Yao, J. Wang and M. Yin, RSC Adv., 2013, 3, 24605–24611 RSC.
- P. Sivakumar, S. Priyatharshni and K. Kumar, Mater. Chem. Phys., 2020, 240, 122167 CrossRef CAS.
- D. Wen, W. Liu, A. K. Herrmann, D. Haubold, M. Holzschuh, F. Simon and A. Eychmüller, Small, 2016, 12, 2439–2442 CrossRef CAS PubMed.
- Y. Lin, C. Chen, C. Wang, F. Pu, J. Ren and X. Qu, Chem. Commun., 2011, 47, 1181–1183 RSC.
- J. Wen, L. Zhou, L. Jin, X. Cao and B.-C. Ye, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1793–1798 CrossRef CAS PubMed.
- K. Jackowska and P. J. A. Krysinski, Anal. Bioanal. Chem., 2013, 405, 3753–3771 CrossRef CAS PubMed.
- U. Amara, S. Riaz, K. Mahmood, N. Akhtar, M. Nasir, A. Hayat, M. Khalid, M. Yaqub and M. H. Nawaz, RSC Adv, 2021, 11, 25084–25095 RSC.
- D. P. Quan, D. P. Tuyen, T. D. Lam, P. T. N. Tram, N. H. Binh and P. H. Viet, Colloids Surf., B, 2011, 88, 764–770 CrossRef CAS PubMed.
- J. Yang, Y. Hu and Y. Li, Biosens. Bioelectron., 2019, 135, 224–230 CrossRef CAS PubMed.
- M. Holzinger, A. Le Goff and S. J. Cosnier, Front. Chem., 2014, 2, 63 Search PubMed.
- M. Terrones, A. R. Botello-Méndez, J. Campos-Delgado, F. López-Urías, Y. I. Vega-Cantú, F. J. Rodríguez-Macías, A. L. Elías, E. Muñoz-Sandoval, A. G. Cano-Márquez, J.-C. Charlier and H. Terrones, Nano Today, 2010, 5, 351–372 CrossRef.
- N. L. Teradal and R. J. Jelinek, Adv. Healthcare Mater., 2017, 6, 1700574 CrossRef PubMed.
- O. C. Compton, Z. An, K. W. Putz, B. J. Hong, B. G. Hauser, L. C. Brinson and S. T. Nguyen, Carbon, 2012, 50, 3399–3406 CrossRef CAS.
- M. Meyyappan, Nano Convergence, 2015, 2, 18 CrossRef.
- E. Rand, A. Periyakaruppan, Z. Tanaka, D. A. Zhang, M. P. Marsh, R. J. Andrews, K. H. Lee, B. Chen, M. Meyyappan and J. E. Koehne, Biosens. Bioelectron., 2013, 42, 434–438 CrossRef CAS PubMed.
- D. Porter and F. Vollrath, Adv. Mater., 2009, 21, 487–492 CrossRef CAS.
- K. Hu, M. K. Gupta, D. D. Kulkarni and V. V. Tsukruk, Adv. Mater., 2013, 25, 2301–2307 CrossRef CAS PubMed.
- J. Melke, S. Midha, S. Ghosh, K. Ito and S. J. Hofmann, Acta Biomater., 2016, 31, 1–16 CrossRef CAS PubMed.
- F. Mottaghitalab, M. Farokhi, M. A. Shokrgozar, F. Atyabi and H. J. Hosseinkhani, J. Controlled Release, 2015, 206, 161–176 CrossRef CAS PubMed.
- S. Mehrotra, D. Chouhan, R. Konwarh, M. Kumar, P. K. Jadi and B. B. Mandal, ACS Biomater. Sci. Eng., 2019, 5, 2054–2078 CrossRef CAS PubMed.
- C. Wang, K. Xia, Y. Zhang and D. L. Kaplan, Acc. Chem. Res., 2019, 52, 2916–2927 CrossRef CAS PubMed.
- A. Benvidi, Z. Abbasi, M. D. Tezerjani, M. Banaei, H. R. Zare, H. Molahosseini and S. Jahanbani, Acta Chim. Slov., 2018, 65, 278–288 CrossRef CAS PubMed.
- A. Abdulkhani, M. D. Sousefi, A. Ashori and G. Ebrahimi, Polym. Test., 2016, 52, 218–224 CrossRef CAS.
- K. Li, P. Li and Y. Fan, J. Mater. Chem. B, 2019, 7, 6890–6913 RSC.
- C. B. Marín, V. Fitzpatrick, D. L. Kaplan, J. Landoulsi, E. Guénin and C. Egles, Front. Chem., 2020, 8, 604398 CrossRef PubMed.
- N. V. Padaki, B. Das and A. Basu, in Advances in Silk Science and Technology, ed. A. Basu, Woodhead Publishing, 2015, pp. 3–16, DOI:10.1016/B978-1-78242-311-9.00001-X.
- M. Jian, C. Wang, Q. Wang, H. Wang, K. Xia, Z. Yin, M. Zhang, X. Liang and Y. Zhang, Sci. China Mater., 2017, 60, 1026–1062 CrossRef CAS.
- S. Tadepalli, H. Hamper, S. H. Park, S. Cao, R. R. Naik and S. Singamaneni, ACS Biomater. Sci. Eng., 2016, 2, 1084–1092 CrossRef CAS PubMed.
- S. Zhao, J. Li, D. Cao, G. Zhang, J. Li, K. Li, Y. Yang, W. Wang, Y. Jin, R. Sun and C.-P. Wong, ACS Appl. Mater. Interfaces, 2017, 9, 12147–12164 CrossRef CAS PubMed.
- Y. Wan, Y. Wang and C. F. Guo, Mater. Today Phys., 2017, 1, 61–73 CrossRef.
- M. A. Gilmartin and J. P. Hart, Analyst, 1995, 120, 1029–1045 RSC.
- X. Liu, W. Xu, D. Zheng, Z. Li, Y. Zeng and X. Lu, J. Mater. Chem. A, 2020, 8, 17938–17950 RSC.
- R. M. Kakhki, Arabian J. Chem., 2019, 12, 1783–1794 CrossRef.
- J. Wu, X. Shen, L. Jiang, K. Wang and K. Chen, Appl. Surf. Sci., 2010, 256, 2826–2830 CrossRef CAS.
- U.-J. Kim, J. Park, H. J. Kim, M. Wada and D. L. Kaplan, Biomaterials, 2005, 26, 2775–2785 CrossRef CAS PubMed.
- F. Li, Y. Hua, C.-B. Qu, H.-M. Xiao and S.-Y. Fu, Composites, Part A, 2016, 89, 47–55 CrossRef CAS.
- Y. Shen, S. Yang, P. Zhou, Q. Sun, P. Wang, L. Wan, J. Li, L. Chen, X. Wang and S. Ding, Carbon, 2013, 62, 157–164 CrossRef CAS.
- N. Zaaba, K. Foo, U. Hashim, S. Tan, W.-W. Liu and C. Voon, Procedia Eng., 2017, 184, 469–477 CrossRef CAS.
- V. Loryuenyong, K. Totepvimarn, P. Eimburanapravat, W. Boonchompoo and A. Buasri, Adv. Mater. Sci. Eng., 2013, 2013 Search PubMed.
- M. Narimani, A. Teimouri and Z. Shahbazarab, Polym. Bull., 2019, 76, 725–745 CrossRef CAS.
- A. Teimouri, R. Ebrahimi, R. Emadi, B. H. Beni and A. N. Chermahini, Int. J. Biol. Macromol., 2015, 76, 292–302 CrossRef CAS PubMed.
- S. Ling, Z. Qi, D. P. Knight, Z. Shao and X. Chen, Polym. Chem., 2013, 4, 5401–5406 RSC.
- S. Sadhukhan, T. K. Ghosh, D. Rana, I. Roy, A. Bhattacharyya, G. Sarkar, M. Chakraborty and D. Chattopadhyay, Mater. Res. Bull., 2016, 79, 41–51 CrossRef CAS.
- M. El Achaby, F. Arrakhiz, S. Vaudreuil, E. Essassi and A. Qaiss, Appl. Surf. Sci., 2012, 258, 7668–7677 CrossRef CAS.
- R. E. Marsh, R. B. Corey and L. Pauling, Biochimica et biophysica acta, 1955, 16, 1–34 CrossRef CAS PubMed.
- Y. Tamada, Biomacromolecules, 2005, 6, 3100–3106 CrossRef CAS PubMed.
- M. Rafi, B. Samiey and C.-H. Cheng, Materials, 2018, 11, 496 CrossRef PubMed.
- S. Asha, Y. Sangappa and G. Sanjeev, J. Opt., 2016, 45, 66–72 CrossRef.
- D. Chen, L. Li and L. Guo, Nanotechnol., 2011, 22, 325601 CrossRef PubMed.
- Y. Yang, Z. Shao, X. Chen and P. Zhou, Biomacromolecules, 2004, 5, 773–779 CrossRef CAS PubMed.
- I. Georgakoudi, I. Tsai, C. Greiner, C. Wong, J. DeFelice and D. Kaplan, Opt. Express, 2007, 15, 1043–1053 CrossRef CAS PubMed.
- J. Zhang, S. Du, A. Kafi, B. Fox, J. L. Li, X. Liu, R. Rajkhowa and X. Wang, RSC Adv., 2015, 5, 1640–1647 RSC.
- J. Chen, G. Cheng, R. Liu, Y. Zheng, M. Huang, Y. Yi, X. Shi, Y. Du and H. J. Deng, J. Colloid Interface Sci., 2018, 523, 208–216 CrossRef CAS PubMed.
- S. Z. Fu, X. H. Wang, G. Guo, S. Shi, M. Fan, H. Liang, F. Luo and Z. Y. J. Qian, J. Biomed. Mater. Res., Part B, 2011, 97, 74–83 CrossRef PubMed.
- H. P. Bei, Y. Yang, Q. Zhang, Y. Tian, X. Luo, M. Yang and X. Zhao, Molecules, 2019, 24, 658 CrossRef CAS PubMed.
- J. Kundu, Y.-I. Chung, Y. H. Kim, G. Tae and S. Kundu, Int. J. Pharm., 2010, 388, 242–250 CrossRef CAS PubMed.
- B. Konkena and S. J. T. Vasudevan, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS PubMed.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- M.-S. Hsu, Y.-L. Chen, C.-Y. Lee and H.-T. Chiu, Appl. Mater. Interfaces, 2012, 4, 5570–5575 CrossRef CAS PubMed.
- A. Ejaz, Y. Joo and S. Jeon, Sens. Actuators, B, 2017, 240, 297–307 CrossRef CAS.
- W. He, R. Liu, P. Zhou, Q. Liu and T. Cui, Bioelectronics, 2020, 167, 112473 CrossRef CAS PubMed.
- Y. Zhou, W. Tang, J. Wang, G. Zhang, S. Chai, L. Zhang and T. Liu, Anal. Methods, 2014, 6, 3474–3481 RSC.
- L. A. Mercante, A. Pavinatto, L. E. Iwaki, V. P. Scagion, V. Zucolotto, O. N. Oliveira Jr, L. H. Mattoso and D. S. Correa, ACS Appl. Mater. Interfaces, 2015, 7, 4784–4790 CrossRef CAS PubMed.
- M. Amiri, H. Eynaki and Y. Mansoori, Electrochim. Acta, 2014, 123, 362–368 CrossRef CAS.
- S.-S. Choo, E.-S. Kang, I. Song, D. Lee, J.-W. Choi and T.-H. Kim, Sensors., 2017, 17, 861 CrossRef PubMed.
- Y.-R. Kim, S. Bong, Y.-J. Kang, Y. Yang, R. K. Mahajan, J. S. Kim and H. Kim, Biosens. Bioelectron., 2010, 25, 2366–2369 CrossRef CAS PubMed.
- R. Chen, Y. Wang, Y. Liu and J. Li, RSC Adv., 2015, 5, 85065–85072 RSC.
- S.-M. Li, S.-Y. Yang, Y.-S. Wang, C.-H. Lien, H.-W. Tien, S.-T. Hsiao, W.-H. Liao, H.-P. Tsai, C.-L. Chang and C.-C. M. Ma, Carbon, 2013, 59, 418–429 CrossRef CAS.
- G. Xu, Z. A. Jarjes, V. Desprez, P. A. Kilmartin and J. Travas-Sejdic, Biosens. Bioelectron., 2018, 107, 184–191 CrossRef CAS PubMed.
- Y.-S. Hsieh, B.-D. Hong and C.-L. Lee, Microchim. Acta, 2016, 183, 905–910 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra05585f |
| This journal is © The Royal Society of Chemistry 2022 |