
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aromatic sensitizers in luminescent hybrid films†
Per-Anders Hansen *,
Joachim Svendsen,
Hanne Nesteng and
Ola Nilsen
*,
Joachim Svendsen,
Hanne Nesteng and
Ola Nilsen
Department of Chemistry, Centre for Materials Science and Nanotechnology, University of Oslo, Sem Sælandsvei 26, 0371 Oslo, Norway. E-mail: p.a.hansen@kjemi.uio.no
First published on 20th June 2022
Abstract
Atomic layer deposition offers a unique set of design possibilities due to the vast range of metal and organic precursors that can be used and combined. In this work, we have combined lanthanides with aromatic aids as strongly absorbing sensitizers to form highly luminescent thin films. Terephthalic acid is used as a base sensitizer, absorbing shorter wavelengths than 300 nm. The absorption range is extended towards the near-UV and blue range by increasing the aromatic system and adding functional groups that have strong red-shifting effects. While terbium and europium provide green and red emission, yttrium allows emission from the sensitizer itself spanning the whole color range from purple, blue and green to red. Many organic dye molecules show very high luminescence quantum yields and several of the molecules and materials investigated in this work show bright luminescence.
Introduction
Atomic layer deposition (ALD) and its organic counterpart molecular layer deposition (MLD) is a remarkable synthesis tool capable of synthesizing a vast array of materials ranging from inorganic1 including quaternary2 and even quinary compounds,3 nanostructures,4 over to organic–inorganic hybrids,5 crystalline MOF films6 and pure organics.7 The layer-by-layer chemistry and low deposition temperatures prevents interdiffusion between layers and enables sub-nanometer scale designs, while the vast array of deposition materials allows combinations and layered nanocomposites of different material classes. We have recently shown how combining ALD and MLD allows designing nanocomposites that combines luminescence properties of inorganic fluorides and optical absorption of organic molecules,8 in addition to controlling and confining movement of excited states and thus reducing concentration quenching effects.9 In addition, deposition by ALD allows on high aspect ration 3D nanostructures like photonic crystals or waveguides, and the (generally) low deposition temperature can be compatible with temperature sensitive substrate materials like textiles and polymers.The inclusion of organic species into hybrid or nanocomposite thin film is an exciting field as the organic component adds very strong optical absorption and an exceptional flexibility in designing the molecules towards specific absorption and emission ranges. Unfortunately, the choice of organic components in MLD is quite limited. The need to get these molecules into the vapour phase while also having sufficient reactivity reduces the practical range to fairly small molecules. This puts strong limits on the range of optical and electronic properties to choose from.
Reviews on ALD and MLD chemistries7,10 reveals that the organic precursors utilized so far are quite limited beyond a single aromatic ring or a short conjugated chain. There are examples of larger conjugated systems like naphthalene and biphenyl6 showing near-UV activity and even visible absorption.11 These optically absorbing dye precursors require sublimation temperatures of around 250 °C or more, approaching the temperature where such organic molecules start to decompose or polymerize. This illustrates well the difficulty in balancing Vis and NIR active optical properties with ALD/MLD compatibility. An exception to this is our recent work on quinizarin molecules, which sublimes efficiently at 130 °C and produces deep pink films with TMA as cation precursor.12
In this work, we explore a range of mono- and bi-aromatic di-carboxylic acids deposited with lanthanides as cations, as shown in Fig. 1. All three lanthanides have very similar deposition chemistries.13 Terbium and europium was chosen due to their well-known and easily identified sharp emission peaks which are easily distinguishable from organic molecule emissions. In addition, these two lanthanides have large energy gaps from the emitting energy level(s) to the next lower energy level. This allows them to be highly luminescent even in high-phonon matrices such has hybrid materials. Yttrium was chosen as an optically inactive ion that allows characterization of the organic molecules emission properties in the same lanthanide-organic hybrid material environment. In previous works, we have investigated the growth and material chemistry of optically active hybrid materials.5,6,12,14 Here, we focus on the hybrid materials optical properties, absorption and luminescence colour.
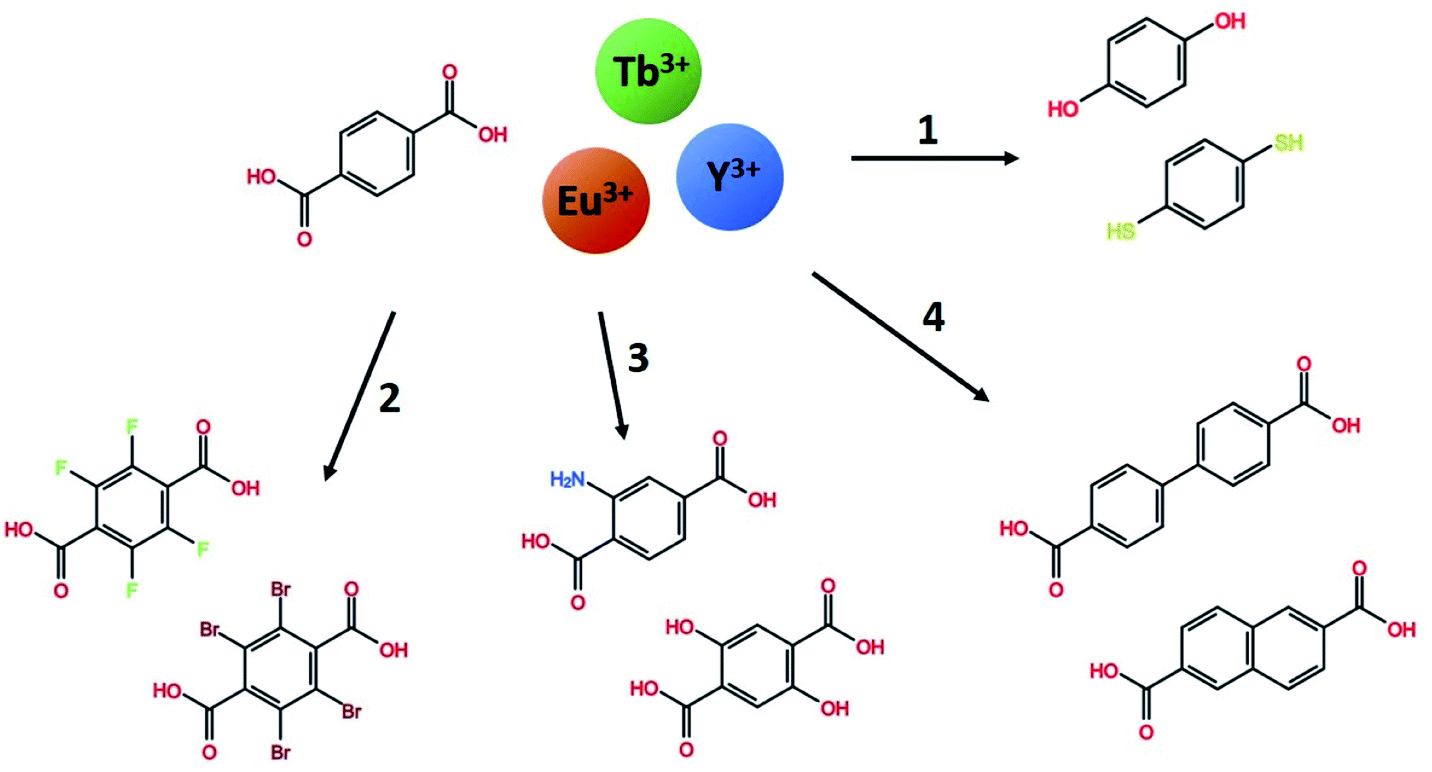 | ||
| Fig. 1 The range of mono- and bi-aromatic di-carboxylic acids investigated in this work, combined with the three lanthanides Tb, Eu and Y. Using terephthalic acid and Tb as a base system, four paths of molecule variations are explored. (1) Other binding group in H2bdo and H2bdt, (2) halogenated H2bdc-F and H2bdc-Br, (3) functionalized H2bdc-NH2 and H2bdc-2OH, and (4) the larger conjugated system H2bpdc and H2ndc. | ||
Experimental
The films were deposited in an F-120 research-type ALD-reactor (ASM Microchemistry Ltd). The deposition temperature were 250 °C except when otherwise noted. Ln-β-diketonate Ln(thd)3 (Strem Chemicals, >99.9%, Ln = Y, Eu, Tb) were used as lanthanide precursors, while a range of different organic precursors were used as optical sensitizers. The full names, abbreviations used in this work and the sublimation temperatures used for all precursors are listed in Table 1. Nitrogen was used as carrier and purge gas, supplied from gas bottles (AGA, 99.999%). p-Type Si(100) substrates were used for all depositions. In addition, 0.5 × 4 cm2 Si(100) strips were placed some 8 cm apart in the gas inlet and exhaust sides of the deposition chamber to monitor thickness gradients. Silicon substrates were used with native oxide intact and its thickness was measured before each deposition. All substrates were dry wiped and dust was removed using pressurized air. Pulse times for Ln(thd)3 and organics were 1.5 and 2 seconds respectively. Purge times were between 1 and 2 seconds. Exceptions to the standard parameters are that H2bpdc was deposited at 275 °C due to its high precursor temperature, H2bdt was deposited at 200, 250 and 300 °C, and H2bto depositions used 3 second lanthanide and organic pulses. In addition, Tb2bdc3 was used as a basis material and was deposited in the 225–350 °C range in steps of 25 °C.| Compound name | Short name | Tsub (°C) | |
|---|---|---|---|
| Ln(2,2,6,6-tetramethyl-3,5-heptanedione)3 | Y(thd)3 | 130 | |
| Eu(thd)3 | 150 | ||
| Tb(thd)3 | 145 | ||
| 1,4-Benzenedicarboxylic acid | H2bdc | 200 | |
| (1) | 2,3,5,6-Tetrafluoro-1,4-benzenedicarboxylic acid | H2bdc-F | 145 |
| 2,3,5,6-Tetrabromo-1,4-benzenedicarboxylic acid | H2bdc-Br | 230 | |
| (2) | 2-Amino-1,4-benzenedicarboxylic acid | H2bdc-NH2 | 210 |
| 2,5-Dihydroxy-1,4-benzenedicarboxylic acid | H2bdc-2OH | 225 | |
| (3) | Biphenyl-4,4′-dicarboxylic acid | H2bpdc | 250 |
| 2,6-Naphthalenedicarboxylic acid | H2ndc | 225 | |
| (4) | Benzenediol | H2bdo | 130 |
| Benzenedithiol | H2bdt | 70 |
In situ quartz crystal microbalance (QCM) analyses were conducted using a 6 MHz AT-cut quartz crystal. The crystal was mounted in a home-made holder and was used to monitor the mass increase, proportional to the change in frequency during the deposition to determine saturation conditions for pulse and purge parameters. The signal was recorded using a Colnatec Eon-LT and processed by averaging over 16 consecutive ALD cycles. The temperature was stabilized for 60 to 90 minutes before any experiments were conducted to ensure a stable temperature and response from the QCM-crystals. Note that the QCM investigations were conducted over an extended timeframe where modifications to the setup have occurred. Thus, the scale of the frequency changes is not necessarily comparable between different materials in this work.
Film thickness and refractive index n(λ) were determined with a J. A. Woollam alpha-SE spectroscopic ellipsometer in the 380–900 nm range. This ellipsometry data was modelled using a Cauchy model. A VASE spectroscopic ellipsometer with range of 280–1000 nm, also from J. A. Woollam, was used to model n(λ) and the extinction coefficient k(λ). These films were modelled with a general oscillator model consisting of several Gaussian peaks. Silicon samples were used for ellipsometry. In both instruments, the sample is modelled by a single layer on top of a thin native silicon oxide layer. This native oxide was measured before each deposition and was generally in the 2–4 nm range. Interface and surface roughnesses were not included in the model as these did not improve the fit. The relation between absorption coefficient α(λ) and k(λ) is given in eqn (1). For readers more accustomed to α(λ), the practical difference between these is a constant scaling factor (2π) a simple wavelength dependent factor (1/λ). FTIR characterization was done using a Bruker VERTEX 70 FTIR spectrometer in transmission mode on single-side polished silicon substrates. Luminescence (PL) and excitation (PLE) measurements was done using an Edinburgh Instruments FLS920 fluorescence spectrometer with a 450 W Xe lamp as excitation source and a Hamamatsu R928 PMT for detection. PL decay measurements utilized an optical parametric oscillator (OPO) system (Opotek HE 355 II) pumped by the third harmonic of a Nd:YAG laser as excitation source. The OPO system was set to 355 nm and a repetition rate of 20 Hz. The detector for decay measurements was the same equipment used for the PLE.
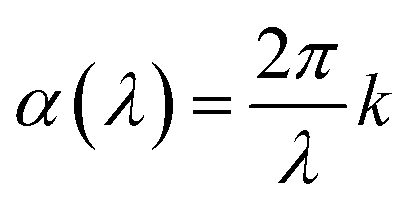 | (1) |
Results and discussion
This work uses Tb2bdc3 as basis material, modifying the luminescence and absorption properties by replacing Tb3+ with other lanthanides and bdc with other aromatic molecules. Thus, the growth and properties of Tb2bdc3 was investigated in more detail than the other materials, while the other materials focus on their differences compared to Tb2bdc3.Tb2bdc3 – growth and optical properties
The saturation conditions for Tb2bdc3 based on QCM experiments is given in Fig. 2. Both lanthanide and acid precursor show clear saturation at around 2 seconds pulses. Purge durations have little effect apart from a gradual growth rate reduction upon long acid purges.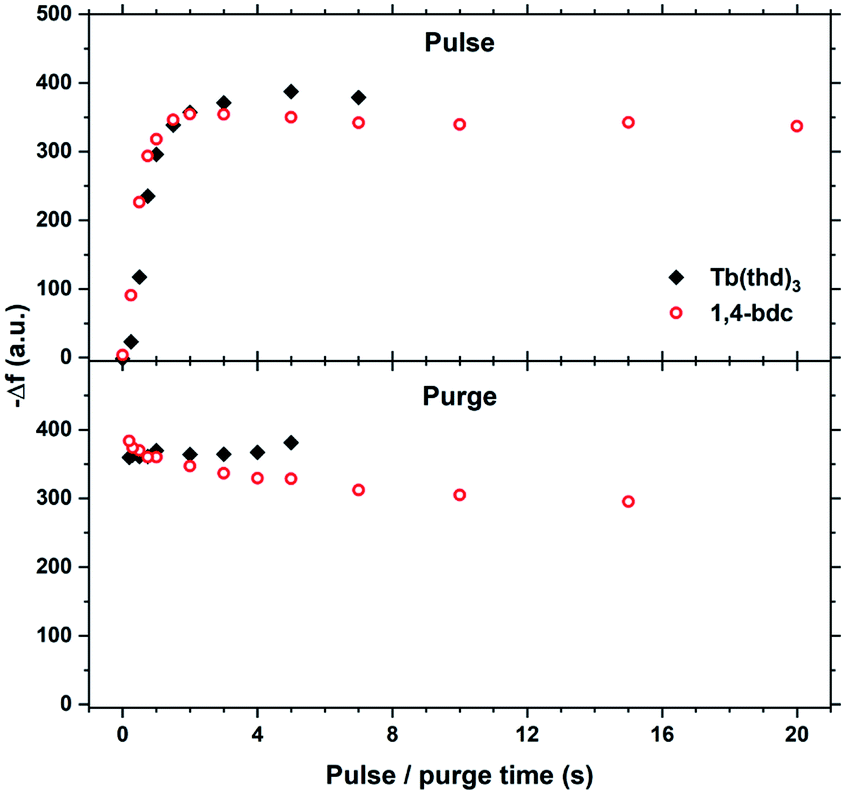 | ||
| Fig. 2 QCM characterization of growth saturation as a function pulse durations for Tb2bdc3. | ||
The QCM response during a cycle with long pulse and purge times is shown in Fig. 3. The two ideal half cycle reactions are given below. For comparison, the masses of the precursors and leaving groups are: Tb(thd)3 = 709, H2bdc = 166, Hthd = 184 g mol−1. Both the lanthanide and acid pulses show initial rapid frequency change of around 2 seconds followed by a slower evolution. The mass decrease during the acid pulse is due to H2bdc being slightly lighter than the leaving molecule Hthd. The ratio between the ideal reactions are Δm1/Δm2 = 16, while the measured ΔmTb/Δmbdc = 3.5. This indicate that either less lanthanide or more acid is deposited during each half cycle than expected from the ideal reactions.
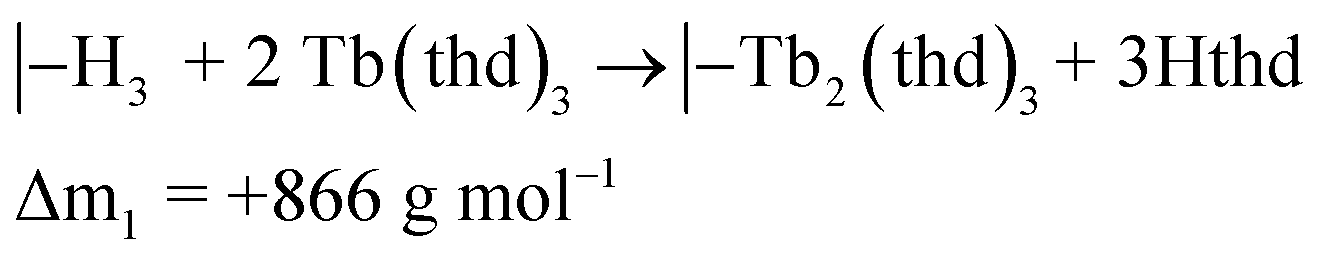 | (2) |
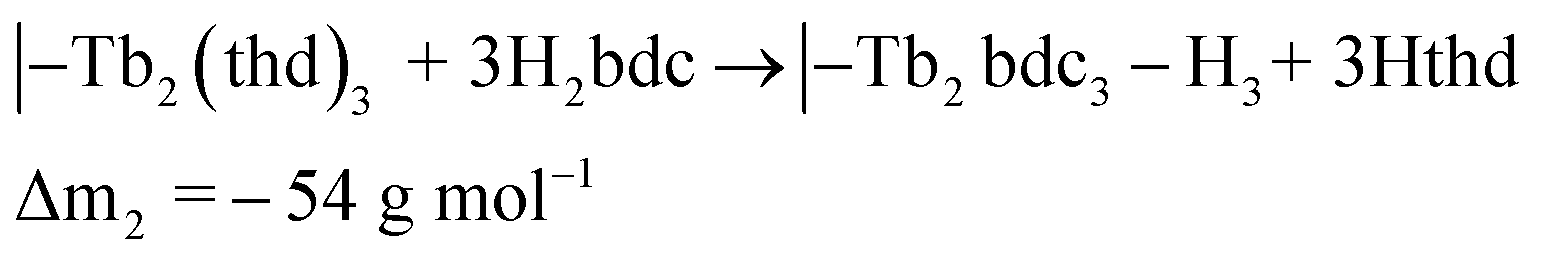 | (3) |
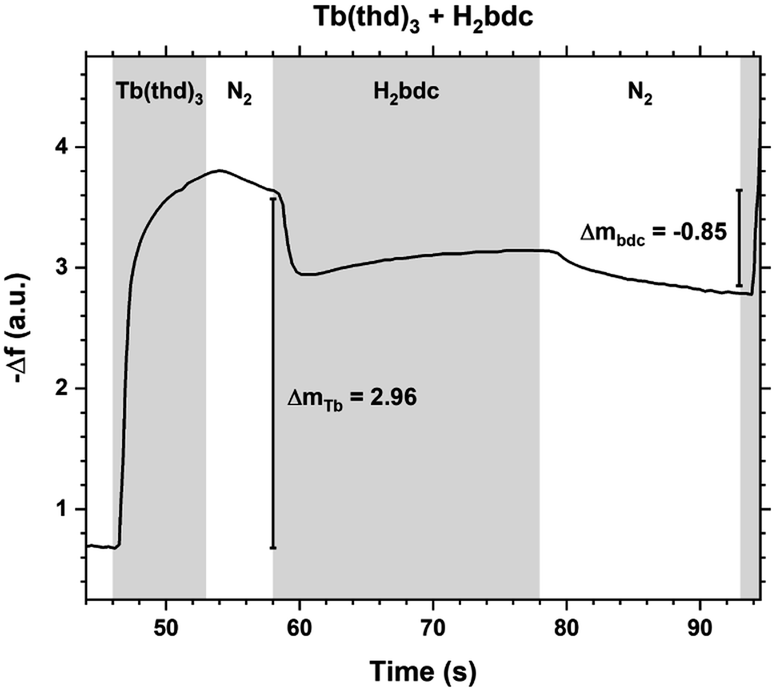 | ||
| Fig. 3 QCM response during a Tb2bdc3 cycle with long pulse and purge times. Tb(thd)3/purge/1,4-bdc/purge = 7/5/20/15. | ||
The growth rate as a function of deposition temperature is shown in Fig. 4. The growth decreases linearly with temperature up until 300 °C, and becomes almost stable up to 350 °C. The films above 300 °C had a brown-ish colour and was non-luminescent indicating thermal decomposition of either H2bdc or Hthd, or both. The luminescence intensity of the films deposited in the 225–275 °C range did not vary significantly. Thus it was decided to keep the deposition temperature at 250 °C for all films, unless otherwise stated.
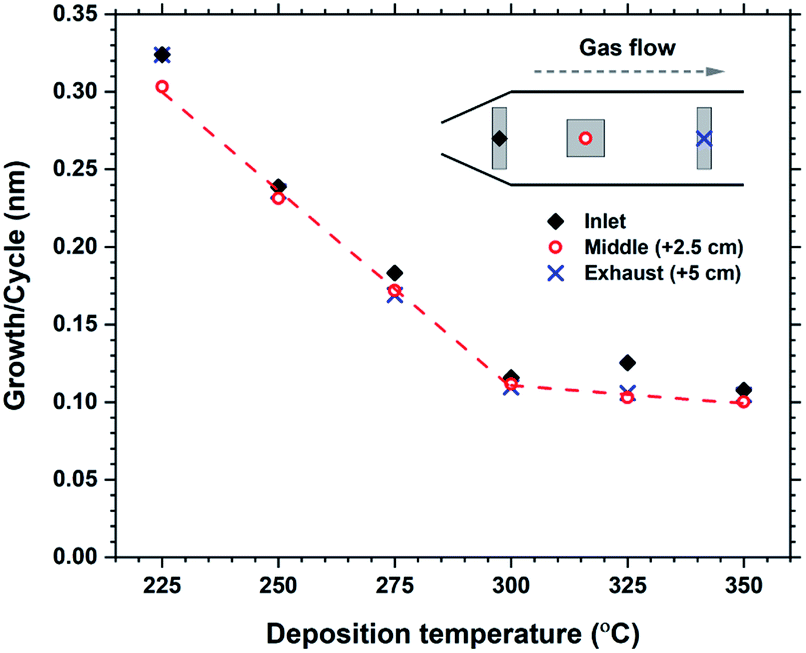 | ||
| Fig. 4 Growth rate as a function of deposition temperature. Measurements from 3 different locations over a 5 cm distance indicates the evenness at each temperature. | ||
Some hybrid materials are not long-term stable in air, either through absorption and release of for example moisture or oxidation reactions. Tb2bdc3 films were characterized by ellipsometry immediately after deposition and 4 additional times up to 3 days to monitor any changes to the films thickness. The results are shown in Fig. 5. The films show a swelling of about 4% after 3 days with no other apparent changes to the films structure or properties. In fact, films deposited over 4 years ago show identical optical and luminescent properties as freshly deposited films.
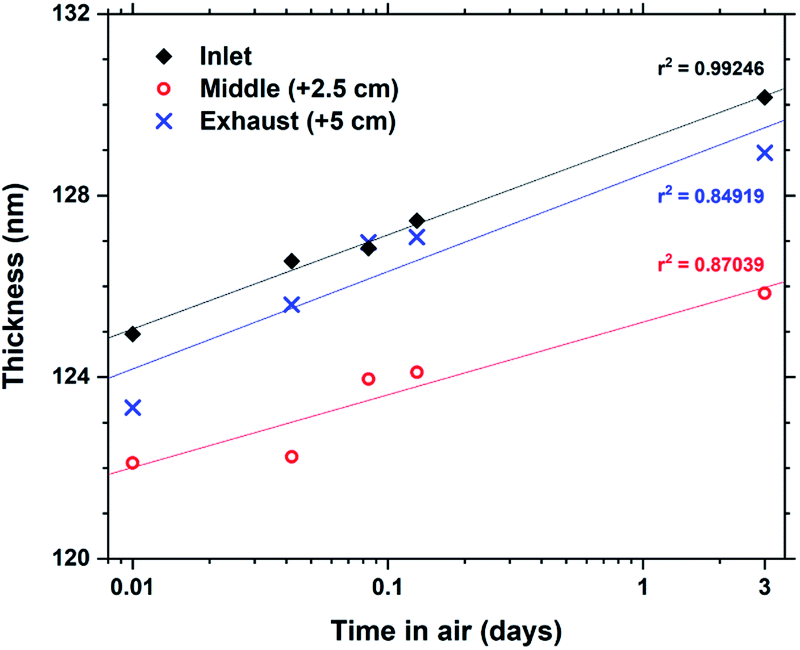 | ||
| Fig. 5 Evolution of film thickness upon air exposure for Tb2bdc3 films. | ||
Fig. 6 shows FTIR spectra from a Tb2bdc3 film taken during the day of deposition and 16 days later. The spectra show that there are no significant differences in number, position or intensities of peaks, indicating no change in bonding during this time. The films show no sign of protonated carboxylic acid groups, which should be clearly visible around 3000 cm−1. FTIR is useful to investigate the type of bonding between the metal and acid group based on the splitting (Δacid) between the asymmetric and symmetric carboxylate stretching peaks.15 A Δacid in the 50–150 cm−1 range indicate bidentate binding, 130–200 cm−1 indicate bridging and >200 cm−1 indicate unidentate binding. The asymmetric carboxylic stretching peak is also split into a double peak, giving two values for Δacid, 151 and 117 cm−1. This indicates that the Tb2bdc3 films contains a mixture of bidentate and bridging binding. An enlarged version zoomed in on these peaks is shown in ESI Fig. 1,† where also the exact wavenumber position of these three peaks are numbered.
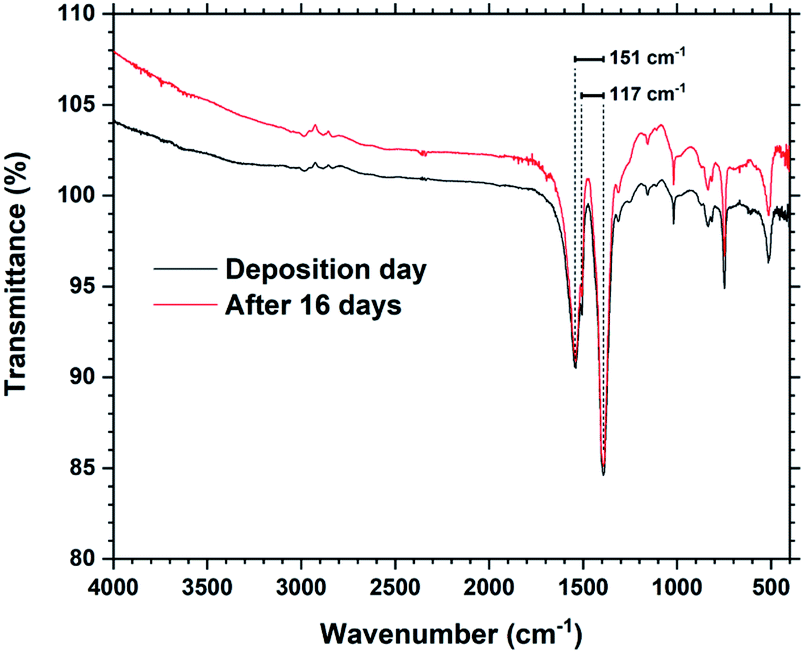 | ||
| Fig. 6 FTIR spectra of a Tb2bdc3 film taken on the day of deposition and after 16 days. The splitting between the asymmetric and symmetric peaks are shown. | ||
The optical absorption and luminescence properties of Tb2bdc3 is shown in Fig. 7, alongside a photograph of a sample under a 280 nm diode. Transmittance data is obtained with a 28 nm film on silica substrate, where the multiplet absorption from the benzene ring of terephthalic acid is clearly seen at around 250 and 300 nm. The PLE spectrum is obtained by monitoring the 5D4 → 7F5 emission. The two first aromatic absorption peaks are clearly seen in the PLE spectrum. The spectrum is not corrected for the decrease in the Xe lamps light intensity towards deep UV, which causes the apparent red-shift of the high energy peak.
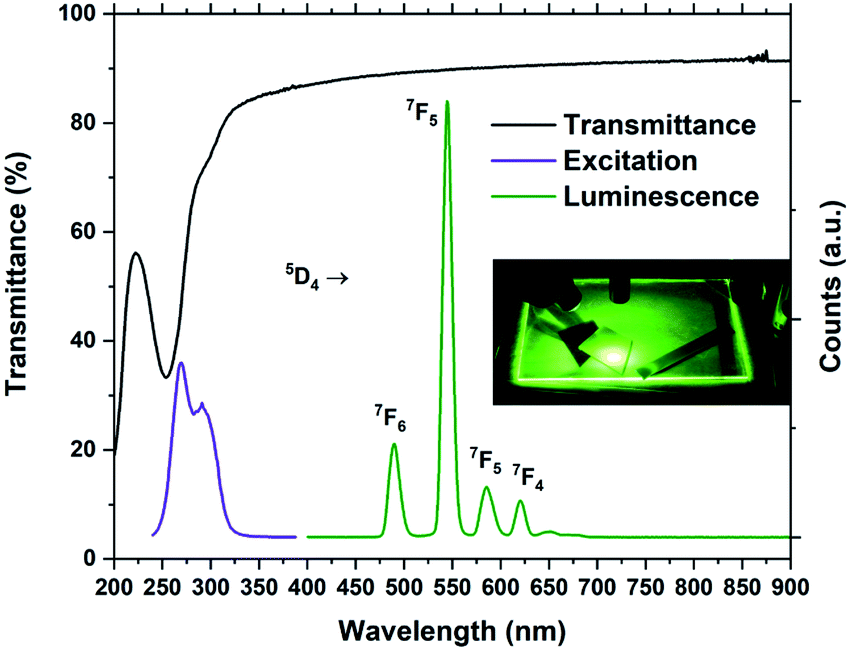 | ||
| Fig. 7 Transmittance, PL and PLE spectra of Tb2bdc3. The inset shows a glass plate and silicon substrates coated with Tb2bdc3 under 280 nm illumination from a diode. | ||
Fig. 7 forms the basis for exploring the other organic sensitizers and lanthanides with the aim to obtain absorption towards the near-UV and blue range and different emission colours. Each of the 4 groups of molecules illustrated in Fig. 1 is compared to Tb2bdc3 individually below, while the deposition parameters and obtained growth rates are summarized in Table 2.
| Organic | Tdep (°C) | g (nm/cycle) | Parameters (s) | |
|---|---|---|---|---|
| a All films showed strong gradients. | ||||
| bdc | 250 | 0.23 | 1.5/1.5/2/1 | |
| (1) | bdo | 175 | 0.12 | 3/1/3/1 |
| bdt | 250 | 0 | 1.5/1.5/2/1 | |
| (2) | bdc-F | 250 | 0.24 | 1.5/1.5/2/1 |
| bdc-Br | 250 | 0.27 | 1.5/1.5/2/1 | |
| (3) | bdc-NH2 | 250 | 0.27 | 1.5/1.5/2/1 |
| bdc-2OH | 250 | ∼1a | 1.5/1.5/2/2 | |
| (4) | bpdc | 275 | 0.26 | 1.5/1/2/1 |
| ndc | 250 | 0.27 | 1.5/1.5/2/2 | |
1,4-Benzenediol and 1,4-benzenedithiol
The aim of these two R–OH and R–SH molecules is to investigate if other binding groups that carboxylic acid can be used to obtain hybrid films with Ln(thd)3, in addition to how these bonds affect the materials optical properties. In particular the sulfur atom can participate in low energy charge transfer transitions and lower the d-shell and create low energy f → d transitions with lanthanides.16Films deposited with Tb(thd)3 and H2bdo showed a growth rate of 0.13 nm per cycle, which is about half of the growth rate of Tb2bdc3. The films were visually transparent like Tb2bdc3, however these films did not luminesce under a 254 nm UV lamp. As enhancing the optical and luminescent properties of lanthanide-hybrid materials is the aim of this work, the H2bdo precursor was not further explored apart from noting that H2bdo and Tb(thd)3 do react and form ALD films. Thus, hydroxy groups can be suitable reaction groups to bind to Ln(thd)3. In fact, H2bdo sublimes at 70 °C lower than H2bdc, which can be a desired property.
Tb(thd)3 and H2bdt did not result in film deposition. Film growth was attempted at 200, 250 and 300 °C. This was somewhat surprising as H2S can be used with Ln(thd)3 to produce lanthanide sulfide films.17
Tb2bdc3: Ln3+ and halogenated bdc
Halogenation of the aromatic centre of H2bdc can affect both the growth and optical properties in addition the physical properties of the precursor. From Table 1 it is seen that H2bdc-F and H2bdc-Br sublimes at 70 °C lower and 30 °C higher than H2bdc, respectively. Table 2 show that both precursors result in similar growth rates as H2bdc. Regarding growth, the main difference between H2bdc, H2bdc-F and H2bdc-Br is a substantial difference in sublimation temperature.Films produced with these three precursors and Tb(thd)3 all produced luminescent with the same green colour and with similar intensity under a 254 nm UV lamp. To further investigate differences in optical properties, the films optical absorption and luminescence decays were characterized. k(λ) and n(λ) of these films is shown in Fig. 8. Compared to Tb2bdc3, fluorination causes a red-shift in absorption and a lower n(λ) while bromination causes a blue-shift with unchanged n(λ). Fig. 9 show decays of the 5D4 → 7F5 emissions of the three materials. All three can be fitted with a single exponential model. The brominated sample show a lower lifetime than Tb2bdc3 and Tb2bdc-F3. This could indicate higher quenching rates in Tb2bdc-Br3. In summary, bromination offers neither improved growth (high sublimation temperature), more near-UV absorption (weaker and blue-shifted) nor improved luminescence yield (lower Tb3+ lifetime). On the other hand, the main advantage of fluorination is the substantially lower sublimation temperature and a small red-shift of the absorption while the growth and material properties are otherwise very similar to the un-halogenated acid.
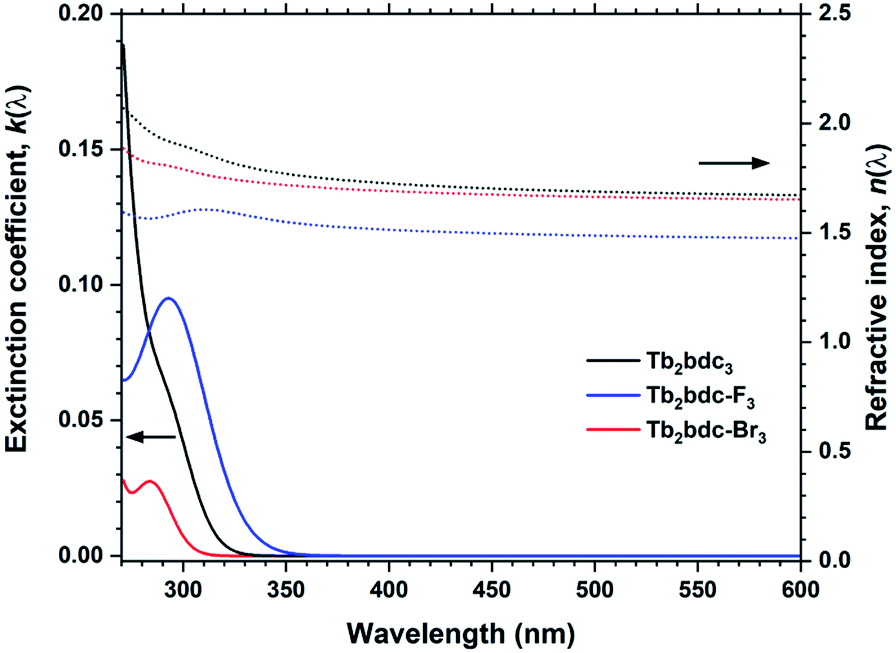 | ||
| Fig. 8 k(λ) and n(λ) for Tb2bdc3, Tb2bdc-F3 and Tb2bdc-Br3 films. | ||
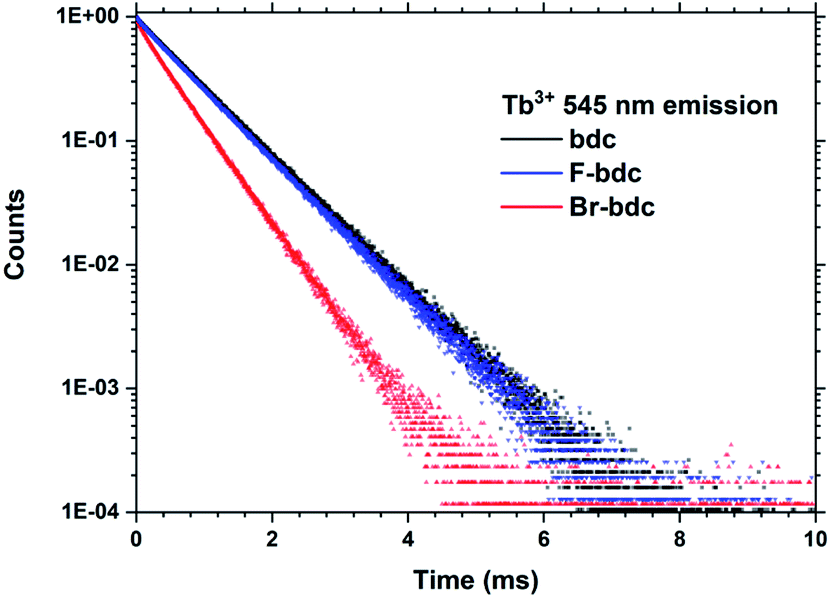 | ||
| Fig. 9 PL decay data from Tb2bdc3, Tb2bdc-F3 and Tb2bdc-Br3 films. | ||
Replacing Tb3+ with Eu3+ and Y3+ results in Ln2bdc3 films with very different emissions, shown in Fig. 10. The PLE spectra are very similar to each other, although the lighter Y3+ shows a small red-shift compared to the heavier Tb3+ and Eu3+. Both Tb2bdc3 and Eu2bdc3 produce their regular Tb3+ and Eu3+ emission spectra, showing that bdc is an efficient sensitizer for these two lanthanides. Y3+ doesn't have a partially filled d or f shell, allowing the sensitizer molecule to emit its own photon. Y2bdc3 emits in the near-UV, partly extending into the blue range. This gives the material a violet emission colour, relatively weak compared to Tb2bdc3 and Eu2bdc3.
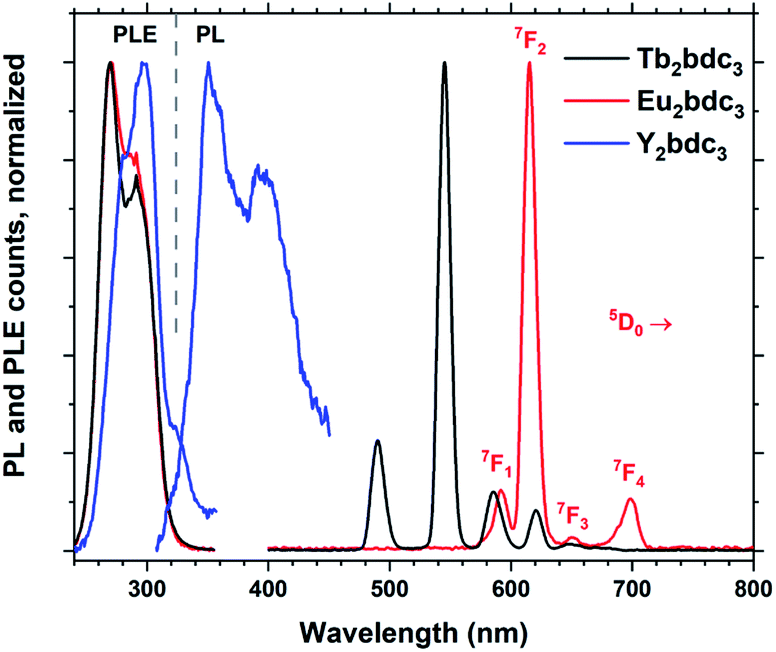 | ||
| Fig. 10 Normalized PLE and PL spectra of Ln2bdc3, Ln = Tb, Eu and Y. The emission lines of Eu3+ are named. | ||
H2bdc-NH2 and H2bdc-OH
These two molecules are interesting from both a chemical and optical perspective. In addition to the two carboxylic acid groups, they have one amine and two hydroxy groups, respectively, which can take part in bonding. Depositions with Tb(thd)3 and H2bdc-NH2 does indeed show a somewhat higher growth rate, while H2bdc-OH resulted a fourfold increase in growth rate and strong gradients throughout the gradients. The large increase in growth rate and gradients were reproducible and varying deposition parameters did not result in more homogeneous films. This indicates that the growth chemistry is more complex that for the other sensitizer acids, in particular for H2bdc-OH.These films show large red-shifts compared to Tb2bdc3, covering the near-UV and even blue range for Tb2(bdc-OH)3 (Fig. 11). However, all depositions with Tb, Eu and Y with these two acids produced non-luminescent samples. The fact that the Y depositions were quenched indicate that the sensitizer acids are quenched due to how they are packed in the structure. Both acids produce strongly luminescent solutions when dissolved. Crystalline MOF structures with H2bdc-NH2 and Tb and Eu has been shown to produce sensitized Ln3+ emission.18 A study by Vogel et al. on MOF structures of Y and H2bdc-OH show that the exact bonding scheme between Ln3+ and the sensitizer acid will have major impacts on the materials optical and luminescent properties.19 Thus, although the films produced in our work are non-luminescent, it is likely that control of the resulting structure and molecule packing can produce luminescent films. We have previously shown that control of crystallinity can be achieved both through in situ modulation by pulsing other species to modify growth mechanics and ex-situ treatments.20,21 However, exploration of how crystallinity and control of growth mechanics can enable luminescent variants is beyond the scope of the current work.
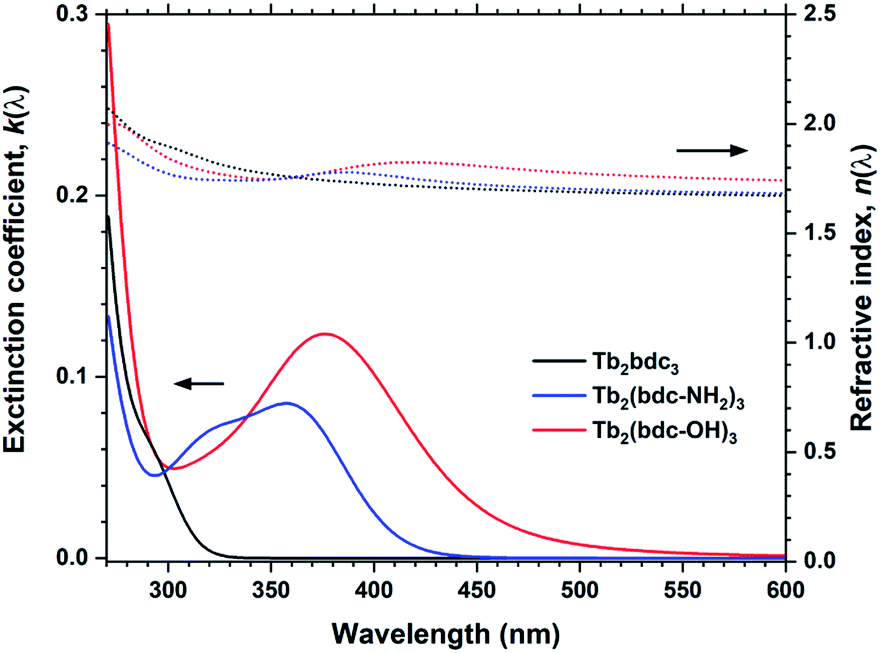 | ||
| Fig. 11 k(λ) and n(λ) for Tb2bdc3, Tb2bdc-NH2 and Tb2bdc-OH films. | ||
H2bpdc and H2ndc
These two biaromatic acids are simpler in terms of not having other functional groups that affect growth. They both produce homogeneous and gradient free films with Y, Tb and Eu, with a slightly larger growth rate than Tb2bdc3 due to their larger size. Interestingly, Y2pbdc3 and Y2ndc3 films show bright violet and blue luminescence from the sensitizer molecule, while absorption covers the near UV range with a tail extending until 400 nm. Thus, due to their good optical properties and even growth, these two sensitizer acids were investigated in more detail.The QCM response per cycle Eu2bpdc3 and Tb2ndc3 is shown in Fig. 12. Tb2ndc3 show clear saturation after 1 and 1.5 s Tb(thd)3 and H2ndc pulses, respectively. Eu2bpdc3 show strong indication of the same saturation values. However, Eu2bpdc3 also show a steady increase with increasing Eu(thd)3 pulse times longer than 1 s, and some growth at 0 s acid pulses. This is likely due to the higher precursor temperature used for H2bpdc. We based the sublimation temperatures on prior sublimations temperatures in ALD-like vacuum conditions. Likely, 250 °C is slightly too warm for this precursor, leading to some precursor vapours seeping into the reaction chamber. This CVD component is quite small for the 2 and 3 s Ln(thd)3 and H2bpdc pulses used for samples, which was also seen in negligible growth gradients.
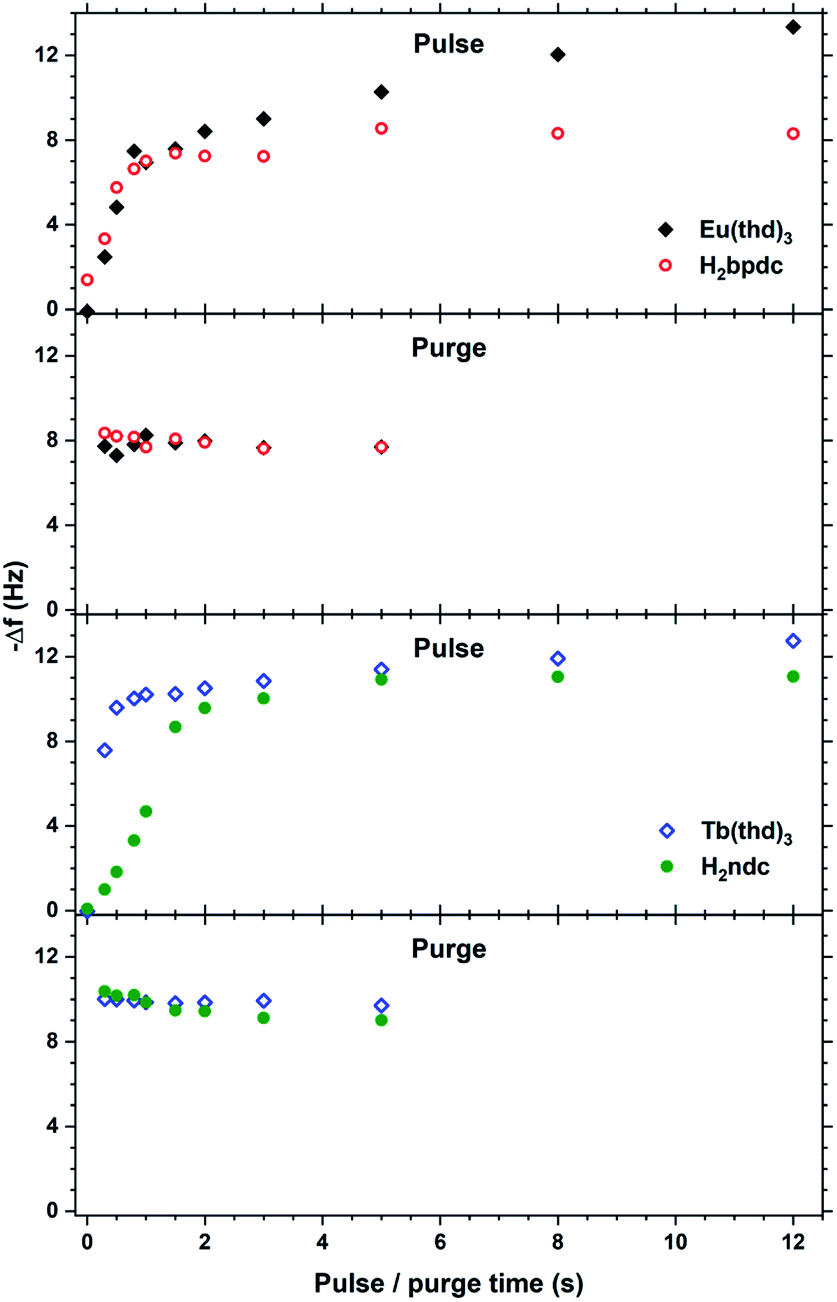 | ||
| Fig. 12 QCM characterization of growth saturation as a function pulse durations for Eu2bpdc3 and Tb2ndc3. | ||
The QCM response during a single cycle of Eu2bpdc3 and Tb2ndc3 using long pulse and purge times is shown in Fig. 13. As for Tb2bdc3 in Fig. 3, both Eu2bpdc3 and Tb2ndc3 show an initial rapid growth for both Ln(thd)3 and acid pulse, indicating surface saturation. Contrary to Tb2bdc3 which experience a mass decrease during the acid pulse, these two depositions experience a mass gain. This is due to the large H2bpdc and H2ndc being heavier than Hthd while H2bdc is lighter, 242 and 216 g mol−1 respectively.
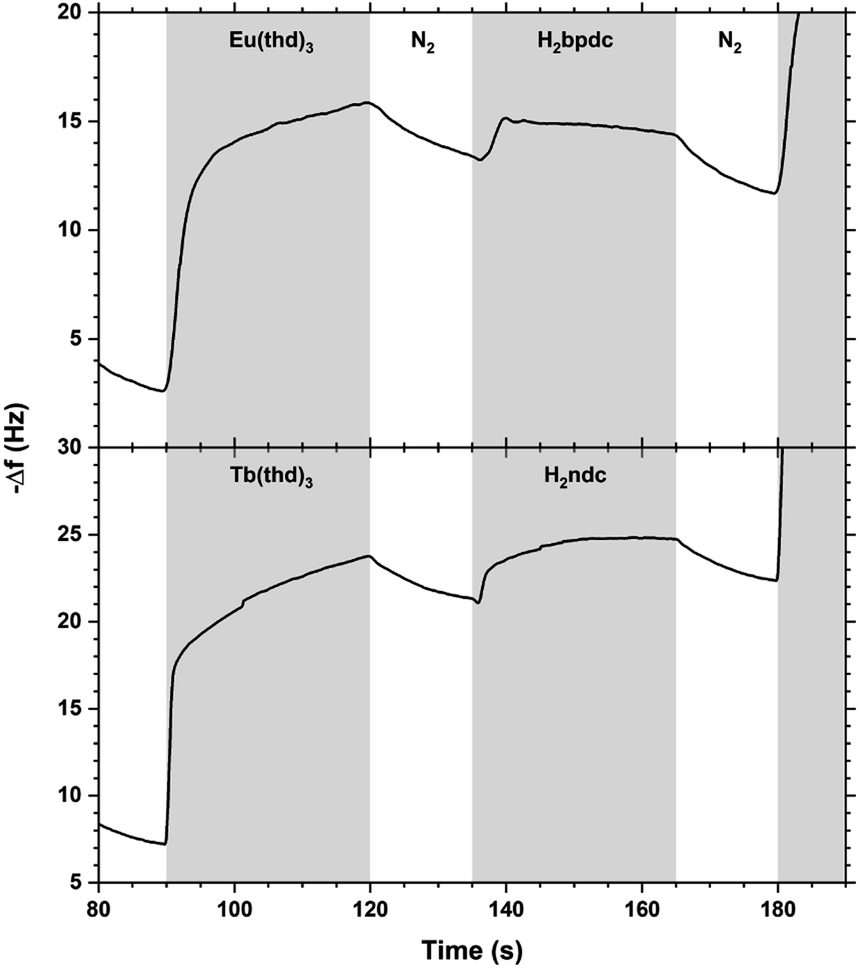 | ||
| Fig. 13 QCM response during a Eu2bpdc3 and Tb2ndc3 cycle with long pulse and purge times. Ln(thd)3/purge/acid/purge = 30/15/30/15. | ||
The perhaps most interesting aspects of Ln2bpdc3 and Ln2ndc3 films are their optical properties. These films have absorptions covering most of near UV while being almost an order of magnitude stronger than Ln2bdc3, Ln2(bdc-NH2)3 and Ln2(bdc-OH)3. Unlike Ln2(bdc-NH2)3 and Ln2(bdc-OH)3, these films also result in highly luminescent films. Fig. 14 summarizes k(λ), n(λ), PLE and optical transmittance of selected films, in addition to the PL spectra originating from the aromatic acids.
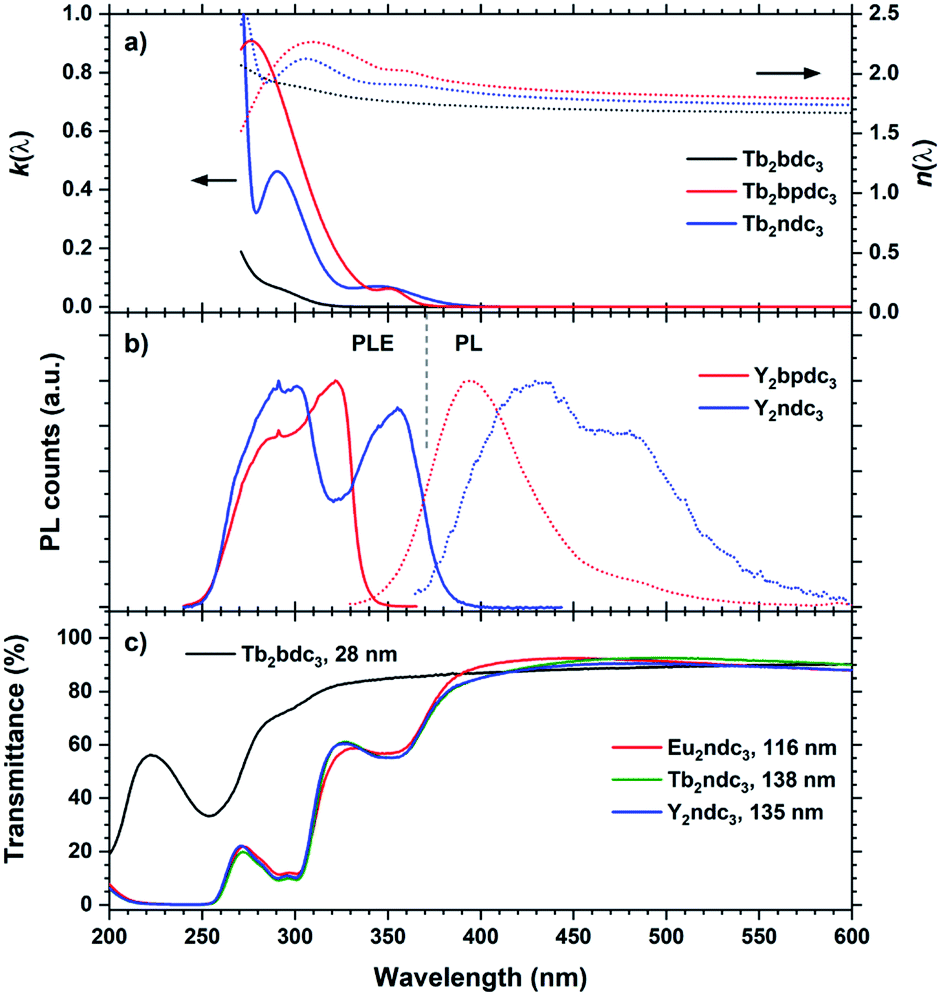 | ||
| Fig. 14 (a) k(λ) and n(λ) of Tb2bpdc3 and Tb2ndc3. Tb2bdc3 is included for comparison. (b) PLE and PL of Y2bpdc3 and Y2ndc3. λex/λem 320/420 nm and 355/475 nm for Y2bpdc3 and Y2ndc3, respectively. (c) Transmission spectra of a selection of thin films from this work, illustrating the strong and near-UV absorption from the organic molecules. | ||
Y2bpdc3 and Y2ndc3 show strong violet and blue emissions. Their emission spectra are shown in Fig. 14(b). Both consist of broad emission peaks which are normal for aromatic emission. The emission from Y2ndc3 consist of two distinguishable but overlapping peaks at 430 and 480 nm, while Y2bpdc3 consist of one major peak at 395 nm and a smaller shoulder at 475 nm. Replacing Y with Tb or Eu fully removed the aromatic emissions, indicating complete energy transfer to these lanthanides in Ln2bpdc3 and Ln2ndc3. However, while both Tb and Eu were luminescent in Ln2bpdc3, only Eu3+ was luminescent in Ln2ndc3. Tb2ndc3 showed neither aromatic nor Tb emission. The lack of emission from bpdc2− and ndc2− means that there is a complete transfer from these two to something else. ESI Fig. 3† show the emission spectra of Y2bpdc3 and Y2ndc3 along with the ground state f–f absorption transitions of Tb3+ and Eu3+. Both lanthanides show overlapping absorption lines with the emission of both sensitizers. Note that for wavelengths shorter than 400 nm, both lanthanides have dense absorption lines that are excluded in the figure for clarity. The lack of Tb3+ emission in Tb2ndc3 indicated that either Tb3+ is fully quenched or that Tb3+ never received the energy. In both cases, a possible explanation is a low energy intervalence charge transfer (IVCT) state that can accept the energy from either the sensitizer, lanthanide or both. This IVCT and its quenching on Tb3+ is well known in d0 metal oxides.22 Both Tb3+ and Eu3+ are redox active (to Tb4+ and Eu2+, respectively), and so is polyaromatic molecules.23 However, a detailed investigation into the quenching mechanisms is beyond the scope of this work.
The Tb and Eu emission spectra were identical to those from Ln2bdc3 in Fig. 10. In addition, the PLE spectra of Tb and Eu were identical to the PLE spectra of the aromatic emission in Y2bpdc3 and Y2ndc3, thus only the PL and PLE spectra of the Y variants are shown in Fig. 14. The modelled k(λ) and n(λ) were also identical between lanthanides. Comparing the modelled k(λ) in (a) and PLE in (b) show that the absorption and excitation peaks matches well for Ln2ndc3. However, even though both Ln2bpdc3 and Ln2ndc3 show similar absorption at 350 nm, this absorption only produces an excitation band in Ln2ndc3.
In (c) the transmittance spectra on all three Ln2ndc3 materials are shown alongside Tb2bdc3 for comparison. All samples are on silica substrates. The absorption bands 350 and 290 nm match well with the modelled k(λ) and PLE spectre.
Summary of absorption properties
One of the defining properties of these mono- and bi-aromatic hybrid films is their strong near-UV absorption. Fig. 15 summarizes the absorption coefficient for all terbium films (note the different y-scale for the two bi-aromatic films). To help visualize how k(λ) and absorption coefficients manifests as absorption in thin films, the film thickness required for 90% absorption according to Beer–Lambert law is plotted as t90. Plotting t90 helps understanding how thick films are required for a given absorption. For example, for 90% absorption at 300 nm, films of 100 and 150 nm are sufficient for Ln2bpdc3 and Ln2ndc3, while 90% absorption at 375 nm requires 550 nm and 1 μm of Ln2(bdc-2OH)3 and Ln2(bdc-NH2)3, respectively. The luminescence from these films span the full range from violet to blue, green and red as shown in Fig. 16 along with the films colour coordinates.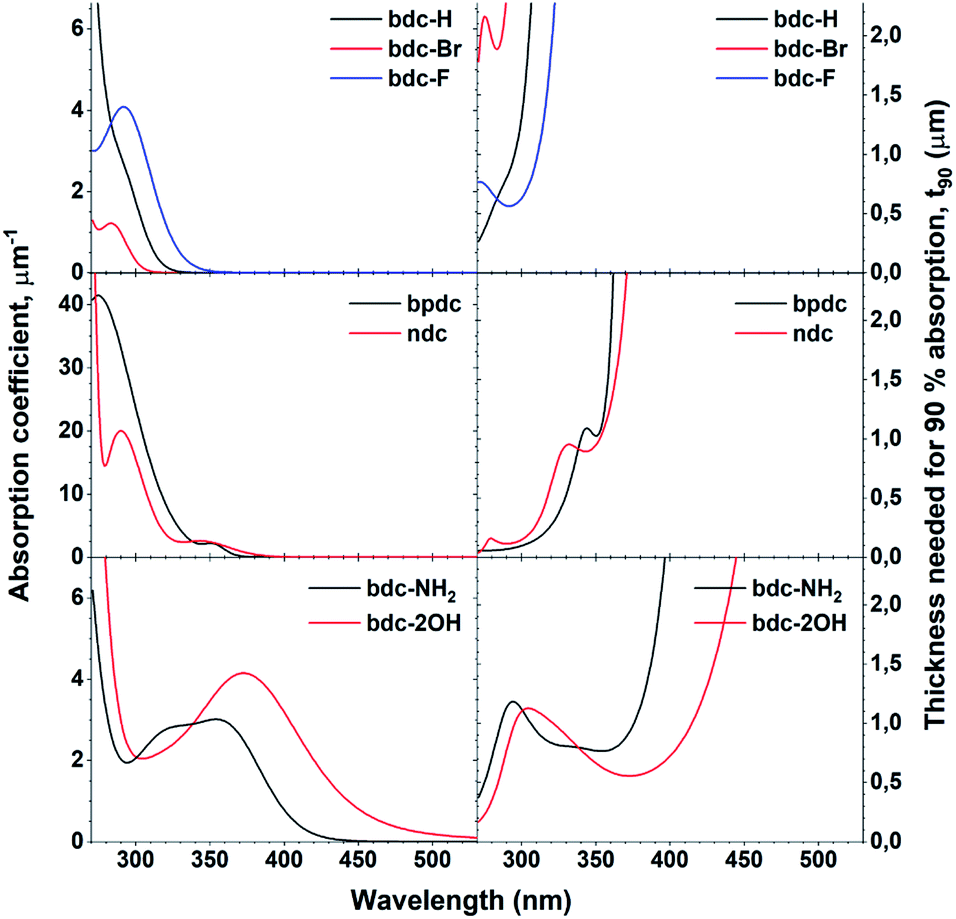 | ||
| Fig. 15 Absorption coefficient of Tb films with all acids, obtained through ellipsometry and the calculated thickness needed for 90% absorption, t90. Note the different y-scale on the absorption coefficients for bpdc and ndc. | ||
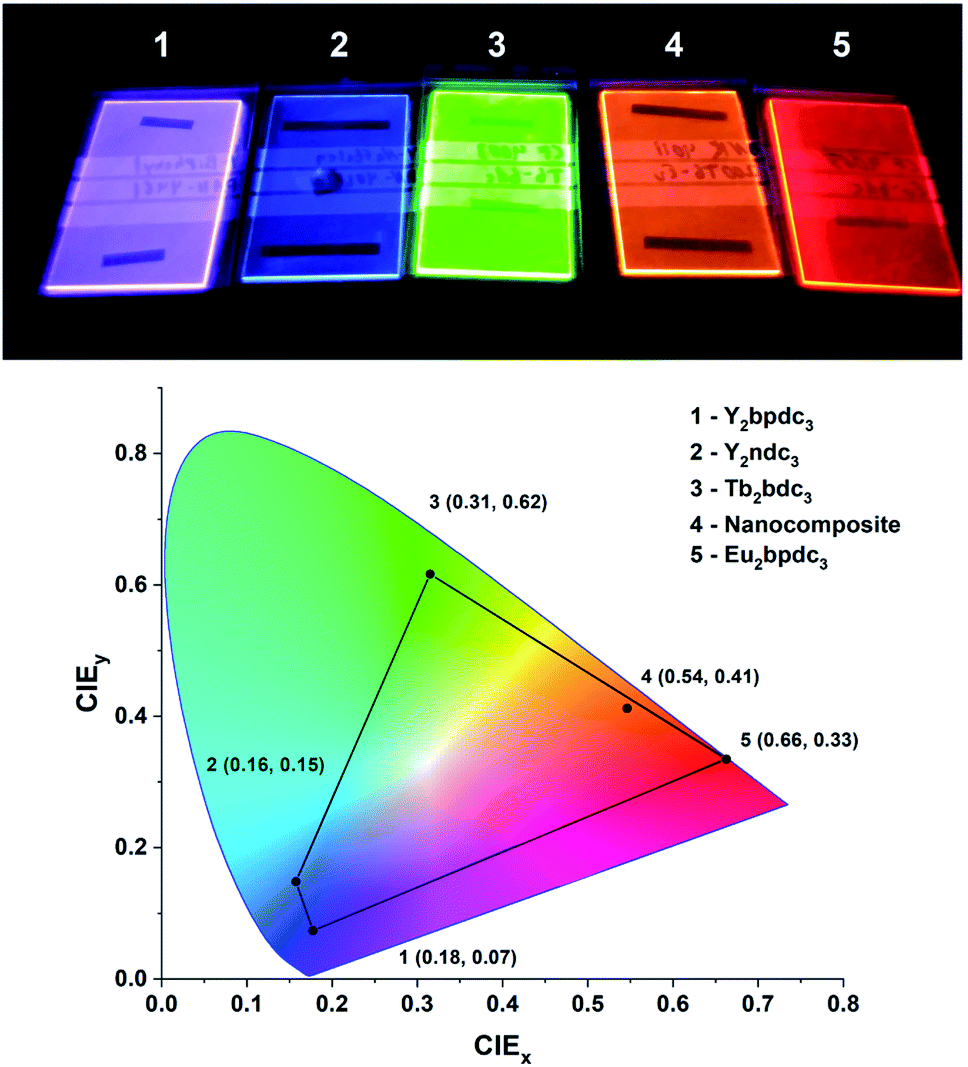 | ||
| Fig. 16 Photo and CIE coordinates of samples from this work under a 254 nm UV lamp. The violet, blue, green and red films as Y2bpdc3, Y2ndc3, Tb2bdc3 and Eu2bdc3. The orange film is a hybrid-fluoride nanocomposite from another recent work,8 showing orange Eu3+ emission sensitized by terephthalic acid layers. | ||
Conclusion
In this work we have deposited hybrid films of lanthanides with 9 different mono- and bi-aromatic di-carboxylic acids, and explored their optical and luminescent properties. The terbium–terephthalic acid system has been used as a base and is explored in more detail, including film growth and air stability. Yttrium has been used as an optically passive cation so that the luminescence properties of the aromatic molecule itself can be investigated. The yttrium films show that the two bi-aromatic acids produce films with bright violet and blue luminescence. Yet with Tb and Eu all bi-aromatic films are non-luminescent except Eu2ndc3. The bi-aromatic films show by far the strongest absorption strength of all the acids. The two acids with added functional groups on the other hand, namely bdc-2OH and bdc-NH2, show the largest red-shift in absorption and even reaches slightly into the blue range for bdc-2OH. In conclusion, the optical properties of hybrid materials made from lanthanides and aromatic di-carboxylic acids show a large variation in both absorption range and strength while their luminescence spans violet, blue, green and red.Author contributions
Hansen: conceptualization, data curation, formal analysis, supervision, writing – original draft. Svendsen: investigation, formal analysis, writing – review & editing. Nesteng: investigation, formal analysis, writing – review & editing. Nilsen: data curation, formal analysis, supervision, writing – review & editing, project administration, funding acquisition.Conflicts of interest
There is no conflict of interest to report.Acknowledgements
This work was performed within “The Norwegian Research Centre for Solar Cell Technology” project number 193829, a Centre for Environment-friendly Energy Research co-sponsored by the Research Council of Norway and research and industry partners in Norway.References
- V. Miikkulainen, et al., Crystallinity of inorganic films grown by atomic layer deposition: Overview and general trends, J. Appl. Phys., 2013, 113(2), 021301 CrossRef.
- A. J. M. Mackus, et al., Synthesis of Doped, Ternary, and Quaternary Materials by Atomic Layer Deposition: A Review, Chem. Mater., 2019, 31(4), 1142–1183 CrossRef CAS.
- H. H. Sønsteby, O. Nilsen and H. Fjellvåg, Atomic layer deposition of (K,Na)(Nb,Ta)O3 thin films, J. Vac. Sci. Technol., A, 2016, 34(4), 041508 CrossRef.
- P.-A. Hansen, et al., Luminescent Properties of Multilayered Eu2O3 and TiO2 Grown by Atomic Layer Deposition, Chemical Vapor Deposition, 2014 Search PubMed.
- K. B. Klepper, et al., Atomic layer deposition of organic–inorganic hybrid materials based on saturated linear carboxylic acids, Dalton Trans., 2011, 40(17), 4636–4646 RSC.
- K. B. Lausund, et al., MOF thin films with bi-aromatic linkers grown by molecular layer deposition, J. Mater. Chem. A, 2020, 8(5), 2539–2548 RSC.
- X. Meng, An overview of molecular layer deposition for organic and organic–inorganic hybrid materials: mechanisms, growth characteristics, and promising applications, J. Mater. Chem. A, 2017, 5(35), 18326–18378 RSC.
- P.-A. Hansen, et al., Single-step approach to sensitized luminescence through bulk-embedded organics in crystalline fluorides, Commun. Chem., 2020, 3(1), 162 CrossRef CAS.
- P.-A. Hansen, et al., Controlling luminescence and quenching mechanisms in subnanometer multilayer structure of europium titanium oxide thin films, J. Lumin., 2019, 215, 116618 CrossRef CAS.
- P. Sundberg and M. Karppinen, Organic and inorganic–organic thin film structures by molecular layer deposition: A review, Beilstein J. Nanotechnol., 2014, 5, 1104–1136 CrossRef PubMed.
- M. Tuomisto, et al., Up-converting ALD/MLD thin films with Yb3+, Er3+ in amorphous organic framework, J. Lumin., 2019, 213, 310–315 CrossRef CAS.
- P.-A. Hansen and O. Nilsen, Quinizarin: a large aromatic molecule well suited for atomic layer deposition, Dalton Trans., 2021, 50(24), 8307–8313 RSC.
- P.-A. Hansen, et al., Structural and optical properties of lanthanide oxides grown by atomic layer deposition (Ln = Pr, Nd, Sm, Eu, Tb, Dy, Ho, Er, Tm, Yb), Dalton Trans., 2013, 42(30), 10778–10785 RSC.
- M. Rogowska, et al., Molecular layer deposition of photoactive metal-naphthalene hybrid thin films, Dalton Trans., 2021, 50, 12896–12905 RSC.
- F. Verpoort, et al., Characterization of a Surface Coating Formed from Carboxylic Acid-Based Coolants, Appl. Spectrosc., 1999, 53(12), 1528–1534 CrossRef CAS.
- P.-A. Hansen, S. Kumar and A. Meijerink, Strong self-sensitized green and NIR emission in NaYS2 doped with Pr3+ and Yb3+ by inducing Laporte allowed and charge transfer transitions, J. Lumin., 2021, 235, 118012 CrossRef CAS.
- M. Tammenmaa, et al., Zinc sulphide thin films doped with rare earth ions, J. Less-Common Met., 1986, 126, 209–214 CrossRef CAS.
- J.-N. Hao and B. Yan, Amino-decorated lanthanide(iii) organic extended frameworks for multi-color luminescence and fluorescence sensing, J. Mater. Chem. C, 2014, 2(33), 6758–6764 RSC.
- D. J. Vogel, T. M. Nenoff and J. M. Rimsza, Tuned Hydrogen Bonding in Rare-Earth Metal–Organic Frameworks for Design of Optical and Electronic Properties: An Exemplar Study of Y–2,5-Dihydroxyterephthalic Acid, ACS Appl. Mater. Interfaces, 2020, 12(4), 4531–4539 CrossRef CAS PubMed.
- K. B. Lausund, V. Petrovic and O. Nilsen, All-gas-phase synthesis of amino-functionalized UiO-66 thin films, Dalton Trans., 2017, 46(48), 16983–16992 RSC.
- K. B. Lausund and O. Nilsen, All-gas-phase synthesis of UiO-66 through modulated atomic layer deposition, Nat. Commun., 2016, 7, 13578 CrossRef CAS PubMed.
- P. Boutinaud, et al., Quenching of Lanthanide Emission by Intervalence Charge Transfer in Crystals Containing Closed Shell Transition Metal Ions, Spectrosc. Lett., 2007, 40(2), 209–220 CrossRef CAS.
- G. Wang, et al., Exploring polycyclic aromatic hydrocarbons as an anolyte for nonaqueous redox flow batteries, J. Mater. Chem. A, 2018, 6(27), 13286–13293 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03360g |
| This journal is © The Royal Society of Chemistry 2022 |