
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecurrent neural network (RNN) model accelerates the development of antibacterial metronidazole derivatives†
Nannan Chen‡
a,
Lijuan Yang‡bc,
Na Dinga,
Guiwen Lia,
Jiajing Caia,
Xiaoli Anb,
Zhijie Wanga,
Jie Qin*a and
Yuzhen Niu *a
*a
aSchool of Life Sciences and Medicine, Shandong University of Technology, Zibo, 255049 Shandong, China. E-mail: 295722387@qq.com; niuyzh12@lzu.edu.cn
bInstitute of Modern Physics, Chinese Academy of Science, Lanzhou, 730000 Gansu, China
cSchool of Physics and Technology, Lanzhou University, Lanzhou 730000, China
First published on 15th August 2022
Abstract
Metronidazole is a specific drug against trichomonas and anaerobic bacteria, and is widely used in the clinic. However, extensive clinical application is often accompanied by extensive side effects, so it is still of great significance to develop metronidazole derivatives with a new skeleton. Compared with other traditional receptor-based drug design methods, the computational model based on a neural network has higher accuracy and reliability. In this work, a Recurrent Neural Network (RNN) model is applied to the discovery of metronidazole drugs with a new skeleton. Firstly, the generation model based on a Gated Recurrent Unit (GRU) is trained to generate an effective Simplified Molecular-Input Line-Entry System (SMILES) string library with high precision. Then, transfer learning is introduced to fine-tune the GRU model, and many molecules with structures similar to known active drugs are generated. After cluster analysis of the structures of the new compounds, 20 small molecular compounds with metronidazole structures of all different categories were selected, of which 19 may not belong to any published patents or applications. Through prediction and personal experience, the difficulty of synthesizing these 20 new structures was analyzed, and compound 0001 was chosen as our synthetic target, and a series of structures (8a–l) similar to compound 0001 were synthesized. Finally, the inhibitory activities of these compounds against bacteria E. coli, P. aeruginosa, B. subtilis and S. aureus were determined. The results showed that compound 8a–l had obvious inhibitory activity against these four bacteria, which proved the accuracy of our compound generation model.
1. Introduction
At present, bacterial infection1 is still the main disease endangering human health, and the systemic and local application of antibiotics is the main way to kill pathogenic bacteria in vivo.2 As the first choice for anti-trichomonas and anti-anaerobic bacteria, metronidazole is usually used in combination with other antibiotics in various clinical fields.3 As a typical representative of nitroimidazole drugs, metronidazole has more extensive clinical application because of its high curative effect, short course of treatment, long half-life and good tolerance.4 However, despite its wide clinical application, metronidazole still has a wide range of side effects. Researchers have conducted clinical trials on metronidazole in patients with oral inflammation in many regions, and found that about 1/3 of patients have adverse reactions, mainly in the digestive tract and nervous system.5 Therefore, the development of metronidazole derivatives with new skeletons is still of great significance.6The starting point of traditional ligand-based drug design methods, such as the 2D/3D Quantitative Structure–Activity Relationship (QSAR) and pharmacophore model,7 is based on the possible common structural basis between a series of ligands with similar structure, the same action type and different activity. Therefore, their common pharmacophores can be found on this structural basis, and new skeleton compounds can be found. The implementation steps of this method include calculating the descriptors of the collected compounds, such as molecular weight, logarithmic p value and hydrogen bond number, donor or recipient, and then creating quantitative models and physiological activity, toxicity and other effects as response values by using these descriptors as features. Although this method is very simple, its effectiveness and wide use must be remembered. It has an inherent limitation, that is, it depends on the interpretation ability of the calculated descriptor, which is directly related to the structure of the compounds in training set.8 Therefore, they may not be able to cover all the factor response values required to explain the problem. Moreover, this kind of method finally obtains the candidate compounds by screening the compound library.9
Recent developments in the field of Artificial Intelligence (AI) and big data10 show that it is possible to fundamentally change the accuracy and reliability of computational models, including drug discovery.11,12 The model generation method based on neural network has been widely used in drug design, such as affinity prediction, compatibility prediction, synthetic route design and protein folding prediction.13,14 Compared with QSAR and other calculation methods, the model shows superior performance in calculation speed and accuracy. In recent years, many deep-seated generative models have been proposed, including variational automatic encoder,15 generative game network16 and terminal RNN.17 They understood the basic data distribution in an unsupervised environment and explored the broad space of pharmaceutical chemistry by encoding molecules into a continuous potential space. The structure of compounds is usually encoded by a molecular input line input system (e.g. the Simplified Molecular-Input Line-Entry System, SMILES) or represented by molecular graphs, having sequence structures similar to natural languages. Therefore, machine learning algorithms (e.g. RNN) for natural language processing (e.g. text generation and machine translation) can be transplanted to the task of small molecule generation. The RNN generation model will simplify the molecular input line input system representation or molecular graph representation used to train the learning characteristics of deep neural network models. Memory enhanced RNN is introduced to improve the efficiency of generating effective molecules.18 RNN has been shown to be fine-tuned by Transfer Learning (TL) to produce molecules with structures similar to similar to drugs with known activity, which are known to be active for specific targets. Therefore, in order to design metronidazole compounds, we apply TL to this work, and combine the generation model with the prediction model to guide the generator to generate new chemical entities with metronidazole compounds.
In this work, we propose a method to redesign metronidazole derivatives using RNN deep learning method. Firstly, we train a gated recurrent unit (GRU)-based generative model to generate libraries of valid SMILES strings with high accuracy. Then we use transfer learning to fine-tune our model, generating molecules that are structurally similar to drugs with known activities. Even with just a few representative molecules for transfer learning training, our approach yielded structures with similar chemical characteristics to known scaffolds. Subsequently, the library activity of our pipeline design is verified. We performed cluster analysis on the structures of the new compounds and selected all small molecular compounds containing metronidazole structures in different categories. Of the 20 selected structures, 19 may not belong to the scope of any published patents or applications. Finally, we analyzed the synthesis difficulty of these 20 new structures through prediction and personal experience, selected compound 0001 as our synthesis target, and synthesized a series of similar structures (8a–l) of 0001. The antibacterial activity test shows that these compounds have obvious inhibitory effects on four kinds of bacteria: E. coli, P. aeruginosa, B. subtilis and S. aureus, which proves the accuracy of our RNN production model.
2. Materials and methods
2.1 Deep learning algorithms
As with the text generation task, the first step in de novo drug design is to train a generator, which aims to learn rules of organic chemistry that define SMILES strings corresponding to realistic chemical structures.19 However, random generation without lead experience is too blind to be generated for specific tasks or targets.20 Therefore, how to endow deep learning methods with specific research experience is the key to molecular design for specific tasks. In our work, deep learning algorithms were employed to generate molecules by a pre-trained generator model coupled with a transfer learning model introducing the existing leading experience (Fig. 1).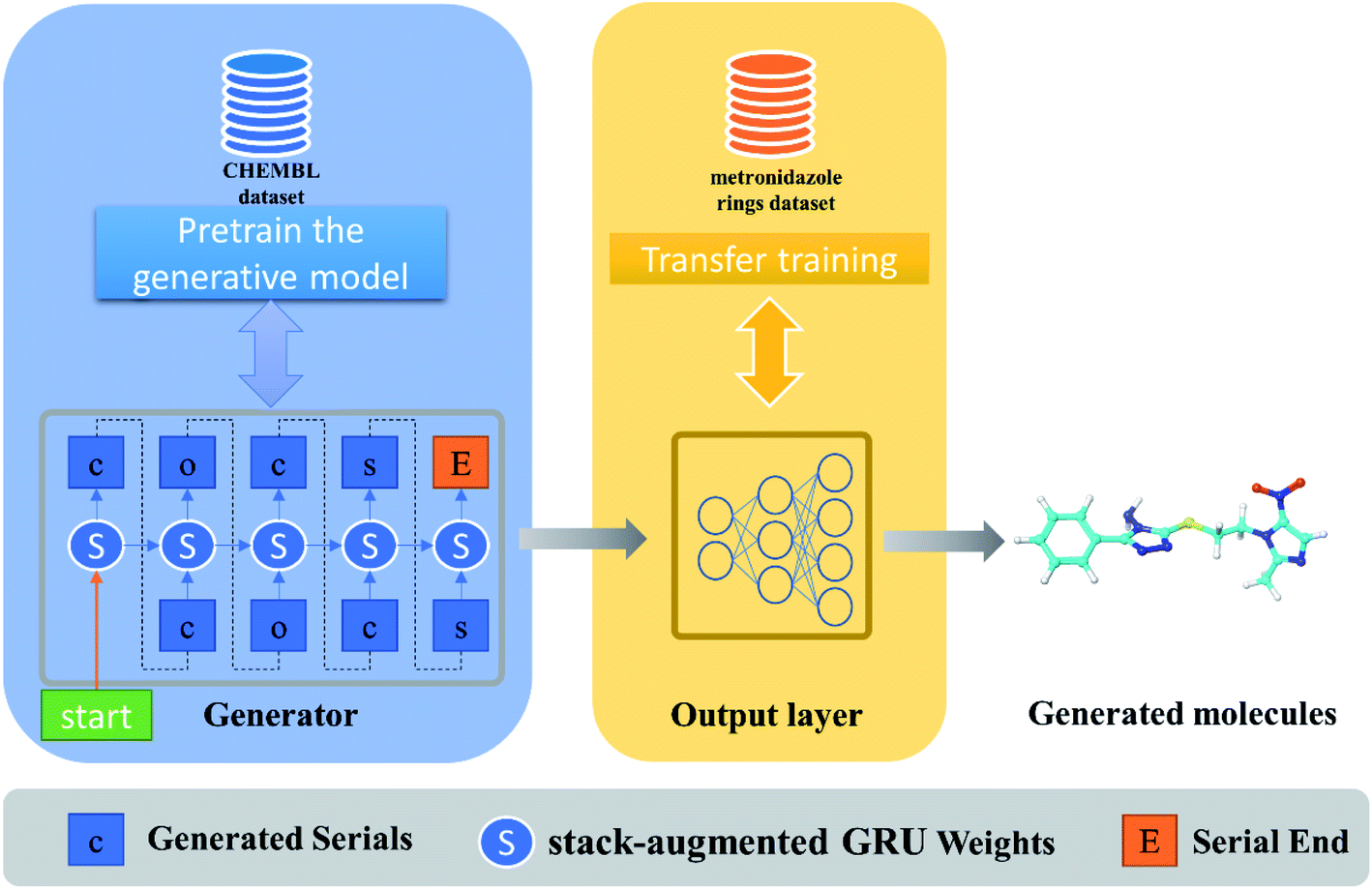 | ||
| Fig. 1 Pipeline of generative model for novel compound generation. | ||
In our model, the dimension of the stack-augmented layer is 1500, and the depth is 200, which means that 200 steps of sequence information can be stored in stack memory. The GRU has 1500 + 1500 units for processing splicing vectors from hidden state and stack memory, and returns a 1500-dimensional vector containing the current token and previous sequence information, and finally returns the predicted probability distribution of the next token through the decoder. The parameters of the model are updated by reducing the cross-entropy loss between the real tokens and the predicted tokens. In the training process, the optimizer we used was AMSGrad, the learning rate was set as 0.0005, and the model tended to converge after 500 iterations of training. In our previous work, the performance of generative models with the above parameters was evaluated in terms of generating effective molecular proportions and generating molecular properties. We found that the 1-layer GRU performs comparable to the 4-layer GRU and can be successfully used for small molecule library design.22 Therefore, we use the above model to accelerate the development of antibacterial drugs, and the trained stack-augmented GRU is saved as a pretrained model to be weighted by a task-specific transfer learning model.
During transfer learning, all the layers of the generative model were frozen, which will not be adjusted in the process of gradient calculation and backward transmission, except for the last decoder module. Only the parameters of the decoder layer are adjusted, which prevent the model forget the molecular features on small samples, so as to generate compounds that are similar to the training set. The weight of the pretrained model were loaded and trained on the small samples, until the training stopped when the sample distribution generated by the model was similar to the data of the training set but the repetition rate was low.
2.2 Cluster analysis and synthesis difficulty analysis of compounds
Cluster analysis of compounds was completed in the canvas of schrödinger2015,32 and the molecular fingerprint types were selected as hashed. Synthetic Accessibility Score (SAS)33 is designed according to concise rules, which can quickly evaluate a large number of compounds. This method34 is based on the “complexity” of molecules, but in order to combine the action of reagents and reactions, complex structures can be constructed immediately, so the assumption that “often occurring substructures are easy to synthesize” is used. The SAS is calculated according to the follow:| SAS = fragmentScore − complexityPenalty | (1) |
“FragmentScore” is to capture “historical synthesis knowledge” by analyzing the common structural characteristics of a large number of synthesized molecules. The score is calculated as the sum of the contributions of all fragments in the molecule divided by the number of fragments in the molecule. The database of contributions has been generated by statistical analysis of substructures in PubChem database35 “ComplexityPenaltyonly” considers factors such as macrocycle and molecular weight. Standardize the value from 1 (simple) to 10 (difficult).
A measure of drug-likeness called weighted Quantitative Estimation of Drug-Likeness Molecules (QEDw)36 was applied in our study.
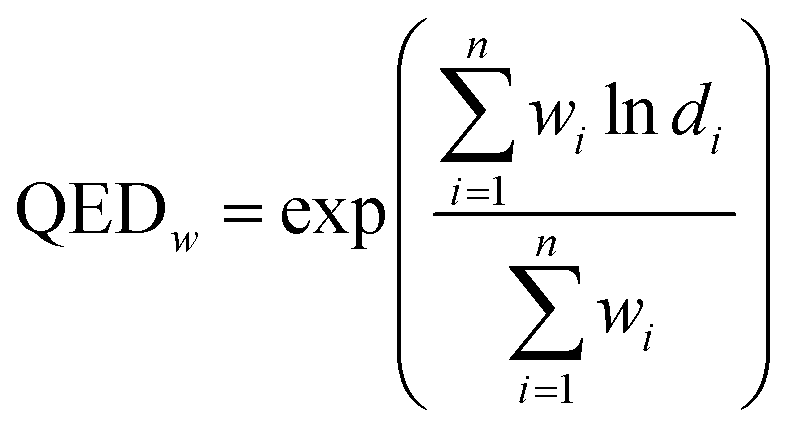 | (2) |
It is calculated as shown in eqn (2), in which d represents the individual desirability functions (including molecular weight, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, topological polar surface area, number of hydrogen bond donors and acceptors, number of aromatic rings and rotatable bonds, harmful chemical functional group distribution and etc.), w is the weight applied to each function and n is the number of descriptors. SAS and QEDw were calculated by the RDKit toolkit [https://www.rdkit.org].
P, topological polar surface area, number of hydrogen bond donors and acceptors, number of aromatic rings and rotatable bonds, harmful chemical functional group distribution and etc.), w is the weight applied to each function and n is the number of descriptors. SAS and QEDw were calculated by the RDKit toolkit [https://www.rdkit.org].
2.3 Materials and measurements
All the starting materials, chemicals and solvents used in the synthesis of metronidazole derivatives were analytical reagent grade and purchased commercially from Aladdin Industrial Corporation (China). All reactions were routinely checked by Thin-Layer Chromatography (TLC) on 0.25 mm silica gel plates (silica GF 254). 1H NMR spectra were carried out at ambient temperature on a Bruke AVANCE III 400 spectrometer using tetramethylsilane (TMS) as an internal standard. Mass spectra were measured with an Autoflex II TM instrument for ESI-MS. Elemental analyses were analyzed on a PerkinElmer model 2400 analyzer. The intermediate 4-amino-3-substituent-1,2,4-triazole-5-thiol (5a–l) was synthesized according to the literature method.372.4 General method of synthesis of target compounds 8a–l
Metronidazole (1.71 g, 10 mmol), 4-toluene sulfochloride (2.28 g, 12 mmol) and trimethylamine (TEA) (1.67 mL, 12 mmol) were dissolved in CH2Cl2 (15 mL). The reaction mixture was stirred at room temperature (298 K) overnight. After the completion of the reaction, as monitored by TLC, the resulted precipitate was filtered and washed with dilute hydrochloric acid and ethanol to obtain white crystal powder 2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethyl 4-methylbenzenesulfonate (6). A solution of 6 (3.25 g, 10 mmol), sodium iodide (2.25 g, 15 mmol), and acetone (25 mL) was refluxed with stirring for 10 h. The reaction mixture was cooled and filtered, then the filtrate was evaporated under reduced pressure to afford 1-(2-iodoethyl)-2-methyl-5-nitro-1H-imidazole (7). 5 (5 mmol) and potassium hydroxide (0.28 g, 5 mmol) were dissolved in methanol (25 mL), stirred for 30 min at room temperature. Later, slowly added 7 (1.40 g, 5 mmol) to the reaction mixture, and heated to reflux for 5–8 h. The reaction progress was detected by using TLC technique. After completion of the reaction, the mixture was concentrated under reduced pressure to afford a crude solid. The acquired crude solid was purified by column chromatography skill eluting with dichloromethane and methanol to obtain pure metronidazole-triazole compounds 8a–l (Fig. 4).2.5 Bioassay conditions
The antibacterial activity of the synthesized compounds was evaluated against two Gram-negative bacterial strains, Escherichia coli ATCC 35218 and Pseudomonas aeruginosa ATCC 27853, and two Gram-positive bacterial strains, Bacillus subtilis ATCC 6633 and Staphylococcus aureus ATCC 6538. The bioassay was conducted using MH medium (Mueller–Hinton medium: casein hydrolysate 17.5 g, soluble starch 1.5 g, beef extract 1000 mL). The IC50 (half minimum inhibitory concentrations) of the test compounds were determined by a colorimetric method using the dye MTT (3-(4,5-dimethylth-iazol-2-yl)-2,5-diphenyl tetrazoliumbromide). Clinically used antibiotics streptomycin was used as reference. The antibacterial activities were evaluated by the method reported before. The procedure of antimicrobial activity was given in detail in the ESI 1.†3. Results and discussion
3.1 Deep learning model accuracy evaluation
To train the generative model, we collected ∼1.6 million small molecules from CHEMBL,38 and each small molecule is represented as SMILES format to train a generator of Seq2Seq. The SMILES dataset was preprocessed by applying sequential filters to remove stereochemistry, salts, and molecules with undesirable atoms (metal atoms) or groups.20 In the end, ∼1.6 million small molecules with a length of 100 or less were retained for training the generative model. In order to evaluate the accuracy of the pretrained generator in generating chemically valid molecules, a total of 100000 compounds were sampled from the generation model for 10 times, 10000 compounds at a time, and an average of 95.6% of the small molecules in all sample batches were chemically valid (removal of redundant and can be parsed by the RDKit library). We predicted the QED and SAS of all the compounds produced by the GRU-based model with existing metronidazole compounds, and the results show that the molecules generated by GRU-based generators have higher drug-likeness properties and lower synthesizable scores (Fig. 2).
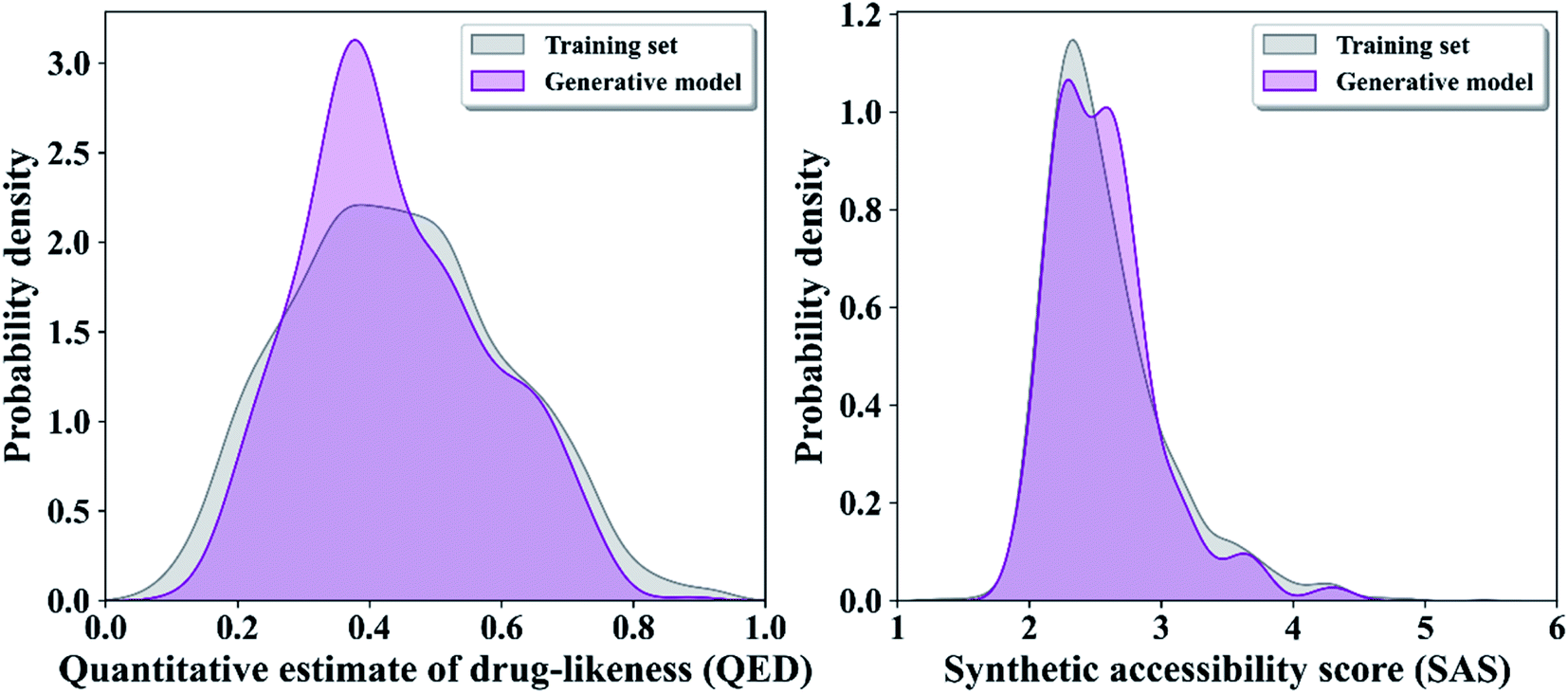 | ||
| Fig. 2 Distribution of the quantitative estimate of drug-likeness scores (QED) (A) and distribution of the synthetic accessibility scores (SAS) in two generator and existing metronidazole compounds. | ||
3.2 Discovery of new metronidazole derivative
After TL, the RNN model generated 3314 small molecular compounds (ESI 2†), of which 321 compounds containing the metronidazole groups were retained, and they were input into canvas to calculate the binary fingerprints of all small molecules. Then hierarchical clustering was carried out based on the tanimoto similarity metric. The 321 compounds are grouped into 20 categories by default, while the merging distance is 0.8, and the representative structure with the lowest SAS are selected from each category shown in Fig. 3.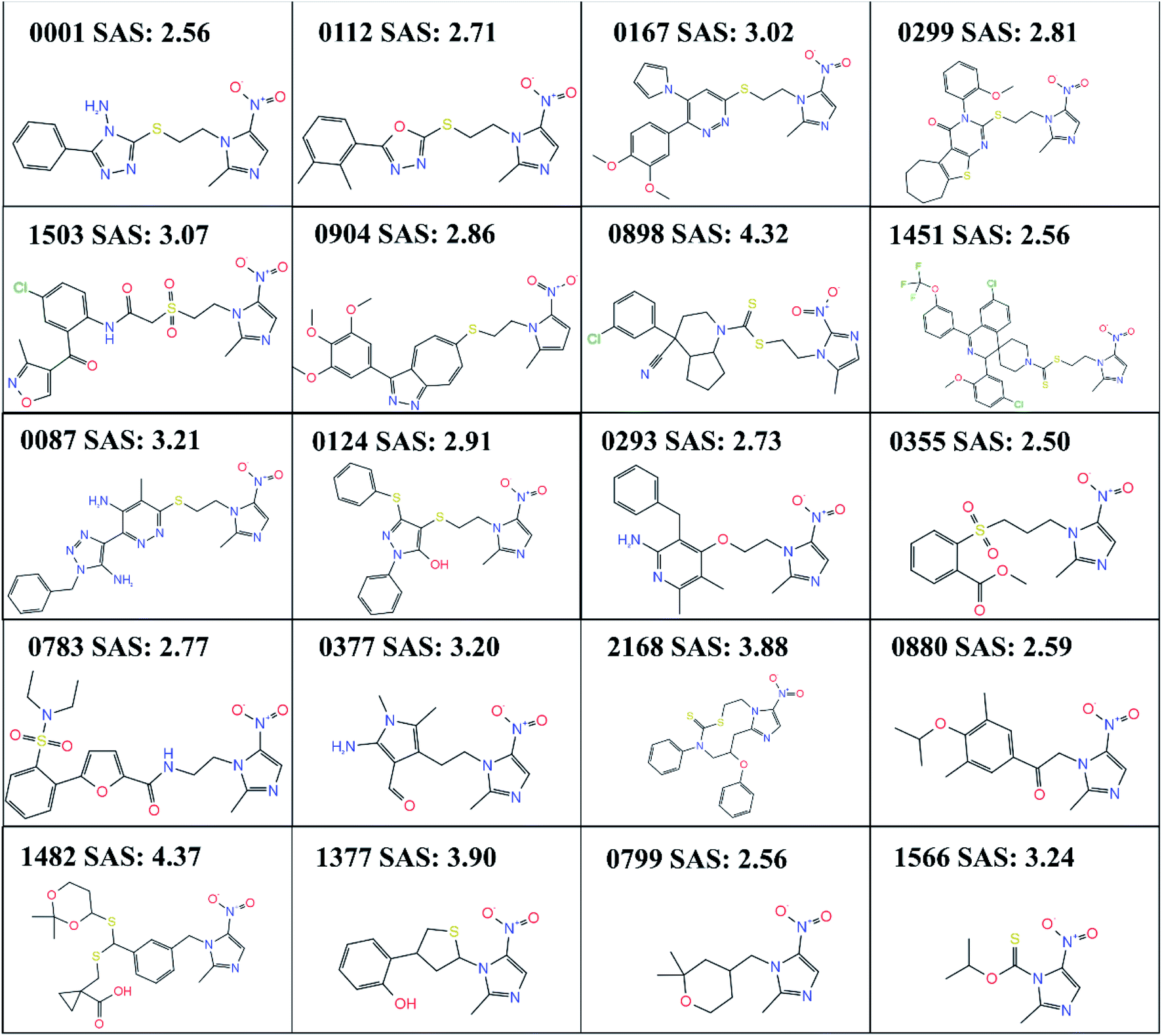 | ||
| Fig. 3 20 compounds with different skeleton obtained by clustering. SAS refers to the synthetic accessibility scores. | ||
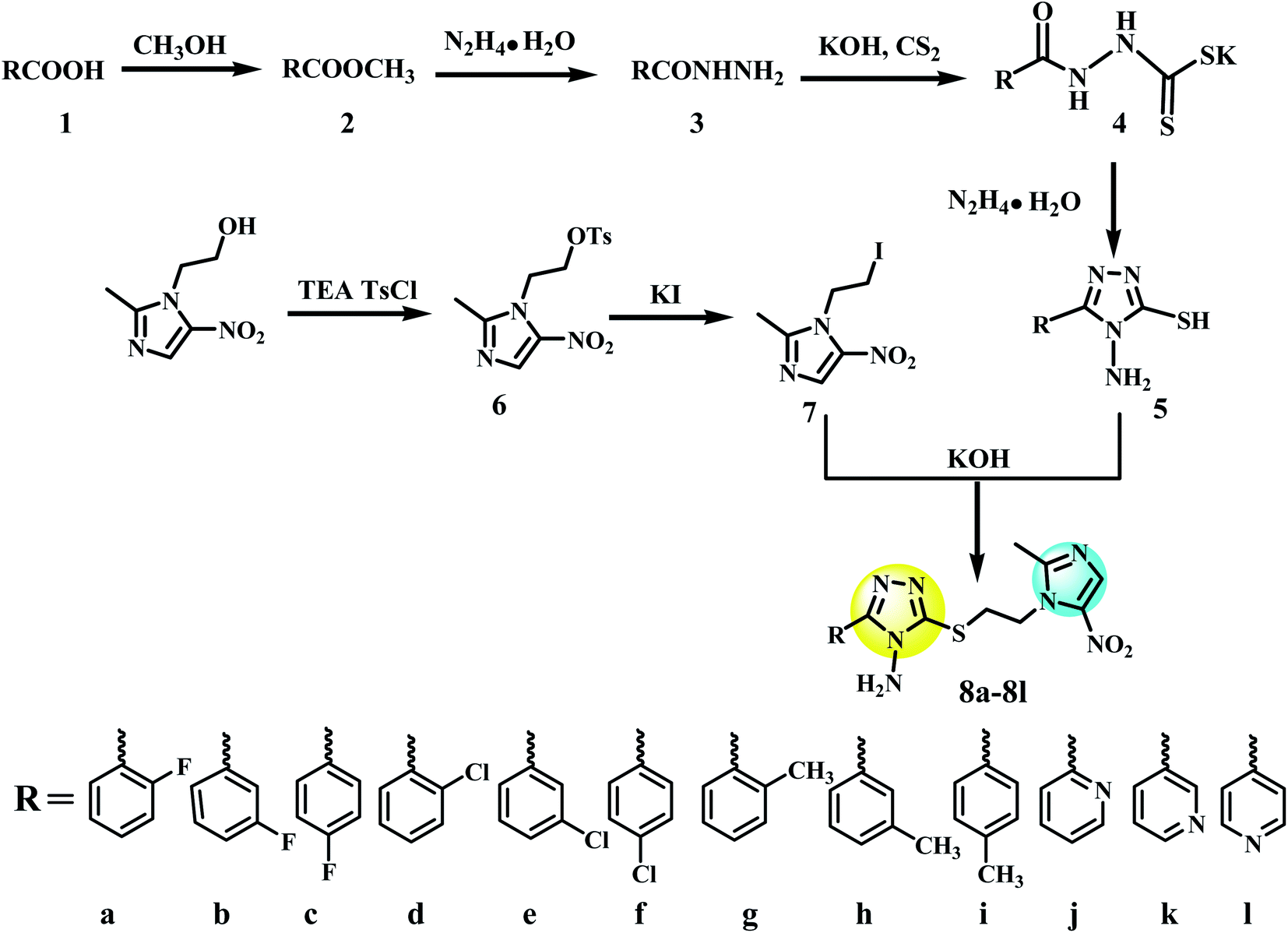 | ||
| Fig. 4 Synthesis process of compound 8a–l. | ||
No matter how to obtain lead compounds, it is very important to evaluate the synthesis difficulty of candidate lead compounds, and it is a priority issue. In any case, “easy synthesis” of compounds must be considered at a certain stage of research. In this case, if other indexes (such as activity) are given priority and “difficulty of synthesis” is considered at the end, compounds with similar chemical types and skeletons tend to be selected. We evaluated the SAS of 20 selected compounds by RDKit, and the results are shown in Fig. 3. Molecules with the high SAS are difficult to synthesize, whereas, molecules with the low SAS values are easily synthetically accessible. Because the lowest SAS are 0355 (SAS: 2.50), 0001 and 0799 (SAS: 2.56) among the 20 compounds, the three compounds are the easiest to synthesize theoretically. However, the software prediction is inevitably mechanical. We also predicted the synthetic route of 0355 and 0799 by using chemical.ai (https://chemical.ai/), as shown in Fig. S1.† For compound 0799, we need to consider the following points when considering the optimization of synthetic route: (1) Raw materials like (2) or (4) are expensive; (2) the compound 0799 has chiral carbon atoms, and it needs to be resolved during the synthesis process, which increases the complexity of the synthesis; (3) it is difficult to obtain high purity of compounds containing cyclic ether structure during purification; however, for compound 0355, its raw materials (6)–(9) can't be purchased directly. As mentioned above, we have more experience in the synthesis of 0001, so we tried to synthesize 0001 firstly. Furthermore, compounds 0001 and 0112 are highly similar in structure, while 0112 has been reported to have obvious antibacterial activity. At the same time, we also found that there are many structures with methyl or chlorine atoms substituted on the benzene ring of 0001 among the 3314 small molecular structures we generated, such as 0004 and 0041. Therefore, we first synthesized 0001 and measured its antibacterial activity, and then continued to try to synthesize 11 other compounds with substituents on the benzene ring of 0001 and measured their antibacterial activity.
3.3 Synthesis of candidate compounds
The synthetic route of target compounds 0001 and its similar structures (8a–l) was showed in Fig. 4. To obtain the designed metronidazole-triazole derivatives, we began with the esterification reaction of aromatic acids (1) in methanol, which resulted in aromatic esters (2) followed by reaction with hydrazine hydrate to get hydrazide derivatives (3). 1,2,4-Triazole derivatives (5) were acquired by fist nucleophilic addition reaction of 3 and carbon disulfide to obtain (4), followed by condensation reaction with hydrazine hydrate. 1-(2-Iodoethyl)-2-methyl-5-nitro-1H-imidazole (7) was obtained by two successive substitution reaction of metronidazole. Finally, nucleophilic substitution reaction between (7) and (5) in the presence of potassium hydroxide yielded 8a–l. The synthesized analogs were characterized by utilizing ESI-MS and 1H-NMR spectroscopic techniques (Fig. S2–25 in ESI 1†).3.4 In vitro antibacterial activity
The antibacterial activity of the synthesized metronidazole derivatives 8a–l was tested using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. The results of IC50 are presented in Table 1. Known antibiotic like streptomycin was used as control drug. As our results show, the tested compounds exhibited various degrees of inhibition against Gram-positive and Gram-negative bacteria. And against the all tested bacteria, metronidazole derivatives 8a–l showed higher antibacterial activity against Gram positive strains, with IC50 values ranging from 7.61 to 18.39 μg mL−1 for B. subtilis, and 2.46 to 13.49 μg mL−1 for S. aureus, respectively. S. aureus was observed to be the most sensitive bacteria, compound 8i with p-methylphenyl group on the 1,2,4-triazole skeleton showed the best antibacterial activity against S. aureus. Compared to the reference drug streptomycin, 8a–l had less potency against Gram negative strains, only compounds 8g, 8h and 8i, with methylphenyl group on the 1,2,4-triazole skeleton had comparable efficacy against P. aeruginosa with IC50 values ranging from 5.74 to 6.71 μg mL−1. Regarding the effect of the substituent on the phenyl moiety of the 1,2,4-triazole skeleton, the substitution with electron-donating methyl group had a better antibacterial activity towards both Gram-positive and Gram-negative bacteria, compared with electron-withdrawing halogen groups at the same position. Moreover, structure–activity relationship studies revealed the substituent at different positions led to different activity, and the potency order was para > meta > ortho.| Compound | IC50 (μM L−1) | |||
|---|---|---|---|---|
| E. coli | P. aeruginosa | B. subtilis | S. aureus | |
| 8a | 72.64 | 99.52 | 45.65 | 28.21 |
| 8b | 70.11 | 78.07 | 37.71 | 37.16 |
| 8c | 61.46 | 69.61 | 29.15 | 20.8 |
| 8d | 74.01 | 42.82 | 33.43 | 24.59 |
| 8e | 60.63 | 49.95 | 37.92 | 19.97 |
| 8f | 51.03 | 40.29 | 33.91 | 16.73 |
| 8g | 59.11 | 15.99 | 26.02 | 11.98 |
| 8h | 48.89 | 16.32 | 21.20 | 9.89 |
| 8i | 42.51 | 18.69 | 18.97 | 6.85 |
| 8j | 30.00 | 28.38 | 37.80 | 23.73 |
| 8k | 38.64 | 37.51 | 53.15 | 25.90 |
| 8l | 41.07 | 53.12 | 48.35 | 22.23 |
| Streptomycin | 22.92 | 12.48 | 16.22 | 8.67 |
4. Conclusions
Ligand-based de novo drug design method has benefited from the development in the machine learning community as applied to natural language processing and machine translation. RNN-based generation model is widely used, and transfer learning is used to further optimize the structure of compounds in order to produce molecules with desired properties. In this work, a series of metronidazole compounds were found by combining RNN neural network generation compound model, transfer learning and chemical synthesis. Collecting ∼1.6 million small molecules from CHEMBL were used to train the generative stack-augmented GRU model. We then use transfer learning to fine-tune our model, generating molecules that are structurally similar to drugs with known activities. Even with just a few representative molecules for transfer learning training, our approach yielded structures with similar chemical characteristics to known scaffolds. Subsequently, the library activity of our pipeline design is verified. We performed cluster analysis on the structures of the new compounds and selected all small molecular compounds containing metronidazole structures in different categories. Of the 20 selected structures, 19 may not belong to the scope of any published patents or applications. Finally, we analyzed the synthesis difficulty of these 20 new structures through prediction and personal experience, selected compound 0001 as our synthesis target, and synthesized a series of similar structures (8a–l) of 0001. The inhibitory activities of these compounds on four bacteria E. coli, P. aeruginosa, B. subtilis and S. aureus were determined. The results showed that compound 8a–l had obvious inhibitory activities on these four bacteria, which proved the accuracy of our compound generation model.Appendix
Abbreviations are listed in Table 2.| Abbreviations | Explanations |
|---|---|
| GRU | Gated recurrent unit |
| SMILES | Simplified molecular-input line-entry system |
| QSAR | Quantitative structure–activity relationship |
| AI | Artificial intelligence |
| TL | Transfer learning |
| RNN | Recurrent neural network |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| SAS | Synthetic accessibility score |
| QEDw | Weighted quantitative estimation of drug-likeness |
| logP |
Oil–water partition coefficient |
| TLC | Thin-layer chromatography |
| TMS | Tetramethylsilane |
Conflicts of interest
The authors declare that there is no conflict of interest in this work.Acknowledgements
This work was supported by the Natural Foundation of Shandong Province (Grant No. ZR2018BB055).References
- M. M. Biernat and T. Wróbel, Int. J. Mol. Sci., 2021, 22 Search PubMed.
- F. Tao, S. Ma, H. Tao, L. Jin, Y. Luo, J. Zheng, W. Xiang and H. Deng, Carbohydr. Polym., 2021, 251, 117063 CrossRef CAS PubMed.
- D. L. Bourque, A. Neumayr, M. Libman and L. H. Chen, J. Trav. Med., 2022, 29, taab120 CrossRef PubMed.
- Y. Nishikawa, N. Sato, S. Tsukinaga, K. Uchiyama, S. Koido, D. Ishikawa and T. Ohkusa, Ther. Adv. Chronic Dis., 2021, 12, 20406223211028790 CAS.
- D. Cicek, B. Kandi, S. Bakar and D. Turgut, J. Dermatol. Treat., 2009, 20, 344–349 CrossRef CAS PubMed.
- O. P. S. Patel, O. J. Jesumoroti, L. J. Legoabe and R. M. Beteck, Eur. J. Med. Chem., 2021, 210, 112994 CrossRef CAS PubMed.
- I. Jabeen, P. Wetwitayaklung, P. Chiba, M. Pastor and G. F. Ecker, J. Comput. Aided Mol. Des., 2013, 27, 161–171 CrossRef CAS PubMed.
- R. Zanni, M. Galvez-Llompart, R. Garcia-Domenech and J. Galvez, Expet Opin. Drug Discov., 2020, 15, 1133–1144 CrossRef CAS PubMed.
- P. Li, Y. Niu, S. Li, X. Zu, M. Xiao, L. Yin, J. Feng, J. He and Y. Shen, Chem. Biol. Drug Des., 2022, 99, 222–232 CrossRef CAS PubMed.
- Z. Dlamini, F. Z. Francies, R. Hull and R. Marima, Comput. Struct. Biotechnol. J., 2020, 18, 2300–2311 CrossRef CAS PubMed.
- G. Hessler and K. H. Baringhaus, Molecules, 2018, 23, 2520 CrossRef PubMed.
- M. Thomas, A. Boardman, M. Garcia-Ortegon, H. Yang, C. de Graaf and A. Bender, Methods Mol. Biol., 2022, 2390, 1–59 CrossRef PubMed.
- H. Öztürk, A. Özgür and E. Ozkirimli, Bioinformatics, 2018, 34, i821–i829 CrossRef PubMed.
- M. Eisenstein, Nature, 2021, 599, 706–708 CrossRef CAS.
- Y. Deng, A. Sander, L. Faulstich and K. Denecke, Artif. Intell. Med., 2019, 93, 29–42 CrossRef PubMed.
- G. Gao, J. Cao, Z. Bu, H.-j. Li and Z. Wu, Expert Syst. Appl., 2018, 96, 450–461 CrossRef.
- J. Fei and Z. Wang, IEEE Access, 2020, 8, 167965–167974 Search PubMed.
- Y. Yu, X. Si, C. Hu and J. Zhang, Neural Comput., 2019, 31, 1235–1270 CrossRef PubMed.
- M. Popova, O. Isayev and A. Tropsha, Sci. Adv., 2018, 4, eaap7885 CrossRef CAS PubMed.
- S. R. Krishnan, N. Bung, G. Bulusu and A. Roy, J. Chem. Inf. Model., 2021, 61 Search PubMed.
- A. Joulin and T. Mikolov, presented in part at the Proceedings of the 28th International Conference on Neural Information Processing Systems - Volume 1, Montreal, Canada, 2015 Search PubMed.
- L. Yang, G. Yang, Z. Bing, Y. Tian, Y. Niu, L. Huang and L. Yang, ACS Omega, 2021, 6, 33864–33873 CrossRef CAS PubMed.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, Nucleic Acids Res., 2012, 40, D1100–D1107 CrossRef CAS PubMed.
- W.-J. Mao, P.-C. Lv, L. Shi, H.-Q. Li and H.-L. Zhu, Bioorg. Med. Chem., 2009, 17, 7531–7536 CrossRef CAS PubMed.
- Y. Luo, Y. Li, K.-M. Qiu, X. Lu, J. Fu and H.-L. Zhu, Bioorg. Med. Chem., 2011, 19, 6069–6076 CrossRef CAS PubMed.
- Y. Qian, H. J. Zhang, Z. Hao, X. Chen, J. Zhao and H. L. Zhu, Bioorg. Med. Chem., 2010, 18, 4991–4996 CrossRef CAS PubMed.
- S. F. Wang, Y. Wang, F. Yin, X. Qiao, S. Wu and L. Zhang, Bioorg. Med. Chem., 2014, 22, 2409–2415 CrossRef CAS PubMed.
- Z. H. Guo, Y. Yin, C. Wang, P. F. Wang, X. T. Zhang, Z. C. Wang and H. L. Zhu, Bioorg. Med. Chem., 2015, 23, 6148–6156 CrossRef CAS PubMed.
- Y. J. Qin, P. F. Wang, J. A. Makawana, Z. C. Wang, Z. N. Wang, Y. Gu, A. Q. Jiang and H. L. Zhu, Bioorg. Med. Chem. Lett., 2014, 24, 5279–5283 CrossRef CAS PubMed.
- L. Yao, Y. Luo, H. Yang, D. D. Zhu, S. Zhang, Z. J. Liu, H. B. Gong and H. L. Zhu, Bioorg. Med. Chem., 2012, 20, 4316–4322 CrossRef PubMed.
- H. J. Zhang, D. D. Zhu, Z. L. Li, J. Sun and H. L. Zhu, Eur. J. Med. Chem., 2011, 19, 4513–4519 CAS.
- H. Alogheli, G. Olanders, W. Schaal, P. Brandt and A. Karlén, J. Chem. Inf. Model., 2017, 57, 190–202 CrossRef CAS PubMed.
- J. C. Baber and M. Feher, Mini Rev. Med. Chem., 2004, 4, 681–692 CrossRef CAS PubMed.
- P. Ertl and A. Schuffenhauer, J. Cheminf., 2009, 1, 8 Search PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2021, 49, D1388–d1395 CrossRef CAS PubMed.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Nat. Chem., 2012, 4, 90–98 CrossRef CAS PubMed.
- Y.-P. Xu, Y.-H. Chen, Z.-J. Chen, J. Qin, S.-S. Qian and H.-L. Zhu, Eur. J. Inorg. Chem., 2015, 2015, 2076–2084 CrossRef CAS.
- G. Anna, H. Anne, N. Michał, B. Patrícia, C. Jon, M. David, M. Prudence, A. Francis, L. J. Bellis and C. U. Elena, Nucleic Acids Res., 2017, D945–D954 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01807a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |