
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe methodology for preparing domperidone: strategies, routes and reaction processes
Lili Yu
abc,
Rixin Shao
abc,
Qingxiang Guo
*abc,
Hailong Hong
 abc and
Ning Zhu
*abc
abc and
Ning Zhu
*abc
aCollege of Chemical Engineering, Inner Mongolia University of Technology, Hohhot 010051, China. E-mail: guoqingxiang@imut.edu.cn; zhuning@imut.edu.cn
bKey Laboratory of CO2 Resource Utilization at Universities of Inner Mongolia Autonomous Region, Hohhot 010051, China
cInner Mongolia Engineering Research Center for CO2 Capture and Utilization, Hohhot 010051, China
First published on 15th August 2022
Abstract
Domperidone is a powerful peripheral dopamine receptor antagonist; however, a systematic review of the synthetic methods and processes of this drug has not been reported so far. This review summarizes the synthetic strategies, synthetic routes and reaction processes of domperidone in detail. Domperidone can be synthesized from the coupling reaction of two benzimidazolone derivatives (intermediates 1 and 2). Intermediate 1 can be prepared by two synthetic routes: the cyclization of o-phenylenediamine with carbonyl reagents followed by coupling with 1,3-dihalopropane, and the coupling reaction of o-halo or o-amino substituted nitrobenzene with 1,3-disubstituted propane followed by reduction and cyclization. The latter route avoids the production of di-substituted by-products and has higher reaction selectivity. Intermediate 2 is synthesized by coupling substituted nitrobenzene with 4-aminopiperidine followed by reduction and cyclization, which is similar to the synthetic route of intermediate 1. Understanding the advantages and drawbacks of these synthetic methodologies would provide insights for the development of new strategies to prepare domperidone. Moreover, the methods used to synthesize domperidone can provide alternative approaches in the preparation of drugs or compounds with similar structure.
 Lili Yu | Lili Yu received her bachelor's degree (2015–2019) from Xinjiang Agricultural University. She completed her master's degree (2019–2021) from College of Chemical Engineering, Inner Mongolia University of Technology and conducted research on carbon dioxide (CO2) resource utilization under the guidance of professor Ning Zhu. |
 Rixin Shao | Rixin Shao completed his bachelor's degree (2016–2020) from College of Chemical Engineering, Inner Mongolia University of Technology and conducted research on carbon dioxide (CO2) resource utilization under the guidance of professor Ning Zhu. He has studied for a master's degree at School of Pharmacy, East China University of Science and Technology from 2021. He is currently working under the supervision of professor Jinxing Ye and his research interest involves continuous flow chemistry. |
 Qingxiang Guo | Qingxiang Guo is an associate professor at Inner Mongolia University of Technology (China). He received his B.S. degree from Ocean University of China (2011), and PhD degree in Biochemistry and molecular biology from Nankai University (2018). Before starting his independent research at Inner Mongolia University of Technology in October 2021, he was a postdoctoral fellow at Institute of Radiation Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College (China). His current research interests include the design and synthesis of small molecule inhibitors/functional nanomaterials and their biomedical applications for tumor targeted therapy/drug delivery. |
 Hailong Hong | Hailong Hong received his bachelor's degree (2002) in Chemical Engineering and Technology, master's degree (2005) in Applied Chemistry and PhD (2008) in Chemical Technology from the Inner Mongolia University of Technology. He became an associate professor (2009) in College of Chemical Engineering, Inner Mongolia University of Techonology. His research mainly focused on the research and development of synthetic process for the fine chemicals, the utilization of CO2 resources and the application of supercritical carbon dioxide technology. |
 Ning Zhu | Ning Zhu received his bachelor's degree (2002) and master's degree (2005) in Applied Chemistry from the Inner Mongolia University of Technology. After he successfully obtained his PhD (2010) in medicinal chemistry in Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, he became a professor (2017) in College of Chemical Engineering, Inner Mongolia University of Techonology. His research mainly focused on the synthetic method of sulfur-containing heterocycles, the research and development of synthetic process for the fine chemicals, the utilization of CO2 resources. |
1. Introduction
Domperidone (5-chloro-1-(1-(3-(2-oxo-2,3-dihydro-1H-benzo[d]imidazole-1-yl)propyl) piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazole-2-one), also named “Motinorm”, is a strong peripheral dopamine receptor antagonist.1 It acts as a prokinetic agent through its regulation on the motility of gastric and small intestinal smooth muscle and has antiemetic activity due to the blockade of dopamine receptors in the chemoreceptor trigger zone.2,3 As domperidone rarely penetrates the blood–brain barrier, it does not cause any adverse neurological symptoms, which is different from the metoclopramide in the central and peripheral effects.4 It can also be made into various suspensions, tablets or suppositories for patients with long-term oral administration.5 In the clinical trials for patients with symptoms of the diabetic gastropathy, some symptoms such as nausea, vomiting, and abdominal pain can be better relieved by domperidone.6 However, domperidone could cause sudden cardiac death and ventricular arrhythmia when used in daily doses >30 mg and in patients aged >60 years. Thus, the use of domperidone have also been alerted by several regulatory agencies (Health Canada, European Medicines Agency, Medicines and Healthcare Products Regulatory Agency in the UK, and Health Sciences Authority in Singapore) since 2012.7Domperidone was first synthesized in 1974,3 but the synthetic methods and processes of this drug have not been systematically reviewed. Herein, the reported synthetic methods and processes are summarized in this review. Domperidone is synthesized by coupling two benzimidazolone derivatives, N-halopropyl-2-benzimidazolone (intermediate 1) and N-(piperidin-4-yl)-6-chloro-2-benzimidazolone (intermediate 2) (Fig. 1). Therefore, the synthesis of domperidone can be divided into three processes, the syntheses of intermediates 1 and 2, and the coupling of the two intermediates. Additionally, the different processes of cyclization, coupling, reduction, protection and deprotection to prepare domperidone are analyzed in details. New possible routes of synthesizing domperidone are also proposed, which may provide more inspirations for the drug application.
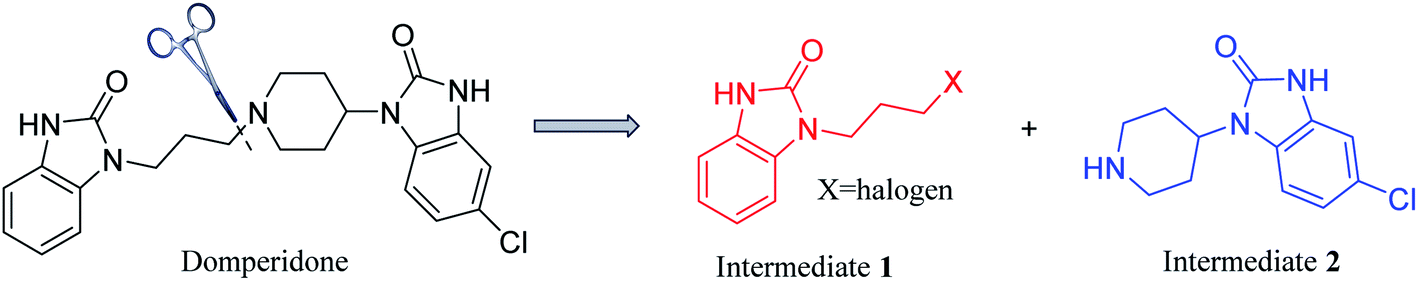 | ||
| Fig. 1 Retrosynthetic analysis process of domperidone. | ||
2. Synthesis of intermediate 1
There are two main synthesis routes of intermediate 1 (Scheme 1).8–13 One synthetic pathway is the cyclization of o-phenylenediamine with carbonyl reagents followed by coupling with 1,3-dihalopropane to afford intermediate 1 (Scheme 1, Route A). However, di-substituted by-products may be formed in the coupling process, resulting in poor reaction selectivity. Another synthetic route is the coupling of the o-halo or o-amino substituted nitrobenzene with 1,3-disubstituted propane followed by reduction and cyclization to afford intermediate 1 (Scheme 1, Route B). These synthetic routes demonstrate that intermediate 1 could be synthesized in at least 2–3 steps.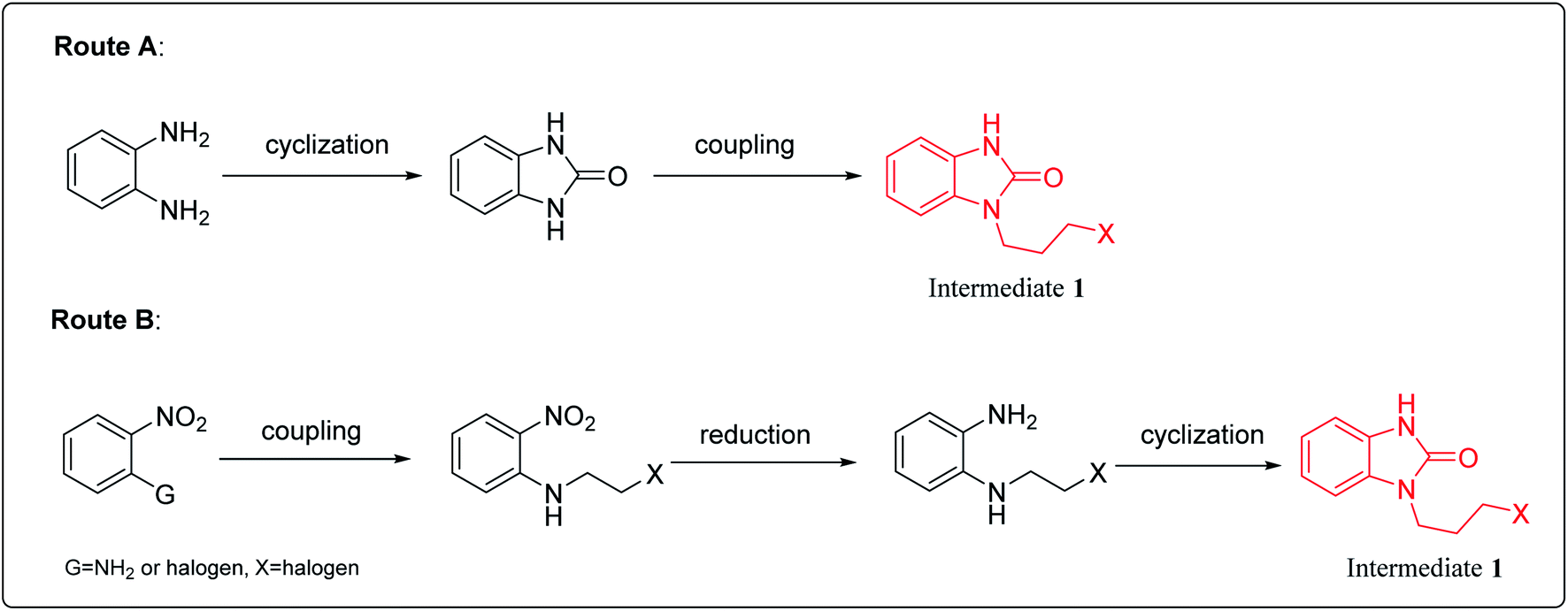 | ||
| Scheme 1 The synthetic routes of intermediate 1. | ||
2.1 Synthesis of benzimidazolone
Benzimidazolone, as a key intermediate product, needs to be prepared first. A host of methods have been developed for the synthesis of benzimidazolone and its derivatives. The synthetic methods of benzimidazolone can be classified into 6 types (Scheme 2).14,15 Firstly, benzimidazolone is commonly synthesized by cyclocarbonylation of ortho-substituted anilines 1 and various carbonyl reagents according to related references. For example, Bhanage et al.16 reported that benzimidazolone was obtained in 98% yields from the reaction of urea with 1,2-diaminobenzene without any catalysts under a reduced pressure. Qi et al.17 developed a selenium-catalyzed carbonylation reaction of 2-nitroanilines with TFBen (benzene-1,3,5-triyl triformate) to synthesize benzimidazolone. Vecchio et al.18 described a mild and efficient approach to synthesize benzimidazolone from o-azidoaniline. Additionally, the transition metal-catalyzed intermolecular 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 or intramolecular 320 ring closure reactions were reported as an alternative procedure. Furthermore, some methods for the synthesis of benzimidazolones using heterocyclic rings have been reported. For example, Li et al.21 reported a method of C–H oxidation reactions for the synthesis of benzimidazolone via copper-catalyzed benzimidazolium salts 4. More convenient methods, benzimidazolone could be synthesized via the Curtius reactions of phthalic anhydrides 5 or anthranilic acid 6 and azide.15 Interestingly, Shingare et al.22 proposed a mild protocol for the synthesis of benzimidazolone through decarbonylation of quinoxalinediones 7. Among them, the cyclization reaction of o-phenylenediamine and carbonyl reagents is the main method to synthesize benzimidazolone.
19 or intramolecular 320 ring closure reactions were reported as an alternative procedure. Furthermore, some methods for the synthesis of benzimidazolones using heterocyclic rings have been reported. For example, Li et al.21 reported a method of C–H oxidation reactions for the synthesis of benzimidazolone via copper-catalyzed benzimidazolium salts 4. More convenient methods, benzimidazolone could be synthesized via the Curtius reactions of phthalic anhydrides 5 or anthranilic acid 6 and azide.15 Interestingly, Shingare et al.22 proposed a mild protocol for the synthesis of benzimidazolone through decarbonylation of quinoxalinediones 7. Among them, the cyclization reaction of o-phenylenediamine and carbonyl reagents is the main method to synthesize benzimidazolone.
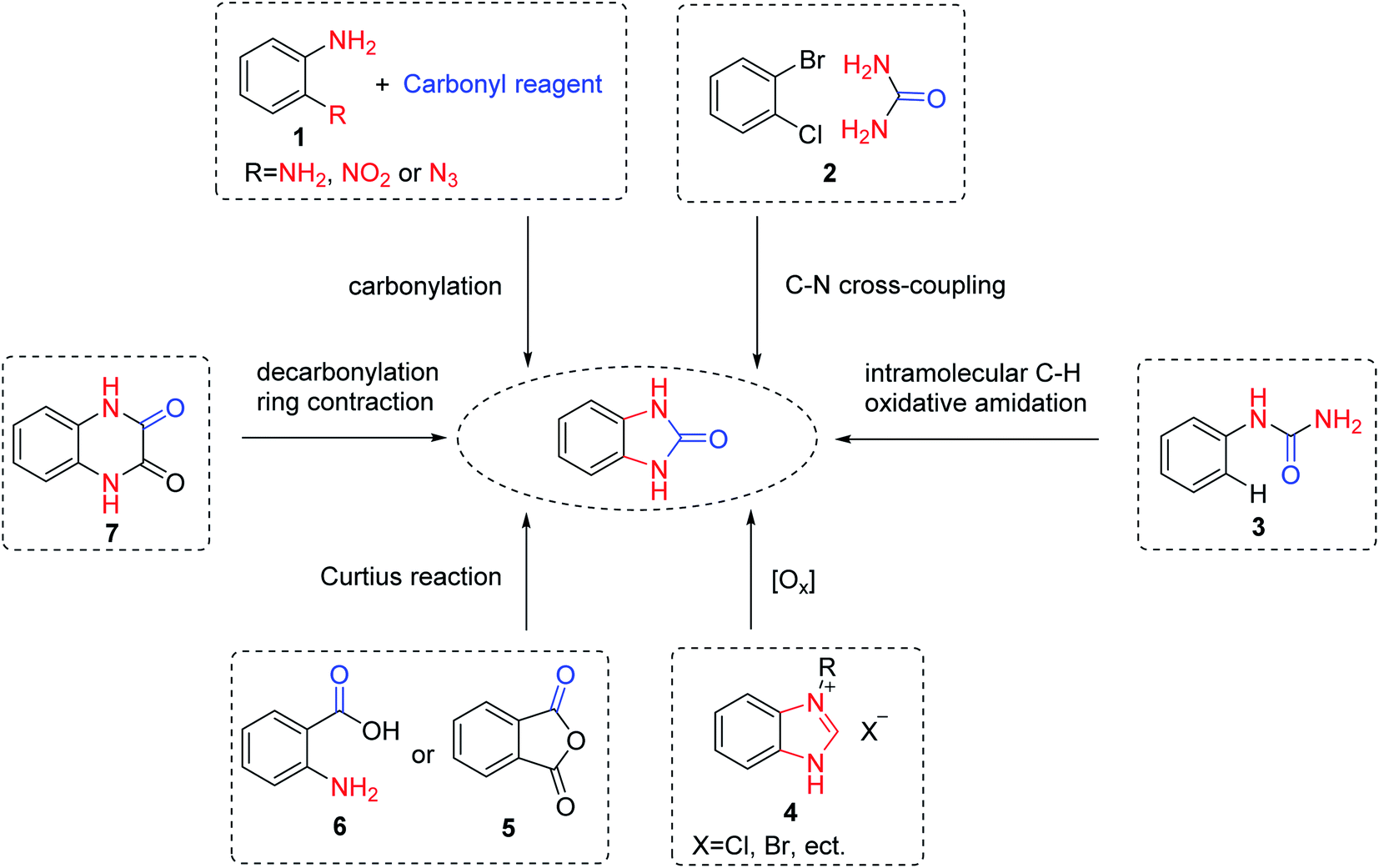 | ||
| Scheme 2 Synthesis of benzimidazolone from different starting materials. | ||
To avoid the use of toxic phosgene in the synthesis of benzimidazolone, various carbonyl reagents have been applied with good to excellent yields (Fig. 2).23–28 However, these catalytic systems have suffered from several drawbacks, such as the use of precious metals (Au, Pb and Ln etc.), and the complex preparation process of catalysts. Moreover, only a carbonyl group is required in the cyclization reaction by comparing the structure of o-phenylenediamine with benzimidazolone. In order to avoid the generation of by-products, improve atomic economy, the carbonyl reagents with simpler structure are more suitable for synthesizing benzimidazolone from o-phenylenediamine, such as CO or CO2.
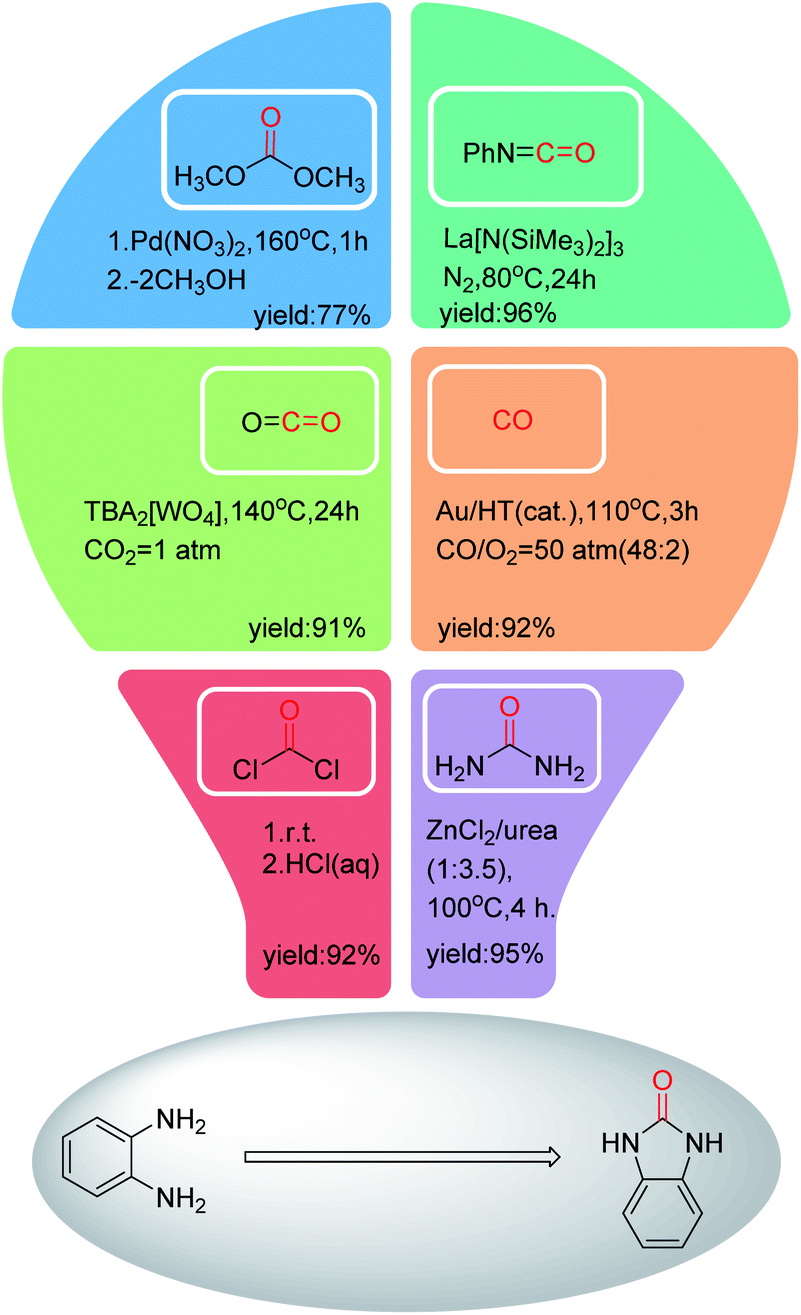 | ||
| Fig. 2 Preparation of benzimidazolone from the reaction of o-phenylenediamine with different carbonyl reagents. | ||
Most of the existing carbonyl reagents can be prepared from carbon monoxide (CO) (Fig. 3). High activity phosgene is obtained by the reaction of CO and chlorine gas,29 and a series of carbonyl reagents such as isocyanate,30 N,N′-carbonyldiimidazole (CDI),31 ethyl chloroformate32 and dimethyl carbonate33 are derived subsequently. Dimethyl carbonate is obtained by the liquid-phase oxidative carbonylation of methanol and CO. The production of urea in the industry is accomplished mainly through the reaction of NH3 and CO2 under high temperature and high pressure.34 Notably, CO2 is produced by the water–gas shift, which needs a great amount of CO. Therefore, CO, as the key starting material to form carbonyl reagents, has high demands in chemical manufacturing. However, CO is obtained through incomplete combustion of coal, which consumes a huge amount of fossil energy for industrial production. CO is a toxic, flammable and explosive gas, which is dangerous for large-scale industrial utilization, storage and transportation. Although the use of CO as a carbonyl reagent has the simplest structure and the highest atomic economy, the carbon–oxygen bond in CO is relatively stable, which needs to be catalyzed by active metal catalysts.24 The expensive and stable precious-metal catalysts also hinder the utilization of CO.
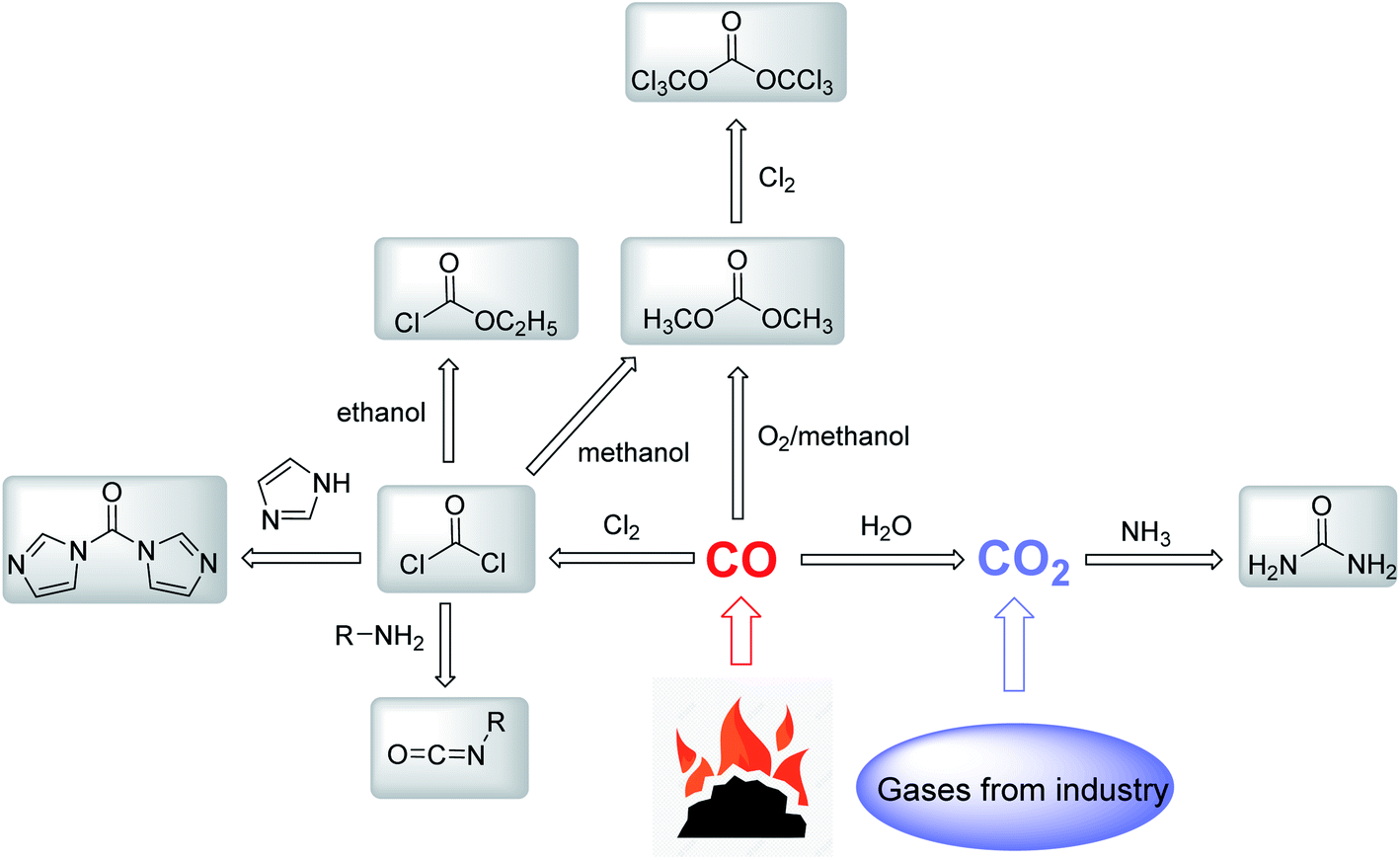 | ||
| Fig. 3 General synthetic approaches of different carbonyl reagents. | ||
CO2 is a kind of abundant and cheap industrial waste gas. More than 30 billion tons of CO2 are released into the atmosphere every year since 2010,35 which cause global warming and climate change. According to the production sources and availability of existing carbonyl reagents as well as their impact on environment, CO2 is suitable as a carbonyl reagent undoubtedly. In recent years, various methods using CO2 as a green carbonyl reagent have been developed for synthesizing benzimidazolone and its derivatives.26,36,37 Liu research group reported that benzimidazolone was synthesized via the reactions of ScCO2 with o-phenylenediamine catalyzed by [DBUH][OAc] ionic liquids under solvent free conditions.36 Notably, CO2 reacts with o-phenylenediamine to produce benzimidazolone and the only water by-product. Therefore, the synthesis of domperidone intermediates using CO2 will become a greener synthesis method in the future.
2.2 Preparation of intermediate 1 from benzimidazolone
Benzimidazolone has two chemically identical amino groups, the hydrogen of which is removed or hydrogen-bonded with a base to enhance the nucleophilicity of the nitrogen atom. When benzimidazolone is directly coupled with 1,3-dihalopropane in a basic system, the intermediate 1 or di-substituted by-product (1,3-bis(3-halogenpropyl)-2-benzimidazolone) could be generated during the coupling reaction process (Fig. 4). Therefore, how to obtain mono-substituted product (intermediate 1) selectively is the key for the synthesis of intermediate 1 by direct N-alkylation of benzimidazolone.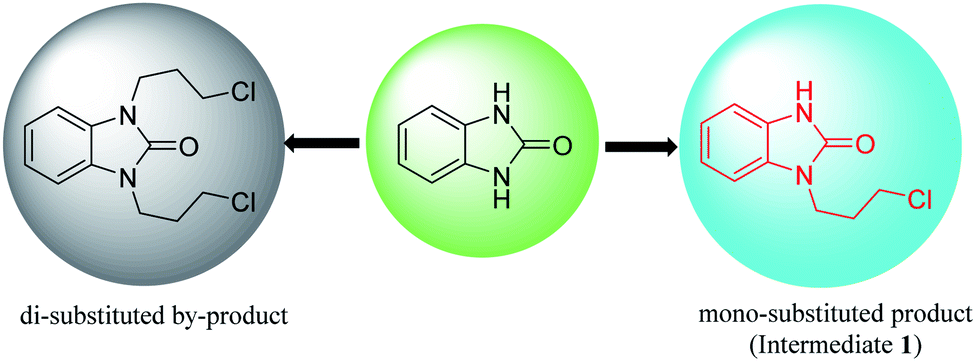 | ||
| Fig. 4 Substitution of benzimidazolone by halide. | ||
A patent10 reported that the intermediate 1 was obtained in 83% yield by direct coupling reaction of benzimidazolone with 1-bromo-3-chloropropane in a two-phase solution consisting of 10% (w/v) NaOH aqueous solution and dichloromethane (Scheme 3). Benzimidazolone has limited solubility in majority of the common organic solvents (alcohols, esters, ketones and alkanes) at room temperature. Solvent polarity and hydrogen bond has important effects on the solubility of benzimidazolone.38 When dichloromethane is used as the solvent, the incompletely dissolved benzimidazolone may be dispersed in the reaction system as solid particles. Although this patented method has the advantages of simple operation and high yield, little is discussed on the role of alkali and whether di-substituted by-product was produced.
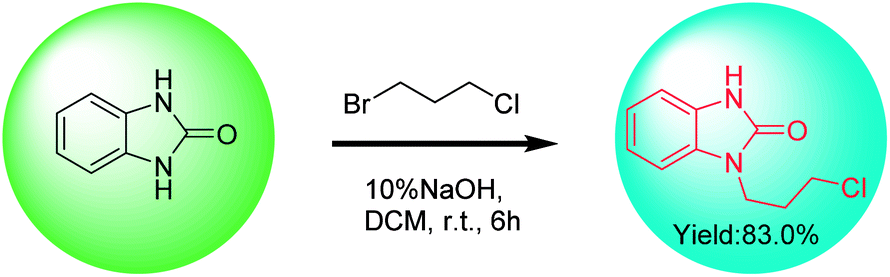 | ||
| Scheme 3 The synthetic process of intermediate 1 from benzimidazolone. | ||
To solve the selectivity problem, the following two strategies of protecting benzimidazolone are mainly used to synthesize intermediates 1. In the first strategy, the mono-N-protected benzimidazolone was prepared by the reaction of benzimidazolone with a N-protecting reagent, followed by the substitution of unsubstituted N-site with 1,3-dihalopropane, and then deprotection of the protecting group yields intermediate 1.
tert-Butylcarbonyl (Boc) is often used as the protecting group of amines in preparing mono-N-protected benzimidazolone.39 The coupling reaction of mono-Boc-protected benzimidazolone and 1,3-dibrompropan is proceeded rapidly by microwave radiation with the catalysis of tetrabutyl ammonium bromide (TBAB), followed by the Boc group removal in the presence of trifluoroacetic acid (TFA) to give intermediate 1 with a yield of more than 92% (Scheme 4). The protection and deprotection strategy are frequently used in the preparation of drug molecules, in which the Boc group is often used to protect amino groups in drug synthesis due to its high stability towards base, nucleophiles or the reaction conditions of catalytic hydrogenolysis. Many methods for N-Boc deprotection in acidic conditions have been reported, such as protic acids (TFA, HCl, H2SO4, HNO3 and aqueous phosphoric acid, etc.) and Lewis acids (FeCl3, BiCl3 AlCl3, TiCl4, SnCl4, ZnBr2, BF3⋅OEt2, TMSI, TMSOTf, Sn(OTf)2 and Ce(NH4)2(NO3)6, etc.).40,41 However, these methods have several disadvantages in drug synthesis, such as the need for cumbersome neutralization in the later stage, generating intractable waste salts, and the use of excessive and unrecoverable catalysts and solvents, etc. Recently, a flow chemistry strategy for N-Boc removal was proposed.42,43 This strategy is a clean, fast and highly selective method. Under the action of high temperature or Lewis acid, nearly quantitative and highly selective N-Boc deprotection products were obtained within minutes to hours in a continuous flow reactor, while avoiding the subsequent process of purification or concentration.
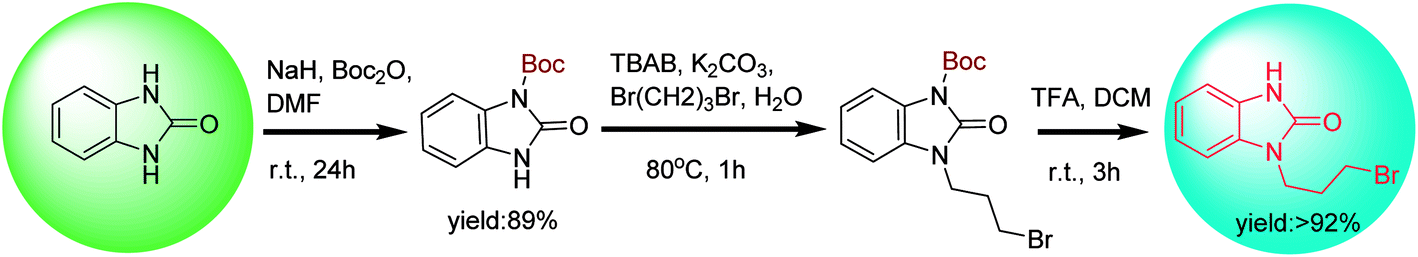 | ||
| Scheme 4 Synthesis of intermediate 1 though N-protected benzimidazolone. | ||
The presence of N-protecting group improves the yield and purity of intermediate 1, avoiding the formation of di-substituted by-product in the coupling process. However, the introduction of protection and deprotection processes increases the lengths and complexity of the synthetic route.
In the second strategy, o-phenylenediamine reacted with acetyl acetate to form N-isopropenyl-2-benzimidazolone (mono-substituted benzimidazolone), in which one N-position of the imidazole ring is substituted with an isopropenyl group.12,44,45
The cyclization and rearrangement of o-phenylenediamine and ethyl acetoacetate to afford N-isopropenyl-benzimidazolone under the alkaline condition, followed by coupling with 1-bromo-3-chloropropane using TBAB as phase-transfer catalysts and the final deprotection of isopropenyl by hydrolysis, afforded intermediate 1 in up to 90% yield and 99% purity (Scheme 5).12 In this strategy, mono-substituted benzimidazolone (N-isopropenyl-benzimidazolone) is prepared by a simple one-step reaction, in which the cyclization and mono-N-protection of benzimidazolone was completed simultaneously. Compared with the first strategy of adding additional protecting groups, it is obvious that the second strategy using acetyl acetate as a cyclizing reagent is a shorter synthetic route.
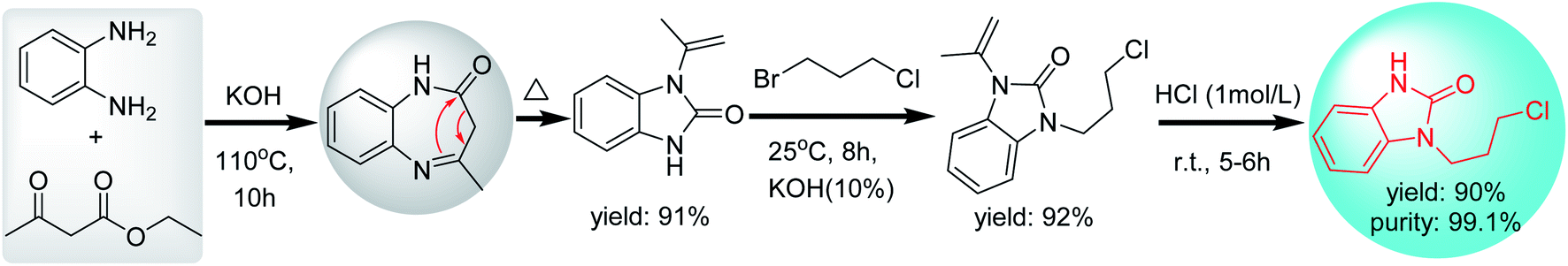 | ||
| Scheme 5 Synthesis of intermediate 1 though N-substituted benzimidazolone. | ||
The aforementioned synthesis process of intermediate 1 (Scheme 1, Route A) was carried out in basic conditions. The base can remove the hydrogen on nitrogen atom of benzimidazolone to enhance the electron-cloud density of the nitrogen atom, which promotes the reaction of benzimidazolone with 1,3-disubstituted propane. Additionally, the base can neutralize the released acid and prevent protecting group from being deprotected by the acid during the coupling reaction of benzimidazolone and 1,3-disubstituted propane. Inorganic bases such as alkaline metal carbonates or hydroxides are generally used in the synthesis of intermediate 1 due to their low cost. However, aqueous alkaline solution and organic reagent could form an oil-water two-phase reaction system, which may prevent the coupling reaction of benzimidazolone and 1,3-disubstituted propane to occur. Thus, TBAB ((C4H9)4N+Br−) as a phase transfer catalyst is required to promote this two-phase reaction system (Fig. 5).46–48
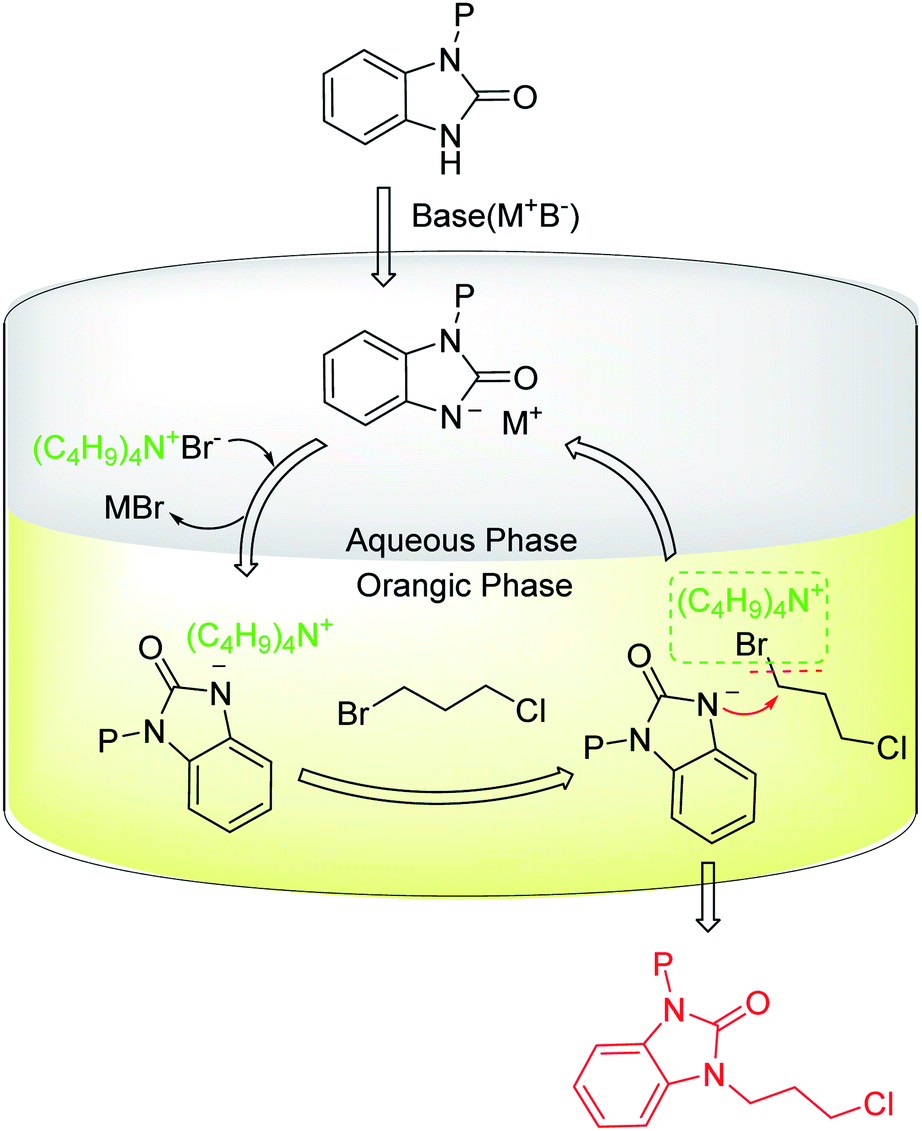 | ||
| Fig. 5 The proposed catalytic process of TBAB as phase transfer catalyst for the synthesis of intermediate 1. | ||
2.3 Synthesis of intermediate 1 through coupling followed by reduction and cyclization
Unlike the synthetic route of intermediate 1 by cyclization followed by coupling (Scheme 1, Route A), intermediate 1 can also be synthesized by coupling followed by reduction and cyclization (Scheme 1, Route B). Firstly, N-hydroxypropyl-2-nitroaniline is synthesized by two methods with different starting materials: (1) reaction of 2-nitroaniline and 3-chloropropan-1-ol; (2) reaction of 2-halogeno-nitrobenzene and 3-aminopropane-1-ol. However, 2-nitroaniline is rarely used due to its conjugation effect, which may reduce the reactivity of amino group and make it difficult to couple with 3-chloropropan-1-ol to give N-hydroxypropyl-2-nitroaniline. Therefore, o-chloronitrobenzene and 3-aminopropane-1-ol are generally selected as starting materials to afford intermediate 1. Generally, a small amount of sodium iodides or potassium iodides are used as catalysts to enhance the coupling reaction. During which, the chloride is replaced by iodide, which makes coupling reaction easier, accelerates reaction rates, shortens reaction time and increases reaction yield. Subsequently, N-hydroxypropyl-2-nitroaniline undergoes reduction, cyclization and halogenation to afford intermediate 1 (Scheme 6).49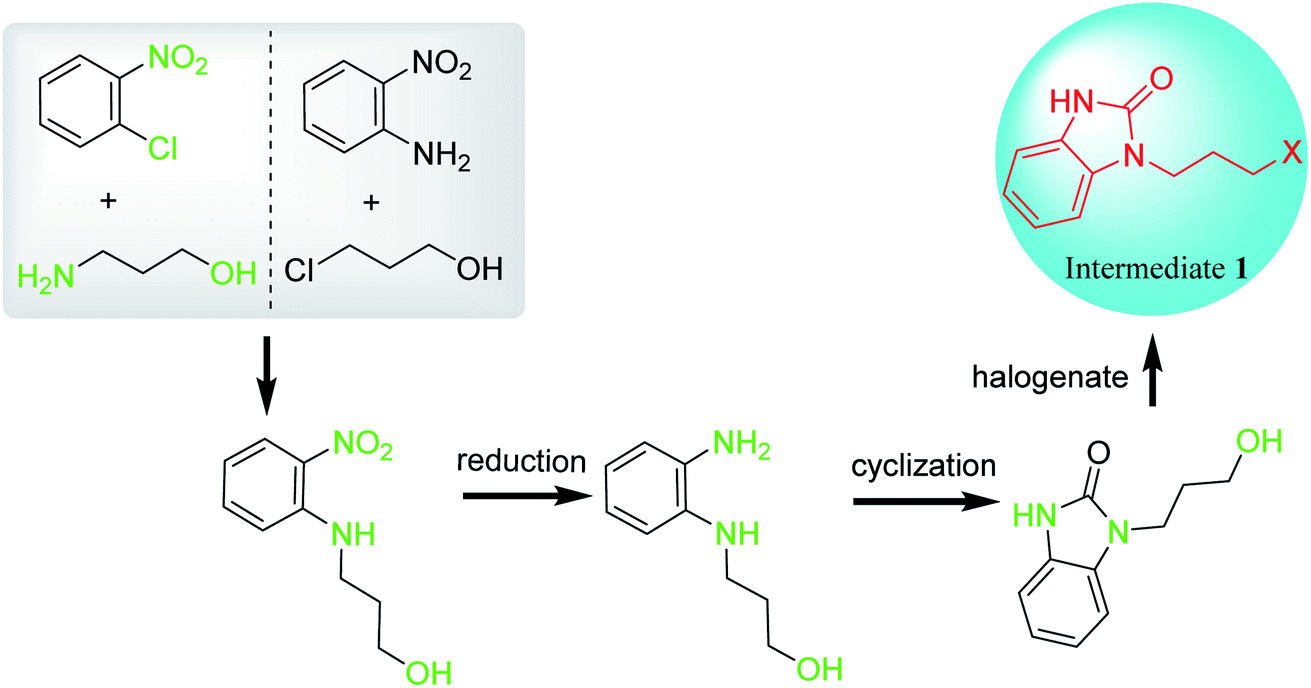 | ||
| Scheme 6 Preparation of intermediate 1 from o-substituted nitrobenzene. | ||
The reduction of nitro group can be divided into heterogeneous catalytic reduction and homogeneous catalytic reduction. Compared with the homogeneous catalysts, supported heterogeneous catalysts (such as RANEY®-Ni, Pd/C, Pt/C, Au/TiO2, Ag/Al2O2) have greater application value in industry due to their easily recyclable and reusable properties.50 In the synthesis of intermediate 1, nitro group is reduced by H2 using RANEY®-Ni or Pd/C (10%) catalysts at room temperature, and N-hydroxypropy-benzene-1,2-diamine is obtained almost quantitatively after the catalyst was simply removed by filtration from the reaction solution. Notably, nitro reduction is a multistep reaction in which a number of hydroxylamine intermediates and by-products of azos and azo oxides are produced (Scheme 7).51,52 Although the residual mutagenic intermediates are rarely controlled, the catalysts for the reduction of nitro groups must be highly selective and reactive, capable of rapidly reducing all intermediates or by-products to amino groups.
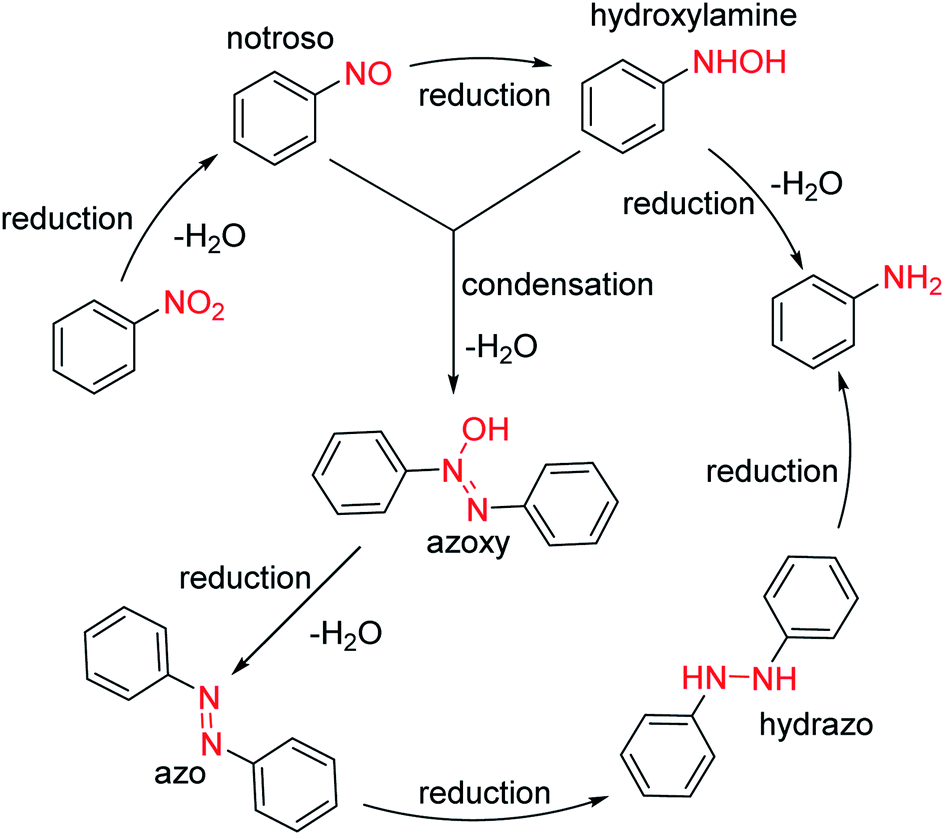 | ||
| Scheme 7 Proposed mechanisms for nitro reduction. | ||
The cyclization reactions of o-phenylenediamine and carbonylation reagents to prepare benzimidazolone are also suitable for preparing N-hydroxypropy-2-benzimidazolone from N-(3-hydroxypropy)-benzene-1,2-diamine. Furthermore, as the hydroxyl group of N-hydroxypropy-2-benzimidazolone is a poor leaving group, the conversion of this group into a better leaving group such as halogen is required during the synthesis of intermediate 1. During the conversion of alcohols into corresponding halides, the protonated hydroxyl groups of alcohols are dehydrated under the catalysis of Brønsted acid, and then react with halide anions to form halides. Notably, water would be formed as the sole by-product. However, dehydrating agents such as sulfuric acid are usually required to promote the dehydration, which causes low levels of chemoselectivity (e.g. functional group tolerance). Additionally, some simple and cheap acid chlorides such as phosgene, thionyl or oxalyl chloride enable the conversion of alcohols to the corresponding halides under the catalysis of organic base. Since the by-product HCl is generated during the reaction, triethylamine or pyridine as an organic base catalyst is usually required to react with HCl to release chlorine atoms, thereby improving the reaction efficiency of the chlorinating reagent.53,54 Therefore, the development of halogenation methods with high atom economy, mild reaction conditions and environmental friendliness is the goal to realize the halogenation of N-hydroxypropyl-benzimidazolones to synthesize intermediate 1.
Among the discussed synthesis methods, the preparation of intermediate 1 directly from benzimidazolone (Route A) has advantages of short reaction route and simple post-processing. However, the oil-water two-phase system requires additional phase transfer catalysts to promote the coupling reaction. Additionally, benzimidazolone with two identical N-alkylation sites may produce the di-substituted by-products. Therefore, the synthesis method of intermediate 1 by route A has poor selectivity, and mono-N-protected benzimidazolone need to be used in the coupling reaction process. In contrast, the synthesis of intermediate 1 through route B is a homogeneous reaction process. To avoid producing by-product, o-chloronitrobenzene is coupled with 3-aminopropane-1-ol to form N-hydroxypropyl-2-nitroaniline, which is successively reduced, cyclized and halogenated to generate intermediate 1. However, the process of reduction and halogenation increases the complexity and length of the route B. Undoubtedly, the synthesis route of intermediate 1 should be improved towards shorter route and higher selectivity in the near future.
3. Synthesis of intermediate 2
Intermediate 2 is synthesized in a manner similar to the route B of intermediate 1. First, N-(4-chloro-2-nitrophenyl)piperidin-4-amine is synthesized by the coupling reaction of 2-halogeno-5-chloro-nitrobenzene with 4-aminopiperidine in weak alkali condition, followed by reduction, cyclization and deprotection to prepare intermediate 2 (Scheme 8).55–58 Notably, N-(4-chloro-2-nitrophenyl)piperidin-4-amine could be also synthesized by the coupling reaction of 4-chlorobenzene-1,2-diamine and 4-halopiperidine. However, the coupling by-product could be obtained due to the limitation of the chloro-substituent on benzene ring. Therefore, there is a unique synthetic route for preparing intermediate 2 from 2-halo-5-chloro-nitrobenzene and 4-aminopiperidine.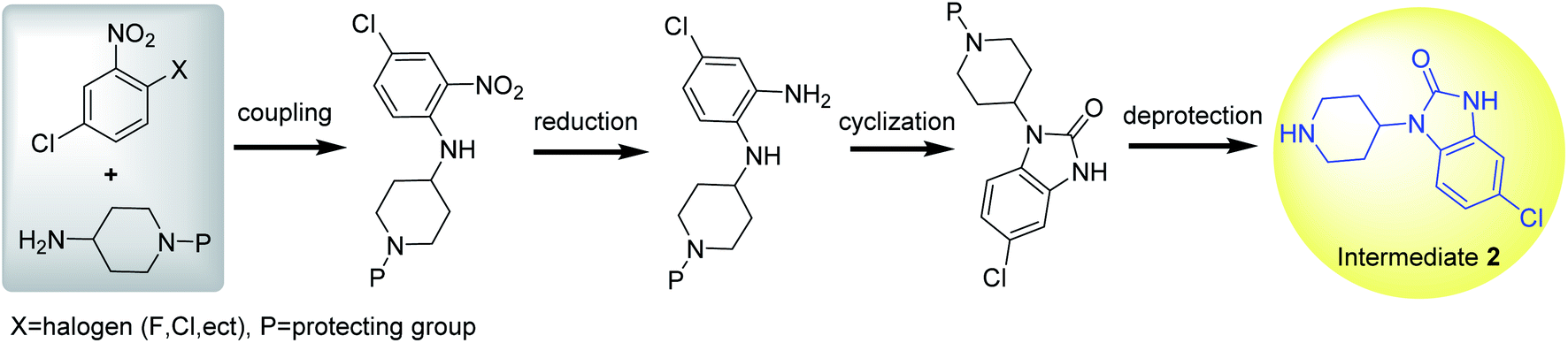 | ||
| Scheme 8 Synthesis of intermediate 2. | ||
To improve the selectivity of coupling reaction, benzyl (Bn), benzylcarbonyl (Cbz),56 alkoxycarbonyl,59,60 and Boc61 have been used as the protective groups of 4-aminopiperidine in the synthesis of intermediate 2. Moreover, different protecting groups have different ways of deprotection. Benzyl or Cbz could be removed by catalytic hydrogenation (20% Pd(OH)2/C, H2) at room temperature with high yield of target product after filtering catalysts (Table 1, entry 1). Boc could be deprotected in 20–35% TFA at room temperature (Table 1, entry 2). Alkoxycarbonyl group could be released by hydrolysis in alkaline or acidic conditions, and the intermediate 2 could be precipitated by adjusting the reaction mixture solution to neutral or weak alkaline after acidic hydrolysis. Therefore, the nitrogen atom in the piperidine ring could accept multiple types of protecting groups, which allows for flexible protection and deprotection.
| Entry | N-protecting group | Deprotection method | Nitro reduction method | ||
|---|---|---|---|---|---|
| 1 | Bn or Cbz | Catalytic hydrogenation | 20% Pd(OH)2/C, H2, r.t. | Bechamp reduction | SnCl2–H2O |
| 2 | Boc | Acidic hydrolysis | TFA (20–35%), r.t. | Catalytic hydrogen transfer reaction | RANEY®-Ni, N2H4 H2O |
| Pd/C (5%), N2H4, MeOH | |||||
| 3 | Ethoxycarbonyl | Alkaline hydrolysis | NaOH (1N), 100 °C | Catalytic hydrogenation | RANEY®-Ni, H2 |
| Zn, HCl, MeOH | |||||
To prepare intermediate 2, the reduction method of nitro group is different from that of intermediate 1. When the protecting group is benzyl, the nitro group is reduced to amino group quantitatively through Bechamp reductive reaction under the catalytic system of SnCl2–H2O (Table 1, entry 1). During which, SnCl2 provides electrons for the reduction reaction, and H2O acts as both proton provider and solvent.56 When the protecting group is Boc, RANEY®-Ni/N2H4 H2O61 or Pd/C (5%)/N2H4/MeOH62 could efficiently reduce nitro group to amino group through catalytic hydrogen transfer reaction, in which the hydrazine provided the protons for the reductive reaction (Table 1, entry 2). When the protecting group is ethoxycarbonyl, RANEY®-Ni/H2 or Zn/HCl/MeOH could reduce nitro group to amino group with high selectivity through catalytic hydrogenation (Table 1, entry 3).10,55,60 Although these reductive methods have strong nitro reduction ability, the greener, lower-cost and higher active nitro reduction systems still need to be developed due to the environmental impact and production cost.
Even though only one unique synthesis route for preparing intermediate 2 had been reported in the literature, there were other four synthetic strategies to prepare the analogues of intermediate 2 that have no 6-chloro substituent on the benzene ring (Scheme 9).63 Among them, (1) arylurea (I) is formed from a range of different starting materials, followed by cyclization and deprotection to produce the analogues of intermediate 2. (2) Condensation of o-phenylenediamine with 3-alkoxycarbony-4-piperidone generated intermediate II, followed by rearrangement, reduction and deprotection to produce the analogues of intermediate 2. (3) Reductive alkylation of o-nitroaniline with an N-protected 4-piperidone could form intermediate III, followed by reduction of the nitro group, cyclization and deprotection to provide the analogues of intermediate 2. (4) Reductive alkylation of anthranilic ester and 4-piperidone as starting materials generated intermediate IV, followed by Curtius rearrangement, cyclization and deprotection to provide the analogues of intermediate 2. These synthetic methodologies provide new routes to synthesize intermediate 2.
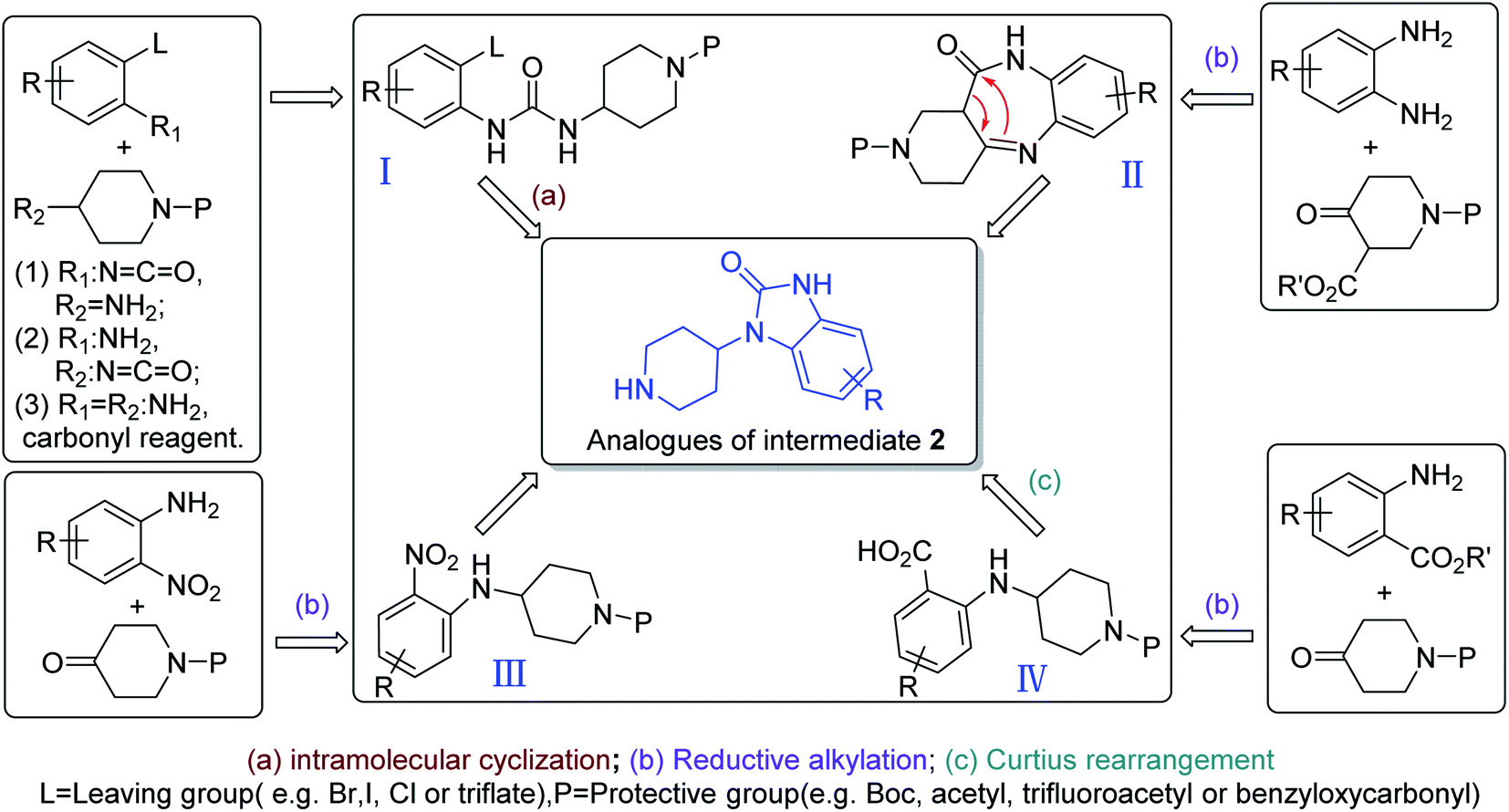 | ||
| Scheme 9 Synthetic strategies to prepare the analogues of intermediate 2. | ||
4. Synthesis of domperidone
Domperidone can be prepared by simple coupling of intermediates 1 and 2 under alkali condition (Scheme 10). This coupling reaction of amine and halide is similar to the reaction to prepare intermediate 1 or 2. Similarly, KI is used as a catalyst to accelerate the coupling reaction. After the reaction was completed, the domperidone product is isolated from the alkaline solution through column separation or precipitation by pH adjustment.8,10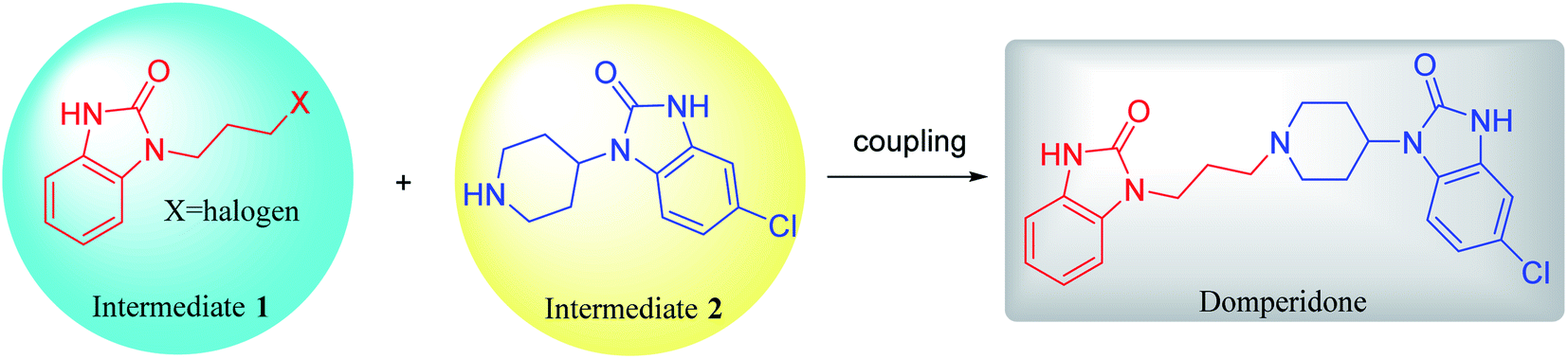 | ||
| Scheme 10 Coupling of intermediates 1 and 2 to form domperidone. | ||
Moreover, the domperidone can also be prepared via uncyclized intermediates according to the synthetic strategies reported in a patent.8 These uncyclized intermediates could be prepared from the coupling and reduction reaction of N-(4-chloro-2-nitrophenyl)piperidin-4-amine with N-protected intermediates 1 or N-halopropyl-2-nitroaniline, and then cyclized to form domperidone (Scheme 11). However, the detailed experimental procedures are not available in the patent, which still need to be explored by some researchers in the near future.
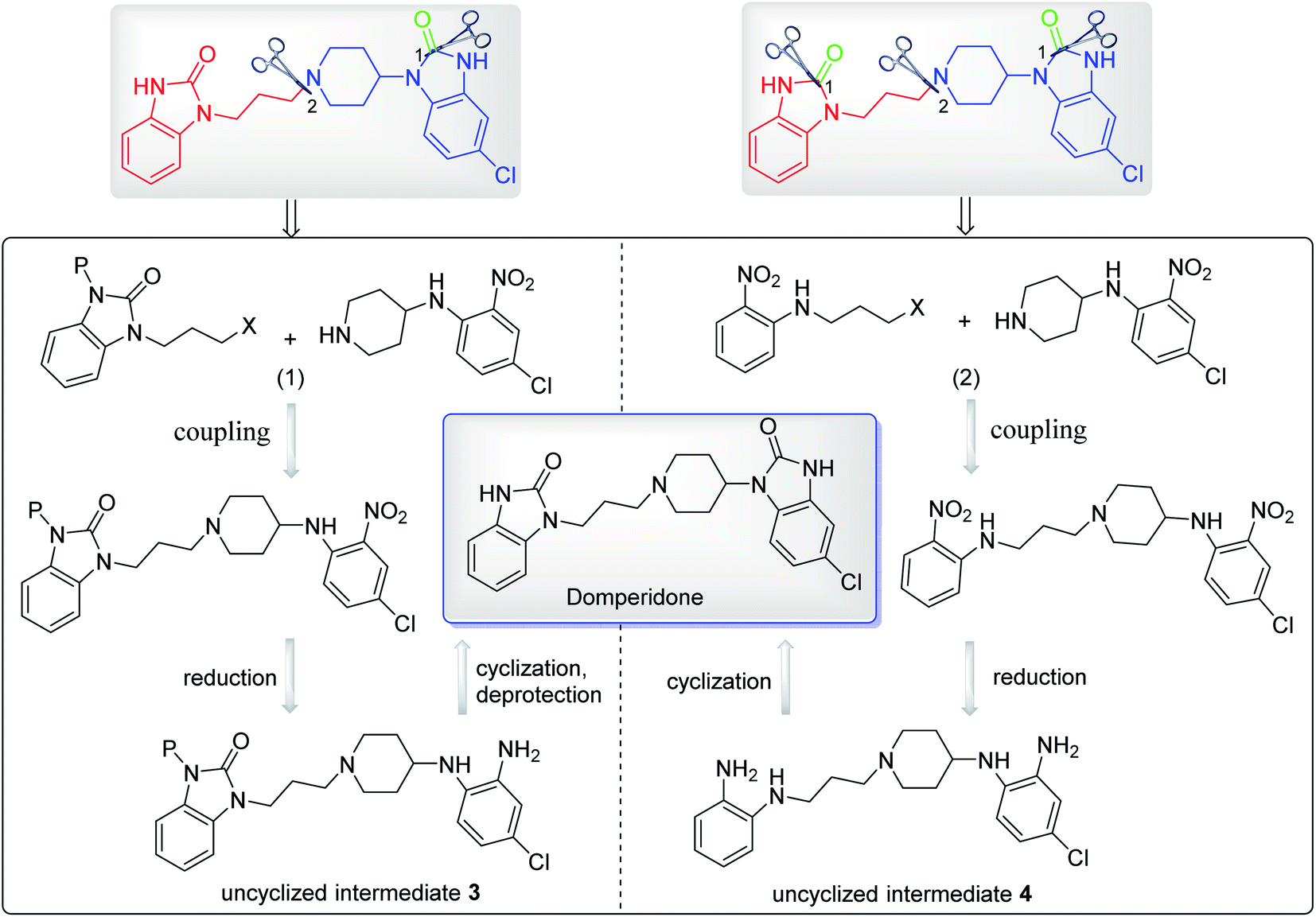 | ||
| Scheme 11 Other synthetic strategies for preparing domperidone. | ||
5. Conclusion and outlook
Domperidone is synthesized by the coupling reaction of N-halopropyl-2-benzimidazolone (intermediate 1) and N-(piperidin-4-yl)-6-chloro- 2-benzimidazolone (intermediate 2). Intermediate 1 can be synthesized by two synthetic routes (Scheme 1). Of which, the coupling of benzimidazolone prepared from the reaction of o-phenylenediamine and different carbonyl reagents with 1,3-dihalopropane is used to produce intermediate 1 (Route A). The o-substituted nitrobenzene reacted with 1,3-disubstituted propane, followed by reduction and cyclization to produce intermediate 1 (Route B). Notably, the coupling reaction of Route B has higher reaction selectivity than that of Route A, which indicated that Route B is more suitable for synthesizing intermediate 1. Additionally, intermediate 2 is synthesized by only one synthetic route from the substituted nitrobenzene, which is similar to the Route B of intermediate 1. Fortunately, several reported synthetic methods for the analogs of intermediate 2 would greatly accelerate the future development of synthetic methodologies of intermediate 2. In addition, an overall understanding of these synthetic routes will provide new approaches for preparing domperidone.Benzimidazolone derivatives exhibit a wide range of biological activities and can be used to synthesize various protein receptor inhibitors or antagonists (Fig. 6).64–68 However, the processes for synthesizing these bioactive compounds face similar problems, such as regioselective substitution, protection-deprotection of amino group and the selection of reaction route. The methods for synthesizing domperidone discussed in this review would provide more insights for preparing compounds with similar structures.
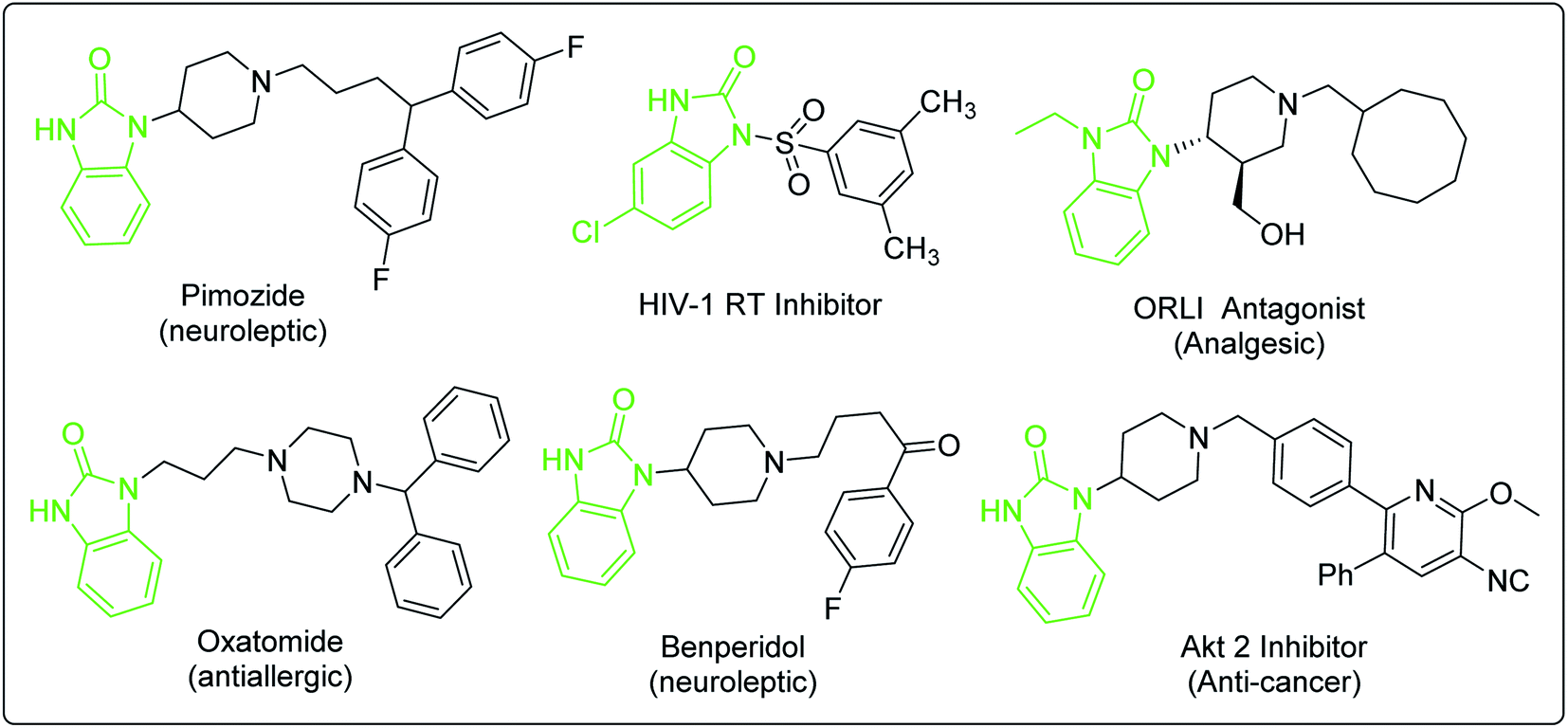 | ||
| Fig. 6 Bioactive compounds with benzimidazolone moiety. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21865020), Natural Science Foundation of Inner Mongolia Autonomous Region (2021ZD02 and 2022QN05004), Inner Mongolia Science and Technology Key Projects (2021GG0335), Program for Grassland Excellent Talents of Inner Mongolia Autonomous Region, the Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (2019), and Innovative Research Team in Universities of Inner Mongolia Autonomous Region (NMGIRT2212), Research Foundation for Introducing High-level Talents in Inner Mongolia Autonomous Region (2021), Doctoral Scientific Research Foundation of Inner Mongolia University of Technology for financial support.References
- G. M. Brown, H. Verhaegan, T. B. Van Wimersma Greidanus and J. Brugmans, Clin. Endocrinol., 1981, 15, 275–282 CrossRef CAS PubMed.
- T. Takahashi, S. Kurosawa, J. W. Wiley and C. Owyang, Gastroenterology, 1991, 101, 703–710 CrossRef CAS.
- S. C. Reddymasu, I. Soykan and R. W. Mccallum, Am. J. Gastroenterol., 2007, 102, 2036–2045 CrossRef CAS PubMed.
- P. M. Laduron and J. E. Leysen, Biochem. Pharmacol., 1979, 28, 2161–2165 CrossRef CAS PubMed.
- M. C. Champion, M. Hartnett and M. Yen, Drug Therapeut. Bull., 1988, 26, 2659–2660 Search PubMed.
- J. A. Barone, Ann. Pharmacother., 1999, 33, 429–440 CrossRef CAS PubMed.
- L. B. Ou, C. Moriello, A. Douros and K. B. Filion, Br. J. Clin. Pharmacol., 2021, 87, 3649–3658 CrossRef CAS PubMed.
- J. Vandenberk, L. E. J. Kennis, M. J. M. C. Van der Aa and A. H. M. Th. Van Heertum, US Pat., 4066772 A, 1978 Search PubMed.
- J. Vandenberk, L. E. J. Kennis, A. H. M. Th. Van Heertum and M. J. M. C. Van der Aa, US Pat., 4254127 A, 1981 Search PubMed.
- S. Li, C. Liu and W. Li, CN 1810805 A, 2006.
- R. Liao, CN 105439960 A, 2016.
- A. Qiu, C. Zhang and Z. Ye, CN 108129396 A, 2018.
- A. F. Fliri, B. T. O'Neill, W. S. Faraci, M. A. Sanner and S. H. Zorn, WO 199534555 A1, 1995.
- H. Zhang, D. Huang, K. Wang, J. Li, Y. Su and Y. Hu, J. Org. Chem., 2017, 82, 1600–1609 CrossRef CAS PubMed.
- R. Armenta and J. Sarmiento-Sánchez, Chem. Heterocycl. Compd., 2016, 52, 1002–1004 CrossRef CAS.
- B. M. Bhanage, S. I. Fujita, Y. Ikushima and M. Arai, Green Chem., 2004, 6, 78–80 RSC.
- X. Qi, R. Zhou, J. Peng, J. Ying and X. Wu, Eur. J. Org. Chem., 2019, 2019, 5161–5164 CrossRef CAS.
- A. Del Vecchio, F. Caillé, A. Chevalier, O. Loreau, K. Horkka, C. Halldin, M. Schou, N. Camus, P. Kessler, B. Kuhnast, F. Taran and D. Audisio, Angew. Chem., Int. Ed., 2018, 57, 9744–9748 CrossRef CAS PubMed.
- J. B. Ernst, N. E. S. Tay, N. T. Jui and S. L. Buchwald, Org. Lett., 2014, 16, 3844–3846 CrossRef CAS PubMed.
- J. Yu, C. Gao, Z. Song, H. Yang and H. Fu, Eur. J. Org. Chem., 2015, 2015, 5869–5875 CrossRef CAS.
- J. Li, H. Yu, M. Xie, G. Qu, H. Niu, H. Wang, B. Qin and H. Guo, J. Org. Chem., 2013, 78, 12629–12636 CrossRef CAS PubMed.
- R. D. Shingare, A. S. Kulkarni, R. L. Sutar and D. S. Reddy, ACS Omega, 2017, 2, 5137–5141 CrossRef CAS PubMed.
- R. L. Clark and A. A. Pessolano, J. Am. Chem. Soc., 1958, 80, 1657–1662 CrossRef CAS.
- A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Green Chem., 2013, 15, 608–611 RSC.
- K. Aghapoor, F. Mohsenzadeh, H. R. Darabi, H. Sayahi and M. R. Jalali, ChemistrySelect, 2019, 4, 11093–11097 CrossRef CAS.
- T. Kimura, K. Kamata and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 6700–6703 CrossRef CAS PubMed.
- Y. Fu, T. Baba and Y. Ono, J. Catal., 2001, 197, 91–97 CrossRef CAS.
- Y. Jing, R. Liu, Y. Lin and X. Zhou, Sci. China: Chem., 2014, 57, 1117–1125 CrossRef CAS.
- A. Bähr, G. Moon, J. Diedenhoven, J. Kiecherer, E. Barth and H. Tüysüz, Chem. Ing. Tech., 2018, 90, 1513–1519 CrossRef.
- P. Wang, S. Liu and Y. Deng, Chin. J. Chem., 2017, 35, 821–835 CrossRef CAS.
- H. A. Staab and K. Wendel, Org. Synth., 1968, 48, 44 CrossRef CAS.
- F. Liang, M. Yanai, Y. Suzuki and A. Tsuda, Org. Lett., 2020, 22, 3566–3569 CrossRef CAS PubMed.
- H. Tan, Z. Wang, Z. Xu, J. Sun, Y. Xu, Q. Chen, Y. Chen and C. Guo, Catal. Today, 2018, 316, 2–12 CrossRef CAS.
- J. Meessen, Chem. Ing. Tech., 2014, 86, 2180–2189 CrossRef CAS.
- IEA, Global Energy Review 2021, IEA, 2021, https://www.iea.org/reports/global-energy-review-2021 Search PubMed.
- B. Yu, H. Zhang, Y. Zhao, S. Chen, J. Xu, L. Hao and Z. Liu, ACS Catal., 2013, 3, 2076–2082 CrossRef CAS.
- M. Brahmayya, S. A. Dai and S. Suen, J. CO2 Util., 2017, 22, 135–142 CrossRef CAS.
- J. Lin, P. Shi, Y. Gao, C. Yu, Y. Liu and S. Wu, J. Chem. Eng. Data, 2020, 65, 2838–2845 CrossRef CAS.
- E. Deau, E. Robin, R. Voinea, N. Percina, G. Satała, A. Finaru, A. Chartier, G. Tamagnan, D. Alagille, A. J. Bojarski, S. Morisset-Lopez, F. Suzenet and G. Guillaumet, J. Med. Chem., 2015, 58, 8066–8096 CrossRef CAS PubMed.
- G. P. Kumar, D. Rambabu, M. V. B. Rao and M. Pal, J. Chem., 2013, 2013, 5 Search PubMed.
- G. Wang, C. Li, J. Li and X. Jia, Tetrahedron Lett., 2009, 50, 1438–1440 CrossRef CAS.
- J. Wu, C. Zheng, B. Li, J. M. Hawkins and S. L. Scott, React. Chem. Eng., 2021, 6, 279–288 RSC.
- A. R. Bogdan, M. Charaschanya, A. W. Dombrowski, Y. Wang and S. W. Djuric, Org. Lett., 2016, 18, 1732–1735 CrossRef CAS PubMed.
- L. Wang, G. Wang, B. Liu and Z. Ji, Chin. J. Pharm., 1994, 25, 196–197 CAS.
- X. Liu, T. Liu and B. Liu, J. Shenyang Pharm. Univ., 2001, 18, 327–328 CAS.
- B. K. Banik, B. Banerjee, R. Kumar, G. Kaur, S. Saroch and R. Kumar, Molecules, 2020, 25, 5918 CrossRef CAS PubMed.
- N. Patel, R. Sood and P. V. Bharatam, Chem. Rev., 2018, 118, 8770–8785 CrossRef CAS PubMed.
- D. E. Jose, U. S. Kanchana, T. V. Mathew and G. Anilkumar, J. Organomet. Chem., 2020, 927, 121538 CrossRef CAS.
- A. Mastitski, A. Abramov, A. Kruve and J. Järv, Proc. Est. Acad. Sci., 2017, 66, 10–17 CrossRef.
- H. Goksu, H. Sert, B. Kilbas and F. Sen, Curr. Org. Chem., 2016, 21, 794–820 CrossRef.
- H. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- C. Xiao, X. Wang, C. Lian, H. Liu, M. Liang and Y. Wang, Curr. Org. Chem., 2012, 16, 280–296 CrossRef CAS.
- M. Hellal, F. C. Falk, E. Wolf, M. Dryzhakov and J. Moran, Org. Biomol. Chem., 2014, 12, 5990–5994 RSC.
- P. H. Huy and I. Filbrich, Chem.–Eur. J., 2018, 24, 7410–7416 CrossRef CAS PubMed.
- P. N. Y. Janssen, DT 2400094, 1974.
- I. Fumitaka, K. Hiroshi, N. Hiroshi, O. Yoriko and Y. Tatsuya, WO 9936421 A1, 1999.
- B. Bang-Andersen, WO 2009124882 Al, 2009.
- T. D. Bannister and L. M. Bohn, WO 2017161017 A1, 2017.
- C. S. Burgey, C. Stump, D. N. Nguyen, J. Z. Deng, A. G. Quigley, B. R. Norton, I. M. Bell, S. D. Mosser, C. A. Salvatore, R. Z. Rutledge, S. A. Kane, K. S. Koblan, J. P. Vacca, S. L. Graham and T. M. Williams, Bioorg. Med. Chem. Lett., 2006, 16, 5052–5056 CrossRef CAS PubMed.
- R. Henning, R. Lattrell, H. J. Gerhards and M. Leven, J. Med. Chem., 1987, 30, 814–819 CrossRef CAS PubMed.
- N. M. Kennedy, C. L. Schmid, N. C. Ross, K. M. Lovell, Z. Yue, Y. Chen, M. D. Cameron, L. M. Bohn and T. D. Bannister, J. Med. Chem., 2018, 61, 8895–8907 CrossRef CAS PubMed.
- B. Budzik, V. Garzya, D. Shi, G. Walker, W. Marie, J. Pardoe, A. Lucas, B. Tehan, R. A. Rivero, C. J. Langmead, J. Watson, Z. Wu, J. Forbes and I. T. Jin, ACS Med. Chem. Lett., 2010, 1, 244–248 CrossRef CAS PubMed.
- B. Budzik, D. G. Cooper, I. T. Forbes, V. Garzya, J. Jin, D. Shi, P. W. Smith and G. Walker, WO 2007036718 A2, 2007.
- H. Kawamoto, S. Ozaki, Y. Itoh, M. Miyaji, S. Arai, H. Nakashima, T. Kato, H. Ohta and Y. Iwasawa, J. Am. Chem. Soc., 1932, 54, 3773–3782 Search PubMed.
- Z. Wu, R. G. Robinson, S. Fu, S. F. Barnett, D. D. Jones, R. E. Jones, A. M. Kral, H. E. Huber, N. E. Kohl, G. D. Hartman and M. T. Bilodeau, Bioorg. Med. Chem. Lett., 2008, 18, 2211–2214 CrossRef CAS PubMed.
- A. M. Monforte, A. Rao, L. Patrizia, S. Ferro, L. D. Luca, M. L. Barreca, N. Iraci, G. Maga, E. Clercq, C. Pannecouque and A. Chimirri, Bioorg. Med. Chem., 2008, 16, 7429–7435 CrossRef CAS PubMed.
- V. Patel, N. Bhatt, P. Bhatt and H. D. Joshi, Med. Chem. Res., 2014, 23, 2133–2139 CrossRef CAS.
- E. Menteşe, O. Güven, N. Çalışkan and N. Baltaş, J. Heterocycl. Chem., 2021, 58, 1259–1267 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |