
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn experimental and theoretical investigation into the self-assembly of a chemically modified high-χ coil–rod diblock copolymer†
Lei Dong *a,
Kevin Wylieb,
Yuta Nabaeb and
Teruaki Hayakawab
*a,
Kevin Wylieb,
Yuta Nabaeb and
Teruaki Hayakawab
aSchool of Materials Science and Engineering, Southeast University, Nanjing 211189, China
bDepartment of Materials Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, S8-813, 2-12-1-S8-36, Ookayama, Meguro-ku, Tokyo, 152-8552, Japan
First published on 17th June 2022
Abstract
A precursor diblock copolymer with a silicon backbone, polystyrene-block-poly(methylvinylsiloxane), was synthesized, and 1H,1H,2H,2H-perfluorodecanethiol was quantitatively introduced into the backbone via a thiol-ene reaction to yield a novel coil–rod diblock copolymer, poly(styrene-block-poly(2-((3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-heptadecafluorodecyl)thio)ethyl)methylsiloxane). The ultra-hydrophobicity of the introduced perfluoroalkyl side chain enhanced the segregation between counter-blocks and significantly increased the χ value, which is essential for minimizing the size of self-assembled domains for lithographic applications. Thus, self-assembled domains with a minimal spacing of approximately 10 nm were formed. A hexagonally packed array with significant anisotropy was observed in the self-assembled morphology by small-angle X-ray scattering and transmission electron microscopy. Such an array was precisely reproduced by modified self-consistent field theory (SCFT) calculation developed for the coil–rod structure. Furthermore, the phase diagram was estimated, and the morphological dependence on the relative scale of the rod unit was investigated by SCFT prediction.
Introduction
The self-assembly of block copolymers (BCPs) could form patterns on the scale of 3–100 nm, which are promising for next-generation lithographic applications. In general, lithography requires BCPs with a high χ parameter, high etching contrast, and orientation control.1,2 Among all reported BCP materials, silicon-based ones are advantageous due to their high χ parameter and etching contrast. The high χ parameter enables minimization of both the self-assembled periodic nanostructure and the interfacial roughness.3–6 For example, one widely studied silicon-based BCP is poly(styrene-b-dimethylsiloxane) (PS-b-PDMS) with χ ≈ 0.2, which is significantly higher than that of poly(styrene-b-methyl methacrylate) (PS-b-PMMA, χ ≈ 0.04).7 As a result, the reported minimum feature size of PS-b-PDMS (∼16 nm (ref. 7 and 8)) is much smaller than that achieved in PS-b-PMMA (∼24 nm (ref. 9)). Additionally, the above-mentioned hybrid Si-containing BCPs possess advantages in the process of transferring patterns, as the Si-containing block within the self-assembled BCP can be directly transformed into silicon oxide via oxygen plasma etching without causing any potential over-etching problems.Segments more hydrophilic than polystyrene have been introduced as counter-blocks to further enhance the χ parameter of silicon-based BCPs. Examples include poly(dimethylsiloxane-b-methyl methacrylate) (PDMS-b-PMMA),10 poly(2-vinylpyridine)-b-poly(dimethylsiloxane),11 poly(4-vinylpyridine)-b-poly(dimethylsiloxane),11 poly(trimethylsilylstyrene-b-D,L-lactide),12 poly(lactide-b-dimethylsiloxane-b-lactide),12 and poly(3-hydroxystyrene)-b-poly(dimethylsiloxane).13 Recently hydroxyl-end groups were introduced into the siloxane-based backbone of BCP,14,15 in order to balance the SFE of the PS and siloxane-based blocks for the desired perpendicular orientation of domains in thin-film states. Methylvinylsiloxane was selected as the precursor because it can be introduced into functional side chains via a thiol-ene reaction or hydrosilylation. The thiol-ene click reaction is particularly attractive because of its use of a metal-free catalyst.16 However, the attempt to balance surface free energy (SFE) via chemical tailoring on BCP also diminishes the strength of block segregation and limits the minimization of feature domain size. Therefore, a silicon-containing BCP of high χ parameter is in demand for lithographic applications. To increase the incompatibility between the counter-blocks and enhance the χ parameter of silicon-containing BCPs, the siloxane-based blocks could be chemically modified to form more hydrophobic segments, instead of replacing the PS block with more hydrophilic ones. Long fluorinated side chains, such as perfluoroalkyl, in BCPs exhibit an extremely low SFE (∼10 mN m−1, depending on the crystal type or liquid crystal phase17–19), that is even lower than that of siloxane (∼20 mN m−1).20 Thus, the introduction of a fluorinated side chain into the backbone of siloxane could effectively enhance the hydrophobicity of the block21 and obtain a higher χ parameter with PS (SFE ≈ 43 mN m−1)20 segments. Additionally, the low friction coefficient of the fluorinated side chain22–24 in combination with the highly mobile siloxane backbone accelerates self-assembly during thermal annealing.
Besides lithography, the self-assembly of coil–rod BCPs is appealing in other application areas, such as photovoltaics25–28 and biomaterials.29 Some coil–rod BCPs can segregate into nematic nanostructures at even lower χN values (χN ≥ 5.0) than the segregation limit for coil–coil BCPs (χN ≥ 10.5),30–33 owing to the different kinds of physics governing their self-assembly. The self-assembly of coil–coil diblock copolymers favors an isotropic morphology in the thermodynamically stable states,34 despite the reported anisotropic fluctuation of cylinders in a poly(styrene-block-isoprene-block-styrene) coil triblock copolymer.35 In contrast, the self-consistent field theory (SCFT) model often indicates the anisotropic packing of cylinders to be the thermodynamically stable state for rod–coil36 or semiflexible–flexible37,38 BCPs. However, experimental observation of such an anisotropic self-assembled morphology has never been reported in diblock copolymer systems. Meanwhile, the pendent bulky perfluoroalkyl side chain attached to the polymer main chains, being highly rigid39–41 with a large free volume,42,43 could be approximated as rods against the flexible coils of PS segments, when the annealing temperature is well above the isotropic transition temperature of the introduced perfluoroalkyl side chain with liquid crystalline behavior. In this situation, as the liquid crystalline ordering of perfluoroalkyl side chains becomes negligible, the modified SCFT could be used to predict the self-assembled morphology of rod–coil BCPs in their thermodynamically stable states.
In this study, we employed a molecular design of a rod–coil diblock copolymer including PS segments against polysiloxane backbone segments with pendent perfluoroalkyl side chains, in order to develop BCP materials with high χ for next-generation lithographic applications. This paper discusses estimation of the χ parameter and related self-assembled morphologies, both before and after the introduction of perfluoroalkyl side chains into the backbone of BCPs. In addition, the experimentally observed self-assembled morphologies, especially the observed anisotropic hexagonal packing of the cylinders, were studied using SCFT. Since the anisotropic array predicted from SCFT is highly dependent on the assumption of rods, possible effects of the scale of the rods on the morphology were further revealed by SCFT calculations (Scheme 1).
 | ||
| Scheme 1 Concept for designing high-χ parameter coil–rod BCPs via introducing perfluoroalkyl side chains and the resulting self-assembled morphology in an anisotropic hexagonal packing array. | ||
Experimental section
Methods
1H NMR spectra were recorded with a JEOL JNM-ECS400 (400 MHz) instrument using chloroform-d as the solvent. Gel permeation chromatography (GPC) analysis was performed on a Shodex GPC-101 instrument equipped with Shodex LF804 columns. The number average molecular weights (Mn) and the molecular weight distributions (Mw/Mn) were determined by GPC (Showa-Denko GPC-101) with polymer/tetrahydrofuran solution at a flow rate of 1.0 mL min−1 at 40 °C and calibrated with polystyrene. The thermal behaviors of the polymers were estimated by differential scanning calorimetry (DSC; Seiko DSC 7020) at a heating rate of 10 °C min−1 under a nitrogen flow. The scanning temperature range was set from −30 °C to 200 °C. The transition temperatures were determined from the second heating and cooling scans. Small angle X-ray scattering (SAXS) measurements were carried out using a Bruker SAXS NanoSTAR instrument (output: 50 kV, 50 mA). After monochromatic processing by a Göbel mirror, focused CuKα radiation (wavelength: 1.5416 Å) was applied to the sample. The scattered X-rays were collected by a 2D-PSPC detector to determine the morphologies in the bulk sample. Bright-field transmission electron microscope (TEM) images of the sample structure were also obtained using a Hitachi H7650 Zero A instrument under an accelerating voltage of 100 kV. For TEM analysis, the bulk samples were first pasted onto epoxy resin for handling, and then microtomed (Reichert-Jung Ultracut E) with a DiATOME diamond knife at room temperature to a preset thickness of 70 nm. The produced sections were then placed onto TEM grids and stained by ruthenium oxide for observation. The value of δ was estimated from the structural formula of the material,44 and the density and volume fraction of the fluorinated block could thus be calculated.Materials
Styrene was washed with 0.1 M sodium hydroxide (NaOH) aqueous solution three times and dried with magnesium sulfide (MgSO4). Calcium hydride (CaH2) was then added and stirred overnight before distillation. After distillation, di-n-butylmagnesium (MgBu2) was added under Ar protection to remove any residual water in the styrene. A trap-to-trap process was further carried out for degassing, using liquid nitrogen as the freezing source. Similarly, 1,3,5-trimethyl-1,3,5-trivinylcyclotrisiloxane (D3v) was distilled over CaH2 and subsequently degassed under argon. Chlorotrimethylsilane (TMSiCl) was degassed under argon. All other materials were reagent grade (purchased from Kanto Chemical Co. Inc., Tokyo Chemical Industry, FUJIFILM Wako Pure Chemical Corporation, and Sigma-Aldrich) and used as received. The detailed synthetic procedures of the homopolymers are described in ESI.†Synthetic procedures
1H NMR (CDCl3, 400 MHz) δ (ppm): 0.14 (s, 3H, CH3), 1.42 (br, 2H, CH2), 1.84 (br, 1H, CH), 5.76–6.04 (br, 3H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 6.31–6.72 (br, 2H, Ar), 6.89–7.22 (br, 3H, Ar).
CH2), 6.31–6.72 (br, 2H, Ar), 6.89–7.22 (br, 3H, Ar).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v), depending on the Mn of the BCP) and repeated twice. The white powder was dried in vacuo under 40 °C overnight to yield the desired product.
:3 v/v), depending on the Mn of the BCP) and repeated twice. The white powder was dried in vacuo under 40 °C overnight to yield the desired product.1H NMR (CDCl3, 400 MHz) δ (ppm): 0.15 (s, 3H, CH3 PSi8F), 0.90 (br, 2H, Si–CH2), 1.41 (br, 2H, CH2 PS), 1.82 (br, 1H, CH PS), 2.29 (t, 2H, Si–CH2–S–CH2 PSi8F), 2.59 (t, 2H, CF2–CH2 PSi8F), 2.69 (t, 2H, CF2–CH2–CH2 PSi8F), 6.31–6.72 (br, 2H, Ar), 6.89–7.22 (br, 3H, Ar).
Estimating the surface free energies of homopolymers Homo-PSi8F
The SFE of the homopolymer Homo-PSi8F was calculated by measuring the water and diiodomethane contact angles (C.A.) on thin polymer films. Because of the poor solubility of Homo-PSi8F in common organic solvents, the thin films were prepared by uniformly placing approximately 20 mg of homopolymer powder on a 1 cm × 1 cm bare silicon wafer, followed by melting at 180 °C for 24 h under vacuum.Bulk sample preparation
The bulk samples used for investigating the bulk morphology and domain spacing (dspacing) in the microphase-separated structures were prepared by slowly evaporating a dilute THF solution of BCP at 30 °C, after filtering through a PTFE membrane syringe filter (0.25 μm pore size). The as-prepared samples were dried under reduced pressure before annealing at 160 °C for 24 h.Morphology prediction by SCFT
Briefly, an incompressible melt of AB linear diblock polymers is placed in a simulation volume V. Each polymer with volume fractions of fA and fB and a degree of polymerization N is modeled as a Gaussian chain characterized by random-walk statistics. The local interactions are governed by the estimated binary interaction parameter χAB = 0.169 at 160 °C, along with the incompressibility condition of fA + fB = 1. The code for performing SCFT was taken from our published paper45 and other literature.46–48The simulated polymers were assumed to be monodispersed with temperature-independent densities. The statistical segment lengths of PS and PSi8F were set at a = 1 and b = 2.3 units, respectively, according to estimation based on the random-walk model (in which the end-to-end distance is governed by the root square value of the molecular weight of each segment). The statistical segment lengths of PSi8F were also set to other arbitrary units to mimic rods of different rigidities. A typical grid size of 64 × 64 was used for the 2D SCFT calculations with 100 contour steps in inner loops. A simulation box of size L × L with L = 16 Rg with periodic boundary conditions and a spatial resolution of 0.25 Rg was used for all simulations. The simulations were performed using the pseudo-spectral method developed by Rasmussen and Kalosakas, starting from random initial conditions followed by Anderson acceleration once a stable morphology could be identified (typically when the change in the free energy ≤1 × 10−10).46,49 Calculations were performed on a broadly accessible Intel Core i7 CPU 10750H 2.60 GHz.
Results and discussion
Synthesis of PS-b-PMVS
Synthesis of the targeted diblock copolymer could be divided into two steps: synthesis of the precursor main chain polymer PS-b-PMVS via sequential anionic living polymerization, and the subsequent post-functionalization to introduce side chains.The synthesis routine for the targeted BCP is illustrated in Scheme 2. All the precursor BCPs (PS-b-PMVS with distinctive molecular weights) were synthesized via sequential anionic polymerization of styrene and 1,3,5-trimethyl-1,3,5-trivinyl cyclotrisiloxane. As shown in Fig. 1, the appearance of characteristic proton signals from the two blocks indicated the successful synthesis of PS-b-PMVS. The composition of vinylsiloxane was kept at low levels because of the bulky side chain introduced in the next step.
 | ||
| Scheme 2 Synthetic scheme of PS-b-PMVS precursor via sequential living anionic polymerization of styrene and cyclic siloxane-based monomer, and post-functionalization with thiols. | ||
 | ||
| Fig. 1 Representative 1H NMR spectra of (a) PS-b-PMVS (SV13.6) and (b) PS-b-PSi8F (SF20.8) in CDCl3 solvent. | ||
Synthesis of PS-b-PSi8F
After synthesis of the BCP precursors, the vinyl groups in the siloxane unit were chemically modified with 1H,1H,2H,2H-perfluorodecanethiol. In Fig. 1b, the disappearance of proton signals from the vinyl groups and the emergence of those from the introduced side chain indicated complete conversion in the modification reaction. The 1H NMR signals are well assigned to each proton in the targeted product without any characteristic signals from residual thiols. Besides, the assigned peak integration is highly proportional to the number of protons in the chemical structure (Fig. S2(b)†), indicating the high purity of the products. Following the thiol-ene modification reaction, the trace of SEC was shifted to the higher Mn direction, together with a near constant narrow dispersity (Fig. S3†). The determined Mn value of PS-b-PSi8F from SEC was in good agreement with the estimated Mn based on the precursor PS-b-PMVS.Bulk morphology
To determine the suitable thermal annealing temperature for inducing self-assembly, DSC characterization was carried out first. The results (Fig. S4†) suggest an obvious glass transition attributed to the PS segments near 72–100 °C, depending on the Mn of the PS block in PS-b-PSi8F. A transition from an ordered liquid crystal phase to an isotropic phase was detected at 50 °C or a lower temperature proportional to the molecular weight of PS-b-PSi8F. Compared to the reported isotropic transition of similar fluorinated side chains attached to a poly-(methyl acrylate)-based50 or poly-(isoprene)-based backbone,18 that of the perfluoroalkyl side chain attached to a siloxane-based backbone is significantly lower because of the intrinsically flexible backbone. The annealing temperature for the bulk sample in this study was set at 160 °C, which far exceeds the isotropic transition temperature. Therefore, the interplay between liquid crystal nano-ordering and self-assembly under thermal annealing should be negligible. Afterwards, the bulk morphologies of the synthesized BCPs were characterized by SAXS, as shown in Fig. 2. | ||
| Fig. 2 SAXS profiles of the synthesized (a) precursor PS-b-PMVS, (b) chemically modified PS-b-PSi8F bulk samples following thermal annealing at 160 °C for 24 h, and (c) collected SAXS scattering signals from the SF7.2 annealed bulk sample. | ||
The formation of a liquid crystal nanostructure from the side chain below the isotropic transition temperature was evidenced by the SAXS profiles (Fig. S5†) of the as-prepared SF20.8 bulk sample evaporated from THF solution at 30 °C, which is below the corresponding isotropic transition temperature detected from thermal analysis. A significant scattering peak at q of 2.0 nm−1 indicates a liquid crystal ordering of 3.14 nm, which is highly consistent with the reported self-assembly scale from similar perfluoroalkyl side chains in the literature.18,50 It is also worth noting that compared to the reported 0.74 nm Gaussian segment length of polystyrene,51 the segment length of a single introduced perfluoroalkyl side chain was estimated to be half of the liquid crystal ordering, i.e., 1.6 nm considering the reported head-to-head regularity of the smectic phase consisting of perfluoroalkyl side chains.18 Therefore, the segment length of PSi8F is estimated to be more than double that of the PS segment, indicating the non-negligible rigidity of the PSi8F segment compared to the PS segment. In comparison, no liquid crystalline ordering was observed in the self-assembled nanostructure of as-prepared SF6.0 or SF7.2 with lower Mn (Fig. S5†), because their isotropic transition temperatures were all below 10 °C according to the DSC analysis (Fig. S4†). Therefore no significant liquid crystal ordering could proceed during solvent evaporation induced self-assembly at 30 °C.
Following thermal annealing, all chemically modified PS-b-PSi8Fs exhibited multiplied scattering peaks, unlike the disordered morphologies of the PS-b-PMVS precursors, indicating the formation of an ordered self-assembly nanostructure. The domain spacing in the formed nanostructure was determined by the position of the primary scattering peak, and the results are summarized in Table 1. The SAXS profiles of SF20.8 reveal a self-assembly morphology of a hexagonally packed cylinder, as a significant scattering peak at  was observed. The self-assembled hexagonally packed cylinder domains were also evidenced by TEM observation (Fig. S6†). Considering the near symmetric volume fraction of counter-blocks and strong segregation driving force (χN) of SF20.8, a self-assembled lamellar morphology should be predicted based on a flexible coil–coil model. This mismatch between experimental observation and theoretical prediction could be attributed to the rigid perfluoroalkyl side chains, which differ from flexible PS segments.
was observed. The self-assembled hexagonally packed cylinder domains were also evidenced by TEM observation (Fig. S6†). Considering the near symmetric volume fraction of counter-blocks and strong segregation driving force (χN) of SF20.8, a self-assembled lamellar morphology should be predicted based on a flexible coil–coil model. This mismatch between experimental observation and theoretical prediction could be attributed to the rigid perfluoroalkyl side chains, which differ from flexible PS segments.
| BCPs | Labela | Mnb (kg mol−1) | Đ | wPSc | fPSd | dspacinge (nm) | Morphologyf |
|---|---|---|---|---|---|---|---|
| a The labels SV and SF refer to PS-b-PMVS and PS-b-PSi8F, respectively, while the following number refers to the number-average molecular weight (Mn) of the polymer.b Mn and dispersity (Đ) were obtained with SEC using THF as the eluent based on PS standards.c PS weight fractions (wPS) of PS-b-PMVS and of PS-b-PSi8F were calculated from 1H NMR data.d PS volume fractions (fPS) were calculated using the bulk densities for each block (1.05 g cm−3 for PS, 1.01 g cm−3 for PMVS, and 1.65 g cm−3 for PSi8F).e The domain spacings (dspacing) were estimated from the position of the first-order scattering peak in the SAXS profile upon thermal annealing at 160 °C for 24 h.f The morphologies in the bulk were determined by SAXS and TEM. | |||||||
| PS-b-PMVS | SV13.6 | 13.6 | 1.22 | 0.83 | 0.82 | 16.1 | Disordered |
| SV4.4 | 4.4 | 1.29 | 0.86 | 0.86 | — | Disordered | |
| SV3.6 | 3.6 | 1.20 | 0.81 | 0.80 | — | Disordered | |
| SV4.0 | 4.0 | 1.35 | 0.93 | 0.93 | — | Disordered | |
| SV6.7 | 6.7 | 1.14 | 0.56 | 0.55 | 10.0 | Disordered | |
| PS-b-PSi8F | SF20.8 | 20.8 | 1.18 | 0.43 | 0.54 | 26.6 | HEX |
| SF7.2 | 7.2 | 1.17 | 0.49 | 0.60 | 12.0/10.7 | Aniso-HEX | |
| SF6.0 | 6.0 | 1.16 | 0.39 | 0.50 | 10.7 | HEX | |
| SF5.8 | 5.8 | 1.28 | 0.62 | 0.72 | 10.1 | LAM | |
When the Mn of BCP was further reduced from 20k to 6k Da, the self-assembled morphology revealed cylinders in isotropic/anisotropic hexagonal packing arrays, depending on the specific volume fraction of the blocks in each BCP. In contrast, all the PS-b-PMVS precursors exhibited disordered morphologies, indicating a significant increase in the χ parameter after chemical modification. Chemically modified SF6.0 and SF7.2 exhibited multiple scattering peaks, compared to the sole primary scattering peak of SV6.7 with a similar molecular weight. Generally, the periodicity of a self-assembly structure is revealed by the number of scattering peaks. Therefore, the different number of scattering peaks also indicated the enhanced χ parameter between blocks after chemical modification.
It is worth noting that two distinctive primary scattering peaks were observed in the SAXS profiles of the SF7.2 annealed bulk sample (Fig. 2b and c), indicating anisotropy in the self-assembled periodic structure. Besides, TEM observation also suggested that SF7.2 seemingly revealed a structure of anisotropic hexagonally packed cylinders (Aniso-HEX), which is different from the conventional isotropic hexagonally packed cylinders (HEX) observed in SF20.8 and SF6.0. The TEM image in Fig. 3 serves as direct evidence for the formation of an anisotropic hexagonal packing array.
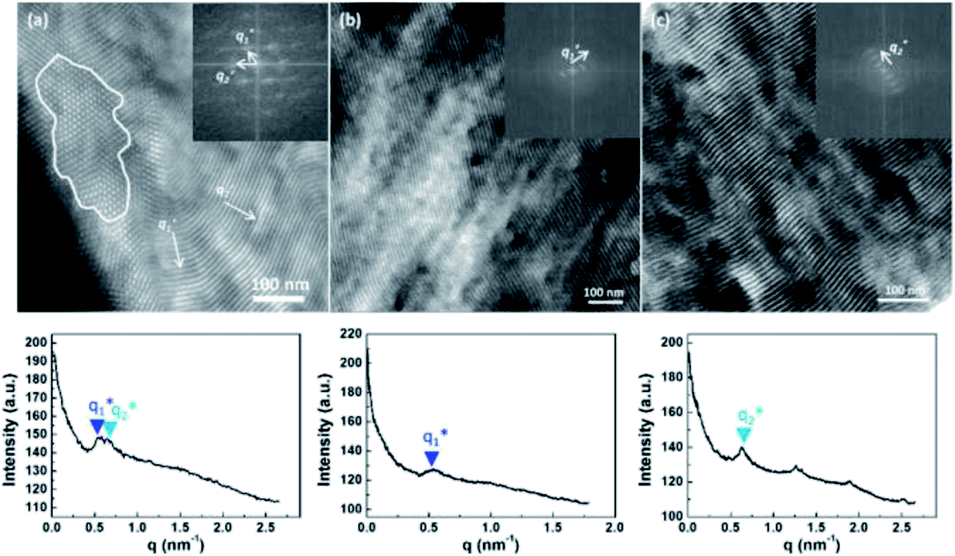 | ||
Fig. 3 Top row: TEM micrographs showing the domains corresponding to (a) the top view of PSi8F cylinders in anisotropic hexagonal packing and (b and c) the striations of two distinctive dimensions at  and and  corresponding to the side view in the annealed SF7.2 bulk sample. The inset displays the fast Fourier-transform (FFT) pattern of the real-space image (a) within the indicated white circle or (b and c) the entire image. Bottom row: integrated signals along the radius to the center of the FFT image. Two distinctive spacings could thus be estimated: d1 = 11.8 nm corresponding to the side view in the annealed SF7.2 bulk sample. The inset displays the fast Fourier-transform (FFT) pattern of the real-space image (a) within the indicated white circle or (b and c) the entire image. Bottom row: integrated signals along the radius to the center of the FFT image. Two distinctive spacings could thus be estimated: d1 = 11.8 nm  and d2 = 10.0 nm and d2 = 10.0 nm 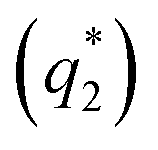 , which correspond to two center-to-center distances , which correspond to two center-to-center distances  d1 = 13.6 nm and d1 = 13.6 nm and  d2 = 11.5 nm. The PS and PSi8F domains appear as dark and bright areas, respectively, due to selective ruthenium tetraoxide staining of the PS segments. d2 = 11.5 nm. The PS and PSi8F domains appear as dark and bright areas, respectively, due to selective ruthenium tetraoxide staining of the PS segments. | ||
Two distinctive periodic spacings at an angle of approximately 60° to each other were indicated from the FFT analysis. Moreover, the split primary scattering peaks at  (d1 = 12.0 nm) and
(d1 = 12.0 nm) and  (d2 = 10.7 nm) in the SAXS profiles are in good agreement with the two estimated spacings
(d2 = 10.7 nm) in the SAXS profiles are in good agreement with the two estimated spacings  (d1 = 11.8 nm) and
(d1 = 11.8 nm) and  (d2 = 10.0 nm) extracted by FFT analysis from the TEM images of the anisotropic packing array. This anisotropic hexagonal packing array was thought to be caused by the mismatch between certain block volume fractions and rigid perfluoroalkyl side chains, which failed to form the isotropic packing array with incompressible chains. Owing to a similar side chain effect, a coil–comb diblock copolymer capable of forming a tetragonally packed cylinder structure was previously reported.52 In contrast with SF7.2 and SF6.0, SF5.8 with a relatively more asymmetric volume fraction (fPS = 0.72) revealed a lamellar morphology with 10.1 nm domain spacing, which indicates a potential narrow half-pitch of 5 nm in width.
(d2 = 10.0 nm) extracted by FFT analysis from the TEM images of the anisotropic packing array. This anisotropic hexagonal packing array was thought to be caused by the mismatch between certain block volume fractions and rigid perfluoroalkyl side chains, which failed to form the isotropic packing array with incompressible chains. Owing to a similar side chain effect, a coil–comb diblock copolymer capable of forming a tetragonally packed cylinder structure was previously reported.52 In contrast with SF7.2 and SF6.0, SF5.8 with a relatively more asymmetric volume fraction (fPS = 0.72) revealed a lamellar morphology with 10.1 nm domain spacing, which indicates a potential narrow half-pitch of 5 nm in width.
Estimation of χeff
The strength of the segregation before and after chemical modification of the fluorinated side chain was quantified using the random-phase approximation method,13,53–55 in terms of the effective Flory–Huggins interaction parameter expressed by χeff = A/T + B, where A/T is the enthalpic contribution, and B is the entropic contribution. Firstly, temperature-dependent SAXS analyses were conducted on the precursor PS-b-PMVS (SV6.7) and the chemically modified PS-b-PSi8F (SF5.8), from 240 °C to 160 °C or from 290 °C to 210 °C in 10 °C decrements (Fig. S7 and S8†). The SAXS profiles of SF5.8 showed a continuous linear change in the Imax−1 vs. T−1 plot between 290 and 240 °C, and a breakthrough point between 240 and 230 °C, as the temperature range of 290–240 °C is beyond the BCP order-to-disorder transition temperatures (ODTs) (Fig. S8(b)†). Therefore, the SAXS profiles in the mean-field disordered state were then analyzed, based on Leibler's mean-field theory and modified to include the effects from the molecular weight dispersity and asymmetry in the segmental volume. Remarkably, since χeff depends on the reference volume used to calculate N, a common reference volume (v0 = 118 Å3) was utilized to compare the χeff with other reported values. For the PS-b-PMVS precursor, χeff was estimated from χeff = 1.12/T + 0.0924 (Table 2). A weak temperature dependence was observed for χPS/PDMS, possibly because of geometrical packing constraints on the differently shaped monomers of the two species56 and the chain polarization/stretching effects near ODTs,57 which have been observed in other systems with a large entropic contribution to χeff.45 The estimated χeff value of PS-b-PMVS at 160–240 °C was consistent with the previously reported value for PS-b-PDMS (0.108 at 160 °C).58,59 The slightly lower χeff value of the former (PS-b-PMVS) was possibly caused by a lower silicon composition in the siloxane-based segments, which reduces the incompatibility with organic PS segments. According to Leibler's theory,32 the driving force for self-assembly is indicated by the magnitude of χN. Based on the estimation results, SV6.7 has χeffN = 8.6, which falls outside the boundary condition for self-assembly of a symmetric diblock copolymer (χeffN ≥ 10.5). In contrast, owing to the enhanced χ parameter after chemical modification, SF6.0 and SF7.2 of similar N values, and even SF5.8 with a lower N value have χeffN ≥ 10.5, which enables the micro-phase separation to proceed. The estimated χeff from chemically modified SF5.8 is expressed by χeff = 2.56/T + 0.163. Compared to the precursor PS-b-PMVS, the introduced long fluorinated side chain obviously contributed to a higher χeff (0.169 at 160 °C), which is almost twice that of the precursor BCPs prior to chemical modification. The super-hydrophobic fluorinated side chain increases the hydrophobicity of the siloxane segments, as evidenced by the measured SFE value of 5.9 mJ m−2 for Homo-PSi8F, which is extremely low compared to the reported value of 20.4 mJ m−2 of polydimethylsiloxane at room temperature.60 Moreover, the reported SFE of counter-block polystyrene is 40.7 mJ m−2.61 Therefore, the increased difference between the SFEs of counter-blocks leads to enhanced segregation, which is further revealed by a higher χeff parameter.| BCP | Label | A/T | B |
|---|---|---|---|
| PS-b-PMVS | SV6.7 | 1.12/T | 0.0924 |
| PS-b-PSi8F | SF5.8 | 2.56/T | 0.163 |
Morphological predictions by SCFT
SCFT calculation based on two-dimensional models failed to precisely predict the self-assembled morphology of SF20.8, as a less ordered morphology consisting of randomly oriented short rods and dots was preserved in the evolution of in situ self-assembly (Fig. 4a and S10†). With certain volume fractions (fPS), the extremely high driving force for self-assembly (χN) leads to the instant formation of discrete aggregated domains. And the morphology even remains trapped in this metastable state following an extended annealing time, as the high energy barrier separating the metastable and stable states prevents the formation of an ultimately stable structure in SCFT prediction. In fact, the following predicted 2-D morphological phase diagram revealed that SF20.8 falls into the transition boundary between lamellar and HEX morphologies.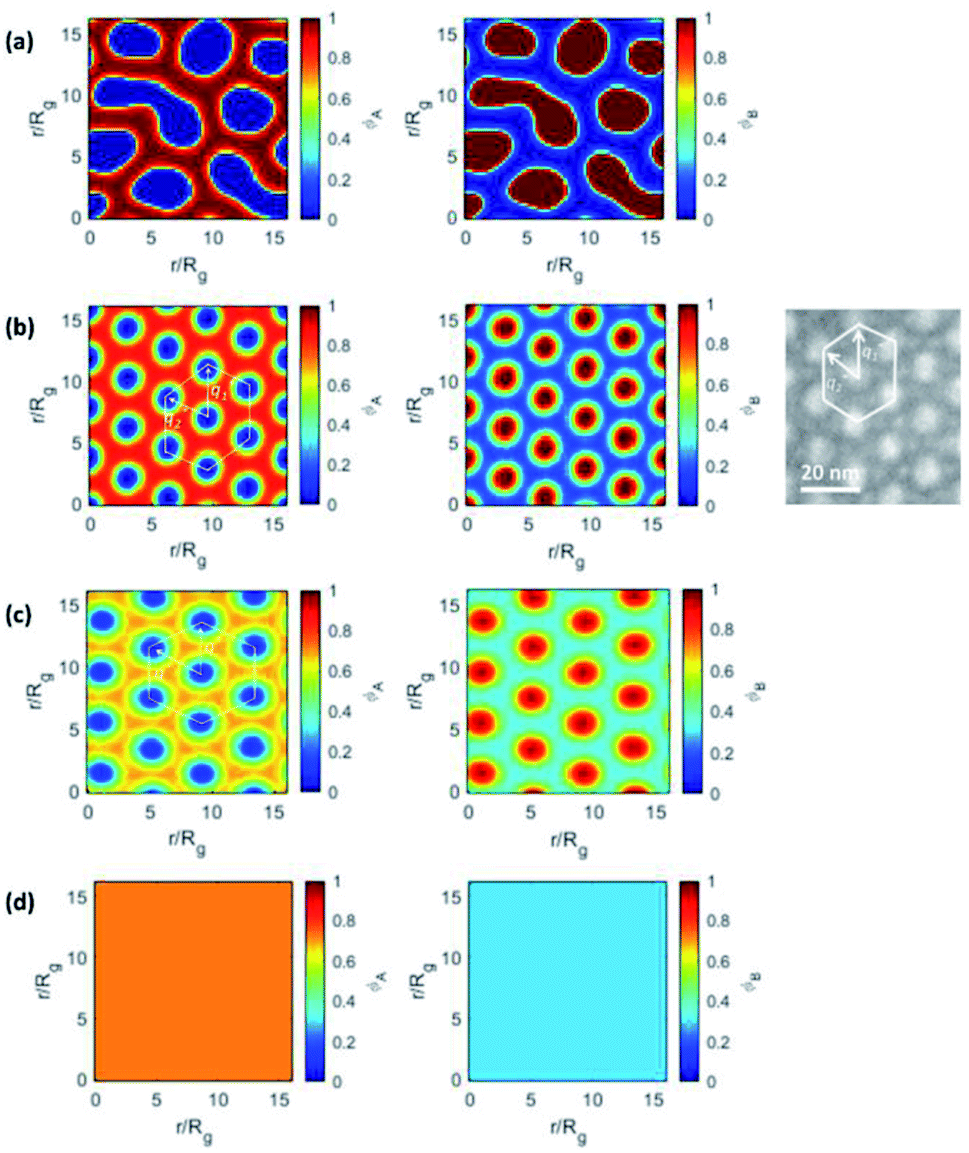 | ||
| Fig. 4 Theoretically predicted 2-D morphologies by SCFT for (a) SF20.8, (b) SF7.2, (c) SF6.0, and (d) SF5.8 by assuming different segment lengths (a = 1, b = 2.3). The red and blue colors indicate PS and PSi8F segments in the left-hand column, and inverted in the right-hand column. The density gradient bar for different segments is given next to each image. In (b), the experimental TEM image of SF7.2 is presented on the right for comparison. The two distinct periodic distances are also marked in both simulated SCFT and experimental TEM results. | ||
As χN becomes lower, SF7.2 and SF6.0 of similar symmetric block volume fractions begin to reveal hexagonally packed cylindrical morphologies. The experimentally observed isotropy and anisotropy in the hexagonal packing array of SF6.0 and SF7.2 are precisely predicted by SCFT. The anisotropic packing array leads to at least two distinct center-to-center periodic distances between adjacent PSi8F cylinders, which match the TEM and SAXS characterization results. The two distinct center-to-center distances from SCFT prediction are d1 = 1.14 units and d2 = 1.00 units, and their ratio is highly consistent with the experimental observation (d1 = 1.12 d2).
In contrast, a typical isotropic hexagonal packing array is suggested for SF6.0, consistent with the experimental observation. This is possibly caused by the difference in block volume fractions. As the molecular weight of PS-b-PSi8F is further reduced to 5.8k Da, SCFT suggests a disordered state at the annealing temperature of 160 °C, in which two different segments are homogeneously mixed. The inconsistency between SCFT and random phase approximation results, which suggests an ODT between 230 °C and 240 °C for SF5.8, originates from the different assumptions regarding the effects of BCP dispersity on the self-assembly.
In addition, SCFT morphological prediction based on equal segment lengths was performed to mimic the flexible coil–coil model. As shown in Fig. S9,† isotropic hexagonal packing of cylinders and lamellae are predicted for SF7.2 and SF6.0, which are obviously inconsistent with the experimental observation. The results indicated that the flexible coil–coil model is not suitable for the studied BCP.
After verifying the ratio of segment lengths, a phase diagram could be further calculated using SCFT for the investigated PS-b-PSi8F (Fig. 5). A minimal molecular weight of 6k Da with a symmetric volume fraction (χeffN = 11.0) was predicted to almost approximate the limit for inducing self-assembly. Upon increasing the volume fraction fPS, there is a transition in morphology from hexagonally packed PS cylinders to lamellae. No axial symmetry was observed at fPS = 0.5 among the predicted morphologies in the left or right halves of the diagram, which is obviously attributed to the assumption of significantly different segment lengths for each block representing rigidity.36,38 Remarkably, the anisotropic hexagonal packing array was only predicted in the vicinity of fPS = 0.6 with relatively low Mn, which correlates with the reported anisotropic fluctuations of hexagonally packed cylinders that mostly occurred in weakly segregated states.62,63
 | ||
| Fig. 5 Theoretically predicted 2-D morphological phase diagram by SCFT using fPS = 0.1/0.2/0.3/0.4/0.5/0.6/0.7/0.8/0.9 under PS-b-PSi8F (Mn = 6000/8000/10000/14000 Da), assuming different segment lengths (a = 1, b = 2.3). A homogenous color inside the square indicates a disordered morphology. The dashed curve is a visual aid to indicate the boundary between ordered and disordered regions. The red and blue colors indicate PS and PSi8F segments, respectively. The density gradient bar for the two different segments is given next to the phase diagram. | ||
To further investigate the effects of varied segment lengths on the self-assembled morphology, the segment length of PSi8F was tuned to 0.5, 0.75, 1.0, 1.5, 2, 3, 4, and 5 times that of the PS segment, as shown in Fig. 6. As the segment length of PSi8F gradually increased, the morphology changed from lamellae to hybrid and then to hexagonally packed cylinders. The anisotropy of the hexagonal packing array becomes more significant when the segment length of PSi8F is more than 1.5 times that of PS segments. The anisotropy is most obvious when this segment length ratio was between 2 and 3, which is consistent with the experimentally and theoretically estimated ratio of 2.3 in this study. However, as the segment length was further increased to more than 3, the anisotropy of the hexagonal packing array seemed to diminish again. These transitions clearly indicate the dependence of the self-assembled morphology on the segment lengths at given block volume fractions and molecular weights.
 | ||
| Fig. 6 Theoretically predicted 2-D morphologies by SCFT for SF7.2, assuming a fixed segment length of a = 1 and varied b: (a) b = 0.5, (b) b = 0.75, (c) b = 1, (d) b = 1.5, (e) b = 2, (f) b = 3, (g) b = 4, and (h) b = 5. The red and blue colors indicate PS and PSi8F segments in the left-hand column, and they are inverted in the right-hand column. The density gradient bar for different segments is given next to each image. | ||
Conclusions
1H,1H,2H,2H-perfluorodecanethiol was introduced into the backbone of Si-containing polystyrene-block-poly(methylvinylsiloxane) via a facile thiol-ene reaction. The super-hydrophobic long fluorinated alkyl chain is meant to boost the segregation between counter-blocks. The estimated χ parameter after modification was determined to be χeff = 2.56/T + 0.163, which is 80% higher than that of the precursor BCP (PS-b-PMVS) at an annealing temperature of 160 °C. Owing to the high rigidity of the introduced perfluoroalkyl side chain, the synthesized coil–rod diblock copolymer could self-assemble into anisotropic hexagonally packed cylinders with two distinct center-to-center distances. The experimental observations were further verified by morphological prediction from self-consistent field theory (SCFT) assuming a coil–rod model. A phase diagram for the investigated coil–rod diblock copolymer was generated using SCFT, and the boundaries between different self-assembled phases were clearly revealed. Additionally, SCFT was used to study the effects of different relative segment lengths, which correspond to varied rigidity or stiffness of the rod segment, on the self-assembled morphologies. A gradual morphological transition from lamellae to anisotropic/isotropic hexagonally packed cylinders was revealed. This paper reports the first experimental observation of the predicted anisotropy in the self-assembly morphology of diblock copolymers, which adds to a fundamental understanding of the unique self-assembly behavior in coil–rod diblock copolymers. Furthermore, by chemically tailoring the BCP to minimize the scale of self-assembly domains, the developed PS-b-PSi8F polymers with a high χ parameter are promising candidates for resists in next-generation BCP lithography.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Ryohei Kikuchi of the Tokyo Institute of Technology, Ookayama Advanced Materials Division for support with TEM measurements. The authors acknowledge the financial support of 2021 Jiangsu Province Shuangchuang (Innovation and Entrepreneurship) Talent Program (No. JSSCBS20210067) and State Key Laboratory of High Performance Civil Engineering Materials Open Fund (No. 2021CEM002).References
- B. L. Zhang, W. C. Liu, L. K. Meng, Z. P. Zhang, L. B. Zhang, X. Wu, J. Y. Dai, G. P. Mao and Y. Y. Wei, RSC Adv., 2019, 9, 3828–3837 RSC.
- M. J. Kim, W. I. Park, Y. J. Choi, Y. K. Jung and K. H. Kim, RSC Adv., 2016, 6, 21105–21110 RSC.
- C. M. Bates, M. J. Maher, D. W. Janes, C. J. Ellison and C. G. Willson, Macromolecules, 2014, 47, 2–12 CrossRef CAS.
- S. J. Jeong, J. Y. Kim, B. H. Kim, H. S. Moon and S. O. Kim, Mater. Today, 2013, 16, 468–476 CrossRef CAS.
- M. P. Stoykovich and P. F. Nealey, Mater. Today, 2006, 9, 20–29 CrossRef CAS.
- D. F. Sunday, M. J. Maher, A. F. Hannon, C. D. Liman, S. Tein, G. Blachut, Y. Asano, C. J. Ellison, C. G. Willson and R. J. Kline, Macromolecules, 2018, 51, 173–180 CrossRef CAS PubMed.
- Y. S. Jung and C. A. Ross, Nano Lett., 2007, 7, 2046–2050 CrossRef CAS PubMed.
- J. G. Son, K. W. Gotrik and C. A. Ross, ACS Macro Lett., 2012, 1, 1279–1284 CrossRef CAS PubMed.
- L. Wan, R. Ruiz, H. Gao, K. C. Patel and T. R. Albrecht, ACS Nano, 2015, 9, 7506–7514 CrossRef CAS PubMed.
- Y. D. Luo, D. Montarnal, S. Kirn, W. C. Shi, K. P. Barteau, C. W. Pester, P. D. Hustad, M. D. Christianson, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Macromolecules, 2015, 48, 3422–3430 CrossRef CAS.
- Y. H. Hur, S. W. Song, J. M. Kim, W. I. Park, K. H. Kim, Y. Kim and Y. S. Jung, Adv. Funct. Mater., 2018, 28, 1800765 CrossRef.
- J. D. Cushen, C. M. Bates, E. L. Rausch, L. M. Dean, S. X. Zhou, C. G. Willson and C. J. Ellison, Macromolecules, 2012, 45, 8722–8728 CrossRef CAS.
- K. Azuma, J. Sung, Y. Choo, Y. Rokhlenko, J. H. Dwyer, B. Schweitzer, T. Hayakawa, C. O. Osuji and P. Gopalan, Macromolecules, 2018, 51, 6460–6467 CrossRef CAS.
- T. Seshimo, R. Maeda, R. Odashima, Y. Takenaka, D. Kawana, K. Ohmori and T. Hayakawa, Sci. Rep., 2016, 6, 19481 CrossRef CAS PubMed.
- S. Yamazaki, R. Odashima, T. Seshimo and T. Hayakawa, J. Photopolym. Sci. Tec., 2017, 30, 191–196 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- S. Perutz, J. Wang, E. J. Kramer, C. K. Ober and K. Ellis, Macromolecules, 1998, 31, 4272–4276 CrossRef CAS.
- J. G. Wang, G. P. Mao, C. K. Ober and E. J. Kramer, Macromolecules, 1997, 30, 1906–1914 CrossRef CAS.
- T. Nishino, Y. Urushihara, M. Meguro and K. Nakamae, J. Colloid Interface Sci., 2005, 283, 533–538 CrossRef CAS PubMed.
- D. K. Owens and R. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- M. Bertolucci, G. Galli and E. Chiellini, Macromolecules, 2004, 37, 3666–3672 CrossRef CAS.
- Y.-S. Kim, J.-S. Lee, Q. Ji and J. E. McGrath, Polymer, 2002, 43, 7161–7170 CrossRef CAS.
- X. M. Li, J. Li, C. X. Wang, Y. Y. Liu and H. Deng, J. Mater. Chem. C, 2019, 7, 2535–2540 RSC.
- C. X. Wang, X. M. Li and H. Deng, ACS Macro Lett., 2019, 8, 368–373 CrossRef CAS PubMed.
- S. H. Li, Y. Li, C. A. Wisner, L. Jin, N. Leventis and Z. H. Peng, RSC Adv., 2014, 4, 35823–35832 RSC.
- A. Yassar, L. Miozzo, R. Gironda and G. Horowitz, Prog. Polym. Sci., 2013, 38, 791–844 CrossRef CAS.
- J. K. Liu, X. G. Zhu, J. L. Li, J. L. Shen and G. L. Tu, RSC Adv., 2016, 6, 61934–61943 RSC.
- D. Zhou, J. L. Liu, L. Chen, H. T. Xu, X. F. Cheng, F. Y. Wu and Y. W. Chen, RSC Adv., 2017, 7, 24345–24352 RSC.
- J. Zhang, X.-F. Chen, H.-B. Wei and X.-H. Wan, Chem. Soc. Rev., 2013, 42, 9127–9154 RSC.
- M. Lee, B.-K. Cho and W.-C. Zin, Chem. Rev., 2001, 101, 3869–3892 CrossRef CAS PubMed.
- M. W. Matsen and C. Barrett, J. Chem. Phys., 1998, 109, 4108–4118 CrossRef CAS.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- L.-Y. Shi, S. Lee, L.-C. Cheng, H. Huang, F. Liao, R. Ran, K. G. Yager and C. A. Ross, Macromolecules, 2019, 52, 679–689 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Ann. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- C. Y. Ryu and T. P. Lodge, Macromolecules, 1999, 32, 7190–7201 CrossRef CAS.
- V. Pryamitsyn and V. Ganesan, J. Chem. Phys., 2004, 120, 5824–5838 CrossRef CAS PubMed.
- J. Gao, P. Tang and Y. L. Yang, Soft Matter, 2013, 9, 69–81 RSC.
- N. A. Kumar and V. Ganesan, J. Chem. Phys., 2012, 136, 101101 CrossRef.
- J. Mu, H. Okamoto, T. Yanai, S. Takenaka and X. Feng, Colloids Surf., A, 2001, 181, 303–313 CrossRef CAS.
- H. Okamoto, N. Yamada and S. Takenaka, J. Fluorine Chem., 1998, 91, 125–132 CrossRef CAS.
- T. Doi, Y. Sakurai, A. Tamatani, S. Takenaka, S. Kusabayashi, Y. Nishihata and H. Terauchi, J. Mater. Chem., 1991, 1, 169–173 RSC.
- U. Cengiz, N. A. Gengec, N. U. Kaya, H. Y. Erbil and A. S. Sarac, J. Fluorine Chem., 2011, 132, 348–355 CrossRef CAS.
- H. Ghassemi, D. A. Schiraldi, T. A. Zawodzinski and S. Hamrock, Macromol. Chem. Phys., 2011, 212, 673–678 CrossRef CAS.
- R. F. Fedors, Polym. Eng. Sci., 1974, 14, 147–154 CrossRef CAS.
- K. Wylie, L. Dong, A. Chandra, Y. Nabae and T. Hayakawa, Macromolecules, 2020, 53, 1293–1301 CrossRef CAS.
- K. Ø. Rasmussen and G. Kalosakas, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 1777–1783 CrossRef CAS.
- M. W. Matsen, J. Phys.: Condens. Matter, 2001, 14, R21–R47 CrossRef.
- D. M. Ackerman, K. Delaney, G. H. Fredrickson and B. Ganapathysubramanian, J. Comput. Phys., 2017, 331, 280–296 CrossRef CAS.
- R. B. Thompson, K. O. Rasmussen and T. Lookman, J. Chem. Phys., 2004, 120, 31–34 CrossRef CAS PubMed.
- M. Al-Hussein, Y. Serero, O. Konovalov, A. Mourran, M. Moller and W. H. de Jeu, Macromolecules, 2005, 38, 9610–9616 CrossRef CAS.
- Y. Miyaki, Y. Einaga and H. Fujita, Macromolecules, 1978, 11, 1180–1186 CrossRef CAS.
- W. S. Chiang, C. H. Lin, C. L. Yeh, B. Nandan, P. N. Hsu, C. W. Lin, H. L. Chen and W. C. Chen, Macromolecules, 2009, 42, 2304–2308 CrossRef CAS.
- Y. Yoshimura, A. Chandra, Y. Nabae and T. Hayakawa, Soft Matter, 2019, 15, 3497–3506 RSC.
- N. Sakamoto and T. Hashimoto, Macromolecules, 1995, 28, 6825–6834 CrossRef CAS.
- Y. Zhao, E. Sivaniah and T. Hashimoto, Macromolecules, 2008, 41, 9948–9951 CrossRef CAS.
- K. F. Freed and J. Dudowicz, J. Chem. Phys., 1992, 97, 2105–2109 CrossRef CAS.
- W. W. Maurer, F. S. Bates, T. P. Lodge, K. Almdal, K. Mortensen and G. H. Fredrickson, J. Chem. Phys., 1998, 108, 2989–3000 CrossRef CAS.
- T. H. Andersen, S. Tougaard, N. B. Larsen, K. Almdal and I. Johannsen, J. Electron. Spectrosc., 2001, 121, 93–110 CrossRef CAS.
- J. G. Kennemur, L. Yao, F. S. Bates and M. A. Hillmyer, Macromolecules, 2014, 47, 1411–1418 CrossRef CAS.
- R. J. Roe, J. Phys. Chem., 1968, 72, 2013–2017 CrossRef CAS.
- S. Wu, J. Phys. Chem., 1970, 74, 632–638 CrossRef CAS.
- C. Yeung, A. C. Shi, J. Noolandi and R. C. Desai, Macromol. Theory Simul., 1996, 5, 291–298 CrossRef CAS.
- S. Qi and Z.-G. Wang, Polymer, 1998, 39, 4639–4648 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, SEC traces and DSC thermograms of all polymers used in this study, SAXS profiles of BCPs from solvent evaporation, TEM images of annealed bulk sample SF20.8, estimation of χ using RPA and 2D SCFT simulations of investigated BCPs (PDF). See https://doi.org/10.1039/d2ra02536a |
| This journal is © The Royal Society of Chemistry 2022 |