
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA large negative magnetoresistance effect in semiconducting crystals composed of an octahedrally ligated phthalocyanine complex with high-spin manganese(III)†
Kosuke Minea,
Masayuki Yamaguchia,
Hiroshi Murakawab,
Noriaki Hanasakib and
Masaki Matsuda *a
*a
aDepartment of Chemistry, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto, 860-8555, Japan. E-mail: masaki@kumamoto-u.ac.jp
bDepartment of Physics, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka 560-0043, Japan
First published on 17th June 2022
Abstract
A design for an octahedrally ligated phthalocyanine complex with high-spin manganese(III) (S = 2) and MnIII(Pc)Cl2 (Pc = phthalocyanine) is presented. The presence of high-spin state MnIII in the fabricated Ph4P[MnIII(Pc)Cl2]2 (Ph4P = tetraphenylphosphonium) semiconducting molecular crystal is indicated by the Mn–Cl distance, which suggests an electronic configuration of (dyz, dzx)2(dxy)1(dz2)1. This was confirmed by the Curie constant (C = 5.69 emu K mol−1), which was found to be significantly larger than that of the isostructural Ph4P[MnIII(Pc)(CN)2]2, where MnIII adopts a low-spin state (S = 1). The magnetoresistance (MR) effects of Ph4P[MnIII(Pc)Cl2]2 at 26.5 K under 9 T static magnetic fields perpendicular and parallel to the c-axis were determined to be −30% and −20%, respectively, which are significantly larger values than those of Ph4P[MnIII(Pc)(CN)2]2. Furthermore, the negative MR effect is comparable to that of Ph4P[FeIII(Pc)(CN)2]2 (S = 1/2), which exhibits the largest negative MR effect reported for [MIII(Mc)L2]-based systems (Mc = macrocyclic ligand, L = axial ligand). This suggests that the spin state of the metal ion is the key to tuning the MR effect.
Introduction
Molecular spintronics, which is the concept of spintronics based on molecular compounds, has attracted significant interest in recent years,1 wherein the magnetoresistance (MR) effect is a typical phenomenon of note. In molecular systems, the magnetic interaction between π-conduction electrons and the local spin is crucial to ensure a correlation between the electrical conductance and the magnetism. In this regard, metal complexes comprised of phthalocyanine (M(Pc)) and its derivatives, where the d-spin of the central metal ion is surrounded by a π-conjugated macrocyclic ligand, are attractive systems. For example, molecular semiconducting crystals have been constructed using [FeIII(Pc)L2] units (L is an axial ligand),2–5 where FeIII adopts a low-spin state (S = 1/2).In an [FeIII(Pc)L2] unit, the highest occupied molecular orbital (HOMO) is the π-orbital of Pc, and the next HOMOs contain the degenerated dyz and dzx orbitals where d-spin exists. Thus, molecular semiconducting crystals, which are mixed-valence compounds composed of the [FeIII(Pc)L2] unit, exhibit large negative MR effects irrespective of the crystal structure.4–7 Although the reported systems were initially expected to possess a 3/4-filled HOMO band, semiconducting behavior caused by the charge-ordered state of the π-conduction electrons has been observed with antiferromagnetic interaction between the d-spins. In the 3/4-filled system, the introduction of antiferromagnetic ordered d-spins means that the insulating state resulting from the charge-ordered state of the π-conduction electrons is stabilized via the intramolecular π–d interaction,8,9 i.e., the magnetic interaction between the π-conduction electrons and the d-spin. It is therefore believed that the large negative MR effects of molecular semiconducting crystals consisting of Fe(Pc)L2 emerge from the suppression of the charge-ordered state of π-conduction electrons by an external magnetic field, which disturbs the antiferromagnetic order of the d-spins.10,11 Furthermore, the MR effect can be tuned through the molecular design, such as the substitution of a macrocyclic ligand,12,13 axial ligands,5 or a central metal ion.14–17
Among the reported systems, the MR ratio of Ph4P[Fe(Pc)(CN)2]2 (Ph4P = tetraphenylphosphonium) was determined to be −99% at 20 K under a 38 T magnetic field.7 When the central metal ion was substituted to give Ph4P[MnIII(Pc)(CN)2]2, where MnIII adopted the low-spin state (S = 1), the MR effect was significantly smaller than those of isostructural FeIII(Pc)L2-based systems.4–7,16 However, it has been shown that not only the kind of the element but also the spin state of the central metal ion significantly affects the MR effect. For example, despite the significantly weaker antiferromagnetic interaction, the MR effect of Ph4P[FeIII(tbp)Br2]2 (tbp = tetrabenzoporpyrin) is similar to that of the isostructural Ph4P[FeIII(tbp)(CN)2]2, where the spin state of FeIII is the high-spin state (S = 5/2) in the former and the low-spin state in the latter.12,18 For the purpose of the current study, we chose to focus on the spin states of the MnIII ion. It is rare for MnIII to adopt a low-spin state,19 and a very strong ligand field is required to induce it.19,20 Therefore, we anticipated that it could be possible to fabricate a Ph4P[MnIII(Pc)L2]2 molecular semiconducting crystal where MnIII adopts a high-spin state (S = 2) by substituting the strong CN ligand with a weaker ligand, such as Cl. Changing the spin state of MnIII would be expected to induce drastic changes in the electrical and magnetic properties of the resulting system.
The spin state of a metal ion in a coordination complex is the key to determining its functionality. For example, heme proteins consisting of iron porphyrin change their functionality when the valence and spin states of iron are altered.21,22 Because their molecular structures resemble heme, metal complexes based on porphyrin and its derivatives, including phthalocyanines, have been widely studied as biomimetic models,23 and various high-spin MnIII complexes with porphyrin or phthalocyanine moieties have been reported.24,25 Prior to the current study, the Gaussian 16 software package26 was used to perform theoretical calculations for a [MnIII(Pc)L2] (L = Cl or CN) unit using the density functional theory (DFT) approach at the B3LYP/6-311G(d) level of theory. The geometry optimization results under D2h symmetry for MnIII(Pc)L2 are shown in Table 1. As indicated, compared with the results for L = CN systems, where the low-spin state is evidently more stable than the high-spin state, the differences between the free energies of the low- and high-spin states of MnIII are relatively small for both MnIII(Pc)Cl2 and [MnIII(Pc)Cl2]−, where Pc is the open shell (oxidized) and the closed shell (not oxidized), respectively. Owing to this small difference in the calculated free energies, we anticipated a possibility that MnIII in the MnIII(Pc)Cl2-based mixed-valence compounds would adopt a high-spin state.
|
||||
|---|---|---|---|---|
| Mn(Pc)Cl2 | Mn(Pc)(CN)2 | |||
| Quartet | Sextet | Quartet | Sextet | |
| Relative free energy (kcal mol−1) | 0 | +8.3 | 0 | +19.2 |
| Mn–N (Å) | 1.969 | 1.979 | 1.968 | 2.039 |
| Mn–N (Å) | 1.969 | 1.980 | 1.968 | 2.039 |
| Mn–L (Å) | 2.269 | 2.510 | 2.015 | 2.036 |

| [Mn(Pc)Cl2]− | [Mn(Pc)(CN)2]− | |||
|---|---|---|---|---|
| Triplet | Quintet | Triplet | Quintet | |
| Relative free energy (kcal mol−1) | 0 | +0.7 | 0 | +12.6 |
| Mn–N (Å) | 1.980 | 1.985 | 1.977 | 2.046 |
| Mn–N (Å) | 1.981 | 1.985 | 1.978 | 2.047 |
| Mn–L (Å) | 2.316 | 2.560 | 2.032 | 2.044 |
Thus, in this study, a semiconducting mixed-valence crystal of Ph4P[MnIII(Pc)Cl2]2 is synthesized, and the high-spin state of MnIII is confirmed. Its molecular and crystal structure, magnetic and electrical transport properties, and MR effect are reported. Notably, this is the first study investigating the effects of changing the spin state of the MnIII center on the electrical transport properties of the resulting system.
Results and discussion
Molecular and crystal structure
The crystal structure and crystal data of Ph4P[MnIII(Pc)Cl2]2 are given in Fig. 1 and Table 2, respectively. As in the cases of Ph4P[FeIII(Pc)L2]2 (FeIII: d5, S = 1/2) and Ph4P[CoIII(Pc)L2]2 (CoIII: d6, S = 0) (L = CN, Cl, or Br), the substitution of axial ligands has little influence on the molecular arrangement. Therefore, Ph4P[MnIII(Pc)Cl2]2, which possesses a tetragonal unit cell with a P42/n space group, is isostructural to Ph4P[MnIII(Pc)(CN)2]2.16 The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of cation:[Mn(Pc)Cl2] units indicates that an average of one electron is oxidized for every two [Mn(Pc)Cl2] units. This means that the one-dimensional regular chains of [Mn(Pc)Cl2] units overlapping two peripheral benzene rings along the c-axis form a one-dimensional electronic system with a 3/4-filled HOMO band. The interplanar distances between benzene rings are 3.47 and 3.40 Å, which are similar to those for Ph4P[MnIII(Pc)(CN)2]2 (i.e., 3.47 and 3.41 Å). Using the extended Hückel calculation,‡ the overlap integral between the HOMOs of Pc in Ph4P[MnIII(Pc)Cl2]2 was estimated to be 9.3 × 10−3, which is slightly larger than that of Ph4P[MnIII(Pc)(CN)2]2 (9.0 × 10−3), thereby indicating that substitution of the axial ligands has little effect on conduction path formation.
:2 ratio of cation:[Mn(Pc)Cl2] units indicates that an average of one electron is oxidized for every two [Mn(Pc)Cl2] units. This means that the one-dimensional regular chains of [Mn(Pc)Cl2] units overlapping two peripheral benzene rings along the c-axis form a one-dimensional electronic system with a 3/4-filled HOMO band. The interplanar distances between benzene rings are 3.47 and 3.40 Å, which are similar to those for Ph4P[MnIII(Pc)(CN)2]2 (i.e., 3.47 and 3.41 Å). Using the extended Hückel calculation,‡ the overlap integral between the HOMOs of Pc in Ph4P[MnIII(Pc)Cl2]2 was estimated to be 9.3 × 10−3, which is slightly larger than that of Ph4P[MnIII(Pc)(CN)2]2 (9.0 × 10−3), thereby indicating that substitution of the axial ligands has little effect on conduction path formation.
 | ||
| Fig. 1 Crystal structure of Ph4P[MnIII(Pc)Cl2]2 viewed along the b-axis. Some molecules have been omitted for clarity. | ||
| CCDC number | 2129804 |
|---|---|
| Chemical formula | C88H52Cl14Mn2N16P |
| Formula weight | 1616.10 |
| Crystal description | Black needle |
| T (K) | 293 |
| Crystal system | Tetragonal |
| Space group | P42/n |
| a (Å) | 21.6778 (4) |
| c (Å) | 7.4426 (2) |
| V (Å3) | 3497.48 (4) |
| Z | 2 |
| dcal (g cm−3) | 1.535 |
| Radiation | MoKα |
| Wavelength (Å) | 0.71073 |
| μ (mm−1) | 0.602 |
| No. of measured reflections | 17320 |
| No. of independent reflection | 3679 |
| No. of observed reflections | 2564 (I > 2σ (I)) |
| R (I > 2σ (I)) | R1 = 0.0420 wR2 = 0.1402 |
| Goodness-of-fit | 0.931 |
| Parameters | 252 |
With respect to the bond length, theoretical calculations predicted that the Mn–Cl distance would be notably larger for high-spin MnIII than for low-spin MnIII, and that the Mn–N distances would be insensitive to the MnIII spin state (Table 1). This suggests that the electronic configuration of MnIII in the high-spin state is (dyz, dzx)2(dxy)1(dz2)1, as reported for porphyrin complexes,27,28 and that in the low-spin state is (dxy)2(dyz, dzx)2.19 The fact that the former possesses occupied dz2 renders the Mn–Cl distance larger in the high-spin MnIII complex. In contrast, since dx2−y2 is unoccupied in both spin states, the Mn–N distance is approximately the same. The observed Mn–Cl distance of 2.555 Å is consistent with the predicted value for a [Mn(Pc)Cl2] unit with high-spin MnIII.
Therefore, it can be concluded that the MnIII center in the obtained Ph4P[Mn(Pc)Cl2]2 adopts a high-spin state.
Magnetic susceptibility
Fig. 2 shows the temperature dependence of the magnetic susceptibility χp of Ph4P[MnIII(Pc)Cl2]2 under a static magnetic field of 1 T. For temperatures greater than 20 K, the χp vs. T plot obeys the Curie–Weiss law with a Curie constant of C = 5.69 emu K mol−1 and a Weiss temperature of θ = −2.4 K. This Curie constant is significantly larger than that reported for Ph4P[MnIII(Pc)(CN)2]2, where MnIII adopts the low-spin state (S = 1) (C = 3.01 emu K mol−1).16 It is therefore apparent that the MnIII center in Ph4P[Mn(Pc)Cl2]2 adopts the high-spin state (S = 2) as expected, although the observed Curie constant of C = 5.69 emu K mol−1 is smaller than the predicted value of 6.00 emu K mol−1 under the assumption of S = 2 and g = 2.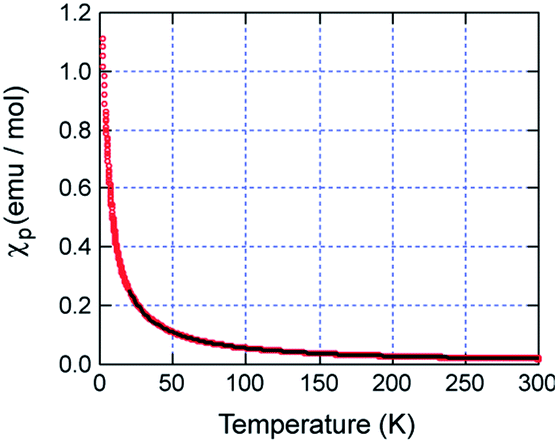 | ||
| Fig. 2 Temperature dependence of the magnetic susceptibility χp for the formula unit Ph4P[MnIII(Pc)Cl2]2 under a static magnetic field of 1 T. The solid line is the best fit to the Curie–Weiss law with the parameters C = 5.69 emu K mol−1 and Θ = −2.4 K. | ||
The negative Weiss temperature indicates that there is an antiferromagnetic interaction between the d-spins of high-spin MnIII. Antiferromagnetic interactions between d-spins are common in the low-spin FeIII and MnIII complexes Ph4P[Fe(Pc)L2]2,2–5 Ph4P[Fe(tbp)(CN)2]2,12 and Ph4P[Mn(Pc)(CN)2]2,16 and the high-spin FeIII complex Ph4P[Fe(tbp)Br2]2.18 According to the mean-field approximation, the Weiss temperature can be expressed as θ = 2zJddS(S + 1)/3 kB, where z, Jdd, and kB are the number of nearest neighbors, an intermolecular magnetic exchange interaction between the d-spins, and the Boltzmann constant, respectively. As summarized in Table 3, the absolute value |Jdd| for Ph4P[MnIII(Pc)Cl2]2 is less than that for Ph4P[MnIII(Pc)(CN)2]2, indicating that substituting the CN axial ligands with Cl weakens the antiferromagnetic interaction.
| Ph4P[Mn(Pc)Cl2]2 | Ph4P[Mn(Pc)(CN)2]2 | Ph4P[Fe(tbp)Br2]2 | Ph4P[Fe(tbp)(CN)2]2 | Ph4P[Fe(Pc)(CN)2]2 | |
|---|---|---|---|---|---|
| a The MR effect at 10.7 K under a 8 T magnetic field is reported to be −8.7%. ⊥ and//indicate perpendicular and parallel alignments, respectively. | |||||
| S | 2 | 1 | 5/2 | 1/2 | 1/2 |
| θ | −2.4 K | −3.1 K | −2.5 K | −8.0 K | −12 K |
| Jdd/kB | −0.30 K | −1.16 K | −0.21 K | −8.0 K | −12.0 K |
| MR effect under 9 T at 30 K | −25% (B⊥c) | N/Aa | −6% (B⊥c) | −14% (B⊥c) | −30% (B⊥c) |
| −18% (B//c) | −8% (B//c) | −9% (B//c) | −18% (B//c) | ||
| Reference | This study | 16 | 18 | 12 | 6 |
Electrical resistance measurements
Fig. 3 shows the temperature dependence of the electrical resistivities of Ph4P[MnIII(Pc)Cl2]2 and Ph4P[MnIII(Pc)(CN)2]2 along their c-axes. Despite the 3/4-filled HOMO band, semiconducting behavior was observed. Such behavior is exhibited by all Ph4P[MIII(Mc)L2]2 systems (Mc = macrocyclic ligand) and has been attributed to the fluctuations in the charge-ordered state of the π-conduction electrons.29 The electrical resistivity of Ph4P[MnIII(Pc)Cl2]2 at room temperature (∼25 °C) is approximately 5 Ω cm, which is one order of magnitude higher than that of Ph4P[MnIII(Pc)(CN)2]2. Furthermore, the activation energy of Ph4P[MnIII(Pc)Cl2]2 in the temperature range of 25–300 K was estimated to be 34 meV, which is significantly larger than that of Ph4P[MnIII(Pc)(CN)2]2 (8.4 meV at <70 K).16 This suggests that the charge-ordered state of the π-conduction electrons in Ph4P[MnIII(Pc)Cl2]2 is more developed than in the case of Ph4P[MnIII(Pc)(CN)2]2. As mentioned above, the charge-ordered state of the π-conduction electrons is stabilized by the antiferromagnetic interaction between the d-spins and the intramolecular π–d interaction. Therefore, the intramolecular π–d interaction in the high-spin Ph4P[MnIII(Pc)Cl2]2 system is believed to be stronger than that in the low-spin Ph4P[MnIII(Pc)(CN)2]2 system. A likely cause of the change in the π–d interaction is the change in the electronic structure resulting from the substitution of the axial ligands. The development of the charge-ordered state in Ph4P[MnIII(Pc)Cl2]2 therefore indicates that the negative MR effect is expected to be larger than that of Ph4P[MnIII(Pc)(CN)2]2. | ||
| Fig. 3 Temperature dependence of the electrical resistivity along the c-axes of Ph4P[MnIII(Pc)Cl2]2 (red) and Ph4P[MnIII(Pc)(CN)2]2 (green). | ||
Magnetoresistance measurements
Fig. 4(a) and (b) show the relationships between the electrical resistance of Ph4P[MnIII(Pc)Cl2]2 and the magnetic fields perpendicular and parallel to the c-axis, respectively. In both cases, a large negative MR effect was observed, which increased as the magnetic field increased or the temperature decreased. At 26.5 K, the MR effect under a 8 T magnetic field perpendicular to the c-axis was −24%, and that under the same field parallel to the c-axis was −17%. These values are significantly larger than the MR effect reported for Ph4P[MnIII(Pc)(CN)2]2 (−8.7% at 10.7 K, under a 8 T magnetic field).16 Table 3 summarizes spins of the d electrons, Jdd, and the MR effects for Ph4P[MnIII(Pc)Cl2]2, Ph4P[MnIII(Pc)(CN)2]2, Ph4P[FeIII(tbp)Br2]2, Ph4P[FeIII(tbp)(CN)2]2, and Ph4P[FeIII(Pc)(CN)2]2. As for the isostructural Ph4P[Fe(Mc)L2]2 with low-spin FeIII (S = 1/2), the negative MR effect reduces as |Jdd| decreases.5,6,12 Therefore, it is notable that a significantly larger negative MR effect was observed for the high-spin Ph4P[MnIII(Pc)Cl2]2 system than for the low-spin Ph4P[MnIII(Pc)(CN)2]2 system, despite the fact that |Jdd| is smaller for Ph4P[MnIII(Pc)Cl2]2. A similar trend was observed for Ph4P[FeIII(tbp)Br2]2 with high-spin FeIII and Ph4P[FeIII(tbp)(CN)2]2 with low-spin FeIII, and the negative MR effect for Ph4P[Fe(tbp)Br2]2 was found to be similar to that of Ph4P[FeIII(tbp)(CN)2]2, despite the fact that |Jdd| is significantly smaller for Ph4P[FeIII(tbp)Br2]2. Considering the mechanism of the negative MR effect, the fact that high-spin systems with a small |Jdd| exhibited negative MR effects larger than or similar to those of low-spin systems implies that the high-spin systems possess stronger intramolecular π–d interactions than the low-spin systems. These results suggest that the high-spin state of MIII in the [M(Mc)L2] unit aids the emergence of the negative MR effect. The MR effects of Ph4P[MnIII(Pc)Cl2]2 at 30 K under 9 T magnetic fields perpendicular and parallel to the c-axis are −25% and −18%, respectively. These values are comparable to those of Ph4P[FeIII(Pc)(CN)2]2, which exhibits the largest MR effect reported in [M(Mc)L2]-based systems. | ||
| Fig. 4 Resistance under the magnetic fields (a) perpendicular and (b) parallel to the c-axis at various temperatures. The resistance values are normalized to the corresponding resistance under a zero magnetic field. | ||
Conclusions
Based on the density functional theory calculations, a Ph4P[MnIII(Pc)Cl2]2 semiconducting molecular crystal where the octahedrally ligated MnIII adopts a high-spin state (S = 2) was designed and fabricated. Although the intermolecular magnetic interaction Jdd between the MnIII ions was estimated to be −0.30 K, and the absolute value was smaller than that of the isostructural Ph4P[MnIII(Pc)(CN)2]2 with low-spin MnIII (S = 1, Jdd = −1.16 K), a large negative magnetoresistance (MR) effect was observed. The MR effects at 26.5 K under 9 T magnetic fields perpendicular and parallel to the c-axis were −30% and −20%, respectively. These MR effects are significantly larger than those observed for the low-spin Ph4P[MnIII(Pc)(CN)2]2 system, and are similar to those of Ph4P[FeIII(Pc)(CN)2]2, which exhibited the largest MR effect among previously reported [MIII(Mc)L2]-based semiconducting crystals. Therefore, this study revealed that the spin state of the metal ion is the key to tuning the MR effect, and the high-spin state aids the emergence of a large negative MR effect. Further research into [MIII(Mc)L2]2-based systems, where MIII adopts a high-spin state, would benefit the development of molecular spintronics.Experimental details
Synthetic procedures
Mn(Pc) was synthesized according to the procedure described by Rutter and McQueen,30 using quinoline as a solvent instead of 1,2-propanediol. The obtained Mn(Pc) (6 mg, 0.01 mmol) was electrochemically oxidized in a mixed solution of DMF:acetone (8 mL, 1:1 v/v, super dehydrated grade, used as purchased from FUJIFILM Wako Chemicals) containing Ph4P·Cl (24 mg, 0.06 mmol, used as purchased from Tokyo Chemical Industry) at 25 °C using an electrocrystallization cell under an Ar atmosphere. The cell was equipped with a glass frit between the two compartments. A constant current of 0.6–1.0 μA was passed between two platinum electrodes immersed in the solutions in each compartment for 2–3 weeks. Black needle-like crystals of Ph4P[MnIII(Pc)Cl2]2 grew on the anode surface and were harvested by filtration.
Crystal structure determination
Crystal data for Ph4P[MnIII(Pc)Cl2]2 were collected at 293 K using an automated Rigaku SuperNova system with monochromated Mo Kα radiation (λ = 0.71073 Å). The structure was solved using a direct method with SHELXT-2014/5 (ref. 31) and refined using a full-matrix least-squares technique with SHELXL-2018/1.32 Anisotropic and isotropic thermal parameters were employed for non-hydrogen and hydrogen atoms, respectively.†Measurements
The electrical resistivity along the c-axis of a Ph4P[MnIII(Pc)Cl2]2 single crystal was measured using a physical property measurement system (PPMS) from Quantum Design with a static magnetic field of up to 9 T over a temperature range of 25–300 K. Gold wires were attached to the sample using gold paste once gold deposition was complete. In the high-temperature region, where the resistance was less than 107 Ω, the standard four-probe method was adopted; in the low-temperature region, where the resistance was greater than 106 Ω, the two-probe method was applied. Static magnetic susceptibility measurements were obtained using a Quantum Design magnetic property measurement system (MPMS) superconducting quantum interference device (SQUID) with a 1 T magnetic field over the temperature range of 2–300 K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported in part by a Grant-in-Aid for Scientific Research (B) (No 19H02691) from the Japan Society for the Promotion of Science. The authors are grateful for the support by the Cooperative Research Program of “New Joint Research Center for Materials and Devices”. We also thank Prof Akira Yoshiasa at Kumamoto University for his help with the X-ray diffraction measurements.Notes and references
- E. Coronado and M. Yamashita, Dalton Trans., 2016, 45, 16553–16555 RSC.
- M. Matsuda, T. Naito, T. Inabe, N. Hanasaki, H. Tajima, T. Otsuka, K. Awaga, B. Narymbetov and H. Kobayashi, J. Mater. Chem., 2000, 10, 631–636 RSC.
- M. Matsuda, T. Naito, T. Inabe, N. Hanasaki and H. Tajima, J. Mater. Chem., 2001, 11, 2493–2497 RSC.
- M. Matsuda, T. Asari, T. Naito, T. Inabe, N. Hanasaki and H. Tajima, Bull. Chem. Soc. Jpn., 2003, 76, 1935–1940 CrossRef CAS.
- D. E. C. Yu, M. Matsuda, H. Tajima, A. Kikuchi, T. Taketsugu, N. Hanasaki, T. Naito and T. Inabe, J. Mater. Chem., 2009, 19, 718–723 RSC.
- N. Hanasaki, H. Tajima, M. Matsuda, T. Naito and T. Inabe, Phys. Rev. B, 2000, 62, 5839–5842 CrossRef CAS.
- N. Hanasaki, M. Matsuda, H. Tajima, E. Ohmichi, T. Osada, T. Naito and T. Inabe, J. Phys. Soc. Jpn., 2006, 75, 033703 CrossRef.
- C. Hotta, M. Ogata and H. Fukuyama, Phys. Rev. Lett., 2005, 95, 216402 CrossRef PubMed.
- Y. Otsuka, H. Seo and Y. Motome, Phys. B, 2010, 405, S317–S320 CrossRef CAS.
- C. Hotta, Phys. Rev. B, 2010, 81, 245104 CrossRef.
- H. Murakawa, A. Kanda, M. Ikeda, M. Matsuda and N. Hanasaki, Phys. Rev. B, 2015, 92, 054429 CrossRef.
- M. Nishi, M. Ikeda, A. Kanda, N. Hanasaki, N. Hoshino, T. Akutagawa and M. Matsuda, Dalton Trans., 2016, 45, 16604–16609 RSC.
- M. Yamaguchi, S. Iwamura, K. Mine, H. Murakawa, N. Hanasaki and M. Matsuda, Dalton Trans., 2021, 50, 5789–5794 RSC.
- Y. Takita, H. Hasegawa, Y. Takahashi, J. Harada, A. Kanda, N. Hanasaki and T. Inabe, J. Porphyrins Phthalocyanines, 2014, 18, 814–823 CrossRef CAS.
- M. Ikeda, T. Kida, T. Tahara, H. Murakawa, M. Nishi, M. Matsuda, M. Hagiwara, T. Inabe and N. Hanasaki, J. Phys. Soc. Jpn., 2016, 85, 064713 CrossRef.
- M. Matsuda, G. Yoshida, J.-i. Yamaura, T. Inabe and H. Tajima, Dalton Trans., 2017, 46, 1892–1897 RSC.
- M. Ikeda, A. Kanda, H. Murakawa, M. Matsuda, T. Inabe, H. Tajima and N. Hanasaki, J. Phys. Soc. Jpn., 2016, 85, 024713 CrossRef.
- M. Nishi, R. Ishii, M. Ikeda, N. Hanasaki, N. Hoshino, T. Akutagawa, M. Sumimoto and M. Matsuda, Dalton Trans., 2018, 47, 4070–4075 RSC.
- A. P. Hansen and H. M. Goff, Inorg. Chem., 1984, 23, 4519–4525 CrossRef CAS.
- M. Matsuda, J.-i. Yamaura, H. Tajima and T. Inabe, Chem. Lett., 2005, 34, 1524–1525 CrossRef CAS.
- H. Chen, M. I. Saito and S. Shaik, J. Am. Chem. Soc., 2008, 130, 14778–14790 CrossRef CAS PubMed.
- Y. Nakahara, K. Kimura and H. Inokuchi, Chem. Phys. Lett., 1977, 47, 251–254 CrossRef CAS.
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561–588 CrossRef CAS PubMed.
- L. J. Boucher, Coord. Chem. Rev., 1972, 7, 289–329 CrossRef CAS.
- A. B. P. Lever, J. Chem. Soc., 1965, 1821–1829 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. L. Hill and M. M. Willamson, Inorg. Chem., 1985, 24, 2836–2841 CrossRef CAS.
- M. G. I. Galinato, E. P. Brocious, F. Paulat, S. Martin, J. Skodack, J. B. Harland and N. Lehnert, Inorg. Chem., 2020, 59, 2144–2162 CrossRef CAS PubMed.
- N. Hanasaki, K. Masuda, K. Kodama, M. Matsuda, H. Tajima, J. Yamazaki, M. Takigawa, J.-i. Yamaura, E. Ohmichi, T. Osada, T. Naito and T. Inabe, J. Phys. Soc. Jpn., 2006, 75, 104713 CrossRef.
- H. A. Rutter, Jr. and J. D. McQueen, J. Inorg. Nucl. Chem., 1960, 12, 361–363 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnotes |
| † CCDC 2129804 for Ph4P[Mn(Pc)Cl2]2. For crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra00188h |
| ‡ Calculations were performed using the CAESAR 2.0 software package (PrimeColor Software, Inc). Default parameters were used. |
| This journal is © The Royal Society of Chemistry 2022 |