
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the catalytic properties of La–Mn perovskite catalyst via variation of A- and B-sites: effect of Ce and Cu substitution on selective catalytic reduction of NO with NH3
Wei Zhang *ab,
Kang Xieab,
Yunhao Tangab,
Shan Chengab,
Mengxia Qingab,
Yanni Xuanab,
Chuan Qinab,
Mengyao Dongab,
Yunhe Zhouab and
Jie Liab
*ab,
Kang Xieab,
Yunhao Tangab,
Shan Chengab,
Mengxia Qingab,
Yanni Xuanab,
Chuan Qinab,
Mengyao Dongab,
Yunhe Zhouab and
Jie Liab
aSchool of Energy and Power Engineering, Changsha University of Science & Technology, Changsha, 410114, China. E-mail: weizhang@csust.edu.cn
bKey Laboratory of Renewable Energy Electric-Technology of Hunan Province, Changsha, 410114, China
First published on 15th August 2022
Abstract
Perovskites with flexible structures and excellent redox properties have attracted considerable attention in industry, and their denitration activities can be further improved with metal substitution. In order to investigate the effect of Ce and Cu substitution on the physicochemical properties of perovskite in NH3-SCR system, a series of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts were prepared by citrate sol-gel method and employed for NO removal in the simulated flue gas, and the physical and chemical properties of the catalysts were studied using XRD, SEM, BET, XPS, DRIFT characterizations. The Ce substitution on A-site cation of LaMnO3 can improve the denitration activity of the perovskite catalyst, and La0.9Ce0.1MnO3 displays NO conversion of 86.7% at 350 °C. The characterization results indicate that the high denitration activity of La0.9Ce0.1MnO3 is mainly attributed to the larger surface area, which contributes to the adsorption of NH3 and NO. Besides, the appropriate Cu substitution on B-site cation of La0.9Ce0.1MnO3 can further improve the denitration activity of perovskite catalyst, and La0.9Ce0.1Mn0.8Cu0.2O3 displays the NO conversion of 91.8% at 350 °C. Although the specific surface area of La0.9Ce0.1Mn0.8Cu0.2O3 is lower than La0.9Ce0.1MnO3, the Cu active sites and the Ce3+ contents are more developed, making many reaction units formed on the catalyst surface and redox properties of catalyst improved. In addition, strong metal interaction (Ce4+ + Mn2+ + Cu2+ ↔ Ce3+ + Mn3+/Mn4+ + Cu+) and high concentrations of chemical adsorbed oxygen and lattice oxygen both strengthen the redox reaction on catalyst surface, thus contributing to the better denitration activity of La0.9Ce0.1Mn0.8Cu0.2O3. Therefore, appropriate cerium and copper substitution will markedly improve the denitration activity of La–Mn perovskite catalyst. We also reasonably conclude a multiple reaction mechanism during NH3-SCR denitration process basing on DRIFT results, which includes the Eley–Rideal mechanism and Langmuir–Hinshelwood mechanism.
1. Introduction
Nitrogen oxide (NOx) is one of the thorniest and most widespread environmental pollutants in combustion flue gas produced from automobile exhaust emissions and industrial processes. NOx is considered a major cause of photochemical smog, ozone depletion, acid rain and the greenhouse effect.1,2 At present, NOx emission is regulated and standardized worldwide to minimize the harm to the environment. Selective catalytic reduction of NOx with NH3 (NH3-SCR) is considered to be the most widely-applied and effective denitration technology.3,4 Typical commercial vanadium-titanium catalysts (V2O5/TiO2 and V2O5-WO3/TiO2) have been commonly used in many industrial activities due to their stability and high efficiency, but are still restrained by their inevitable drawbacks such as vanadium toxicity, narrow and high operation temperature window, poisoning and deactivation of the catalysts by deposited alkaline metals.5–7 Moreover, there is an increasing requirement for limiting NOx emission from industrial applications, which has made the development of NH3-SCR catalysts with excellent denitration activity a hot issue for decades.In recent years, transition metal and rare earth metal catalysts have been widely researched for NH3-SCR systems. Transition metal oxides possess high denitration activity due to their various oxidation states, and rare earth metal oxides have excellent oxygen storage capacity and redox properties. Moreover, perovskite oxides also exhibit good structure stability beyond the above superiorities, thus they have received much attention in the field of NH3-SCR catalysts. Perovskite oxides have a typical chemical formula of ABO3, where A is a lanthanide or alkaline cation and B is a transition metal cation. The B cation is six-fold coordinated with the oxygen atoms to form BO6 octahedron, while A cations occupy the voids created by the BO6 octahedrons.8 Owning to the presence of BO6 octahedron, perovskite is capable to maintain the structure stability when A and B cations are partially substituted by other metal ions. This could induce the lattice distortion and symmetry reduction,9 causing the BO6 octahedral units change from ideal cubic symmetry to the rhombohedral symmetry or orthorhombic symmetry. The cubic symmetry can provide a larger diffusion space than the rhombohedral or orthorhombic symmetry, making the oxide ions diffuse more easily.10 While the lattice distortion leads to the formation of abundant oxygen vacancies, promoting the surface oxygen species adsorption and increasing the lattice oxygen migration.11 Furthermore, lanthanide metal cation substitutes in A-site cation will produce lattice defects and oxygen vacancies, and transition metal cation substitutes in B-site cation will directly changes its oxidation valence state, thus affecting the redox performance of perovskite composite oxides. Therefore, A- and B-sites can be substituted by designated cations with different valences or radius to improve perovskite physicochemical properties for industrial application.12
Mn-based catalysts have the multivalent oxidation states mainly including Mn2+, Mn3+ and Mn4+, and the mutual conversion of three valence states is crucial to the denitration activity of catalyst in NH3-SCR reaction.13,14 Among the Mn-containing catalysts, LaMnO3 has attracted interest with its Mn3+/Mn4+ coexisted oxidation state and over-stoichiometric oxygen,15,16 excessive oxygen always formed to guarantee the lattice electroneutrality of the perovskite due to increasing Mn4+ content. The research also shows that copper substitution can promote the generation of oxygen vacancies, so that NO adsorbed on the surface oxygen vacancies can obtain more electrons and be easier to be reduced. Cu partially substitutes Mn ions can lead to the synergistic effect of Cu2+ and Mn3+, meanwhile improve the denitration performance of the catalyst.16 Cerium oxide is usually adopted in the NH3-SCR catalysts to enhance their denitration activities due to the excellent oxygen storage via the redox shift between Ce4+ and Ce3+.17 It has also been reported that chemisorbed oxygen, Mn4+ content, NH3 adsorption capacity and reducibility can be further increased with cerium substitution.13 Besides, NO oxidation over LaMnO3+δ catalysts also can be improved by selectively removing the La atoms.17 Basing on the above information, it is promising and logical to develop NH3-SCR catalysts by partially replacing La in with cerium, partially replacing Mn with copper and combining the prominent properties of Ce, Cu and perovskite oxides.
In this paper, we have synthesized various La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts with citrate sol-gel method, and the efficiency of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts on the NO removal in simulated flue gas was carried to evaluate the NH3-SCR catalytic activity. We also investigated the inherent relationships between structure, morphology, surface element composition and catalytic activity of the catalysts. The structure, morphological and redox properties of the catalysts were studied by XRD, SEM, BET and XPS. Besides, the mechanism of denitration reaction on perovskite catalyst surface was discussed with DRIFT results. This study focused on the effects of Ce and Cu substitution on the physicochemical properties and denitration activity of the La–Mn perovskite catalysts and would provide a possibility of controlling the catalytic properties of NH3-SCR catalysts for industrial application by changing the composition of A- and B-sites of La–Mn perovskite catalysts.
2. Materials and methods
2.1 Catalyst preparation
The series of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts were synthesized by the citrate sol-gel method. The chemicals of La(NO3)3·6H2O (99%, Macklin, China), Ce(NO3)3·6H2O (99.95%, Macklin, China), Mn(NO3)2 (50 wt% aqueous solution, Macklin, China) and Cu(NO3)2·3H2O (99%, Xilong scientific, China) with specific ratios were dissolved in deionized water and mixed with magnetic stirrer. The citric acid (99.5%, Macklin, China) was used as a chelating agent and added into the above solution, where the mole ratio of citric acid to total metal ions was 1.2. Next, the solution was mixed at 90 °C in a magnetic stirrer until yellow viscous gel was formed. Then the gel was dried at 120 °C for 12 h at an oven, and the spongy precursor was obtained. After that, the precursor was ground and calcined at 700 °C for 5 h in a muffle furnace to form the perovskite structure. Additionally, the heating ramp rates of evaporation and calcination were 5 °C min−1 and 10 °C min−1, respectively. Finally, the samples were ground and sieved using a 40–60 mesh sieve for catalytic activity tests.2.2 Catalyst characterization
The X-ray Diffraction (XRD) patterns of the samples were measured in an X'Pert PRO MPD diffractometer (PANalytical, Netherlands) with Cu Kα radiation (λ = 0.15406 nm, 45 kV, 40 mA). The diffraction angle was selected in the 2θ range from 10° to 80° with the scanning rate of 10°/min. The average crystal size was calculated from the Scherrer equation.6The Scanning Electron Microscopy (SEM) images were taken with a Zeiss sigma 300 field emission scanning electron microscope (Carl Zeiss AG, China) to study the morphology of catalysts.
The Brunauer Emmert Teller (BET) surface area and Barrett Joyner Halenda (BJH) pore size distribution of the catalysts were determined by N2 adsorption-desorption isotherms at −196 °C on a Micromeritics ASAP 2460 surface area and porosity analyzer (Micromeritics, USA) to study the textural properties. Before measurement, the samples were degassed at 300 °C for 6 h in vacuum.
The X-ray Photoelectron Spectroscopy (XPS) measurements were performed on a ESCALAB Xi+ spectrometer (Thermo Fischer, USA) equipped with Al Kα radiation serve as excitation source. The binding energies were corrected by C 1s peak at 284.6 eV to compensate the charging effect of all catalysts.
The Diffuse Reflaxions Infrared Fourier Transformations spectroscopy (DRIFT) were carried on Nicolet IS 50 spectrometer (Thermo Fischer, USA) equipped with intelligent collector and MCT/A detector, the scanning range was 4000–400 cm−1.
2.3 Denitration performance experiments
The denitration performance experiments were carried with a fixed-bed reactor in the temperature range of 150–400 °C. Here, 200 mg catalyst (40–60 mesh) was fitted in a quartz tube reactor (i.d. = 12 mm) in each test. The reaction conditions were presented as follows: 500 ppm NO, 500 ppm NH3, 5% O2 and N2 balanced. The total flow rate of the feeding gas was kept at 1000 mL min−1, corresponding to a gas hourly space velocity (GHSV) of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. During each temperature point, NO concentrations in the inlet and outlet gas were collected with a professional flue gas analyzer (Madur Photon & PGD-100, Austria) after NH3-SCR reaction was stable for 10 min. NO conversion was calculated according to the following equation:
000 h−1. During each temperature point, NO concentrations in the inlet and outlet gas were collected with a professional flue gas analyzer (Madur Photon & PGD-100, Austria) after NH3-SCR reaction was stable for 10 min. NO conversion was calculated according to the following equation:where XNO represents NO conversion, CNO(in) and CNO(out) are the NO concentrations at the inlet and outlet of the quartz tube reactor, respectively.
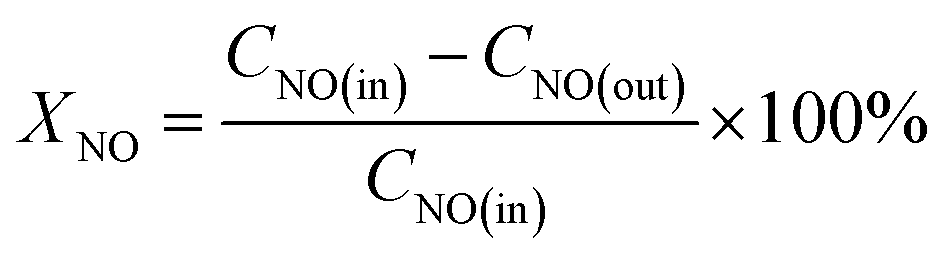
3. Results and discussion
3.1 Textural analysis basing on XRD results
The phase structures of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts were characterized by XRD instrument. As shown in Fig. 1, the diffraction peaks at 23.1°, 32.9°, 40.3°, 47.0°, 52.7°, 58.4°, 68.9°, 78.1° of the catalysts are assigned to the rhombohedral structure of LaMnO3.15 (JCPDS #50-0299), meaning that the LaMnO3+δ perovskite phases with over-stoichiometric oxygen were formed instead of the stoichiometric LaMnO3.18 The main diffraction peak (2θ = 32.9°) of the catalysts shifts to lower values with the addition of cerium and copper, which indicates that the cell volume become smaller due to the lattice distortion. When La3+ (1.17 Å) substituted by Ce4+ (0.92 Å) or Ce3+ (1.03 Å) with a smaller radius, the average radius of A-site cation of perovskite phase reduced, causing the lower tolerance factor value of crystal structure and leading to the decreased symmetry of perovskite (i.e., lattice distortion).19 Besides, the radius of Mn ions with +2, +3 and +4 valence are 0.83 Å, 0.65 Å and 0.54 Å, respectively. The radius of Cu ions with +1 and +2 valence are 0.96 Å and 0.73 Å, respectively. Therefore, Mn ions are replaced by Cu ions with higher average ion radius, which also causing the decrease of tolerance factor value and perovskite symmetry. In addition, the intensity of characteristic diffraction peak corresponding to LaMnO3.15 perovskite phase in La0.9Ce0.1Mn1−yCuyO3 (y = 0, 0.05, 0.1, 0.2, 0.4) catalysts strengthens with the increasement of Cu substitution, indicating that Cu substitution can improve the crystallinity and content of perovskite phase in La0.9Ce0.1Mn1−yCuyO3 (y = 0, 0.05, 0.1, 0.2, 0.4) catalysts. The diffraction peak (2θ = 28.2°) assigned to the structure of Ce2O3 (JCPDS #81-0792) can be observed in La0.9Ce0.1Mn0.95Cu0.05O3 and La0.9Ce0.1Mn0.9Cu0.1O3 catalysts,20 it is reported that Ce2O3 has prominent oxygen storage capacity and high reducibility owing to the redox shifting between Ce4+, Ce3+ and oxygen defects, which provides an important promotion effect on the NH3-SCR activity.21 However, with the increasement of Cu substitution ratio in the catalyst, the characteristic peak of Ce2O3 decreases gradually. The phenomena indicates that there may be strong interaction between Cu and Ce, and the interaction is affected by the amount of Cu substitution in the sample. The characteristic diffraction peak of copper oxide (JCPDS #89-2529) for La0.9Ce0.1Mn0.6Cu0.4O3 catalyst appears at 2θ = 35.6 and 38.7°,22 indicating that a small amount of CuO impurities were formed in La0.9Ce0.1Mn0.6Cu0.4O3 catalyst. Therefore, it is reasonable to conclude that lattice distortion caused by cerium and copper substitution will reduce the symmetry of perovskite phase, thus promoting the formation of oxygen vacancies and the improvement of denitration activity. However, excessive lattice distortion will shrink the diffusion space, making it is unfavorable to the diffusion of oxide ions, which may cause the substantial reduction in denitration activity of catalysts.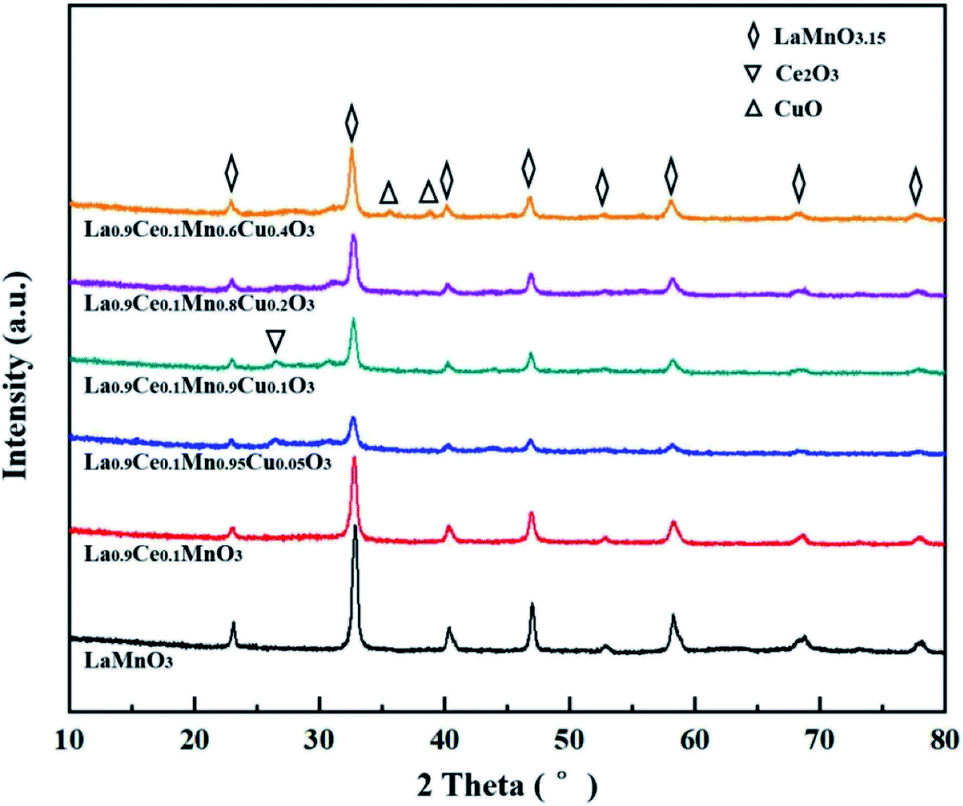 | ||
| Fig. 1 XRD patterns of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts. | ||
In order to further explore the crystal structure of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts, the unit cell volume and average crystal size shown in Table 1, which are obtained from XRD results dealt with MDI Jade 6.0 program. It can be observed that the cerium and copper substitution both will lead to the variability of the unit cell volume and average crystal size of catalysts, which are correlated with the lattice distortion and variation of average valence state and radius of A-site and B-site cations in catalysts. On the one hand, the cerium substitution will cause the lattice distortion and reduce the unit cell volume and average crystal size. On the other hand, the copper substitution can cause the change of the average valence state and average radius of Ce, Cu and Mn. Moreover, according to the previous analysis, the increase of average valence state of Ce or the decrease of average valence state of Cu and Mn will reduce the tolerance factor value and aggravate the degree of lattice distortion, and vice versa. Therefore, the cerium and copper substitution will change the average valence state and radius of A-site and B-site cations of catalysts, then change the lattice distortion, the unit cell volume and average crystal size of catalysts, thus affecting the denitration performance of catalysts.
| Catalysts | Va (Å3) | Db (nm) |
|---|---|---|
| a Determined and calculated from XRD patterns (MDI Jade 6.0 program).b Calculated from XRD patterns with the Scherrer equation. | ||
| LaMnO3 | 347.21 | 17.46 |
| La0.9Ce0.1MnO3 | 346.88 | 17.20 |
| La0.9Ce0.1Mn0.95Cu0.05O3 | 349.84 | 18.27 |
| La0.9Ce0.1Mn0.9Cu0.1O3 | 353.25 | 19.41 |
| La0.9Ce0.1Mn0.8Cu0.2O3 | 352.85 | 16.71 |
| La0.9Ce0.1Mn0.6Cu0.4O3 | 348.94 | 20.99 |
3.2 Morphological analysis basing on SEM and BET results
The morphology of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts was characterized by SEM images, as shown in Fig. 2. The LaMnO3 sample displays a fluffy porous structure with rod-like particles. However, it can be seen that the catalyst particles become smaller and the distances between particles become narrower in La0.9Ce0.1MnO3 compared to LaMnO3. With the partial substitution of Mn ions by Cu ions at B site, the surface morphology of the catalyst still presents porous wool structure in La0.9Ce0.1Mn0.95Cu0.05O3, the metal particles on the catalyst surface gather to a certain extent in La0.9Ce0.1Mn0.9Cu0.1O3, and the aggregation degree of metal particles on the catalyst surface is further intensified in La0.9Ce0.1Mn0.8Cu0.2O3. When the Cu substitution amount reaches 0.4, the surface morphology of the catalyst changes significantly, the porous wool structure basically disappears, and the catalyst particles obviously gather in local areas, which reduces the dispersion of active sites, thus decreasing the denitration efficiency of catalyst.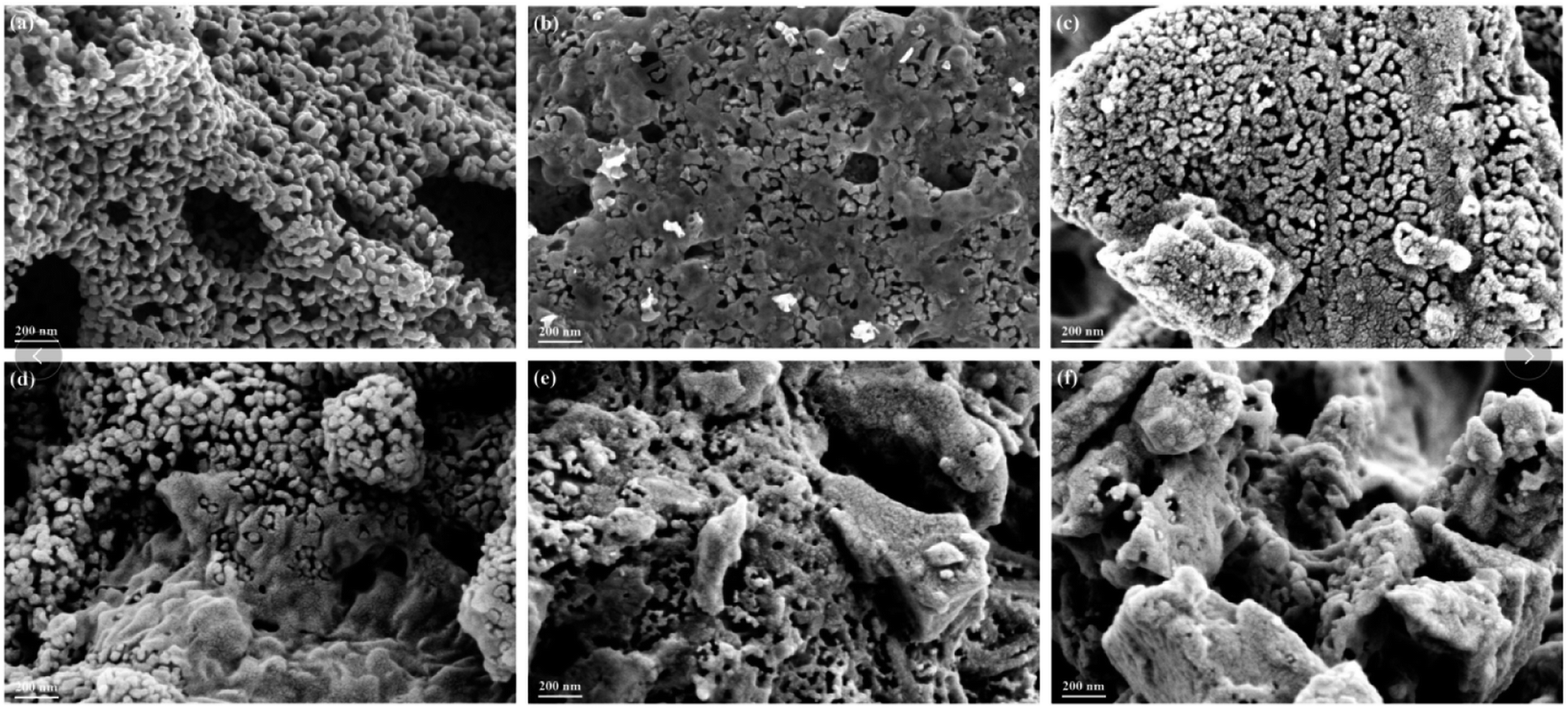 | ||
| Fig. 2 SEM images of (a) LaMnO3, (b) La0.9Ce0.1MnO3, (c) La0.9Ce0.1Mn0.95Cu0.05O3 and (d) La0.9Ce0.1Mn0.9Cu0.1O3, (e) La0.9Ce0.1Mn0.8Cu0.2O3, (f) La0.9Ce0.1Mn0.6Cu0.4O3. | ||
The N2 adsorption-desorption isotherms and BJH pore size distributions of catalysts are shown in Fig. 3 and 4, respectively. It is possible to observe that LaMnO3 and La0.9Ce0.1MnO3 catalysts all present typical type IV isotherms with type H4 hysteresis loop at P/P0 = 0.6–1.0. According to IUPAC classifications, type IV isotherms are exhibited by the mesoporous materials, and type H4 hysteresis loop can be attributed to the capillary condensation of nitrogen molecules in the mesoporous structure.23,24 Besides, pore size distributions obtained by BJH method show mesopores mainly in the range of 2–3 nm. Therefore, LaMnO3 and La0.9Ce0.1MnO3 catalysts both have many mesoporous structures. The BET surface area and pore structure of the catalysts are summarized in Table 2. Compared with the LaMnO3 catalyst, the surface area and pore structure of the La0.9Ce0.1MnO3 catalyst increase, which may be related to the decrease of crystal size caused by lattice distortion. With the addition of Cu substitution, the N2 adsorption desorption isotherms of La0.9Ce0.1Mn0.95Cu0.05O3 and La0.9Ce0.1Mn0.9Cu0.1O3 catalysts are type IV curves with H4 hysteresis loops, indicating that these two catalysts have many mesoporous structures. However, when the Cu substitution ratio reaches 0.2 and 0.4, the hysteresis loop area is significantly reduced and even disappeared, indicating that the high Cu substitution content leads to the reduce in mesoporous structure of catalysts, which is consistent with the SEM results. The reason is that high Cu substitution amount will lead to obvious aggregation of metal particles on the catalyst surface, and the wool porous structure will be transformed into sharp catalyst agglomerates. Therefore, high Cu substitution will significantly destroy the mesoporous structure and reduce the dispersion of active sites on catalysts surface. In addition, the average pore size of the La0.9Ce0.1Mn1−yCuyO3 (y = 0, 0.05, 0.1, 0.2, 0.4) catalyst sample first decreases and then increases with the increase of Cu substitution amount, which is related to the cluster phenomenon on the catalyst surface. Therefore, Cu partially substitute in La0.9Ce0.1MnO3 catalyst will change the average pore size of the catalyst, and then affect the denitration performance of perovskite catalysts.
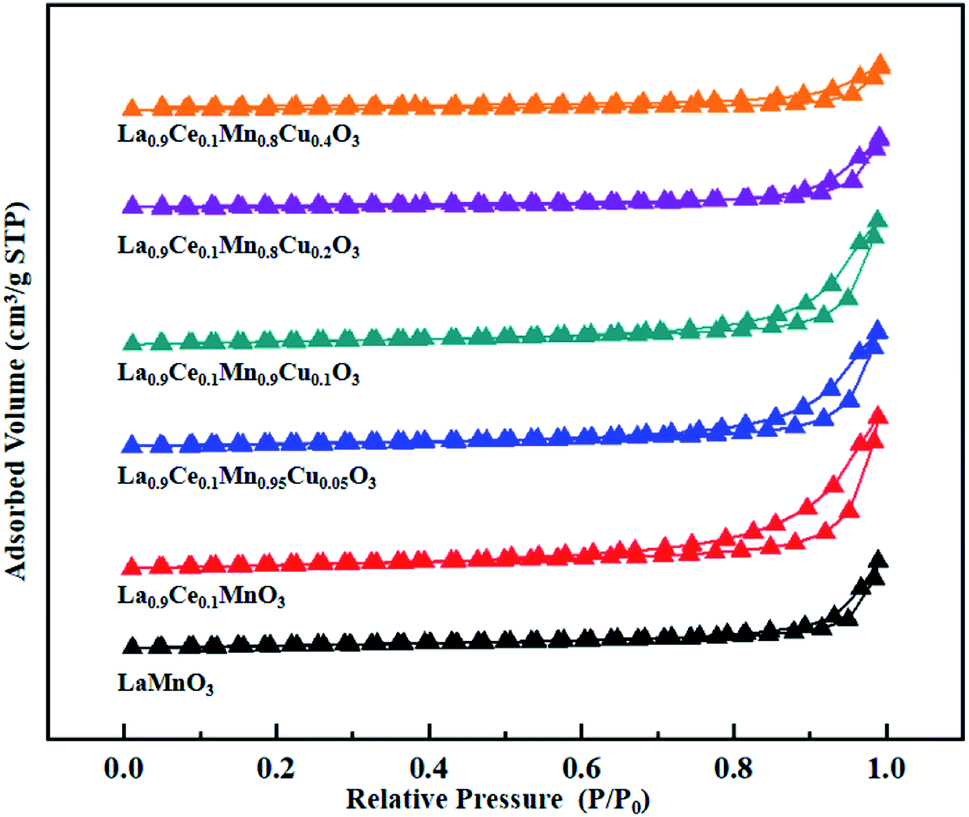 | ||
| Fig. 3 N2 adsorption-desorption isotherms of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts. | ||
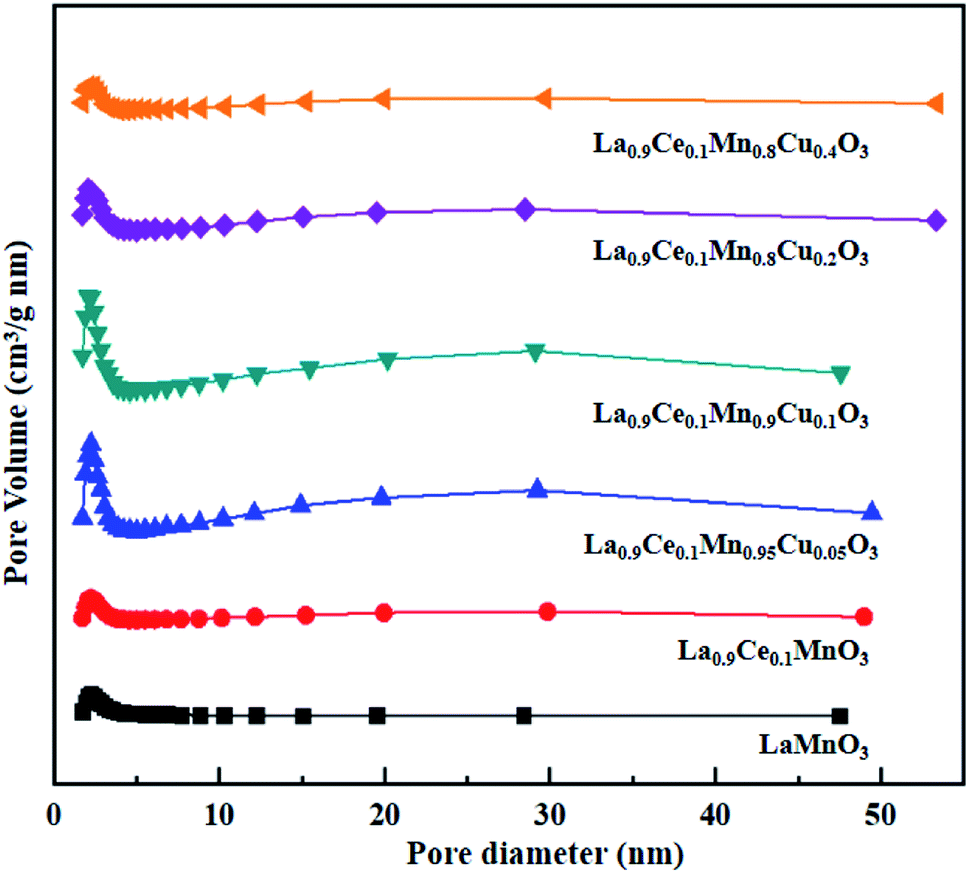 | ||
| Fig. 4 BJH pore size distributions of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts. | ||
| Catalysts | BET surface area (m2 g−1) | Pore volumn (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|
| LaMnO3 | 20.7 | 0.04 | 3.63 |
| La0.9Ce0.1MnO3 | 29.5 | 0.07 | 4.83 |
| La0.9Ce0.1Mn0.95Cu0.05O3 | 22.7 | 0.06 | 9.94 |
| La0.9Ce0.1Mn0.9Cu0.1O3 | 24.7 | 0.06 | 9.43 |
| La0.9Ce0.1Mn0.8Cu0.2O3 | 11.8 | 0.03 | 9.93 |
| La0.9Ce0.1Mn0.6Cu0.4O3 | 7.0 | 0.02 | 9.94 |
In summary, the increase of Cu substitution leads to the reduction in mesoporous structure content, decreases the dispersion of active sites and increases the amount of Cu active sites on catalyst surface. La0.9Ce0.1Mn0.9Cu0.1O3 catalyst and La0.9Ce0.1Mn0.8Cu0.2O3 catalyst both have many mesoporous structure, large specific surface area and high Cu active sites content, which can promote the adsorption of NH3, NO and other reaction gases on catalyst surface, thus improving the denitration activity of perovskite catalyst.
3.3 Redox properties analysis basing on XPS results
The XPS characterization was used to explore the metallic chemical state and active oxygen species on the perovskite surface. La 3d curves of the catalysts shown in Fig. 5(a), the La 3d5/2 peaks are located at 833.9 and 838.2 eV, and the La 3d3/2 peaks are located at 850.7 and 855.0 eV. Therefore, the spin-orbit splitting of the La 3d level is 16.8 eV for all samples, the value observed for the La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts is very closed to the metallic values of pure lanthanum oxide (La2O3), indicating that the La ions are exhibited in trivalent state for all catalysts.25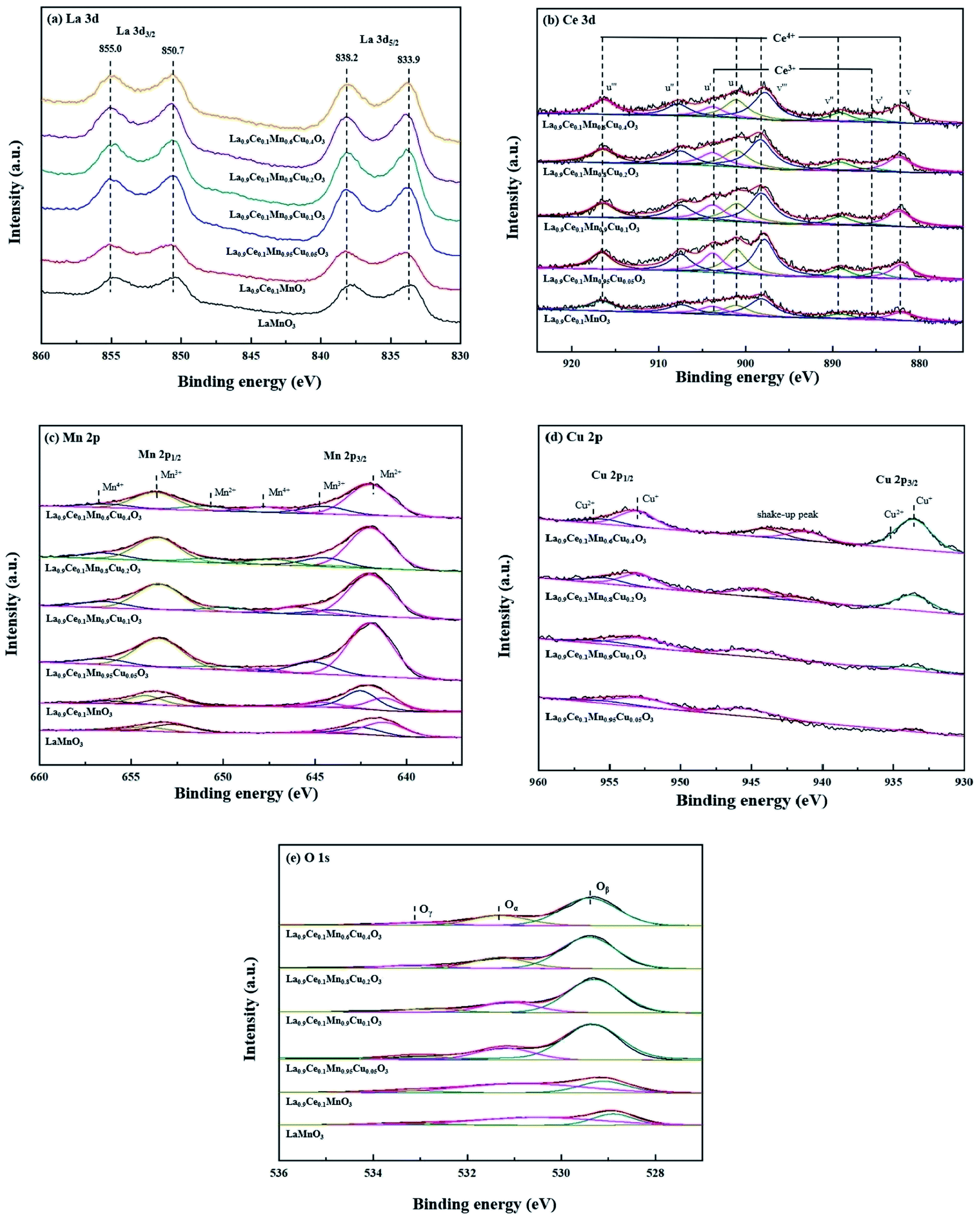 | ||
| Fig. 5 XPS spectra of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts: (a) La 3d, (b) Ce 3d, (c) Mn 2p, (d) Cu 2p, (e) O 1s. | ||
The obtained Ce 3d spectra are represented in Fig. 5(b). Eight components can be obtained by fitting the curves, the four characteristic peaks at 882.1 eV, 885.0 eV, 889.1 eV and 898.1 eV are represented by v, v′, v′′and v′′′, while the other four characteristic peaks at 901.2 eV, 903.8 eV, 907.8 eV and 916.4 eV are represented by u, u′, u′′ and u′′′.26 The peaks denoted as v′, u′ are assigned to Ce3+ species with 3d10 4f1 initial electronic configuration, while the peaks denoted as u, u′′, u′′′, v, v′′ and v′′′ are attributed to the 3d10 4f0 electronic state corresponding to Ce4+.27 It shows that Ce3+ and Ce4+ coexist on cerium containing catalysts surfaces, and the formed Ce4+/Ce3+ redox couple is promotional for the improvement of denitration activity.
Fig. 5(c) shows the Mn 2p spectra of the six catalysts, two main peaks due to Mn 2p3/2 and Mn 2p1/2 are found in the figure, and both of them can be well fitted into three peaks assigned to Mn2+, Mn3+ and Mn4+ species. Among these six peaks, the peaks at 640–641 and 652–653 eV are denoted as Mn2+ ions, the peaks at 641–642 and 653–654 eV are identified as Mn3+, and the peaks at 644–645 and 656–657 eV are attributed to Mn4+.17 Besides, it can be seen from the figure that the peak areas corresponding to different Mn valence states change with the increase of the Cu substitution proportion, indicating that there is a synergistic effect between Cu and Mn on catalyst surface.
Fig. 5(d) shows the Cu 2p spectra of La0.9Ce0.1Mn1−yCuyO3 (y = 0, 0.05, 0.1, 0.2, 0.4) catalysts, two main peaks of Cu 2p3/2 and Cu 2p1/2 are found in the figure. The characteristic peaks near 933.6 eV and 953.1 eV belong to Cu+, the characteristic peaks near 935.4 eV and 955.7 eV belong to Cu2+, and the vibrational satellite peaks between 940-947 eV also confirm the existence of Cu2+ and polycrystalline Cu2O substances in the four catalyst samples.28,29 In addition, it can be seen from the Fig. 5(d) that along with the Cu substitution increases, the width and height of the above characteristic peaks gradually increase, indicating that the Cu content increases significantly. At the same time, the characteristic peak areas corresponding to Cu2+ and Cu+ also change in different degrees, indicating that the change of Cu average valence state.
The O 1s spectra of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts are shown in Fig. 5(e). The peak at 529.4 eV belongs to the lattice oxygen (Oβ) of the catalyst, the peak at 531.3 eV represents the surface chemically adsorbed oxygen (Oα), while the peak at 533.0 eV belongs to the bulk lattice oxygen (Oγ),30 and their promotion degrees on the denitration activity of the catalyst are different. Therefore, it also can be seen from the Fig. 5(e) that the corresponding characteristic peaks areas of the three active oxygen species are different, leading to the different denitration efficiency of the four catalysts.
The chemical valence content of surface elements of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts are shown in Table 3. It can be observed that the oxidation valence states of Ce, Mn and Cu in La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts change along with the increase of Cu substitution. When the Cu substitution ratio at the range of 0.05–0.2, the addition of Cu substitution is accompanied with the decrease in the proportion of Mn2+ and the increase in the proportion of Ce3+ and Cu+. However, La0.9Ce0.1Mn0.6Cu0.4O3 has higher Mn2+ concentration and lower Ce3+ concentration than that of La0.9Ce0.1Mn0.6Cu0.4O3. The above phenomena shows that the decrease of the average valence state of Ce and Cu is accompanied by the increase of the average valence state of Mn. Therefore, there may be exist a synergetic effect among Ce, Mn and Cu, and the effect is inhibited at the moment of excessive Cu substitution. The probable synergetic effect as follows:31
| Ce4+ + Mn2+ + Cu2+ ↔ Ce3+ + Mn3+ + Cu+ |
| Ce4+ + Mn2+ + Cu2+ ↔ Ce3+ + Mn4+ + Cu+ |
| Catalysts | Ce (%) | Mn (%) | Cu (%) | O (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ce3+ | Ce4+ | Mn2+ | Mn3+ | Mn4+ | Cu+ | Cu2+ | Oα | Oβ | Oγ | |
| LaMnO3 | — | — | 60.6 | 31.6 | 7.8 | — | — | 62.3 | 29.0 | 8.7 |
| La0.9Ce0.1MnO3 | 15.7 | 84.3 | 38.5 | 47.9 | 13.6 | — | — | 59.5 | 32.4 | 8.1 |
| La0.9Ce0.1Mn0.95Cu0.05O3 | 17.9 | 82.1 | 53.4 | 39.0 | 7.6 | 77.9 | 22.1 | 23.7 | 68.8 | 7.5 |
| La0.9Ce0.1Mn0.9Cu0.1O3 | 32.4 | 67.6 | 51.2 | 34.2 | 14.6 | 79.9 | 20.1 | 21.7 | 70.5 | 7.8 |
| La0.9Ce0.1Mn0.8Cu0.2O3 | 33.3 | 66.7 | 50.5 | 36.4 | 13.1 | 82.7 | 17.3 | 23.4 | 69.8 | 6.8 |
| La0.9Ce0.1Mn0.6Cu0.4O3 | 32.4 | 67.6 | 51.3 | 35.6 | 13.1 | 89.8 | 10.2 | 24.6 | 68.5 | 6.9 |
According to the chemical valence content of metal elements on the catalyst surface in Table 3, it is reasonable to conclude the above synergetic effect among Ce, Mn and Cu affected by the Cu substitution. When the Cu substitution proportion reaches 0.2, the interaction is the strongest, and the number of electrons transferred between metal elements is the largest, which is conducive to the redox reaction of the gas adsorbed on the catalyst surface. Besides, it can be seen from table that the La0.9Ce0.1Mn0.8Cu0.2O3 has the highest Ce3+ content. Ce3+ has been reported that it can originate oxygen vacancies, charge imbalance and unsaturated chemical bonds,32 thus promoting to improve the redox capacity and the denitration efficiency of the catalyst. The research shows that the high proportion of high Mn valence state can promote the redox reaction on the catalyst surface, and then significantly improve the low-temperature denitration activity of the catalyst.33 It can be seen from Table 3 that the order of (Mn3+ + Mn4+)/Mnn+ of Cu-doped catalysts is La0.9Ce0.1Mn0.8Cu0.2O3 > La0.9Ce0.1Mn0.9Cu0.1O3 > La0.9Ce0.1Mn0.6Cu0.4O3 > La0.9Ce0.1Mn0.95Cu0.05O3, the average valence state of Mn ions in La0.9Ce0.1Mn0.8Cu0.2O3 catalyst is the highest, which is conducive to the enhancement of its denitration activity.
In addition, the surface chemically adsorbed oxygen and the lattice oxygen are the important active oxygen of NH3-SCR denitration reaction, which both assist to enhance the low-temperature denitration activity of the catalyst. It is reported that O2 takes part in the NH3-SCR reaction via taking up the oxygen vacancies of catalyst surface,34 and the lattice oxygen and chemical adsorbed oxygen are compensated in the process. Therefore, the oxygen vacancy also has important role in the denitration activity, and we can utilize the sum of Oα and Oβ to quantify oxygen vacancies in catalysts. It can be seen that cerium and copper substitution both can increase the proportion of (Oα + Oβ), and La0.9Ce0.1Mn0.8Cu0.2O3 has the highest proportion of (Oα + Oβ), which may be helpful to the NH3-SCR denitration activity. Besides, the proportion of (Oα + Oβ) is related to the content of Ce3+, (Mn3+ + Mn4+) and Cu+ in varying degrees, indicating that the proportion of (Oα + Oβ) has a correlation with the synergetic effects among Ce, Mn and Cu, thus jointly promoting the improvement of denitration efficiency.
In conclusion, there is synergetic effect among Ce, Mn and Cu metal ions in the process of Cu substitution, which reduces the average valence state of Ce and Cu and increases the average valence state of Mn, and promotes the formation of the chemical adsorbed oxygen and lattice oxygen, thus affecting the redox ability and denitration performance of perovskite catalysts.
3.4 NH3-SCR activity
NO conversions of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts in the simulated flue gas at the temperature between 150 °C and 400 °C are presented in Fig. 6. It can be seen from the figure that La0.9Ce0.1MnO3 displays higher denitration activity compared with LaMnO3, and NO conversion reaches 86.7% at 350 °C, which shows the Ce substitution can promote to strengthen the denitration activity of La–Mn perovskite catalyst. In addition, the denitration efficiencies of La0.9Ce0.1Mn0.95Cu0.05O3 and La0.9Ce0.1Mn0.6Cu0.4O3 catalysts decrease in varying degrees compared with La0.9Ce0.1MnO3 catalyst, indicating that over lowing or excessive high Cu substitution are both not conducive to improving the denitration performance of the catalyst. Besides, the NO conversion of La0.9Ce0.1Mn0.8Cu0.2O3 catalysts is the highest among the catalysts and reaches 91.8% at 350 °C, indicating that the saturation point of the Cu substitution ratio is around 0.2. Therefore, appropriate cerium and copper substitution will markedly improve the denitration activity of La–Mn perovskite catalyst.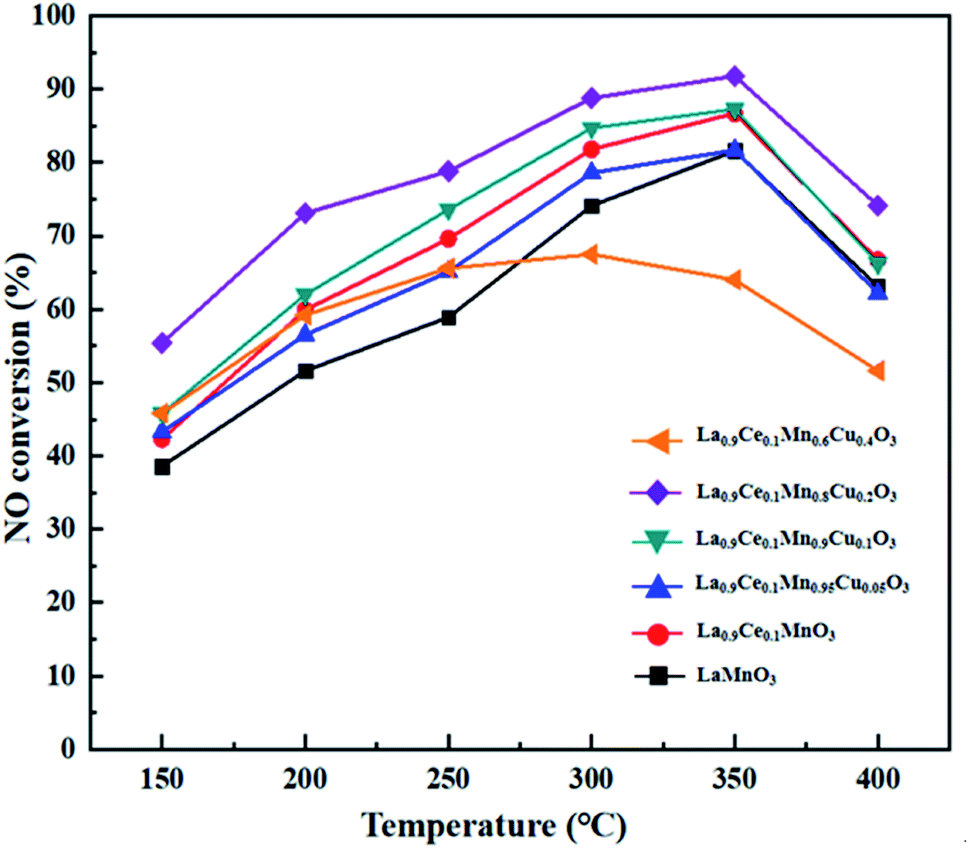 | ||
| Fig. 6 NO conversion of the La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts. | ||
According to the above characterization results, it is reasonable to conclude existence of the inherent relationships between structure, morphology, surface element composition and catalytic activity of the La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) perovskite catalysts. The high denitration activity of La0.9Ce0.1MnO3 is mainly attributed to its many mesoporous structure and large surface area, which contribute to the adsorption of NH3 and NO. La0.9Ce0.1Mn0.8Cu0.2O3 catalyst has high Cu active sites content, highest proportion of Ce3+ and high Mn valence state, strong metal interaction and high concentration of chemical adsorbed oxygen and lattice oxygen, so it has the best denitration activity in the La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) perovskite catalysts.
The comparison of the denitration performance of modified La–Mn perovskite catalysts prepared in this paper with copper-based catalysts, cerium-based catalysts and other reported La–Mn perovskite catalysts have been studied in Table 4. It can be seen that whether copper oxide and cerium oxide alone as catalyst, copper oxide and cerium oxide supported with other catalyst or copper oxide and cerium oxide combine with other materials to form mixture as catalyst, they all show worse denitration efficiency compared with La0.9Ce0.1Mn0.8Cu0.2O3, indicating that the catalyst with perovskite phase has preferable denitration property. Besides, the NH3-SCR activity of La–Mn perovskite catalyst can be improved by supported with other catalyst, and the A-site and B-site of La–Mn perovskite catalyst substituted with other metal element also can achieve high denitration efficiency. Therefore, we can take other modification methods to further improve the denitration performance of La–Mn perovskite catalysts, thus providing a prospect for further enhancing the denitration efficiency of NH3-SCR catalysts in industrial denitration application.
| Catalysts | Best NO conversion | Ref. |
|---|---|---|
| La0.9Ce0.1Mn0.8Cu0.2O3 | 91.8% (350 °C) | This work |
| CuO | <70% (200 °C) | 35 |
| CeO2 | <60% (350 °C) | 36 |
| Cu/TiO2–ZrO2 | <80% (400 °C) | 2 |
| Ce/TiO2–ZrO2 | <85% (400 °C) | 2 |
| Cu–Ce/Al2O3 | 87.9% (250 °C) | 37 |
| Cu4AlOx | 91.1% (200 °C) | 37 |
| CeNTs | 85% (350 °C) | 38 |
| LaMnO3/TiO2 | >90% (300 °C) | 39 |
| La0.9Sr0.1MnO3 | <90% (300 °C) | 39 |
| LaMn0.95V0.05O3 | 90% (250 °C) | 39 |
3.5 NH3-SCR reaction mechanism analysis basing on DRIFT results
The La0.9Ce0.1Mn0.8Cu0.2O3 catalyst with the best denitration activity was selected for in situ DRIFT characterization to identify the active sites, the adsorbed species and the intermediates on the catalyst surface in the denitration process, aiming at figuring out the reaction mechanism of La–Mn perovskite catalyst in low temperature NH3-SCR system.The La0.9Ce0.1Mn0.8Cu0.2O3 catalyst was pre-adsorbed with 500 ppm NH3 on the surface at 200 °C for 30 min, and then exposed at 500 ppm NO and 5% O2 for 30 min. The DRIFT spectrum of the catalyst is shown in Fig. 7. The vibrational peaks located at 1042, 1087, 1123, 1234 and 1626 cm−1 in the figure are attributed to the coordinating NH3 on the Lewis acid site, while the vibrational peak at 1397 cm−1 belongs to the NH4+ adsorption on the Brønsted acid site.40 The peak intensities of the above vibration peaks decrease with time after the introduction of NO and O2, indicating that both coordinated NH3 and NH4+ can participate in the NH3-SCR denitration reaction. In addition, the vibration peak located at 1028, 1503, 1610, 1846 and 1908 cm−1 are not reflected in the spectrum after NH3 pre-adsorption for 30 min, but appear after NO and O2 purged into the catalyst surface, the peak intensity increases and tends to be stable. Among them, the vibration peak at 1028 cm−1 belongs to the cis-N2O22− species,41 and the vibration peak at 1503 cm−1 belongs to the amide species (–NH2).42 The above vibration peaks correspond to the species belong to the reduction product of NO and the oxidation product of NH3, respectively, which are important intermediate products of the NH3-SCR denitration reaction. Meanwhile, the vibrational peak at 1610 cm−1 represents NO2 adsorbed on the catalyst surface,43 while the vibrational peaks at 1846 and 1908 cm−1 represent the adsorbed NOx species.44 The vibrational peak at 1340 cm−1 is attributed to the monodentate nitrate,40 and the peak intensity increases significantly after NO and O2 introduced, indicating that the NH3-SCR denitration reaction mechanism on the catalyst surface follows the L–H mechanism. Besides, three vibrational peaks appear at 3124, 3267 and 3322 cm−1, which belong to the N–H vibrational region (3000–3500 cm−1), and these peaks also correspond to the coordination NH3 on the Lewis acid site.41 The peak intensities of the three vibration peaks weaken along with the NO and O2 purging, indicating that the NH3-SCR denitration reaction mechanism on the catalyst surface follows E–R mechanism. Moreover, many Lewis acid sites on the catalyst surface can adsorb a large amount of reaction gas to participate in the NH3-SCR denitration reaction, which is beneficial to improve the denitration efficiency of lanthanum manganese perovskite catalysts.
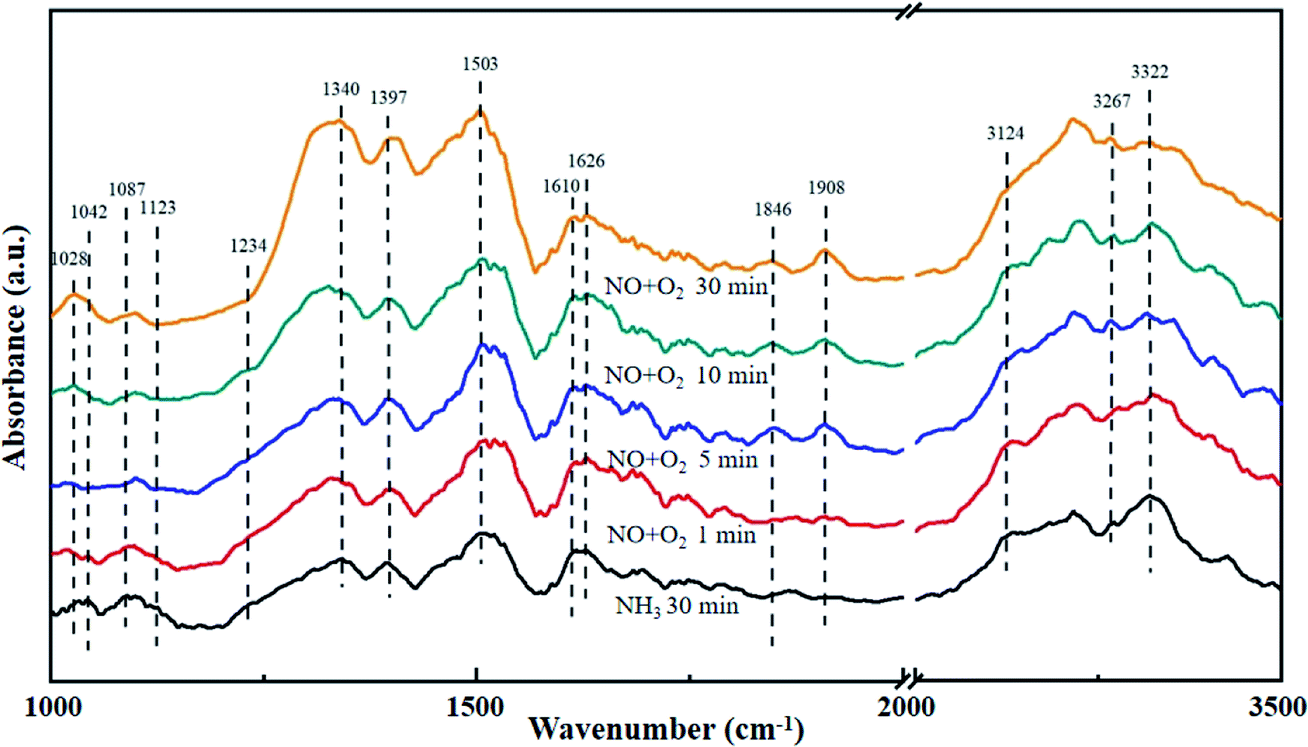 | ||
| Fig. 7 DRIFT spectra of La0.9Ce0.1Mn0.8Cu0.2O3 exposed to NO at various temperatures. Reaction condition: [NH3] = [NO] = 500 ppm, [O2] = 5%, and N2 balance. | ||
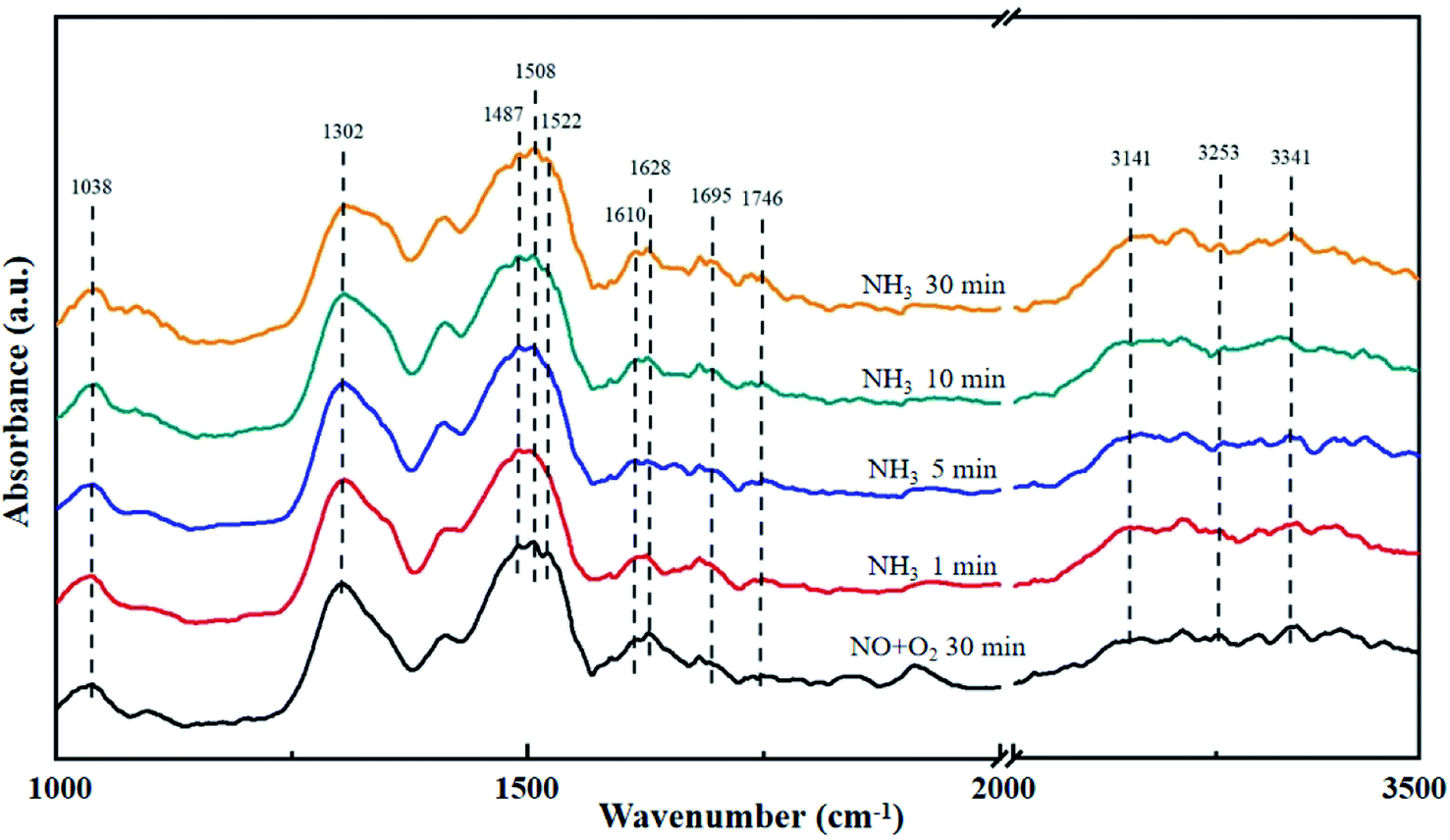
The La0.9Ce0.1Mn0.8Cu0.2O3 catalyst was pre-adsorbed with 500 ppm NO and 5% O2 on the surface at 200 °C for 30 min, and then exposed at 500 ppm NH3 for 30 min. The DRIFT spectrum of the catalyst is shown in Fig. 8. The vibrational peak at 1038 cm−1 represents the cis-N2O22− species,41 the vibrational peak at 1628 cm−1 represents the NO2 adsorbed on the catalyst surface,45 and the vibrational peaks at 1695 and 1746 cm−1 represent NO (M–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) adsorbed on the catalyst surface with covalent bond formation,41 all above peaks represent the intermediate products formed by NO adsorption on the catalyst surface. In addition, the vibrational peaks at 1302, 1508 and 1610 cm−1 belong to monodentate nitrate,43,46 the vibrational peak at 1522 cm−1 belongs to bidentate nitrate, the characteristic peak at 1487 cm−1 belongs to NH4+ adsorbed on the Brønsted acid site, while the characteristic peaks at 3141, 3253 and 3341 cm−1 belong to the coordinated NH3 adsorbed on the Lewis acid site.46 The peak intensity of the above characteristic peaks increases with the increase of NH3 purging time, indicating that there are many Brønsted acid sites and Lewis acid sites can participate in the denitration reaction on the catalyst surface, thereby improving the denitration efficiency of the lanthanum manganese perovskite catalyst.
O) adsorbed on the catalyst surface with covalent bond formation,41 all above peaks represent the intermediate products formed by NO adsorption on the catalyst surface. In addition, the vibrational peaks at 1302, 1508 and 1610 cm−1 belong to monodentate nitrate,43,46 the vibrational peak at 1522 cm−1 belongs to bidentate nitrate, the characteristic peak at 1487 cm−1 belongs to NH4+ adsorbed on the Brønsted acid site, while the characteristic peaks at 3141, 3253 and 3341 cm−1 belong to the coordinated NH3 adsorbed on the Lewis acid site.46 The peak intensity of the above characteristic peaks increases with the increase of NH3 purging time, indicating that there are many Brønsted acid sites and Lewis acid sites can participate in the denitration reaction on the catalyst surface, thereby improving the denitration efficiency of the lanthanum manganese perovskite catalyst.
 | ||
| Fig. 8 DRIFT spectra of La0.9Ce0.1Mn0.8Cu0.2O3 exposed to NH3 at various temperatures. Reaction condition: [NH3] = [NO] = 500 ppm, [O2] = 5%, and N2 balance. | ||
According to the DRIFT characterization results of NH3 pre-adsorption and NO + O2 adsorption experiments as well as NO + O2 pre-adsorption and NH3 adsorption experiments on the surface of La0.9Ce0.1Mn0.8Cu0.2O3 catalyst, it can be seen that the adsorption of NH3 on the catalyst surface is very strong at 200 °C, mainly generate coordinating NH3 on Lewis acid site and NH4+ on Brønsted acid site, which can be dehydrogenated into amide species (–NH2). Besides, NO not only can react with –NH2 directly, but also can be adsorbed on the catalyst surface and converted into monodentate and bidentate nitrates, which are further combined with NH4+ adsorbed on the acid site and finally converted into N2O and H2O for the purpose of denitration. In conclusion, the low-temperature NH3-SCR denitration reaction mechanism on the surface of La0.9Ce0.1Mn0.8Cu0.2O3 catalyst includes E–R mechanism and L–H mechanism.
The adsorption and conversion process of NH3 on the catalyst surface are as follows:
| NH3(g) → NH3(a) (Lewis acid sites) → –NH2(a) |
| NH3(g) → NH4+(a) (Brønsted acid sites) → –NH2(a) |
The adsorption and conversion process of NO on the catalyst surface are as follows:
| NO(g) + O(a) → NO2(a) |
| M(n−1)+ + O(a) → Mn+–O |
| Mn+–O + NO2(a) → M(n−1)+ + NO3− |
The Eley–Rideal denitration reaction mechanism is as follows:
| –NH2(a) + NO(g) → NH2NO(a) → N2 + H2O |
The Langmuir–Hinshelwood denitration reaction mechanism is as follows:
| NH4+ + NO3− → NH4NO3 → N2O + H2O |
As the final summarize comment, the multiple reaction mechanism herein proposed. The multiple reaction mechanism (shown in Fig. 9) on catalyst surface includes Eley–Rideal reaction mechanism and Langmuir–Hinshelwood reaction mechanism, which both promote to improve the denitration activity of La–Mn perovskite catalyst.
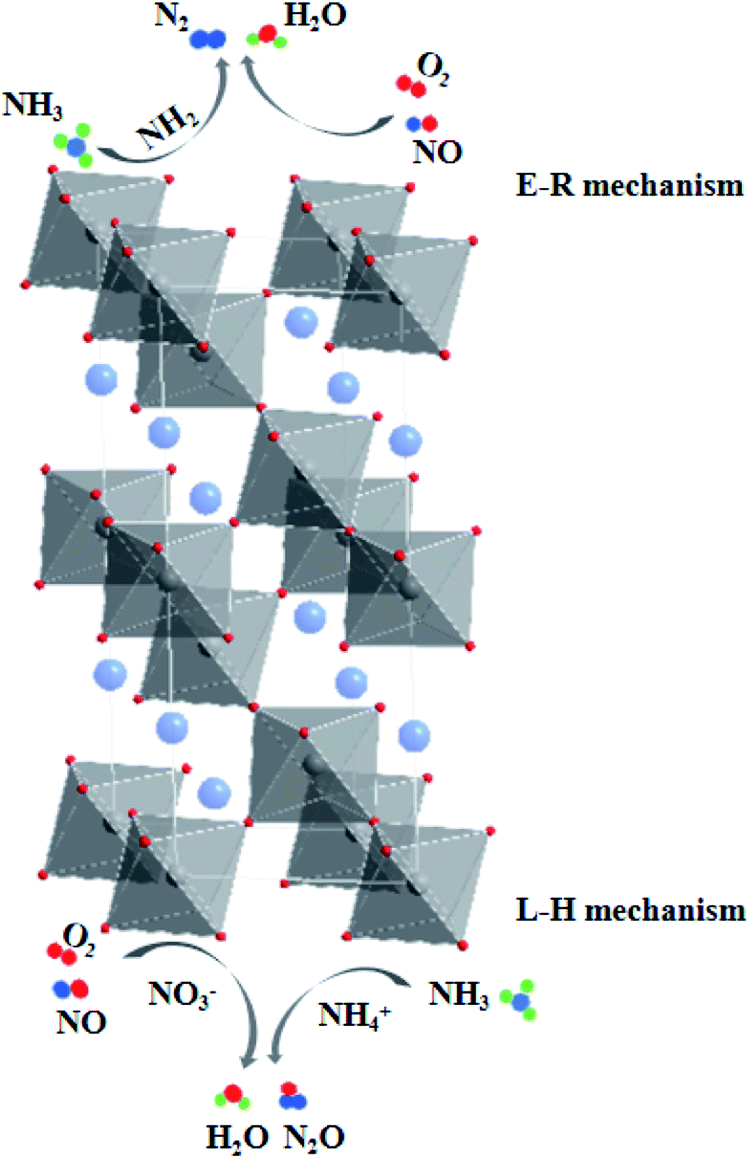 | ||
| Fig. 9 Mechanism of NH3-SCR Denitrification on Lanthanum–Manganese Perovskite Catalyst Surface at 200 °C. | ||
4. Conclusions
In the present work, a series of La1−xCexMn1−yCuyO3 (x = 0, 0.1, y = 0, 0.05, 0.1, 0.2, 0.4) catalysts were synthesized by the citrate sol-gel method and characterized by XRD, SEM, BET, XPS and DRIFT techniques. The catalytic activity tests of selective catalytic reduction of NO with NH3 were carried and the effects of catalyst properties on NH3-SCR denitration activity were studied. The results show that Ce substitution has a positive impact on improving the denitration activity of LaMnO3 perovskite catalyst, and La0.9Ce0.1MnO3 displays the activity with a maximum NO conversion of 86.7% at 350 °C. The high denitration activity of La0.9Ce0.1MnO3 is mainly attributed to the larger surface area, which promotes to the adsorption of NH3 and NO. Moreover, the La0.9Ce0.1MnO3 perovskite catalyst was modified with copper substitution. It is found that when the Cu substitution ratio reaches 0.2, the catalyst has the strongest denitration activity, and the NO conversion at 350 °C reaches 91.8%. Although the specific surface area of La0.9Ce0.1Mn0.8Cu0.2O3 is lower than La0.9Ce0.1MnO3, the Cu active sites and the Ce3+ content are more developed, making many reaction units formed on the catalyst surface and redox properties of catalyst improved. In addition, strong metal interaction (Ce4+ + Mn2+ + Cu2+ ↔ Ce3+ + Mn3+/Mn4+ + Cu+) and high concentrations of chemical adsorbed oxygen and lattice oxygen both assist to strengthen the redox reaction on catalyst surface, thus further improve the denitration activity of perovskite catalyst. Therefore, appropriate cerium and copper substitution will markedly improve the denitration activity of La–Mn perovskite catalyst. We also reasonably conclude a multiple reaction mechanism during NH3-SCR denitration process, which includes the Eley–Rideal mechanism and Langmuir–Hinshelwood mechanism.Author contributions
Wei Zhang: conceptualization, methodology, software, resources, data curation, supervision, project administration, funding acquisition. Kang Xie: formal analysis, writing-original draft, writing-review & editing, funding acquisition. Yunhao Tang: investigation, visualization, validation. Shan Cheng: writing-review & editing, funding acquisition. Mengxia Qing: writing-review & editing, funding acquisition. Yanni Xuan: writing-review & editing, funding acquisition. Chuan Qin: investigation, visualization. Mengyao Dong: investigation, validation. Yunhe Zhou: investigation, validation. Jie Li: investigation, validation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National natural science foundation of China (52006016, 52104391), State scholarship fund awarded by China scholarship (201808430112), Natural science foundation of Hunan province (2020JJ4098, 2021JJ40573), Key projects of scientific research project of Hunan Provincial Department of Education (21A0216), Changsha Municipal Natural Science Foundation (kq2014104), Young teachers growth plan project of CSUST (2019QJCZ044), Key laboratory of renewable energy electric technology of Hunan province (2016ZNDL005, 2018ZNDL004, 2020ZNDL001), 2022 Graduate research and innovation project at Changsha University of Science and Technology (CXCLY2022092).References
- D. Wang, B. Huang, Z. Shi, H. Long, L. Li, Z. Yang and M. Dai, RSC Adv., 2021, 11, 18458, 10.1039/d1ra02352g
.
- W. Zhang, Y. Tang, W. Xiao, M. Ruan, Y. Yin, Q. Song, K. Xie, C. Qin, M. Dong, Y. Zhou and J. Li, RSC Adv., 2022, 12, 378, 10.1039/d1ra06325a
- P. Gong, J. Xie, D. Fang, X. Liu, F. He and F. Li, Chem. Eng. J., 2019, 356, 598–608, DOI:10.1016/j.cej.2018.09.042
- W. Shi, J. Zhang, X. Li, J. Zhou, Y. Pan, Q. Liu, Z. Xu and G. Qian, Energy Fuels, 2017, 31, 11258–11265, DOI:10.1021/acs.energyfuels.7b02230
- K. Liu, H. He and B. Chu, Chem. Eng. J., 2021, 423, 130128, DOI:10.1016/j.cej.2021.130128
- X. Sun, Q. Liu, S. Liu, X. Zhang and S. Liu, RSC Adv., 2021, 11, 22780, 10.1039/d1ra03845a
- S. Xiong, Y. Peng, D. Wang, N. Huang, Q. Zhang, S. Yang, J. Chen and J. Li, Chem. Eng. J., 2020, 387, 124090, DOI:10.1016/j.cej.2020.124090
- W. Wen, X. Wang, S. Jin and R. Wang, RSC Adv., 2016, 6, 74046–74052, 10.1039/C6RA18273A
- J. Zhang, D. Tan, Q. Meng, X. Weng and Z. Wu, Appl. Catal., B, 2015, 172, 18–26, DOI:10.1016/j.apcatb.2015.02.006
- C. Zhou, Z. Feng, Y. Zhang, L. Hu, R. Chen, B. Shan, H. Yin, W. Guo and A. Huang, RSC Adv., 2015, 5, 28054–28059, 10.1039/C5RA02344K
- J. Yao, H. Lu, Y. Xiao, B. Hou, D. Li and L. Jia, Catal. Commun., 2019, 128, 105718, DOI:10.1016/j.catcom.2019.105718
- F. Zhang, X. Zhang, G. Jiang, N. Li, Z. Hao and S. Qu, Chem. Eng. J., 2018, 348, 831–839, DOI:10.1016/j.cej.2018.05.050
- H. Wu, M. He, W. Liu, L. Jiang, J. Cao, C. Yang, J. Yang, J. Peng, Y. Liu and Q. Liu, J. Environ. Chem. Eng., 2021, 9, 105426, DOI:10.1016/j.jece.2021.105426
- J. Yang, S. Ren, T. Zhang, Z. Su, H. Long, M. Kong and L. Yao, Chem. Eng. J., 2020, 379, 122398, DOI:10.1016/j.cej.2019.122398
- J. A. Onrubia-Calvo, B. Pereda-Ayo, U. De-La-Torre and J. R. González-Velasco, Appl. Catal., B, 2017, 213, 198–210, DOI:10.1016/j.apcatb.2017.04.068
- Y. Wu, H. Liu, G. Li, L. Jin, X. Li, X. Ou, L. Dong, G. Jin and B. Li, Appl. Surf. Sci., 2020, 508, 145158, DOI:10.1016/j.apsusc.2019.145158
- B. Zhao, R. Ran, L. Sun, X. Guo, X. Wu and D. Weng, RSC Adv., 2016, 6, 69855–69860, 10.1039/C6RA12308B
- C. Zhang, W. Hua, C. Wang, Y. Guo, Y. Guo, G. Lu and A. Baylet, Appl. Catal., B, 2013, 134–135, 310–315, DOI:10.1016/j.apcatb.2013.01.031
- Y. Shao, X. Wang, M. Ao, C. Gong, G. Fan and H. Chen, Front. Mater. Sci., 2012, 6, 304–310, DOI:10.1007/s11706-012-0179-z
- G. Xiao, S. Xin, H. Wang, R. Zhang, Q. Wei and Y. Lin, Ind. Eng. Chem. Res., 2019, 58, 5388–5396, DOI:10.1021/acs.iecr.8b05674
- T. Wang, C. Li, L. Zhao, J. Zhang, S. Li and G. Zeng, Appl. Surf. Sci., 2017, 400, 227–237, DOI:10.1016/j.apsusc.2016.12.192
- H. Shu, Y. Liu and Y. Jia, Sci. Total Environ., 2022, 820, 152984, DOI:10.1016/j.scitotenv.2022.152984
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069, DOI:10.1515/pac-2014-1117
- G. Zhang, B. Lei, S. Chen, H. Xie and G. Zhou, J. Environ. Chem. Eng., 2021, 9, 105387, DOI:10.1016/j.jece.2021.105387
- C. Zhang, C. Wang, W. Zhan, Y. Guo, Y. Guo, G. Lu, A. Baylet and A. Giroir-Fendler, Appl. Catal., B, 2013, 129, 509–516, DOI:10.1016/j.apcatb.2012.09.056
- G. Kim, S. Lee, K. Nam and S. Hong, Appl. Surf. Sci., 2020, 507, 145064, DOI:10.1016/j.apsusc.2019.145064
- S. Ni, X. Tang, H. Yi, F. Gao, C. Wang, Y. Shi, R. Zhang and W. Zhu, J. Rare Earths, 2022, 40, 268–278, DOI:10.1016/j.jre.2020.12.015
- X. Wang, Y. Shi, S. Li and W. Li, Appl. Catal., B, 2018, 220, 234–250, DOI:10.1016/j.apcatb.2017.08.021
- Sl. Ali, L. Chen, Z. Li, T. Zhang, R. Li, S. Bakhtiar, X. Leng, F. Yuan, X. Niu and Y. Zhu, Appl. Catal., B, 2018, 236, 25–35, DOI:10.1016/j.apcatb.2018.05.014
- Q. Guo, W. Jing, Y. Hou, Z. Huang, G. Ma, X. Han and D. Sun, Chem. Eng. J., 2015, 270, 41–49, DOI:10.1016/j.cej.2015.01.086
- S. Feng, J. Liu and B. Gao, Chem. Eng. J., 2022, 429, 132302, DOI:10.1016/j.cej.2021.132302
- B. Shen, Y. Wang, F. Wang and T. Liu, Chem. Eng. J., 2014, 236, 171–180, DOI:10.1016/j.cej.2013.09.085
- Z. Wang, X. Xu, Y. Zhu, H. He, N. Wang, X. Yang and L. Liu, Microporous Mesoporous Mater., 2022, 333, 111720, DOI:10.1016/j.micromeso.2022.111720
- T. Wang, C. Li, L. Zhao, J. Zhang, S. Li and G. Zeng, Appl. Surf. Sci., 2017, 400, 227–237, DOI:10.1016/j.apsusc.2016.12.192
- M. Xing, Q. Sun, C. Zeng, H. Wang, D. Zhao, N. Zhang and S. Hong, RSC Adv., 2017, 7, 18830, 10.1039/c7ra00581d
- P. Gong, J. Xie, D. Fang, X. Liu, F. He and F. Li, Chem. Eng. J., 2019, 356, 598–608, DOI:10.1016/j.cej.2018.09.042
- Q. Yan, Y. Gao, Y. Li, M. A. Vasiliades, S. Chen, C. Zhang, R. Gui, T. Zhu and A. M. Efstathiou, Appl. Catal., B, 2019, 255, 117749, DOI:10.1016/j.apcatb.2019.117749
- P. Wang, S. Chen, S. Gao, J. Zhang, H. Wang and Z. Wu, Appl. Catal., B, 2018, 231, 299–309, DOI:10.1016/j.apcatb.2018.03.024
- R. Zhang, W. Yang, N. Luo, P. Li, Z. Lei and B. Chen, Appl. Catal., B, 2014, 146, 94–104, DOI:10.1016/j.apcatb.2013.04.047
- J. Wang, Z. Yan, L. Liu, Y. Chen, Z. Zhang and X. Wang, Appl. Surf. Sci., 2014, 313(13), 660–669, DOI:10.1016/j.apsusc.2014.06.043
- Q. Zhang, J. Fan, P. Ning, Z. Song, X. Liu, L. Wang, J. Wang, H. Wang and K. Long, Appl. Surf. Sci., 2018, 435, 1037–1045, DOI:10.1016/j.apsusc.2017.11.180
- L. Chen, J. Li and M. Ge, Environ. Sci. Technol., 2010, 44, 9590–9596, DOI:10.1021/es102692b
- S. Xie, L. Li, L. Jin, Y. Wu, H. Liu, Q. Qin, X. Wei, J. Liu, L. Dong and B. Li, Appl. Surf. Sci., 2020, 515, 146014, DOI:10.1016/j.apsusc.2020.146014
- F. Gao, C. Yang, X. Tang, H. Yi and C. Wang, J. Environ. Sci., 2022, 113, 204–218, DOI:10.1016/j.jes.2021.05.032
- Y. Shi, H. Yi, F. Gao, S. Zhao, Z. Xie and X. Tang, J. Hazard. Mater., 2021, 413, 125361, DOI:10.1016/j.jhazmat.2021.125361
- L. Chen, Z. Si, X. Wu and D. Weng, ACS Appl. Mater. Interfaces, 2014, 6, 8134–8145, DOI:10.1021/am5004969
| This journal is © The Royal Society of Chemistry 2022 |