
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective hydroconversion of coconut oil-derived lauric acid to alcohol and aliphatic alkane over MoOx-modified Ru catalysts under mild conditions†
Rodiansono *ab,
Heny Puspita Dewiab,
Kamilia Mustikasaria,
Maria Dewi Astutia,
Sadang Husainc and
Sutomod
*ab,
Heny Puspita Dewiab,
Kamilia Mustikasaria,
Maria Dewi Astutia,
Sadang Husainc and
Sutomod
aDepartment of Chemistry, Faculty of Mathematics and Natural Sciences, Lambung Mangkurat University, Jl. A. Yani Km 36.0, Banjarbaru, South Kalimantan, Indonesia
bCatalysis for Sustainable Energy and Environment (CATSuRe), Lambung Mangkurat University, Indonesia. E-mail: rodiansono@ulm.ac.id; Fax: +625114773112; Tel: +625114773112
cDepartment of Physics, Faculty of Mathematics and Natural Sciences, Lambung Mangkurat University, Indonesia
dDepartment of Pharmacy, Faculty of Mathematics and Natural Sciences, Lambung Mangkurat University, Indonesia
First published on 3rd May 2022
Abstract
Molybdenum oxide-modified ruthenium on titanium oxide (Ru–(y)MoOx/TiO2; y is the loading amount of Mo) catalysts show high activity for the hydroconversion of carboxylic acids to the corresponding alcohols (fatty alcohols) and aliphatic alkanes (biofuels) in 2-propanol/water (4.0/1.0 v/v) solvent in a batch reactor under mild reaction conditions. Among the Ru–(y)MoOx/TiO2 catalysts tested, the Ru–(0.026)MoOx/TiO2 (Mo loading amount of 0.026 mmol g−1) catalyst shows the highest yield of aliphatic n-alkanes from hydroconversion of coconut oil derived lauric acid and various aliphatic fatty acid C6–C18 precursors at 170–230 °C, 30–40 bar for 7–20 h. Over Ru–(0.026)MoOx/TiO2, as the best catalyst, the hydroconversion of lauric acid at lower reaction temperatures (130 ≥ T ≤ 150 °C) produced dodecane-1-ol and dodecyl dodecanoate as the result of further esterification of lauric acid and the corresponding alcohols. An increase in reaction temperature up to 230 °C significantly enhanced the degree of hydrodeoxygenation of lauric acid and produced n-dodecane with maximum yield (up to 80%) at 230 °C, H2 40 bar for 7 h. Notably, the reusability of the Ru–(0.026)MoOx/TiO2 catalyst is slightly limited by the aggregation of Ru nanoparticles and the collapse of the catalyst structure.
Introduction
The catalytic hydroconversion of fatty acids and their esters into fatty alcohols and aliphatic alkanes is receiving increased attention in the context of upgrading of bio-based feedstocks by using heterogeneous mono- or bi-metallic catalysts.1–4 Both fatty alcohols and aliphatic alkanes are basic building blocks in organic synthesis, living organisms, energy, fuels, surfactants, lubricants, plasticizers, coatings, polymers, and the materials industry.5,6 The catalytic hydroconversion of fatty acids can be classified into three reactions: hydrodeoxygenation (HDO), hydrodecarbonylation (HDCO), and hydrodecarboxylation (HDCO2), and a number of comprehensive reviews have been reported in the last decade.7–12 HDCO and HDCO2 yield hydrocarbons with one carbon atom less than the fatty acid precursor, while HDO gives hydrocarbons with the same chain length as the starting compounds.13 Among these hydroconversion approaches, HDO, producing hydrocarbons without C–C bond cleavage of fatty acids, is more atom-efficient than the other methods (HDCO and HDCO2) with C–C bond cleavage of the fatty acids. Moreover, the reaction rate of HDCO or HDCO2 is slow and needs to be carried out at higher reaction temperatures.14The development of effective heterogeneous catalyst systems (in the form of supported reduced or sulfided metals or bimetallic) for the hydroconversion of fatty acids has been long-standing industrial target by researchers.15–18 The literature shows that heterogeneous supported platinum group metals (PGM) (e.g., Pt, Pd, Rh, Ru, Ni) catalysts showed high selectivity toward HDCO or HDCO2 rather than HDO products even under H2 atmosphere.19–23 To improve the HDO activity rather than HDCO and HDCO2 of fatty acids, the modification of those PGM-based catalysts is necessary, i.e., the addition of more electropositive metals24,25 or the use of oxide supports that strongly interact with the active metals,26–28 or direct modification with the metal oxide species.29 The addition of second metals (e.g., Sn and B) to Ru enhanced the dispersion of Ru and improved the electron density of Ru. The change of electron density Ru enhanced the affinity of Ru towards C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of fatty acids which facilitated the reaction hydrogenation.30–32 Several bimetallic or alloy-based catalysts (e.g., Pd–M (M = Cu, Co, Ni),33 Ni–Sn/TiO2,34 Pd–Nb2O5,35 Ru–Sn,31 Rh–Sn,32 and Pd–Sn/C,36) have shown superior performance for the selective hydrogenation of fatty acids compared with their single metal counterpart. Luo et al. reported the hydrogenation of coconut oil to fatty alcohols using nanocluster Ru3Sn7/SiO2 catalysts at 240 °C, 40 bar. They claimed that Ru3Sn7 and SnOx were active species in the bimetallic Ru–Sn catalyst as indicated by the conversion and product selectivity, and computational modelling calculation.37,38 In the case of direct modification of PGM with metal oxides, oxophilic metal oxides (e.g., ReOx, MoOx, and WOx)-modified PGM-based catalysts showed excellent performance for the catalytic HDO of biomass-derived oxygenates into chemicals and fuels.39–45 The presence of oxophilic metal oxides played the bifunctional catalytic roles, whereas the metal sites can catalyse the hydrogen uptake, dissociation, and spill-over onto the metal-oxide in vicinity.46,47 It was believed that H2 spill-over facilitated the partial reduction of metal-oxide species and generated a new site active in interface of metal–metal oxide. Moreover, the reduced metal–oxide species can act as Lewis acid sites for the C–O bond cracking via dehydration reaction.29,48–51
O bond of fatty acids which facilitated the reaction hydrogenation.30–32 Several bimetallic or alloy-based catalysts (e.g., Pd–M (M = Cu, Co, Ni),33 Ni–Sn/TiO2,34 Pd–Nb2O5,35 Ru–Sn,31 Rh–Sn,32 and Pd–Sn/C,36) have shown superior performance for the selective hydrogenation of fatty acids compared with their single metal counterpart. Luo et al. reported the hydrogenation of coconut oil to fatty alcohols using nanocluster Ru3Sn7/SiO2 catalysts at 240 °C, 40 bar. They claimed that Ru3Sn7 and SnOx were active species in the bimetallic Ru–Sn catalyst as indicated by the conversion and product selectivity, and computational modelling calculation.37,38 In the case of direct modification of PGM with metal oxides, oxophilic metal oxides (e.g., ReOx, MoOx, and WOx)-modified PGM-based catalysts showed excellent performance for the catalytic HDO of biomass-derived oxygenates into chemicals and fuels.39–45 The presence of oxophilic metal oxides played the bifunctional catalytic roles, whereas the metal sites can catalyse the hydrogen uptake, dissociation, and spill-over onto the metal-oxide in vicinity.46,47 It was believed that H2 spill-over facilitated the partial reduction of metal-oxide species and generated a new site active in interface of metal–metal oxide. Moreover, the reduced metal–oxide species can act as Lewis acid sites for the C–O bond cracking via dehydration reaction.29,48–51
Lauric acid is one of typical bio-based aliphatic fatty acids with medium carbon length (C12) that mainly constituent of coconut oil or palm kernel oil (∼50%),52,53 which can be transformed into lauryl alcohol (dodecane-1-ol) and aliphatic alkanes (e.g., n-dodecane or undecane). We have developed bimetallic catalysts (e.g., bimetallic Ni–Sn/TiO2, Pd–Fe/TiO2, Pd–Sn/C and Ru–Fe/TiO2 catalysts) and showed high catalytic performances in the hydrogenation of typical biomass-derived levulinic acid to γ-valerolactone,54–56 lauric acid to dodecane-1-ol,34,57 and stearic acid to octadecanol.36 In the present paper, we describe our studies on the hydroconversion of lauric acid into lauryl alcohol using molybdenum oxide-modified ruthenium supported on titanium oxide (denoted as Ru–(y)MoOx/TiO2; y = loading amount of Mo, mmol g−1) catalysts. The addition of Mo (∼0.026 mmol; Mo/Ru = 0.5) to Ru/TiO2 catalyst (denoted as Ru–(0.026)MoOx/TiO2) greatly improved the hydrodeoxygenation of lauric acid and produced n-dodecane (∼72% yield) at 190 °C, 40 bar H2, and a reaction time of 7 h. An increase in reaction temperature to 200–230 °C significantly enhanced the degree of hydrodeoxygenation of lauric acid and yield of n-dodecane reached to maximum (80%) (Scheme 1). Therefore, the effect of solvent used, reaction temperature, initial H2 pressure, and reaction time on the yields of desired products during the hydroconversion of lauric acid are discussed systematically.
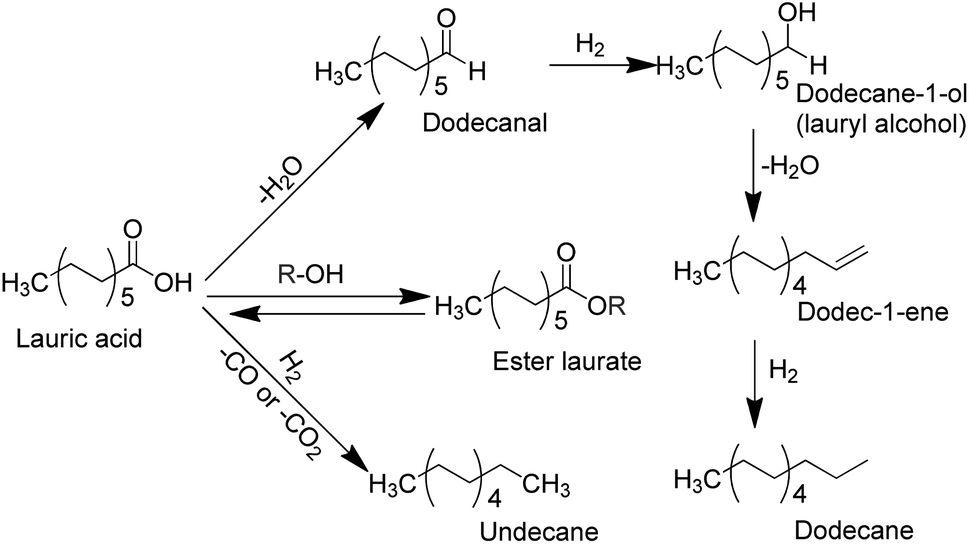 | ||
| Scheme 1 Possible reaction pathways for the hydroconversion of lauric acid to alcohols and aliphatic alkanes. | ||
Results and discussion
Catalytic reactions
| Entry | Solventb | Dielectric constant of solvent (ε) | Donor number (DN) | Conversionc (%) | Yieldc(%) | ||
|---|---|---|---|---|---|---|---|
| Dodecane-1-ol | n-Dodecane | Esters | |||||
| a Reaction conditions: catalyst, 0.065 g; lauric acid, 3.2 mmol; solvent, 5 mL, 170 °C; H2 40 bar, 7 h.b The value in the parenthesis is volume ratio of solvent.c Conversion and yield were determined by GC using an internal standard technique.d Methyl, ethyl, and propyl laurate are included as the esterification lauric acid with the solvent as identified by using GC-MS analysis.e Dodecyl dodecanoate is included as the esterification of lauric acid and dodecane-1-ol. The carbon balance was 93–96% for all the catalyst. | |||||||
| 1 | Methanol | 32.7 | 19.0 | 74 | 17 | 0 | 57d |
| 2 | Ethanol | 24.5 | 19.2 | 82 | 22 | 0 | 59d |
| 3 | 1-Propanol | 21.8 | 19.8 | 92 | 43 | 0 | 49d |
| 4 | 2-Propanol | 19.9 | 21.1 | 91 | 87 | 0 | 4e |
| 5 | H2O | 80.1 | 18.0 | 52 | 32 | 17 | 3e |
| 6 | Methanol/H2O (4.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0 v/v) :1.0 v/v) |
— | — | 84 | 43 | 11 | 30d |
| 7 | Ethanol/H2O (4.0:1.0 v/v) |
— | — | 95 | 59 | 12 | 24d |
| 8 | 1-Propanol/H2O (4.0:1.0 v/v) |
— | — | >99 | 53 | 20 | 26d |
| 9 | 2-Propanol/H2O (4.0:1.0 v/v) |
— | — | >99 | 61 | 38 | <0.1e |
Interestingly, a remarkable difference was observed in H2O, the products were distributed to dodecane-1-ol (32% yield) and n-dodecane (17% yield) at 52% conversion of lauric acid (entry 5), indicating that the hydrodeoxygenation lauric acid to aliphatic alkanes occurred in H2O. The importance of H2O media in the catalytic hydrothermal deoxygenation (330 °C) of fatty acids had been noticed to improve the yield of alipathic alkanes in the presence of supported PGM catalysts.2,58,59 Moreover, under hydrothermal conditions, the presence of molecular water promoted the HDCO2 reaction generating less one carbon of aliphatic alkanes and CO2 as side product.60 In our reaction system, the presence of external H2 prevent the decarboxylation reaction and allow the hydroconversion of lauric acid under milder reaction conditions. However, the dodecyl dodecanoate (3%) product of esterification between lauric acid and dodecan-1-ol was obviously observed in H2O. This results let us to further investigation the effect of solvent to enhance conversion and yield, particularly, in alcohol and H2O mixture solvents. In methanol/H2O (4.0:1.0 volume ratio), the conversion of lauric acid was 84% and the yield of dodecane-1-ol significantly increased by approximately three times (43%) while yields of dodecane and methyl laurate were 11% and 30%, respectively (entry 6). The catalytic reactions in ethanol/H2O, 1-propanol/H2O, and 2-propanol/H2O solvent mixtures significantly enhanced the hydrodeoxygenation of lauric acid reaction as indicated by the increase of n-dodecane or the decrease of ester laurate yields (entries 7–9). It can be seen that catalytic reactions in 2-propanol or 2-propanol/H2O are superior which can be attributed to the relatively higher solubility degree of lauric acid than that other solvents.61–63 The synergistic effect between intrinsic properties of solvent (e.g., dielectric constant and donation number) and the Brønsted acidity of catalysts during hydroconversion of lauric acid to lauryl alcohol and alkane might pronounce the stabilization of the acidic proton relative to the protonated transition states, leading to accelerated reaction rates for these acid-catalyzed biomass conversion reactions.64,65 The catalytic reactions in various solvents other than alcohols and H2O such as 1,4-dioxane, tetrahydrofuran (THF) and its mixture with H2O were carried, however the yield of targeted products were insufficient under the reaction conditions.57 Moreover, the highest yield (38%) of n-dodecane was achieved in 2-propanol/H2O without the formation of ester laurate at >99% conversion of lauric acid (entry 9). Therefore, we conclude that the optimised solvent system for the hydroconversion of lauric acid to dodecane-1-ol or n-dodecane using Ru–MoOx catalysts was in 2-propanol/H2O (4.0:1.0 volume ratio).
| Entry | Catalysta | Conversionb (%) | Yieldb(%) | ||
|---|---|---|---|---|---|
| Dodecane-1-ol | n-Dodecane | Othersc | |||
| a The amount of Mo was arround 0.025 mmol (1 wt% to Ru metal based on the amount of precursor). Reaction conditions: catalyst, 0.065 g; lauric acid, 3.2 mmol; solvent, 2-propanol: H2O, 5 ml (4.0:1.0 volume ratio), 170 °C; H2 40 bar, 7 h.b Conversion and yield were determined by GC using an internal standard technique.c Others include dodecyl dodecanoate as the esterification of lauric acid and dodecane-1-ol and products with smaller carbon number according to GC-MS profiles.d The catalysts were prepared using physical mixing at room temperature, dried at 110 °C for 5 h, followed by reduction with H2 at 500 °C for 3 h.e The catalyst was prepared using one-pot hydrothermal of RuCl3 and (NH4)6Mo7O24·4H2O mixture solutions at 150 °C for 24 h, followed by reduction with H2 at 500 °C for 3 h. The carbon balance was 93–96% for all the catalyst. |
|||||
| 1 | Ru/TiO2 | 73 | 65 | 4 | 4 |
| 2 | Ru–(0.026)MoOx/TiO2(A) | >99 | 61 | 38 | Trace |
| 3 | Ru–(0.026)MoOx/TiO2(R) | >99 | 82 | 10 | 8 |
| 4d | Ru/TiO2 + (NH4)6Mo7O24·4H2O | >99 | 45 | 9 | 46 |
| 5d | Ru/TiO2 + MoOx | 97 | 42 | 12 | 43 |
| 6e | Ru@MoOx | 98 | 59 | 7 | 32 |
A relatively high yield of others (46%) (mainly contain dodecyl dodecanoate ester and others smaller carbon number of compounds) was obtained, suggesting that (NH4)6Mo7O24·4H2O (may be as Mo+6) promoted the further reaction of alcohol and acid to form ester or decomposition of reactant/product into small molecule (entry 4). The Ru/TiO2 + MoOx catalyst was also active for the hydroconversion of lauric acid (97% conversion) and the products were distributed to dodecane-1-ol (42%), n-dodecane (12%), and others (43%) (entry 5). Moreover, Ru@MoOx exhibited high conversion of lauric acid (98%) towards dodecane-1-ol as the main product (59%), while yields of n-dodecane and others were 7% and 32% (respectively) (entry 6). The yield of others obtained over this catalyst was smaller than that of the Ru/TiO2 + (NH4)6Mo7O24·4H2O system. These results suggested that the presence of both Mo6+ and MoOx showed notable promotion effect on the dodecane-1-ol and n-dodecane formation, which are literally different between with and without the addition of (NH4)6Mo7O24·4H2O or MoOx powder. The high conversion of lauric acid can be attributed to the presence of Mo, which can interact strongly with water solvent to contribute to construct and maintain the active species of Mo–OH in the form of HxMoOx. The total acidity (obtained from NH3 temperature programmed desorption (NH3-TPD) and pyridine adsorption confirmed the presence of Brønsted and Lewis acid sites (Tabel S1 and Fig. S4 and S5, in the ESI†). The HxMoOx species acts as the Brønsted sites and helps to stabilise the transition state via hydrogen bonding during the hydrogenolysis of tetrahydrofurfuryl alcohol to 1,5-pentanediol.50 It has been reported that MoO3 is one of the reducible oxide supports which is commonly used as a co-promotor to enhance the activity and selectivity of PGM in the hydrogenation of carboxylic acids. The activity increase caused by reducible oxide supports is attributed to oxygen interaction with metallic ions of the support and/or oxygen vacancies on the metal-support interface. These electrophilic groups promote hydrogenation of the carboxylic acid by interacting with the carbonyl oxygen and weakening the CO bond.66,67 Alternatively, the MoOx species in the Ru–MoOx/TiO2 would be partially reduced to MoOx (0 ≤ x ≤ 3) by activated hydrogen atoms migrated via spill over from Ru nanoparticles considering the readily dissociation of H2 on Ru.67–69 Shimizu and co-workers reported that the presence of MoOx co-loaded Pt/TiO2 catalyst showed higher selectivity to CH3OH from CO2 hydrogenation than that without the presence of MoOx.70
Ru–MoOx on various supports. Six types of supports (γ-Al2O3, active carbon (C), C–TiO2, ZrO2, and ZnO) were employed for the preparation of the supported Ru–MoOx catalysts using a procedure similar to that used for the synthesis of Ru–MoOx/TiO2 (XRD patterns of Ru–MoOx on various supports are shown in Fig. S6, in the ESI†). Ru–MoOx supported on carbon (Ru–MoOx/C) gave high yield of dodecan-1-ol (89%) at >99% conversion of lauric acid with small amount of n-dodecane yield (9%) (entry 1). These results are very consistent with previous reports of Takeda et al. that the presence of MoOx species in Ru–MoOx/C catalyst enhanced the selectivity of diols or primary alcohols from aqueous phase hydrogenation of lactic acid and various low carbon number of carboxylic acids.71 Interestingly, Ru–MoOx supported on carbon doped-titanium oxide (Ru–MoOx/C–TiO2), which was obtained from one-pot hydrothermal of TiCl4 and glucose solutions at 150 °C for 24 h as reported previously (XRD patterns are shown in Fig. S7, in the ESI†),72,73 gave dodecane-1-ol (68%), n-dodecane (23%), and others (8%) (entry 2). Ru–MoOx supported on γ-Al2O3 and SiO2 catalysts showed lower lauric acid conversion (72–80%) and produced dodecane-1ol (52–63%), n-dodecane (10–12%), and others (∼8%) (entries 3–4). Moreover, Ru–MoOx/ZrO2 catalyst produced only dodecane-1-ol (53%) without the formation of n-dodecane under the same reaction conditions (entry 5). The effectiveness of TiO2 or C–TiO2 supported Ru–MoOx catalysts in the hydroconversion of lauric acid to alcohol and alkane were superior than the other supported catalysts. These results can be attributed to the synergistic action between MoOx species and reducible support (e.g. TiO2). Intimate interaction between Ru–MoOx and support with different energy of electron affinity may affect to the electron state of Ru.13,74 In addition, our best result of alkane yield was obtained using Ru–(0.026)MoOx/TiO2 catalyst at 230 oC, 40 bar 7 h (80% in yield) (entry 6), which comparable to the results obtained by using Pt–MoOx/C (entries 7 and 8)13 and much higher than that of Ni3Sn2/TiO2 catalyst (entry 9)35 (Table 3).
| Entry | Catalysta | Conversionb (%) | Yieldb(%) | Ref. | ||
|---|---|---|---|---|---|---|
| Dodecane-1-ol | n-Dodecane | Othersc | ||||
| a The amount of Mo was arround 0.025 mmol (based on the precursor). Reaction conditions: catalyst, 0.065 g; lauric acid, 3.2 mmol; solvent, 2-propanol: H2O, 5 mL (4.0:1.0 volume ratio), 170 °C; H2 40 bar, 7 h.b Conversion and yield were determined by GC using an internal standard technique.c Dodecyl dodecanoate is included as the esterification of lauric acid and dodecane-1-ol. The carbon balance was 93–96% for all the catalysts.d At 230 °C, 40 bar, 7 h.e At 180 °C, 80 bar, 4 h.f At 160 °C, 30 bar 20 h. |
||||||
| 1 | Ru–MoOx/C | >99 | 89 | 9 | 2 | This work |
| 2 | Ru–MoOx/C–TiO2 | 99 | 68 | 23 | 8 | This work |
| 3 | Ru–MoOx/γ-Al2O3 | 80 | 63 | 10 | 7 | This work |
| 4 | Ru–MoOx/SiO2 | 72 | 52 | 12 | 8 | This work |
| 5 | Ru–MoOx/ZrO2 | 55 | 53 | 0 | 2 | This work |
| 6d | Ru–MoOx/TiO2 | >99 | 20 | 80 | 0 | This work |
| 7e | Pt–MoOx/TiO2 | >99 | 2 | 86 | 8 | 13 |
| 8e | Pt/Nb2O5 | >99 | 7 | 60 | 21 | 74 |
| 9f | Ni3Sn2/TiO2 | 85 | 80 | 3 | 2 | 34 |
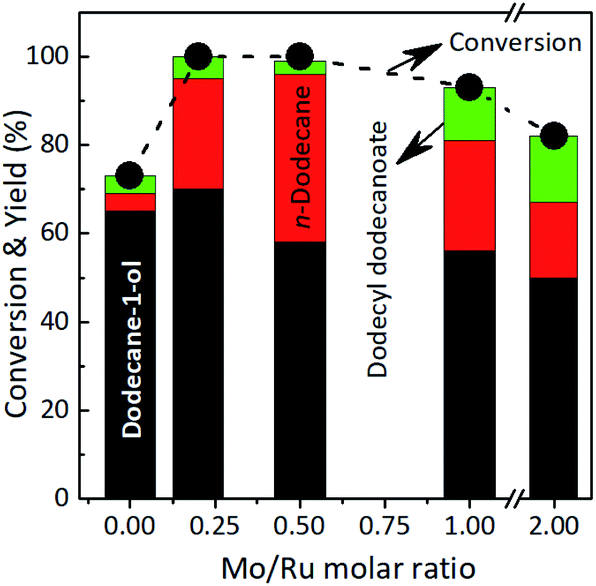 | ||
| Fig. 1 Results of product distribution (yield) from selective hydroconversion of lauric acid using Ru–MoOx/TiO2 catalysts with different Mo loading amounts. Reaction conditions: catalyst, 0.065 g; lauric acid, 3.2 mmol; solvent, 2-propanol: H2O, 5 mL (4.0:1.0 volume ratio); H2 40 bar, 170 °C, 7 h. | ||
Effect of the temperature reduction. To get the insight into the role of MoOx species during the reaction, the catalytic reactions using Ru–(0.026)MoOx/TiO2 catalyst was reduced with hydrogen different reduction temperatures, ca. 300 °C, 400 °C, 500 °C, and 600 °C (The XRD patterns of those catalysts are shown in Fig. S8, in the ESI†) and the results are shown in Fig. 2. By using Ru–(0.026)MoOx/TiO2 300 °C catalyst, the main product was dodecane-1-ol (58%) while n-dodecane and ester were 12% and 30%, respectively at complete reaction. When the catalyst was reduced at 400 °C, the yield of dodecane-1-ol reach to maximum (61%) while n-dodecane remarkably increased to 38%. Over this catalyst, the esterification reaction of lauric acid and dodecane-1-ol to formation dodecyl dodecanoate was inhibited as indicated by only small amount of ester (1%). Further increase the temperature reduction to 500 °C and 600 °C, yield of dodecyl dodecanoate slightly increased to 3% and 5%. However, the maximum yield of n-dodecane (44%) was obtained over Ru–(0.026)MoOx/TiO2 500 °C catalyst at full conversion of lauric acid. From these results, we fixed the catalytic hydroconversion of lauric acid with the Ru–(0.026)MoOx/TiO2 catalyst reduced at 400–500 °C as the most effective catalyst system.
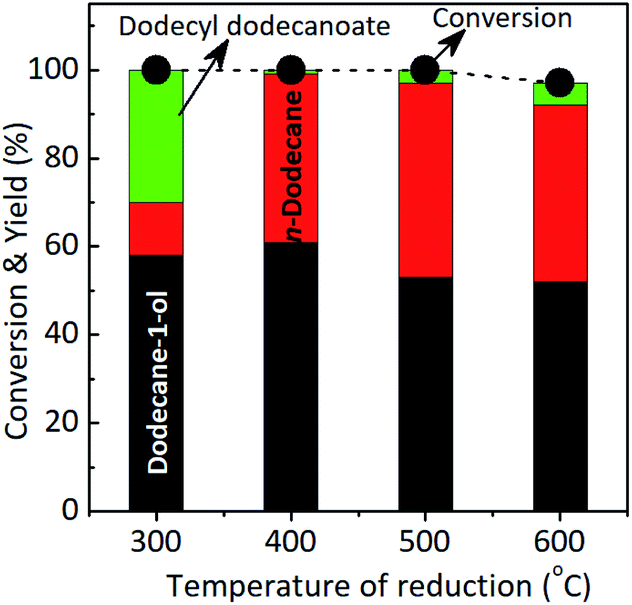 | ||
| Fig. 2 Results of product distribution (yield) from selective hydroconversion of lauric acid using Ru–(0.026)MoOx/TiO2 catalyst with different Mo loading amounts. Reaction conditions: catalyst, 0.065 g; lauric acid, 3.2 mmoL; solvent, 2-propanol: H2O, 5 ml (4.0:1.0 volume ratio); H2 40 bar, 170 °C, 7 h. | ||
 | ||
| Fig. 3 Effect of reaction temperature on the conversion and yield in the hydroconversion of lauric acid over Ru–(0.026)MoOx/TiO2 catalyst. Reaction conditions: catalyst, 0.065 g; lauric acid, 3.2 mmol; solvent, 2-propanol: H2O, 5.0 mL (4.0:1.0 volume ratio); H2 40 bar, 7 h. | ||
The conversion of lauric acid increased gradually as the reaction temperature was increased from 130 °C and completed reaction (100% conversion) was achieved at 170 °C. The maximum yield of dodecane-1-ol was obtained at 150 °C then gradually decreased at 160 to 230 °C which is proportional with the increase of n-dodecane yield. The hydrodeoxygenation (via dehydration-hydrogenation) of lauric acid took place firstly to form dodecane-1-ol at lower temperature ≤150 °C and at the same time, the esterification of lauric acid and dodecane-1-ol to form dodecyl dodecanoate was also occurred at that of reaction temperature. At the reaction temperatures of 170–230 °C, the direct hydrodeoxygenation of lauric acid or subsequently take place the dehydration–hydrogenation reaction of formed dodecane-1-ol which are preferable occurred to yield n-dodecane. There no undecane product was observed, suggesting that decarbonylation/decarboxylation reaction did not proceed over the studied catalysts under current reaction conditions. Additionally, the esterification of lauric acid and formed dodecane-1-ol was also totally disappeared at that of the reaction temperatures. These results are good consistent with the report of Kon et al. who reported the hydrodeoxygenation of lauric acid using Pt–MoOx/TiO2 catalyst and produced dodecane (86% yield) at 100% conversion of lauric acid without the formation of dodecyl dodecanoate at 180 °C, 80 bar H2 and 3 h.13
 | ||
| Fig. 4 Effect of initial H2 pressure on the conversion and yield in the hydroconversion of lauric acid over Ru–(0.026)MoOx/TiO2 catalyst. Reaction conditions: catalyst, 0.065 g; lauric acid, 3.2 mmol; solvent, 2-propanol: H2O, 5.0 ml (4.0:1.0 volume ratio); 170 °C, 7 h. | ||
 | ||
| Fig. 5 (a) Time profiles of the hydroconversion of lauric acid; (b) yield of dodecane-1-ol as a function of time profiles of the hydroconversion of lauric acid; (c) yield of dodecane as a function of time profiles of the hydroconversion of lauric acid; and (d) yield of dodecyl dodecanoate as a function of time profiles of the hydroconversion of lauric acid over the Ru–(0.026)MoOx/TiO2 catalyst. Reaction conditions: catalyst, 0.065 g; lauric acid, 3.2 mmol; solvent, 2-propanol: H2O, 5 ml (4.0:1.0 volume ratio); 130–190 °C, H2 40 bar, 0–21 h. | ||
Fig. 5a shows the profile of lauric acid conversion as a function of reaction time at different reaction temperatures ca. 130 °C, 150 °C, 170 °C, and 190 °C. At 130 °C, the conversion of lauric acid increased smoothly as function of reaction time and the conversion achieved to 100% after 20 h. At 150 °C, the completed reaction was achieved after 10 h indicating that the conversion of lauric acid took place faster (approximately two times) than that of 130 °C. As the reaction temperatures were increased to 170 °C and 190 °C, the reaction rate of lauric acid conversion become faster as indicated by the completed reaction after 7 h and 5 h, respectively.
Fig. 5b displays the profile of dodecane-1-ol yield as function of reaction time at different temperatures. At 130 °C, the formation of dodecane-1-ol was observed firstly after 7 h, then increased gradually to reach maximum yield of 73% at 100% conversion lauric acid after 21 h. The highest yield of dodecane-1-ol (75%) was obtained at reaction temperature of 150 °C after 11 h and the amount of dodecane-1-ol slightly decreased to 70% when the reaction time was prolonged to 15 h. The decrease of dodecane-1-ol yield can be attributed to due to the further reaction of dodecane-1-ol (via dehydration/hydrogenation) to n-dodecane or esterification with lauric acid to form dodecyl dodecanoate. Kon et al. suggested that the esterification between dodecane-1-ol and lauric acid (dodecanoate) gave dodecyl dodecanoate in the presence of Pt/Nb2O5 or Rh–MoOx/TiO2 catalysts at 180 °C.13 However, in our Ru–(0.026)MoOx/TiO2 catalyst, the significant formation of dodecyl dodecanoate is favourable occurred at reaction temperature of ≤150 °C (Fig. 3) or at low initial H2 pressure (Fig. 4). The yield of dodecyl dodecanoate (Fig. 5c) increased to reach maximum yield of 39% at reaction temperature 130 °C after 9 h, then getting decrease as reaction time was prolonged up to 21 h. On the other hand, the amount of ester product not more than 18% when the reaction temperature increased to 150–190 °C, suggesting that further hydrogenation of ester was favourable occurred at high reaction rate. Results of catalytic performance of various Ru–MoOx catalysts confirm that the amount of remained ester product <0.1 and 0 at the reaction temperature of 170 °C and 190 °C, respectively. Moreover, the profiles of n-dodecane yield (Fig. 5d) also confirmed that the proposed reaction pathway in Scheme 1 is basically consistent with those for the hydrodeoxygenation of fatty acids by Pt/Nb2O574 and Pt–MoOx/TiO213 catalysts.
Table 4 compares the results for hydrogenation of possible intermediates (methyl laurate, dodecanal, dodecane-1-ol, dodecyl dodecanoate) and lauric acid under the same reaction conditions. We obtained that the reaction rate of oxygenate conversion changed in the following order: 1-dodecanal (9.17 mmol gcat−1 min−1) > dodecane-1-ol (9.17 mmol gcat−1 min−1) > methyl laurate (6.46 mmol gcat−1 min−1) ≈ lauric acid (6.17 mmol gcat−1 min−1)> >> dodecyl dodecanoate (0.91 mmol gcat−1 min−1). Lauric acid showed highest conversion to produce 34% yield of dodecane-1-ol, 27% yield n-dodecane and 8% ester (dodecyl dodecanoate (entry 1). The formation of ester both in alcoholic or 2-propanol/H2 mixture solvents took place faster than that of n-dodecane as indicated by the high yield of ester (Table 1, entries 1–3) and (Table 4, entry 1). Notably, lauric acid underwent esterification with dodecane-1-ol at low reaction temperature (Fig. 3) or in low initial H2 pressure (Fig. 4). Dodecane-1-ol can also undergo esterification with lauric acid to give dodecyl dodecanoate, which is then hydrogenated to n-dodecane via dodecane-1-ol. Therefore, it can concluded that the hydroconversion of lauric acid in the presence of Ru–(y)MoOx/TiO2 catalysts follows the proposed reaction pathway as shown in Scheme 1.
| Entry | Substrate | Conversionb (%) | Yieldb (%) | Rate of reactant reaction (rR) (mmol gcat−1 min−1) | Rate of product formation (rP) (mmol gcat−1 min−1) | ||
|---|---|---|---|---|---|---|---|
| Dodecane-1-ol | n-Dodecane | Esterc | |||||
| a Reaction conditions: catalyst, 0.065 g; lauric acid, 3.2 mmol; solvent, 2-propanol: H2O, 5.0 ml (4.0:1.0 volume ratio); H2 40 bar; 170 °C, 3 h.b Conversion and yield were determined by GC using an internal standard technique.c Ester was dodecyl dodecanoate. The carbon balance was more than 96% for all the reactions. |
|||||||
| 1 | Lauric acid | 69 | 34 | 27 | 8 | 6.17 | 0.14 |
| 2 | Methyl laurate | 50 | 36 | 14 | 0 | 6.46 | 0.07 |
| 3 | 1-Dodecanal | 67 | 37 | 30 | 0 | 9.17 | 0.27 |
| 4 | Dodecane-1-ol | 53 | 0 | 53 | 0 | 8.98 | 0.23 |
| 5 | Dodecyl dodecanoate | 7 | 0 | 5 | 0 | 0.91 | 0.02 |
| Entry | Substrate | Time (h) | Conversionb (%) | Main product | Selectivityb (%) | |
|---|---|---|---|---|---|---|
| Alcohol | Alkanes | |||||
| a Reaction conditions: catalyst, 0.065 g; lauric acid, 3.2 mmol; solvent, 2-propanol/H2O, 5.0 ml (4.0:1.0 volume ratio); H2 40 bar; 170 °C, 5–15 h.b Conversion and yield were determined by GC using an internal standard technique.c Reaction temperature was 110 °C. The carbon balance was more than 93–96% for all the reactions. 1,4-PeD = 1,4-Pentanediol. 1,4-BeD = 1,4-Butanediol. GVL = γ-Valerolactone. GBL = γ-Butyrolactone. |
||||||
| 1 | Octadecanoic acid (stearic acid) | 15 | 92 | Octadecanol | 79 | 21 |
| 2 | Hexadecanoic acid (palmitic acid) | 9 | 83 | Hexadecanol | 80 | 20 |
| 3 | Methyl palmitate | 9 | 74 | Hexadecanol | 82 | 18 |
| 4 | Tetradecanoic acid (myristic acid) | 5 | 76 | Tetradecanol | 74 | 26 |
| 5 | Dodecanoic acid (lauric acid) | 7 | >99 | Dodecan-1-ol | 61 | 39 |
| 6 | Nonanoic acid | 5 | 89 | Nonanol | 89 | 11 |
| 7 | Octanoic acid | 5 | 79 | Octanol | 87 | 13 |
| 8 | Hexanoic acid | 3 | 78 | Hexanol | 91 | 9 |
| 9c | Valeric acid | 3 | 83 | 1-Pentanol | 94 | 6 |
| 10c | Levulinic acid | 3 | >99 | 1,4-PeD(GVL) | 81(19) | — |
| 11c | Succinic acid | 3 | 73 | 1,4-BeD(GBL) | 63(37) | — |
The reusability of Ru–(0.026)MoOx/TiO2 catalyst was studied with the recovered catalyst regenerated by washing with acetone following by pre-reduction in H2 at 400 °C for 2 h and then the hydroconversion of lauric acid repeated. The regenerated Ru–(0.026)MoOx/TiO2 catalyst showed loss in the hydrogenation activity with an associated in reaction rate. A further loss in the hydrogenation activity was observed on recycling the catalyst for a second time. Some of the decrease in activity is likely to be due to aggregation of active metal during the reaction or the collaps of catalyst structures (XRD patterns and TEM images of recovered Ru–(0.026)MoOx/TiO2 catalyst are shown in Fig. S9 and S10, in the ESI†).
Experimental
Catalyst preparation
A typical procedure for the synthesis of Ru–(0.026)MoOx/TiO2 (0.026 is Mo loading amount Mo) described as follows:75 RuCl3 (0.049 mmol) was dissolved in deionized water (denoted as solution A) and (NH4)6Mo7O24·4H2O (0.026 mmol) was dissolved in ethanol/ethylene glycol (2.0/1.0 volume ratio) (denoted as solution B) at room temperature. Solutions A, B, and TiO2 (as support material; 1.0 g) were mixed at room temperature; the temperature was subsequently raised to 50 °C under gentle stirring for 12 h. The pH of the mixture was adjusted to 12 through the dropwise addition of an aqueous solution of NaOH (3.1 M). The mixture was then placed into a sealed-Teflon autoclave for the hydrothermal process at 150 °C for 24 h. The resulting black precipitate was filtered, washed with distilled water, and then dried under vacuum overnight. Prior to the catalytic reaction, the obtained black powder was treated under hydrogen at 500 °C for 3 h.Catalyst characterization
The X-ray diffraction (XRD) analysis was performed on a Miniflex 600 Rigaku instrument with Cu as monochromatic source of CuKα radiation (λ = 0.15444 nm). The XRD was operated at 40 kV and 15 mA with a step width of 0.02°, a scan speed of 4° min−1 (α1 = 0.154057 nm, α2 = 0.154433 nm), solar slit 1.25°, and using a Ni Kβ filter. ICP measurements were performed on an SPS 1800H plasma spectrometer by Seiko Instruments Inc. Japan (Ni: 221.7162 nm and Sn: 189.898 nm).
The BET surface area (SBET) and pore volume (Vp) were measured using N2 physisorption at −196 °C on a Belsorp Max (BEL Japan). The samples were degassed at 200 °C for 2 h to remove physisorbed gases prior to the measurement. The amount of nitrogen adsorbed onto the samples was used to calculate the Brunauer–Emmett–Teller (BET) surface area via the BET equation. The pore volume was estimated to be the liquid volume of nitrogen at a relative pressure of approximately 0.995 according to the Barrett–Joyner–Halenda (BJH) approach based on desorption data.76
The NH3–TPD was carried out on a Belsorp Max (BEL Japan). The samples were degassed at elevated temperature of 100–200 °C for 2 h to remove physisorbed gases prior to the measurement. The temperature was then kept at 200 °C for 2 h while flushed with He gas. NH3 gas (balanced NH3, 80% and He, 20%) was introduced at 100 °C for 30 min, then evacuated by helium gas to remove the physisorbed also for 30 min. Finally, temperature programmed desorption was carried out at temperature of 100–800 °C and the desorbed NH3 was monitored by TCD. SEM images of the synthesised catalysts were taken on a JEOL JSM-610 SEM after the samples were coated using a JEOL JTC-1600 auto fine coater. The TEM images were taken on Hitachi H7650 at 19 kV.
The H2 uptake was determined through irreversible H2 chemisorption. After the catalyst was heated at 100 °C under vacuum for 30 min, it was heated at 400 °C under H2 for 30 min. The catalysts were subsequently cooled to room temperature under vacuum for 30 min. The H2 measurement was conducted at 0 °C, and H2 uptake was calculated according to the method described in the literature.77
Catalytic reaction
The calibration curve was performed using known concentrations of internal standard, reactants and products in order to determine the correct response factors. The conversion of dodecanoic acid, yield and selectivity of the products were calculated according to the following equations:
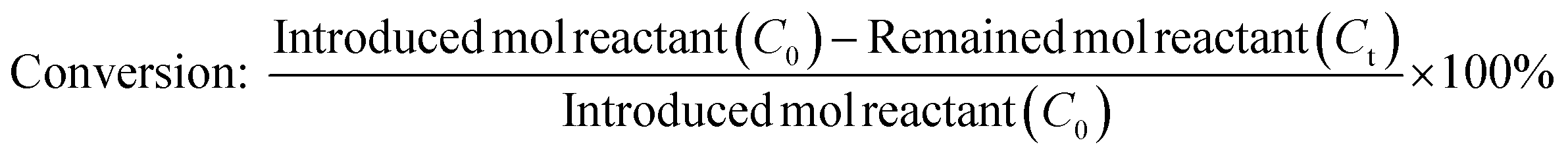
where Co is the introduced mol reactant (dodecanoic acid), Ct is the remaining mol reactant, and ΔC is the consumed mol reactant (introduced mol reactant – remained mol reactant), which are all obtained from GC analysis using an internal standard technique.
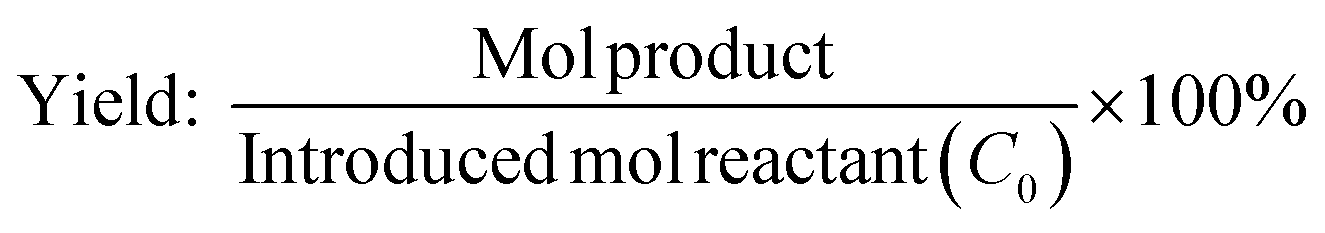
The apparent reaction rates were calculated using the following equation:
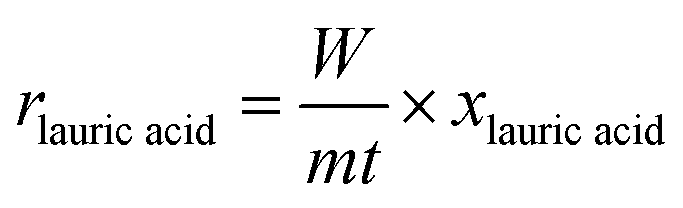
The formation rates of the products were calculated using the following equation:

Conclusions
We described the selective hydroconversion of coconut oil derived lauric acid to lauryl alcohol and n-dodecane in 2-propanol/water (4.0/1.0 v/v) mixture solvent using molybdenum oxide-modified ruthenium on titanium oxide (Ru–(y)MoOx/TiO2; y = Mo loading amount, mmol g−1) catalysts. Among the Ru–(y)MoOx/TiO2 catalysts tested, Ru–(0.026)MoOx/TiO2 (Mo loading amount of 0.026 mmol g−1) shows the highest yields of n-alkanes for hydroconversion of coconut oil derived lauric acid and hydroconversion of various aliphatic fatty acids C8–C18 precursor at 170–230 °C, 30–40 bars for 7–20 h. At lower reaction temperatures (130 ≥ T ≤ 150 °C), the main product was dodecane-1-ol and dodecyl dodecanoate as the results of hydrogenation of lauric acid and esterification of lauric acid and corresponding alcohols. The presence of MoOx/TiO2 might played as Lewis acid sites which cooperated with Ru nanoparticles as H2 dissociation sites, led to high activity of Ru–(y)MoOx/TiO2 catalysts, and suppressed the HDCO2 or HDCO reactions, thus produced high aliphatic alcohols and aliphatic alkanes with same carbon number. Over the Ru–(0.026)MoOx/TiO2 as the best catalyst, an increase in reaction temperature up to 230 °C significantly enhanced the degree of hydrodeoxygenation of lauric acid and produced n-dodecane with maximum yield (up to 80%) at 230 °C, H2 40 bar for 7 h. Ru–(0.026)MoOx/TiO2 is also effective for the hydroconversion of various dicarboxylic acids to the corresponding lactones and diols.Author contributions
Rodiansono: conceptualization, methodology, investigation, writing – original draft, writing – review & editing. Heny Puspita Dewi, Kamilia Mustikasari, Maria Dewi Astuti, Sadang Husain, and Sutomo: formal analysis, investigation, and visualization.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the RISET DASAR FY 2019–2021 (contract number DIPA-042.06-1.401516/2020) and RISET DASAR FY 2021–2023 (119/E4.1/AK.04.PT/2021) from Ministry of Education, Culture, Research and Technology, Indonesian Government for financially supported this work. These works were partially supported by BPDP-Sawit, Ministry of Finances through GRK-2016 under contract number PRJ-49/2016.Notes and references
- N. Hongloi, P. Prapainainar and C. Prapainainar, Mol. Catal., 2021, 111696 Search PubMed
.
- X. Yao, T. J. Strathmann, Y. Li, L. E. Cronmiller, H. Ma and J. Zhang, Green Chem., 2021, 23, 1114–1129 RSC
- H. I. Mahdi, A. Bazargan, G. McKay, N. I. W. Azelee and L. Meili, Chem. Eng. Res. Des., 2021, 174, 158–187 CrossRef CAS
- M. A. Sánchez, G. C. Torres, V. A. Mazzieri and C. L. Pieck, J. Chem. Technol. Biotechnol., 2017, 92, 27–42 CrossRef
- U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed
- K. Noweck and W. Grafahrend, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2006, vol. 14, pp. 117–141 Search PubMed
- S. Lestari, P. Mäki-Arvela, J. Beltramini, G. Q. M. Lu and D. Y. Murzin, ChemSusChem, 2009, 2, 1109–1119 CrossRef CAS PubMed
- M. W. Schreiber, D. Rodriguez-Niño, O. Y. Gutiérrez and J. A. Lercher, Catal.: Sci. Technol., 2016, 6, 7976–7984 RSC
- G. J. S. Dawes, E. L. Scott, J. Le Nôtre, J. P. M. Sanders and J. H. Bitter, Green Chem., 2015, 17, 3231–3250 RSC
- K. C. Sembiring and S. Saka, J. Jpn. Pet. Inst., 2019, 62, 157–172 CrossRef CAS
- M. Tamura, Y. Nakagawa and K. Tomishige, Asian J. Org. Chem., 2020, 9, 126–143 CrossRef CAS
- M. Mohammad, T. Kandaramath Hari, Z. Yaakob, Y. Chandra Sharma and K. Sopian, Renewable Sustainable Energy Rev., 2013, 22, 121–132 CrossRef CAS
- K. Kon, T. Toyao, W. Onodera, S. M. A. H. Siddiki and K. I. Shimizu, ChemCatChem, 2017, 9, 2822–2827 CrossRef CAS
- Ł. Jȩczmionek and K. Porzycka-Semczuk, Fuel, 2014, 131, 1–5 CrossRef
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. J. Kim, G. W. Huber and J. Lee, Green Chem., 2019, 21, 3715–3743 RSC
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC
- J. Pritchard, G. A. Filonenko, R. Van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed
- P. Mäki-Arvela, M. Snåre, K. Eränen, J. Myllyoja and D. Y. Murzin, Fuel, 2008, 87, 3543–3549 CrossRef
- J. Zhang, X. Huo, Y. Li and T. J. Strathmann, ACS Sustainable Chem. Eng., 2019, 7, 14400–14410 CrossRef CAS
- J. Wu, J. Shi, J. Fu, J. A. Leidl, Z. Hou and X. Lu, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed
- L. Yang, K. L. Tate, J. B. Jasinski and M. A. Carreon, ACS Catal., 2015, 5, 6497–6502 CrossRef CAS
- I. Simakova, O. Simakova, P. Mäki-Arvela and D. Y. Murzin, Catal. Today, 2010, 150, 28–31 CrossRef CAS
- N. Chen, Y. Ren and E. W. Qian, J. Catal., 2016, 334, 79–88 CrossRef CAS
- R. Rodiansono, M. I. Pratama, M. D. Astuti, A. Abdullah, A. Nugroho and S. Susi, Bull. Chem. React. Eng. Catal., 2018, 13, 311–319 CrossRef CAS
- K. Kon, W. Onodera, S. Takakusagi and K. I. Shimizu, Catal.: Sci. Technol., 2014, 4, 3705–3712 RSC
- F. Liu, J. Ftouni, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemCatChem, 2019, 11, 2079–2088 CrossRef CAS
- X. Cao, F. Long, Q. Zhai, J. Zhao, J. Xu and J. Jiang, Fuel, 2021, 298, 120829 CrossRef CAS
- K. Tomishige, Y. Nakagawa and M. Tamura, Green Chem., 2017, 19, 2876–2924 RSC
- V. M. Deshpande, K. Ramnarayan and C. S. Narasimhan, J. Catal., 1990, 182, 174–182 CrossRef
- T. Miyake, T. Makino, S. ichi Taniguchi, H. Watanuki, T. Niki, S. Shimizu, Y. Kojima and M. Sano, Appl. Catal., A, 2009, 364, 108–112 CrossRef CAS
- V. M. Deshpande, W. R. Patterson and C. S. Narasimhan, J. Catal., 1990, 121, 165–173 CrossRef CAS
- C. Huang, H. Zhang, Y. Zhao, S. Chen and Z. Liu, J. Colloid Interface Sci., 2012, 386, 60–65 CrossRef CAS PubMed
- R. Rodiansono, M. I. Pratama, M. D. Astuti, A. Abdullah, A. Nugroho and S. Susi, Bull. Chem. React. Eng. Catal., 2018, 13, 311–319 CrossRef CAS
- Y. Shao, Q. Xia, X. Liu, G. Lu and Y. Wang, ChemSusChem, 2015, 8, 1761–1767 CrossRef CAS PubMed
- R. Rodiansono, E. Hayati, A. S. Azzahra, M. D. Astuti, K. Mustikasari, S. Husain and S. Sutomo, Bull. Chem. React. Eng. Catal., 2021, 16, 888–903 CrossRef CAS
- Z. Luo, Q. Bing, J. Kong, J. Y. Liu and C. Zhao, Catal.: Sci. Technol., 2018, 8, 1322–1332 RSC
- M. J. Hidajat, G. N. Yun and D. W. Hwang, Mol. Catal., 2021, 512, 111770 CrossRef CAS
- S. Liu, W. Zheng, J. Fu, K. Alexopoulos, B. Saha and D. G. Vlachos, ACS Catal., 2019, 9, 7679–7689 CrossRef CAS
- T. Asano, Y. Nakagawa, M. Tamura and K. Tomishige, ACS Sustainable Chem. Eng., 2019, 7, 9601–9612 CrossRef CAS
- T. Arai, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2016, 9, 1680–1688 CrossRef CAS PubMed
- Y. Takeda, T. Shoji, H. Watanabe, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ChemSusChem, 2015, 8, 1170–1178 CrossRef CAS PubMed
- T. Asano, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2016, 4, 6253–6257 CrossRef CAS
- Y. Cao, H. Zhang, K. Liu, Q. Zhang and K. J. Chen, ACS Sustainable Chem. Eng., 2019, 7, 12858–12866 CrossRef CAS
- Y. Yang, Q. Liu, D. Li, J. Tan, Q. Zhang, C. Wang and L. Ma, RSC Adv., 2017, 7, 16311–16318 RSC
- Y. Zheng, Z. Tang and S. G. Podkolzin, Chem.–Eur. J., 2020, 26, 5174–5179 CrossRef CAS PubMed
- S. Liu, W. Zheng, J. Fu, K. Alexopoulos, B. Saha and D. G. Vlachos, ACS Catal., 2019, 9, 7679–7689 CrossRef CAS
- S. Koso, H. Watanabe, K. Okumura, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2012, 111–112, 27–37 CrossRef CAS
- K. I. Shimizu, S. Kanno and K. Kon, Green Chem., 2014, 16, 3899–3903 RSC
- J. Guan, G. Peng, Q. Cao and X. Mu, J. Phys. Chem. C, 2014, 118, 25555–25566 CrossRef CAS
- K. Tomishige, Y. Nakagawa and M. Tamura, Curr. Opin. Green Sustainable Chem., 2020, 22, 13–21 CrossRef
- J. A. Bezard, Lipids, 1971, 6, 630–634 CrossRef CAS
- J. Bezard, M. Bugaut and G. Clement, J. Am. Oil Chem. Soc., 1971, 48, 134–139 CrossRef CAS
- R. Rodiansono, M. D. Astuti, T. Hara, N. Ichikuni and S. Shimazu, Catal. Sci. Technol., 2016, 6, 2955–2961 RSC
- R. Rodiansono, M. D. Astuti, A. Ghofur and K. C. Sembiring, Bull. Chem. React. Eng. Catal., 2015, 10, 192–200 CAS
- A. P. Damayanti, H. P. Dewi, I. Ibrahim and R. Rodiansono, in IOP Conference Series: Materials Science and Engineering, 2020, vol. 980, p. 012013 Search PubMed
- I. Ibrahim, M. Riski and R. Rodiansono, in IOP Conference Series: Materials Science and Engineering, 2020, vol. 980, p. 012012 Search PubMed
- D. R. Vardon, B. K. Sharma, H. Jaramillo, D. Kim, J. K. Choe, P. N. Ciesielski and T. J. Strathmann, Green Chem., 2014, 16, 1507–1520 RSC
- J. Zhang, X. Huo, Y. Li and T. J. Strathmann, ACS Sustainable Chem. Eng., 2019, 7, 14400–14410 CrossRef CAS
- J. Fu, X. Lu and P. E. Savage, ChemSusChem, 2011, 4, 481–486 CrossRef CAS PubMed
- E. A. Cepeda, R. Bravo and B. Calvo, J. Chem. Eng. Data, 2009, 54, 1371–1374 CrossRef CAS
- K. Yui, Y. Itsukaichi, T. Kobayashi, T. Tsuji, K. Fukui, K. Maeda and H. Kuramochi, J. Chem. Eng. Data, 2017, 62, 35–43 CrossRef CAS
- A. B. Gonçalves Bonassoli, G. Oliveira, F. H. Bordón Sosa, M. P. Rolemberg, M. A. Mota, R. C. Basso, L. Igarashi-Mafra and M. R. Mafra, J. Chem. Eng. Data, 2019, 64, 2084–2092 CrossRef
- M. A. Mellmer, C. Sener, J. M. R. Gallo, J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Angew. Chem., Int. Ed., 2014, 53, 11872–11875 CrossRef CAS PubMed
- M. A. Mellmer, C. Sener, J. M. R. Gallo, J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Angew. Chem., 2014, 126, 12066–12069 CrossRef
- T. Asano, Y. Nakagawa, M. Tamura and K. Tomishige, ACS Sustainable Chem. Eng., 2019, 7, 9601–9612 CrossRef CAS
- H. G. Manyar, C. Paun, R. Pilus, D. W. Rooney, J. M. Thompson and C. Hardacre, Chem. Commun., 2010, 46, 6279–6281 RSC
- T. Mizugaki, Y. Nagatsu, K. Togo, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, Green Chem., 2015, 17, 5136–5139 RSC
- J. Cui, J. Tan, Y. Zhu and F. Cheng, ChemSusChem, 2018, 11, 1316–1320 CrossRef CAS PubMed
- T. Toyao, S. Kayamori, Z. Maeno, S. M. A. H. Siddiki and K. Shimizu, ACS Catal., 2019, 9, 8187–8196 CrossRef CAS
- Y. Takeda, T. Shoji, H. Watanabe, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ChemSusChem, 2015, 8, 1170–1178 CrossRef CAS PubMed
- H. P. Dewi, J. Santoso, N. F. Trianda and R. Rodiansono, J. Kim. Sains Apl., 2019, 22, 299–304 CrossRef CAS
- K. Putri, A. Annisa, S. Husain and R. Rodiansono, Bull. Chem. React. Eng. Catal., 2020, 15, 35–42 CrossRef CAS
- K. Kon, W. Onodera, S. Takakusagi and K. I. Shimizu, Catal.: Sci. Technol., 2014, 4, 3705–3712 RSC
- R. Rodiansono, S. Khairi, T. Hara, N. Ichikuni and S. Shimazu, Catal.: Sci. Technol., 2012, 2, 2139–2145 RSC
- S. Lowell, J. E. Shields, M. A. Thomas and M. Thommes, Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density, Springer Netherlands, Dordrecht, 2004, vol. 16 Search PubMed
- C. H. Bartholomew and R. B. Pannell, J. Catal., 1980, 65, 390–401 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation of catalyst materials: physico-chemical properties, XRD, NH3-TPD, pyridine adsorption, TEM images. See https://doi.org/10.1039/d2ra02103j |
| This journal is © The Royal Society of Chemistry 2022 |