DOI:
10.1039/D2RA00990K
(Paper)
RSC Adv., 2022,
12, 14492-14501
Synthesis and evaluation of new pirfenidone derivatives as anti-fibrosis agents†
Received
15th February 2022
, Accepted 16th April 2022
First published on 13th May 2022
Abstract
Two series of new pirfenidone derivatives, in which phenyl groups or benzyl groups are attached to the nitrogen atom of the pyridin-2(1H)-one moiety were synthesized and evaluated as anti-fibrosis agents. Among them, compound 5d, with a (S)-2-(dimethylamino) propanamido group in the R2 position (series 1) exhibited 10 times the anti-fibrosis activity (IC50: 0.245 mM) of pirfenidone (IC50: 2.75 mM). Compound 9d (series 2) gave an IC50 of 0.035 mM against the human fibroblast cell line HFL1. The mechanism of the optimal compound inhibiting fibrosis was also studied.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive lung disease associated with high morbidity and mortality and included in the group of interstitial lung diseases of unknown etiology.1 Usually, patients with IPF have a median survival of only 3–5 years, worse than many common cancers.2 The worldwide prevalence of IPF may be close to 60 cases per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 inhabitants and predominantly affects males over 65 years of age;3 especially, since the outbreak of the COVID-19 pneumonia in late 2019, hundreds of millions of people have been infected and millions of people have died worldwide. Lung injury in severe COVID-19 pneumonia can rapidly evolve to established pulmonary fibrosis, with prognostic implications in the acute phase of the disease and long-lasting impact on the quality of life of COVID-19 survivors.4
000 inhabitants and predominantly affects males over 65 years of age;3 especially, since the outbreak of the COVID-19 pneumonia in late 2019, hundreds of millions of people have been infected and millions of people have died worldwide. Lung injury in severe COVID-19 pneumonia can rapidly evolve to established pulmonary fibrosis, with prognostic implications in the acute phase of the disease and long-lasting impact on the quality of life of COVID-19 survivors.4
Pirfenidone (5-methyl-1-phenyl-2[1H]-pyridone) exhibits anti-fibrotic, anti-oxidant and anti-inflammatory effects to reduce lung collagen synthesis and deposition.5–7 In multinational phase III clinical trials, pirfenidone reduced disease progression as reflected by pulmonary function, exercise tolerance, and progression-free survival in patients with IPF,8,9 and was approved by the US FDA in October 2014. Pirfenidone is also one of the top 200 drugs sold in the world in 2020. Except for IPF, in vitro and in vivo studies have also confirmed its effect on the inhibition of myofibroblast differentiation, proliferation, and profibrotic cytokine release in liver,10 heart,11 and renal tissues.12 Liver fibrosis is often a result of wound-healing response to chronic liver injuries. Recently, pirfenidone was reported to inhibit liver fibrosis by targeting the small oxidoreductase glutaredoxin-1.13 The heart usually suffers from various causes. A new trial suggests that pirfenidone may also reduce myocardial fibrosis in patients with heart failure, having a preserved left ventricular ejection fraction.14 It was estimated that there were more than 500 million people suffering from fibrosis caused by renal diseases.15 Pirfenidone on renal protection, inhibition of renal fibrosis, and its mechanisms, signaling pathways, and preclinical evidence have been reported.16,17
The most commonly reported adverse reactions of pirfenidone were gastrointestinal and skin-related events. In clinical trials, a 51.7% incidence of photo-sensitivity was noted after the administration of pirfenidone.18–20 As there are only two drugs approved for the treatment of IPF at present, nintedanib and pirfenidone, it is of great importance to develop new drugs with better anti-fibrotic activity and lower side effects, both academically and practically. In 2012, Hu et al. reported the synthesis and evaluation of 5-substituent of 2(1H)-pyridone derivatives as anti-fibrosis agents; very good in vitro activity was obtained compared to pirfenidone.21 Inspired by the novel scaffold and hope to improve its poor water-solubility and high toxicity,22 herein, we disclose our preliminary study of new pirfenidone derivatives as anti-fibrosis agents.
Results and discussion
Chemistry
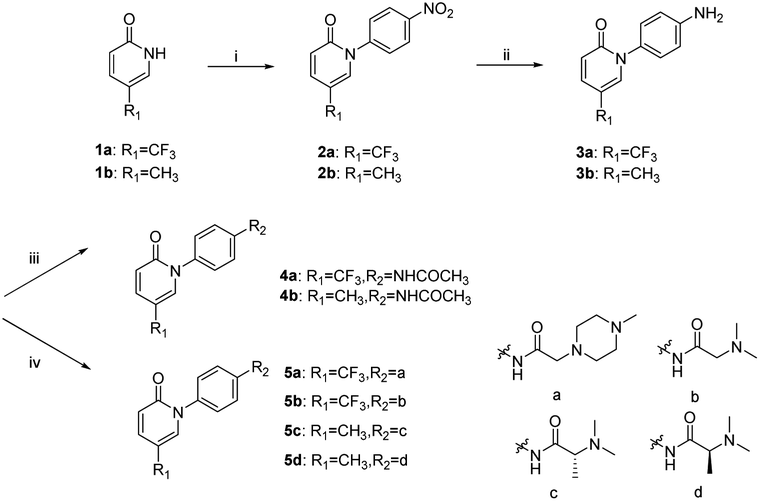
The synthetic route to 4a–4b and 5a–5d is shown in Scheme 1. For example, in the presence of potassium carbonate, coupling reaction of 5-(trifluoromethyl)pyridin-2(1H)-one and 1-fluoro-4-nitrobenzene gave compound 2a, which was reduced by tin dichloride to provide compound 3a. Compound 4a was obtained by the reaction of 3a with acetic anhydride. On the other hand, compound 5a was obtained by the reaction of 3a with 2-(4-methylpiperazin-1-yl)acetic acid in the presence of a dehydrating agent HATU.
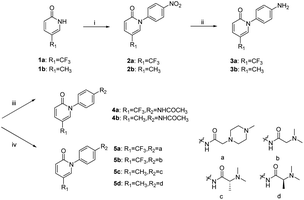 |
| | Scheme 1 Synthetic route to compounds 4a–4b and 5a–5d. Reagents and conditions: (i) MeCN, K2CO3, 85 °C. (ii) SnCl2·2H2O, conc. HCl, 40 °C. (iii) Ac2O, 125 °C. (iv) Acid derivative, HATU, DIPEA, 0–25 °C. | |
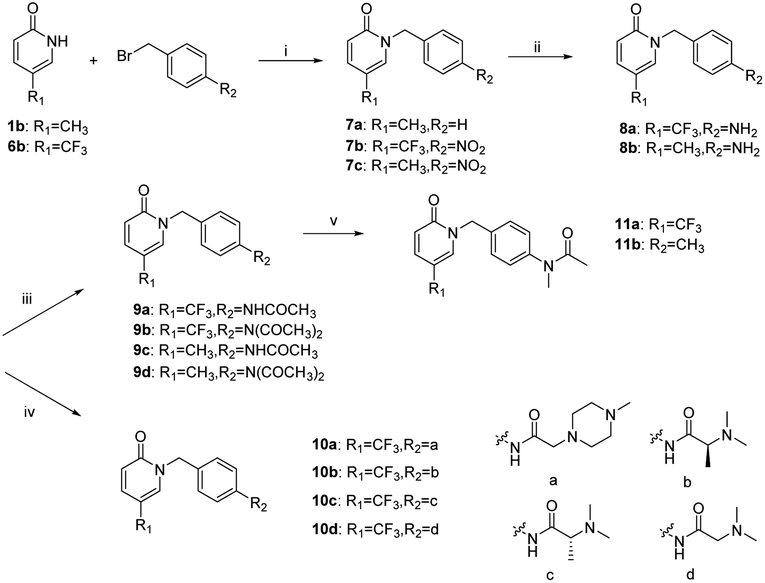
The synthetic route to 9–11 (the benzyl group is attached to the nitrogen atom of the pyridin-2(1H)-one) is shown in Scheme 2. Target compounds 9a–9d and 10a–10d were obtained by a method similar to the synthesis of compounds 4–5. Compounds 11a–11b were obtained by the reaction of 9a–9b with methyl iodide in the presence of sodium hydride.
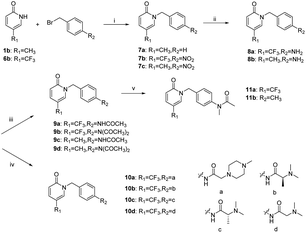 |
| | Scheme 2 Synthetic route to compounds 9–11. Reagents and conditions: (i) MeCN, K2CO3, 105 °C. (ii) SnCl2, conc. HCl, 40 °C. (iii) Ac2O, 125/135 °C. (iv) Acid derivatives, HATU, DIPEA, 0–25 °C. (v) DMF, NaH, MeI. | |
In vitro anti-fibrotic activity test
In searching for potential compounds that display better anti-fibrotic activity, the series in which phenyl groups are attached to the nitrogen atom of the pyridin-2(1H)-one moiety, compounds 4a–4b, 5a–5d were first synthesized and evaluated. The mouse fibroblast cell line (NIH3T3 cells) was used for the screening by CCK8 assay and the results are listed in Table 1. Pirfenidone exhibited 35.7% inhibition rate at a concentration of 1 mM that is consistent with the IC50 value of 2.75 mM reported in the literature.21 Compounds 4a–4b, with CF3 instead of the CH3 or an acetamido substitution in the para position of the benzene ring of pirfenidone, even give lower inhibition rate at the same concentration. It seems that the aliphatic amine structure on the side chain of the benzene ring is beneficial for anti-fibrotic activity. Compound 5b, with a 2-(dimethylamino)acetamido at the R2 position, provided 34.2% inhibition rate at a concentration of 0.5 mM which is nearly the same as that of pirfenidone at the concentration of 1.0 mM. Furthermore, compounds 5a, 5c, and 5d, with 2-(dimethylamino)propanamido or 2-(4-methylpiperazin-1-yl)acetamido at the R2 position, show better results. Compound 5d, with (S)-2-(dimethylamino)propanamido at the same position, gives an IC50 value of 0.246 mM, which is much better than the 2.75 mM of the pirfenidone. The chiral structure of the side chain R2 also has a certain influence on the activity; compound 5c, the enantiomer of 5d, with a (R)-2-(dimethylamino)propanamido group at the R2 position, provided only the half activity (0.490 mM) compared to its isomer. Compounds 7a, 9a–9d, 10a–10d, and 11a–11b, where the benzyl groups are attached to the nitrogen atom of the pyridin-2(1H)-one, show more potent activities with the IC50 values ranging from 0.069 to 0.441 mM besides 7a (with 40.95% inhibition rate at a concentration of 1 mM). Further research on the relationship between structure and activity showed that both the R2 group and the chirality of the R2 group have a great influence on the activity. Compounds 9a, 9c, 11a and 11b with acetamido or N-methylacetamido at the R2 position gave IC50 values of 0.132–0.145 mM that were much better than that of 4a, 4b, with the same group at the same position in a previous compound series (compounds 1–7). On the other hand, activity changes are not as pronounced as in the previous series when R2 groups were changed to 2-(dimethylamino)propanamido or 2-(4-methylpiperazin-1-yl)acetamido (compounds 10a–10c, 0.273 to 0.441 mM). It is worth pointing out that the change of chiral configuration still exists for the activity; compound 10b, with a (S)-2-(dimethylamino)propanamido group at the R2 position, shows an IC50 of 0.310 mM, which is better than its enantiomer 10c (0.441 mM). It is a delight to us that 9b and 9d, with the CF3 or CH3 at the R1 position and an N-acetylacetamido group at the R2 position, exhibited very good activities with IC50 values of 0.087 and 0.069 mM, respectively; especially, compared with 11a and 11b, compounds 9b and 9d have good solubility in water, and have the prospect of further druggability research.
Table 1 Anti-proliferative activities of compounds 1–17a
|
| Entry |
Comp. |
R1 |
R2 |
Inhibition% @ 1 mM |
Inhibition% @ 0.5 mM |
IC50 (μM) |
| Values are averages (±SD) of at least three independent determinations. Values “greater than” indicate that half-maximum inhibition was not achieved at the highest concentration tested. |
| 1 |
BFD |
CH3 |
H |
35.7 |
17.9 |
>1000 |
| 2 |
4a |
CF3 |
Acetamido |
32.59 |
10.4 |
>1000 |
| 3 |
4b |
CH3 |
Acetamido |
25.7 |
1.3 |
>1000 |
| 4 |
5a |
CF3 |
2-(4-Methylpiperazin-1-yl)acetamido |
62.35 |
51.8 |
340.8 ± 7.3 |
| 5 |
5b |
CF3 |
2-(Dimethylamino)acetamido |
45.8 |
34.2 |
>500 |
| 6 |
5c |
CH3 |
(R)-(2-(Dimethylamino)propanamido) |
52.9 |
26.8 |
490.2 ± 2.7 |
| 7 |
5d |
CH3 |
(S)-(2-(Dimethylamino)propanamido) |
60.9 |
58.6 |
246.7 ± 3.8 |
| 8 |
7a |
CH3 |
H |
40.95 |
20.58 |
>500 |
| 9 |
9a |
CF3 |
Acetamido |
86.23 |
68.36 |
132 ± 2.3 |
| 10 |
9b |
CF3 |
N-Acetylacetamido |
90.46 |
80.57 |
87.5 ± 0.7 |
| 11 |
9c |
CH3 |
Acetamido |
88.96 |
72.54 |
113 ± 1.9 |
| 12 |
9d |
CH3 |
N-Acetylacetamido |
98.7 |
88.4 |
69.1 ± 2.7 |
| 13 |
10a |
CF3 |
2-(4-Methylpiperazin-1-yl)acetamido |
87.69 |
66.52 |
246.3 ± 1.8 |
| 14 |
10b |
CF3 |
(S)-(2-(Dimethylamino)propanamido) |
64.2 |
49.4 |
310.2 ± 3.7 |
| 15 |
10c |
CF3 |
(R)-(2-(Dimethylamino)propanamido) |
60.23 |
37.5 |
441.6 ± 4.8 |
| 16 |
10d |
CF3 |
2-(Dimethylamino)acetamido |
71.3 |
52.4 |
273.1 ± 1.5 |
| 17 |
11a |
CF3 |
N-Methylacetamido |
78.5 |
67.9 |
145.8 ± 5.5 |
| 18 |
11b |
CH3 |
N-Methylacetamido |
79.4 |
66.7 |
139.6 ± 9.8 |
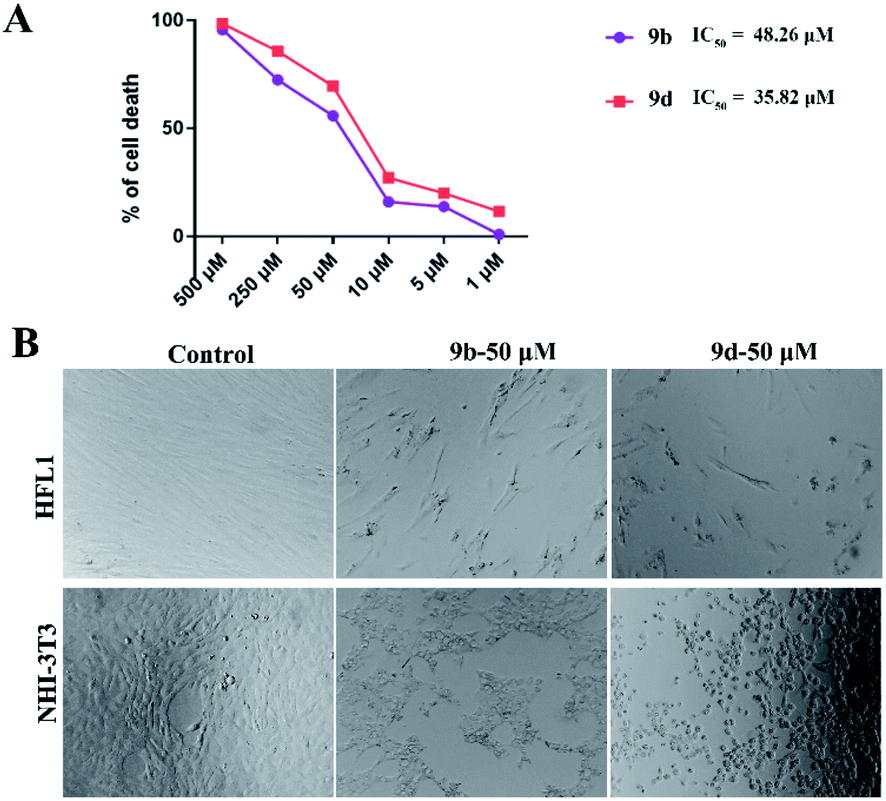
Antiproliferative activity of compounds 9b and 9d against the human fibroblast cell line
To further study the anti-fibrotic activity of synthesized compounds, compounds 9b and 9d were used for the study of antiproliferative activity against the human fibroblast cell line HFL1 with CCK8 assay and the results are shown in Fig. 1A; compounds 9b and 9d displayed better antiproliferative activity with IC50 values of 0.048 and 0.035 mM compared with the use of NHI3T3 cell lines, respectively. The fixed cell morphology showed the cells in the untreated group with a long spindle shape with a clear outline, while the cells began to shrink and become round after addition of the compound 9b or 9d, which shows that the proliferation of HFL1 and NHI3T3 was effectively inhibited when the cell lines were treated with 9b and 9d at a concentration of 0.050 mM for 24 h (Fig. 1B). Compound 9d showed excellent performance and was selected as the optimal compound for the further evaluation of drug availability.
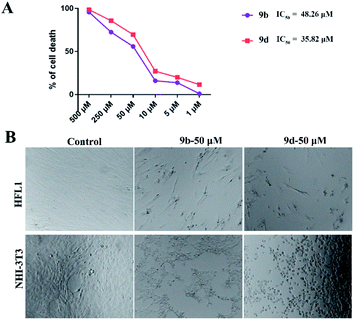 |
| | Fig. 1 Antiproliferative activities of compounds 9b and 9d toward the human fibroblast cell line HFL1 and cell morphology observation of the fibroblast cell line. (A) The cell lines HFL1 were treated with 9b and 9d for 48 h, and then analyzed with GraphPad Prism Software version 5.02 (GraphPad Inc., La Jolla, CA, USA). (B) The cell lines HFL1 and NHI3T3 were treated with 9b and 9d for 24 h; the detection of the fixed cell morphology was performed with a microscope. All images were collected with a 10× objective. | |
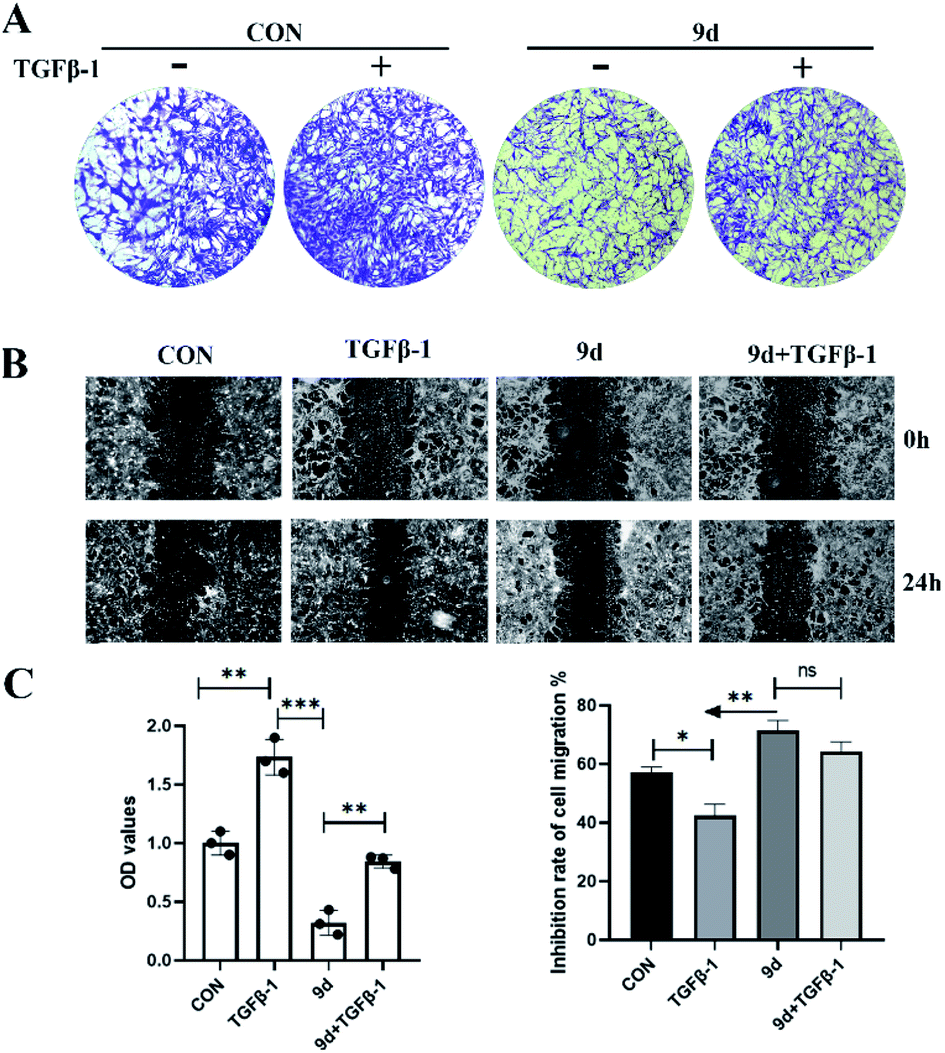
Compound 9d inhibition of NHI3T3 cell migration
In pulmonary fibrosis, the key fibrogenic factor TGF-β1 induces the occurrence of epithelial–mesenchymal transition (EMT). As compound 9d exhibited the best results in inhibition of antiproliferative activity against both the mouse fibroblast NIH3T3 cell line and human fibroblast cell line HFL1, we evaluated the effects of 9d on TGF-β1-induced EMT in NHI3T3 cells. First of all, a transwell assay was performed to evaluate the influence of 9d on the migration capacity of 3T3 cells. As shown in Fig. 2A, treatment with 9d markedly inhibited 3T3 cell migration. The data in Fig. 2C showed that TGF-β1 can effectively promote cell migration with an OD value of about 1.7 (the OD value of the control is 1.0). In contrast, compound 9d provided an OD value of about 0.35, and an OD value of about 0.8 was obtained in 9d + TGF-β1, indicating that compound 9d can inhibit the cell migration effectively. Similar results were also obtained for the cell migration inhibition rates of B and C in Fig. 2. Therefore, compound 9d could attenuate pulmonary fibrosis by inhibiting TGF-β1-induced EMT.
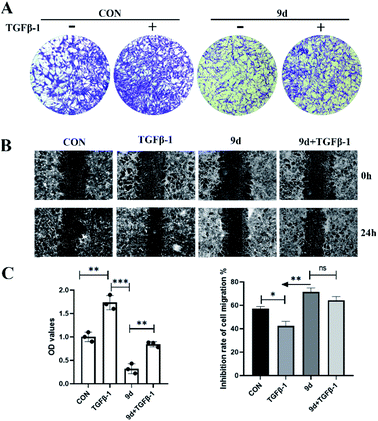 |
| | Fig. 2 Effect of 9d on the migration of NHI3T3 cells. NHI3T3 cells (30 × 104 cells) suspended in free serum DMEM containing 9d (50 μM) for 48 h (A) or 24 h (B) were photographed under phase contrast microscopy (magnification: 4× objective). (C) All results were expressed as the mean ± SD of at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the control group. | |
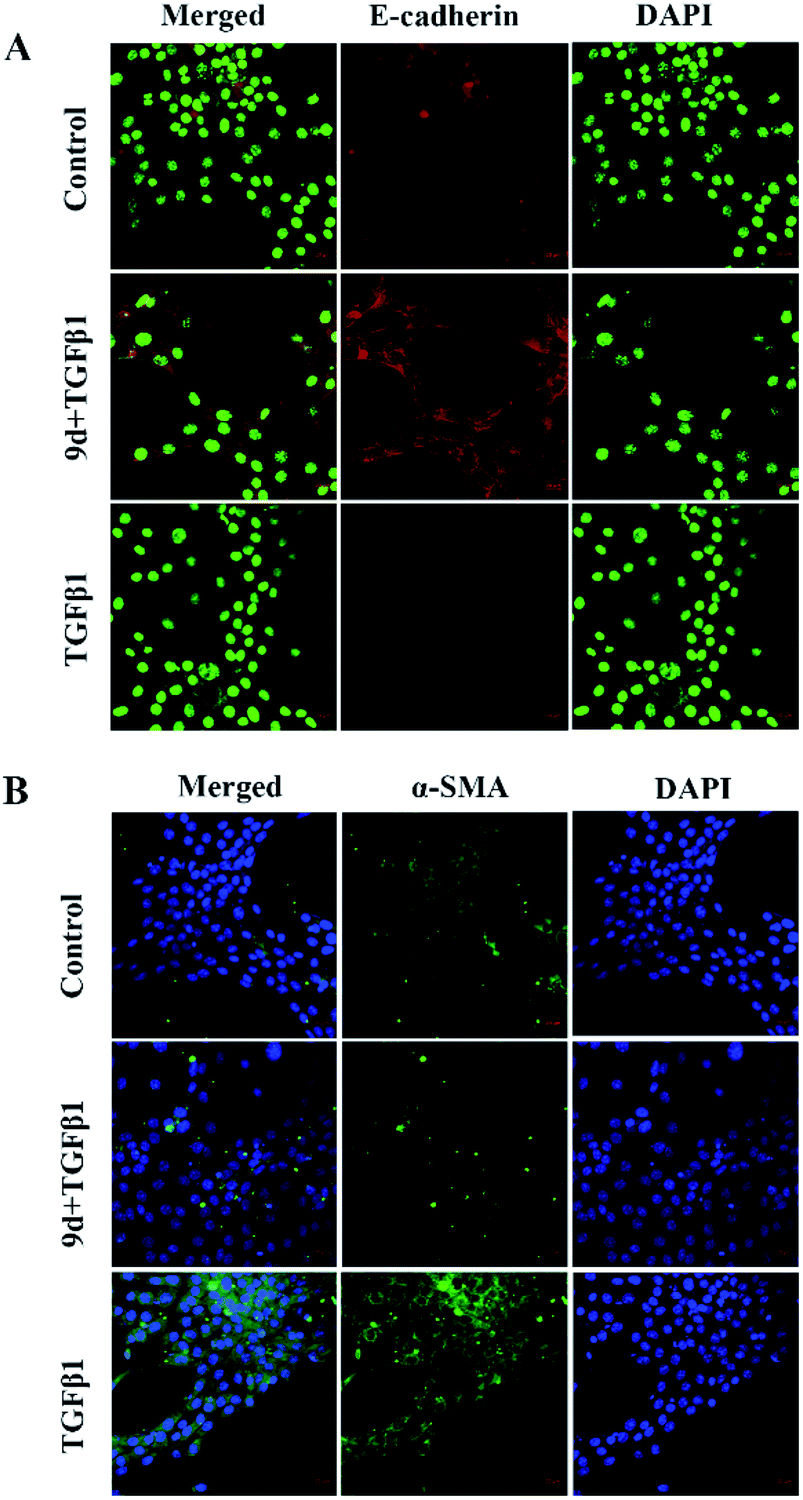
Expression of E-cadherin and α-SMA in NHI3T3 cells treated with 9d by immunofluorescence assay
Loss of E-cadherin expression is a hallmark of epithelial–mesenchymal transition (EMT) and is associated with an increased risk of metastases from mutant cells such as cancer cells. E-cadherin deletion not only helps tumor cells to separate from each other by disrupting cell-to-cell junctions, but also induces intracellular signaling events that produce a mesenchymal state and metastatic phenotype. Mutations of gene in α-smooth muscle actin (α-SMA) cause a variety of vascular diseases, such as thoracic aortic disease, coronary artery disease, stroke, moyamoya disease, and multisystem smooth muscle dysfunction syndrome. α-SMA is currently considered a hallmark of myofibroblasts. Therefore, we chose to study the expression of E-cadherin and α-SMA in NHI3T3 cells treated with TGF-β1 and compound 9d. As shown, after 48 hours of stimulation with recombinant TGF-β1 in NHI3T3 cells treated with DMSO or 9d, immunofluorescence staining showed that the positive staining of E-cadherin decreased in the TGF-β1 group compared with the 9d-treated group and the control group, and the expression of E-cadherin was reversed after 9d treatment, higher than the control group (Fig. 3A). On the other hand, the positive staining of α-SMA increased in the TGF-β1 group compared with the control group and the 9d-treated group, and the expression of α-SMA decreased after the 9d-treatment (Fig. 3B), indicating that 9d can stifle the activation and proliferation of fibroblasts.
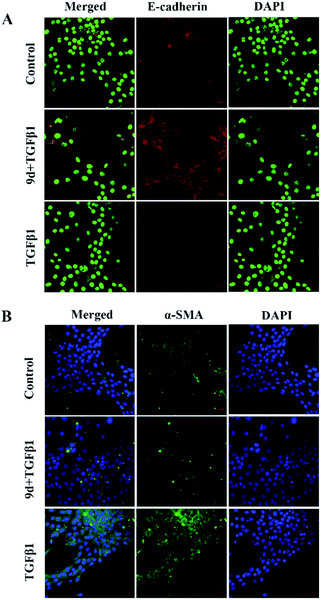 |
| | Fig. 3 Expression of E-cadherin and α-SMA in the cells of each group. NHI3T3 cells were grown on coverslips and treated with TGF-β1 (10 ng mL−1) and 9d (50 μM) for 24 h. Cells were fixed and stained with E-cadherin (A) and α-SMA (B) antibodies followed by incubation with fluorescently tagged secondary antibodies. DAPI was used to stain nuclei. α-SMA (green), E-cadherin (red) and nuclei (blue/green) were visualized by confocal fluorescence microscopy. The experiments were performed with two technical replicates and three biological repeats. | |
Acute toxicity experiment and histopathologic examination
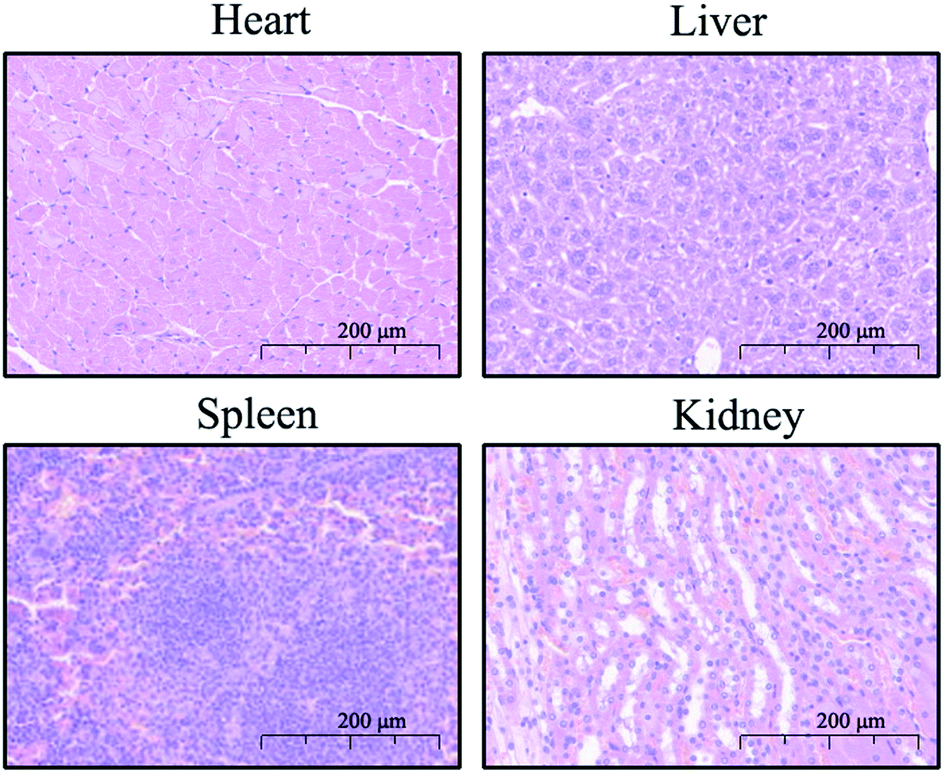
In clinical practice, the biosafety of drugs is the primary concern, so a toxicology assessment was conducted for the preliminary evaluation of the safety of compound 9d. The KM mice, originally from Switzerland, introduced into Kunming Central Epidemic Prevention Office of China from Haffkine Institute of India in 1946, and widely used in the evaluation of drug safety and effectiveness at present, were used for the evaluation.24–26 The mice (5 male and 5 female in each group) were randomly divided into experimental group and control group, and were caudal vein injected with 9d at a dose of 100/150/300 mg kg−1, and the control group was caudal vein injected with physiological saline. The results showed no death and obvious toxicological reactions in mice within 24 hours after administration. Subsequently, the mice were euthanized and histologically analyzed to assess damage of the liver, kidney, heart, and spleen. As shown in Fig. 4, no pathological abnormalities such as inflammation, injury and necrosis were observed in the treatment group.
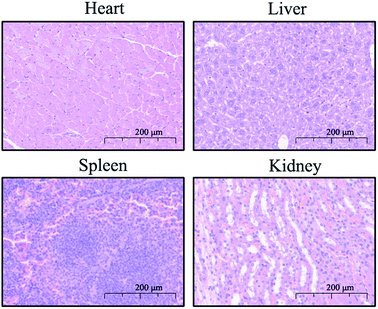 |
| | Fig. 4 Representative image of H&E staining (200×) of the heart, liver, spleen, and kidney of mice in the combination group. | |
Molecular docking
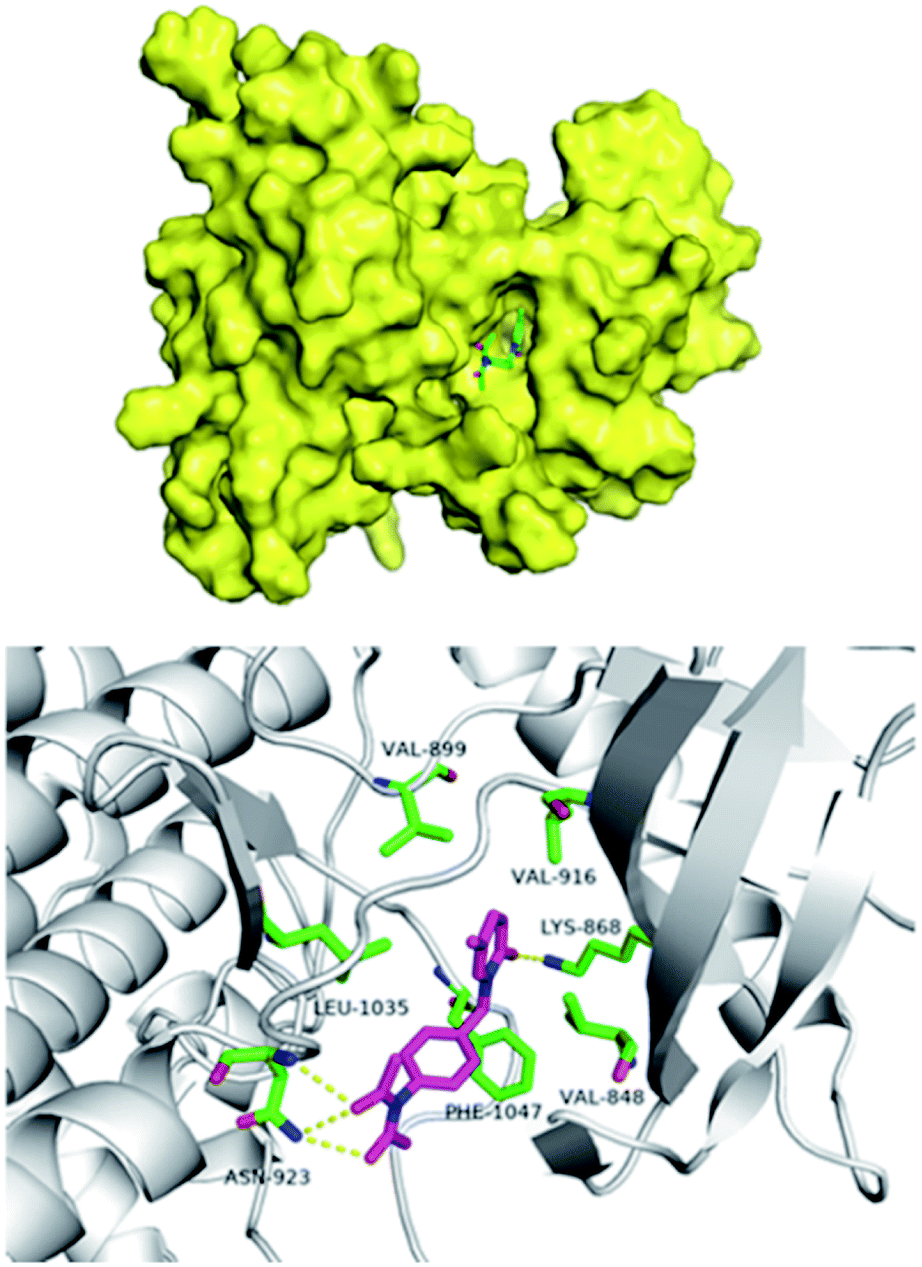
The pathogenesis of IPF is complex and the etiology is not fully clarified. It mainly includes immune inflammatory injury, abnormal repair and fibrosis, oxidative stress, angiogenesis and remodeling. Studies23 showed that inhibiting the expression of VEGF/VEGFR-2 can reduce microvessel density, regulate protein kinase ERK1/2 by inhibiting the extracellular signal, downregulate the levels of TNF-α and TNF-β, and inhibit the apoptosis of type II alveolar epithelial cells, so as to play an inhibitory role in the formation of pulmonary fibrosis. In order to study the possible interaction of compound 9d with VEGFR, we docked compound 9d at the active site of kinase using Schrodinger Suites 2018 software (PDB Code: 3c7q). As shown in Fig. 5, compound 9d is in the hydrophobic active cavity composed of hydrophobic amino acids that is similar to nintedanib. The pyridinone moiety of 9d forms a hydrogen bond with lys868, and the diacetylamine moiety forms a hydrogen bond with asn923, which has high affinity with the kinase. Therefore, the docking results showed that the two ends of 9d formed hydrogen bonds with kinase, which was conducive to the matching with VEGFR-2 active sites.
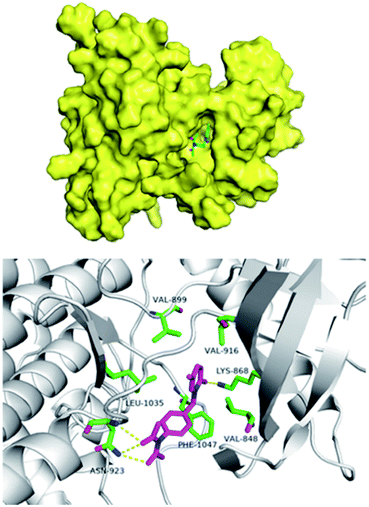 |
| | Fig. 5 The docking results of compound 9d with VEGFR. | |
Conclusions
Pirfenidone is one of the two drugs approved by the FDA for the treatment of IPF at present. As pirfenidone has a poor therapeutic effect on patients with severe pulmonary fibrosis and owing to its side effects on patients, it is necessary to develop new anti-fibrosis drugs. Herein, sixteen new pirfenidone derivatives were synthesized and evaluated against the mouse fibroblast cell line (NIH3T3 cells) and human fibroblast cell line HFL1. By the screening at the cell level, two optimal compounds that can be further studied were obtained. Compound 5d, with the same ring skeleton as pirfenidone, but a (S)-2-(dimethylamino)propanamido group in the R2 position, exhibited 10 times anti-fibrosis activity of pirfenidone. Compound 9d, with benzyl groups attached to the nitrogen atom of the pyridin-2(1H)-one moiety and a N-acetylacetamido group at the R2 position, gave more than 40 times anti-fibrosis activity of pirfenidone. In addition, the synthetic route to the optimal compound in this manuscript is simple and the target compound product is easy to purify. Considering the safety limits of impurities, relevant analytical methods, including appropriate limit of quantitation (LOQ) and limit of detection (LOD), will be developed. The anti-fibrosis assay in vivo of two optimal compounds is in progress.
Experimental
Chemistry
All reagents were analytically pure without further purification, and all chemistry solvents were of reagent-grade and were dried and freshly distilled before the necessary step. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance III spectrometer, and chemical shifts are reported as parts per million (ppm) with tetramethylsilane as the internal standard. High-resolution mass spectra (HR-MS) were recorded using an Agilent LC-MS 6120 instrument. High-performance liquid chromatography (HPLC) was run on a TC-C18 column (4.6 × 250 mm, 5 μm) with two different solvent gradients (methanol/water = 85:15), and a flow rate of 0.50 mL min−1. All synthesized compounds were determined to have at least 95% of purity by HPLC.
1-(4-Nitrophenyl)-5-(trifluoromethyl)pyridin-2(1H)-one (2a)
To a solution of 5-(trifluoromethyl)pyridin-2(1H)-one 1a (1.63 g, 10 mmol) in DMF (20 mL), K2CO3 (2.07 g, 15 mmol) and 1-fluoro-4-nitrobenzene (1.48 g, 10.5 mmol) were added. The reaction mixture was stirred at 105 °C for 3 h under nitrogen. After cooling to room temperature, the reaction mixture was diluted with water and extracted with ethyl acetate. The combined organic layers were washed with brine and then dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography on silica gel to give 2a as a brown solid (2.30 g, 80% yield). 1H NMR (400 MHz, CDCl3) δ: 8.41–8.38 (m, 2H), 7.74 (s, 1H), 7.65–7.61 (m, 2H), 7.56 (dd, J = 9.7, 2.6 Hz, 1H), 6.76 (d, J = 9.7 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ: 161.0, 147.9, 144.9, 136.6 (q, J = 5.4 Hz), 136.0 (d, J = 2.1 Hz), 127.8, 125.1, 123.0.
5-Methyl-1-(4-nitrophenyl) pyridin-2(1H)-one (2b)
To a solution of 5-methylpyridin-2(1H)-one 1b (1.09 g, 10 mmol) in MeCN (20 mL), K2CO3 (2.07 g, 15 mmol) and 1-fluoro-4-nitrobenzene (1.48 g, 10.5 mmol) were added. The reaction mixture was stirred at 85 °C for 5 h under nitrogen. After cooling to room temperature, the reaction mixture was concentrated with a rotary evaporator. Then the residue was diluted with water and extracted with ethyl acetate. The combined organic layers were washed with brine and then dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography on silica gel to give 2b as a brown solid (1.61 g, 70% yield). 1H NMR (400 MHz, CDCl3) δ: 8.27–8.23 (m, 2H), 8.05 (d, J = 1.8 Hz, 1H), 7.60 (dd, J = 8.3, 2.4 Hz, 1H), 7.23–7.20 (m, 2H), 6.95 (d, J = 8.3 Hz, 1H), 2.33 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 160.4, 160.2, 147.7, 143.7, 141.0, 129.9, 125.6, 120.2, 112.6, 17.7.
1-(4-Aminophenyl)-5-methylpyridin-2(1H)-one (3a)
Concentrated hydrochloric acid (4.5 mL) was added to a solution of 1-(4-nitrophenyl)-5-(trifluoromethyl)pyridin-2(1H)-one 2a (0.57 g, 2 mmol) in MeOH (15 mL), and then SnCl2·2H2O was added at 0 °C. The reaction mixture was stirred for 1 h at 40 °C. After cooling to room temperature, a saturated sodium carbonate solution was added. The mixture was filtrated and the filtrate was extracted with ethyl acetate (15 mL × 3). The combined organic layers were washed with brine and then dried over Na2SO4. After filtration, the solvent was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel to give 3a (0.4 g, 79% yield). 1H NMR (500 MHz, CDCl3) δ: 7.73 (d, J = 0.75, 1H), 7.49 (dd, J = 9.6, 2.7 Hz, 1H), 7.13–7.10 (m, 2H), 6.73–6.68 (m, 3H). 13C NMR (125 MHz, CDCl3) δ: 162.1, 147.4, 138.3 (q, J = 5.3 Hz), 135.2 (d, J = 2.2 Hz), 130.50, 127.2, 122.1, 115.2.
1-(4-Aminophenyl)-5-methylpyridin-2(1H)-one (3b)
Compound 3b was obtained by the same method as 3a (0.36 g, 90% yield). 1H NMR (500 MHz, CDCl3) δ: 7.23 (dd, J = 9.3, 2.6 Hz, 1H), 7.12–7.09 (m, 3H), 6.71–6.68 (m, 2H), 6.58 (d, J = 9.3 Hz, 1H), 2.08 (d, J = 0.8 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 162.2, 146.7, 142.4, 136.1, 131.9, 127.3, 121.2, 115.2, 114.6, 17.0.
N-(4-(2-oxo-5-(Trifluoromethyl)pyridin-1(2H)-yl)phenyl)acetamide (4a)
Under nitrogen, compound 3a (0.51 g, 2 mmol) was added in a solution of acetic anhydride (3 mL). The mixture was kept at 125 °C for 2 h and the excess acetic anhydride was removed under reduced pressure. The residue was purified by column chromatography on silica gel to give 4a (0.48 g, 81% yield). 1H NMR (500 MHz, DMSO-d6) δ: 10.15 (s, 1H), 8.21 (s, 1H), 7.74 (d, J = 9.6 Hz, 1H), 7.69 (d, J = 8.5 Hz, 2H), 7.38 (d, J = 8.6 Hz, 2H), 6.63 (d, J = 9.6 Hz, 1H), 2.08 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ: 168.6, 160.9, 139.5 (q, J = 5.0 Hz), 135.6, 134.5, 127.2, 124.8, 122.6, 121.2, 119.1, 24.0. HRMS (ESI) m/z: calculated for C14H11F3N2O2 [M + Na]+ 319.0665; found 319.0666.
N-(4-(5-Methyl-2-oxopyridin-1(2H)-yl)phenyl)acetamide (4b)
Compound 4b was obtained by the same method as 4a (0.39 g, 80% yield). 1H NMR (500 MHz, DMSO-d6) δ: 10.11 (s, 1H), 7.67 (d, J = 8.5 Hz, 2H), 7.40 (s, 1H), 7.37 (d, J = 9.3 Hz, 1H), 7.30 (d, J = 8.5 Hz, 2H), 6.41 (d, J = 9.3 Hz, 1H), 2.07 (s, 3H), 2.04 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ: 168.5, 160.5, 142.9, 138.9, 136.2, 135.7, 127.0, 120.1, 119.1, 113.9, 24.0, 16.3. HRMS (ESI) m/z: calculated for C14H14N2O2 [M + H]+ 243.1128; found 243.1140.
2-(4-Methylpiperazin-1-yl)-N-(4-(2-oxo-5-(trifluoromethyl)pyridin-1(2H)-yl)phenyl)acetamide (5a)
To a solution of 2-(4-methylpiperazin-1-yl)acetic acid in DMF (3 mL), N-ethyl-N-isopropylpropan-2-amine (DIPEA, 0.65 g, 5 mmol) was added at 0 °C. After the mixture was stirred for ten minutes, HATU (0.57 g, 1.5 mmol) was added, and then 3a (0.25 g, 1 mmol) was added after another ten minutes. After the reaction rises to room temperature, DMF was removed under reduced pressure. The residue was purified by column chromatography on silica gel to give 5a (0.32 g, 81% yield). 1H NMR (500 MHz, CD3OD) δ: 8.10 (s, 1H), 7.81 (d, J = 8.8 Hz, 2H), 7.75 (dd, J = 9.6, 2.6 Hz, 1H), 7.38 (d, J = 8.8 Hz, 2H), 6.72 (d, J = 9.6 Hz, 1H), 3.31 (s, 2H), 3.21 (s, 4H), 2.85 (s, 4H), 2.78 (s, 3H). 13C NMR (125 MHz, CD3OD) δ: 170.6, 164.0, 140.2–140.1 (m), 137.7, 137.0, 128.3, 122.5, 122.0, 61.8, 54.9, 51.7, 44.1. HRMS (ESI) m/z: calculated for C19H21F3N4O2 [M + H]+ 395.1689; found 395.1684.
2-(Dimethylamino)-N-(4-(2-oxo-5-(trifluoromethyl)pyridin-1(2H)-yl)phenyl)acetamide (5b)
In addition to replacing 2-(4-methylpiperazin-1-yl)acetic acid with dimethylglycine, compound 5b was obtained by the same method as 5a (0.25 g, 75% yield). 1H NMR (500 MHz, CD3OD) δ: 8.10 (s, 1H), 7.78 (d, J = 8.8 Hz, 2H), 7.77–7.75 (m, 1H), 7.40 (d, J = 8.8 Hz, 2H), 6.73 (d, J = 9.6 Hz, 1H), 3.74 (s, 2H), 2.75 (s, 6H). 13C NMR (125 MHz, CD3OD) δ: 167.2, 164.0, 140.0 (q, J = 6.3 Hz), 137.7, 137.2, 128.4, 122.5, 121.7, 61.6, 45.1. HRMS (ESI) m/z: calculated for C16H16F3N3O2 [M + H]+ 340.1267; found 340.1276.
(R)-2-(Dimethylamino)-N-(4-(5-methyl-2-oxopyridin-1(2H)-yl)phenyl)propanamide (5c)
In addition to replacing 2-(4-methylpiperazin-1-yl)acetic acid with (R)-dimethylalanine, compound 5c was obtained by the same method as 5a (0.21 g, 70% yield). 1H NMR (500 MHz, CD3OD) δ: 7.77 (d, J = 8.8 Hz, 2H), 7.51 (dd, J = 9.3, 2.5 Hz, 1H), 7.40 (s, 1H), 7.36–7.33 (m, 2H), 6.58 (d, J = 9.3 Hz, 1H), 2.45 (s, 6H), 2.15 (s, 3H), 1.37 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CD3OD) δ: 173.5, 164.0, 145.4, 139.8 (q, J = 6.3 Hz), 138.0, 137.6, 128.2, 121.7, 121.0, 118.4, 66.3, 42.4, 16.8, 13.9. HRMS (ESI) m/z: calculated for C17H21N3O2 [M + H]+ 300.1707; found 300.1708.
(S)-2-(Dimethylamino)-N-(4-(5-methyl-2-oxopyridin-1(2H)-yl)phenyl)propenamide (5d)
To a solution of (S)-dimethylalanine (0.26 g, 2.2 mmol) in DCM (10 mL), N-ethyl-N-isopropylpropan-2-amine (DIPEA, 1.03 g, 8.0 mmol) was added at 0 °C. After the mixture was stirred for ten minutes, HATU (0.91 g, 2.38 mmol) was added, and then 3b (0.32 g, 1.6 mmol) was added after another ten minutes. After the reaction rises to room temperature, DCM was removed under reduced pressure. The residue was purified by column chromatography on silica gel to give 5a (0.33 g, 70% yield). 1H NMR (500 MHz, CD3OD) δ: 7.77 (d, J = 8.8 Hz, 2H), 7.51 (dd, J = 9.3, 2.4 Hz, 1H), 7.40 (s, 1H), 7.34 (d, J = 8.8 Hz, 2H), 6.57 (d, J = 9.3 Hz, 1H), 3.36 (q, J = 6.9 Hz, 1H), 2.48 (s, 6H), 2.14 (s, 3H), 1.38 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CD3OD) δ: 173.2, 163.9, 145.4, 139.8, 138.0, 137.5, 128.2, 121.7, 121.0, 118.3, 66.2, 42.4, 16.8, 13.9. HRMS (ESI) m/z: calculated for C17H21N3O2 [M + H]+ 300.1707; found 300.1706.
1-Benzyl-5-(trifluoromethyl)pyridin-2(1H)-one (7a)
To a solution of 5-methylpyridin-2(1H)-one in DMF (6 mL), K2CO3 (0.41 g, 3 mmol) and benzyl bromide (0.34 g, 2 mmol) were added. The reaction was carried out at 105 °C for 2 h and then the DMF was removed under reduced pressure. Water (10 mL) was added to the residue and the mixture was extracted with ethyl acetate (2 × 10 mL). The combined organic phase was washed with brine, and dried over sodium sulfate, and then the solvents were removed under reduced pressure. The residue was purified by column chromatography on silica gel to give 7a (0.34 g, 85% yield). 1H NMR (400 MHz, DMSO-d6) δ: 7.56 (dd, J = 1.4, 0.8 Hz, 1H), 7.35–7.25 (m, 6H), 6.37 (d, J = 9.2 Hz, 1H), 5.05 (s, 2H), 2.00 (d, J = 0.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ: 160.6, 142.5, 137.6, 136.2, 128.5, 127.6, 127.4, 119.6, 114.0, 50.9, 16.4. HRMS (ESI) m/z: calculated for C13H13NO [M + H]+ 200.1070; found 200.1063.
1-(4-Nitrobenzyl)-5-(trifluoromethyl)pyridin-2(1H)-one (7b)
To a solution of 5-(trifluoromethyl)pyridin-2(1H)-one in 35 mL of DMF, K2CO3 (2.05 g, 15 mmol) and 1-(bromomethyl)-4-nitrobenzene (2.15 g, 10 mmol) were added. After stirring at 105 °C for 2 h, DMF was removed under reduced pressure. Water (10 mL) was added to the residue and the mixture was extracted with ethyl acetate (2 × 30 mL), and then with the same procedure as compound 7a to give 7b (2.25 g, 76% yield). 1H NMR (500 MHz, CDCl3) δ: 8.22 (d, J = 8.7 Hz, 2H), 7.74 (s, 1H), 7.51–7.48 (m, 3H), 6.70 (d, J = 9.6 Hz, 1H), 5.25 (s, 2H). 13C NMR (125 MHz, CDCl3) δ: 161.6, 147.9, 142.4, 136.8 (q, J = 5.0 Hz), 135.6 (d, J = 2.5 Hz), 128.7, 124.2, 122.0, 52.4.
5-Methyl-1-(4-nitrobenzyl)pyridin-2(1H)-one (7c)
To a solution of 5-methylpyridin-2(1H)-one 1b (1.09 g, 10 mmol) in MeCN (6 mL), K2CO3 (2.07 g, 15 mmol) and 1-(bromomethyl)-4-nitrobenzene (2.27 g, 10.5 mmol) were added. The reaction was carried out at 85 °C for 5 h and then the MeCN was removed under reduced pressure. Water (10 mL) was added to the residue and the mixture was extracted with ethyl acetate (2 × 20 mL). The combined organic phase was washed with brine, and dried over sodium sulfate, and then the solvents were removed under reduced pressure. The residue was purified by column chromatography on silica gel to give 7c (1.6 g, 65% yield). 1H NMR (400 MHz, CDCl3) δ: 8.19–8.17 (m, 2H), 7.45 (d, J = 8.8 Hz, 2H), 7.24 (dd, J = 9.3, 2.5 Hz, 1H), 7.07 (s, 1H), 6.58 (d, J = 9.3 Hz, 1H), 5.19 (s, 2H), 2.07 (d, J = 0.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 161.8, 147.5, 144.0, 142.7, 134.5, 128.5, 124.0, 121.1, 115.9, 51.6, 17.1.
1-(4-Aminobenzyl)-5-(trifluoromethyl)pyridin-2(1H)-one (8a)
To a solution of 7b (1.41 g, 4.7 mmol) in 25 mL of methanol, concentrated hydrochloric acid (8 mL) and SnCl2·2H2O (5.30 g, 23.5 mmol) were added at 0 °C, and then at 40 °C for 1 h. After the reaction temperature dropped to room temperature, saturated sodium carbonate was added until the solution was neutral. The mixture was extracted with ethyl acetate (20 mL × 3), and the combined organic phase was washed with brine, and dried over sodium sulfate. The solvents were removed under reduced pressure and the residue was purified by column chromatography on silica gel to give 8a (1.00 g, 79% yield). 1H NMR (500 MHz, CDCl3) δ: 7.62 (s, 1H), 7.40 (dd, J = 9.6, 2.6 Hz, 1H), 7.12 (d, J = 8.3 Hz, 2H), 6.67–6.63 (m, 3H), 5.01 (s, 2H). 13C NMR (125 MHz, CDCl3) δ: 162.2, 147.0, 136.6 (q, J = 5.0 Hz), 134.9 (d, J = 2.5 Hz), 130.2, 124.6, 121.5, 115.5, 52.3.
1-(4-Aminobenzyl)-5-methylpyridin-2(1H)-one (8b)
Replacing 7b with compound 7c, compound 8b was obtained by the same method as 8a (0.64 g, 75% yield). 1H NMR (400 MHz, CDCl3) δ: 7.14–7.09 (m, 3H), 7.00 (d, J = 0.5 Hz, 1H), 6.63–6.61 (m, 2H), 6.52 (d, J = 9.2 Hz, 1H), 4.97 (s, 2H), 1.99 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 162.0, 146.4, 141.9, 134.5, 129.7, 126.3, 120.6, 115.2, 115.0, 51.3, 17.1.
N-(4-((2-oxo-5-(Trifluoromethyl)pyridin-1(2H)-yl)methyl)phenyl)acetamide (9a)
Under nitrogen, acetic anhydride (3 mL) was added to the reaction bottle containing 8a (0.54 g, 2 mmol) and the reaction was kept for 2 h at 125 °C. After the excess acetic anhydride was removed under reduced pressure, the residue was purified by column chromatography on silica gel to give 9a (0.53 g, 85% yield). 1H NMR (500 MHz, DMSO-d6) δ: 9.96 (s, 1H), 8.51 (s, 1H), 7.67 (dd, J = 9.6, 2.6 Hz, 1H), 7.54 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 6.56 (d, J = 9.6 Hz, 1H), 5.10 (s, 2H), 2.03 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ: 168.8, 161.4, 139.8 (q, J = 5.0 Hz), 139.4, 135.8 (d, J = 2.5 Hz), 131.5, 128.9, 125.3, 123.2, 121.1, 119.6, 51.9, 24.4. HRMS (ESI) m/z: calculated for C15H13F3N2O2 [M + Na]+ 333.0821; found 333.0822.
N-Acetyl-N-(4-((2-oxo-5-(trifluoromethyl)pyridin-1(2H)-yl)methyl)phenyl)acetamide (9b)
Under nitrogen, acetic anhydride (5 mL) was added to the reaction bottle containing 8a (0.54 g, 2 mmol) and the reaction was kept for 4 h at 135 °C. After the excess acetic anhydride was removed under reduced pressure, the residue was purified by column chromatography on silica gel to give 9b (0.54 g, 76% yield). 1H NMR (400 MHz, CDCl3) δ: 7.73 (s, 1H), 7.48 (dd, J = 9.6, 2.6 Hz, 1H), 7.41 (d, J = 8.4 Hz, 2H), 7.17–7.14 (m, 2H), 6.69 (d, J = 9.6 Hz, 1H), 5.18 (s, 2H), 2.28 (s, 6H). 13C NMR (100 MHz, CDCl3) δ: 173.0, 161.9, 139.6, 137.1 (q, J = 5.0 Hz), 136.2, 135.5 (d, J = 2.0 Hz), 129.5, 122.0, 52.5, 27.1. HRMS (ESI) m/z: calculated for C17H15F3N2O3 [M + Na]+ 375.0927; found 375.0929.
N-(4-((5-Methyl-2-oxopyridin-1(2H)-yl)methyl)phenyl)acetamide (9c)
Replacing 8a with compound 8b, compound 9c was obtained by the same method as 9a (0.37 g, 73% yield). 1H NMR (400 MHz, DMSO-d6) δ: 9.99 (s, 1H), 7.53–7.51 (m, 3H), 7.29–7.22 (m, 3H), 6.36 (d, J = 9.2 Hz, 1H), 4.98 (s, 2H), 2.02 (s, 3H), 2.00 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ: 168.4, 160.7, 142.4, 138.7, 136.1, 132.1, 128.3, 119.6, 119.1, 114.1, 50.5, 24.0, 16.5.
N-Acetyl-N-(4-((5-methyl-2-oxopyridin-1(2H)-yl)methyl)phenyl)acetamide (9d)
Under nitrogen, acetic anhydride (5 mL) was added to the reaction bottle containing 8b (0.43 g, 2 mmol) and the reaction was kept for 4 h at 135 °C. After the excess acetic anhydride was removed under reduced pressure, the residue was purified by column chromatography on silica gel to give 9d (0.42 g, 70% yield). 1H NMR (500 MHz, CDCl3) δ: 7.38 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 9.2 Hz, 1H), 7.11 (t, J = 8.0 Hz, 3H), 6.59 (d, J = 9.3 Hz, 1H), 5.15 (s, 2H), 2.27 (s, 6H), 2.06 (s, 3H). 13C NMR (125 MHz, CDCl3) δ: 172.9, 162.0, 142.5, 138.9, 137.5, 134.8, 129.2, 120.9, 115.7, 51.4, 27.0, 17.1. HRMS (ESI) m/z: calculated for C17H18N2O3 [M + Na]+ 321.1210; found 321.1212.
2-(4-Methylpiperazin-1-yl)-N-(4-((2-oxo-5-(trifluoromethyl)pyridin-1(2H)-yl)methyl)phenyl)acetamide (10a)
To a solution of 2-(4-methylpiperazin-1-yl)acetic acid in DMF (3 mL), N-ethyl-N-isopropylpropan-2-amine (DIPEA, 0.65 g, 5 mmol) was added at 0 °C. After the mixture was stirred for 10 minutes, HATU (0.57 g, 1.5 mmol) was added. After stirring for 10 minutes, 8a (0.27 g, 1 mmol) was added and the reaction was raised to room temperature. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel to give 10a (0.29 g, 70% yield). 1H NMR (500 MHz, CD3OD) δ: 8.24 (s, 1H), 7.65 (dd, J = 9.5, 2.6 Hz, 1H), 7.60 (d, J = 8.5 Hz, 2H), 7.32 (d, J = 8.6 Hz, 2H), 6.65 (d, J = 9.5 Hz, 1H), 5.18 (s, 2H), 3.22 (s, 2H), 2.91 (s, 4H), 2.74 (s, 4H), 2.56 (s, 3H). 13C NMR (125 MHz, CD3OD) δ: 169.2, 162.7, 138.3 (q, J = 8.5 Hz), 137.7, 135.8 (d, J = 1.6 Hz), 131.9, 128.4, 127.1, 124.5, 122.4, 120.4, 60.8, 53.8, 52.1, 51.3, 43.5. HRMS (ESI) m/z: calculated for C20H23F3N4O2 [M + H]+ 409.1846; found 409.1833.
(S)-2-(Dimethylamino)-N-(4-((2-oxo-5-(trifluoromethyl)pyridin-1(2H)-yl)methyl)phenyl)propenamide (10b)
In addition to replacing 2-(4-methylpiperazin-1-yl)acetic acid with (S)-dimethylalanine, compound 10b was obtained by the same method as 10a (0.24 g, 65% yield). 1H NMR (400 MHz, CD3OD) δ: 8.25 (s, 1H), 7.65 (dd, J = 9.5, 2.5 Hz, 1H), 7.59 (d, J = 8.5 Hz, 2H), 7.33 (d, J = 8.5 Hz, 2H), 6.65 (d, J = 9.5 Hz, 1H), 5.18 (s, 2H), 3.28 (q, J = 6.9 Hz, 1H), 2.43 (s, 6H), 1.35 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ: 172.0, 162.7, 138.3 (q, J = 5.0 Hz), 137.9, 135.8 (d, J = 2.5 Hz), 131.8, 128.5, 120.5, 120.2, 64.8, 52.1, 41.0, 12.6. HRMS (ESI) m/z: calculated for C18H20F3N3O2 [M + H]+ 368.1580; found 368.1568.
(R)-2-(Dimethylamino)-N-(4-((2-oxo-5-(trifluoromethyl)pyridin-1(2H)-yl)methyl)phenyl)propenamide (10c)
In addition to replacing 2-(4-methylpiperazin-1-yl)acetic acid with (R)-dimethylalanine, compound 10c was obtained by the same method as 10a (0.37 g, 68% yield). 1H NMR (500 MHz, CD3OD) δ: 8.25 (s, 1H), 7.69–7.62 (m, 1H), 7.59 (d, J = 8.5 Hz, 2H), 7.33 (d, J = 8.1 Hz, 2H), 6.65 (d, J = 9.5 Hz, 1H), 5.18 (s, 2H), 3.16 (q, J = 6.9 Hz, 1H), 2.35 (d, J = 0.9 Hz, 6H), 1.30 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CD3OD) δ: 174.2, 164.1, 139.6 (q, J = 5.0 Hz), 137.2, 133.1, 129.8, 125.9, 123.8, 121.9, 121.6, 66.2, 53.4, 42.4, 13.8. HRMS (ESI) m/z: calculated for C18H20F3N3O2 [M + H]+ 368.1580; found 368.1570.
2-(Dimethylamino)-N-(4-((2-oxo-5-(trifluoromethyl)pyridin-1(2H)-yl)methyl)phenyl)acetamide (10d)
In addition to replacing 2-(4-methylpiperazin-1-yl)acetic acid with dimethylglycine, compound 10d was obtained by the same method as 10a (0.24 g, 69% yield). 1H NMR (500 MHz, CH3OD) δ: 8.25 (s, 1H), 7.66 (dd, J = 9.6, 2.6 Hz, 1H), 7.58 (d, J = 8.5 Hz, 2H), 7.33 (d, J = 8.5 Hz, 2H), 6.65 (d, J = 9.6 Hz, 1H), 5.18 (s, 2H), 3.47 (s, 2H), 2.58 (s, 6H). 13C NMR (125 MHz, CD3OD) δ: 168.6, 164.0, 139.6 (q, J = 5.0 Hz), 139.1, 137.2 (q, J = 2.5 Hz), 133.3, 129.9, 121.9, 121.5, 62.6, 53.4, 45.4. HRMS (ESI) m/z: calculated for C17H18F3N3O2 [M + H]+ 354.1424; found 354.1421.
N-Methyl-N-(4-((2-oxo-5-(trifluoromethyl)pyridin-1(2H)-yl)methyl)phenyl)acetamide (11a)
To a solution of compound 9a in DMF (10 mL), NaH (50 mg, 1.2 mmol) was added at 0 °C. After the mixture was stirred for 15 minutes, methyl iodide (170 mg, 1.2 mmol) was added. After the reaction rose naturally to room temperature, water (1 mL) was added. DMF was removed under reduced pressure and water was added to the residue. The mixture was extracted with ethyl acetate (10 mL × 2) and the combined organic phase was washed with brine, and dried over sodium sulfate. The solvents were removed under reduced pressure and the residue was purified by column chromatography on silica gel to give 11a (0.24 g, 75% yield). 1H NMR (500 MHz, CDCl3) δ: 7.77 (s, 1H), 7.48 (d, J = 9.5 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 7.8 Hz, 2H), 6.69 (d, J = 9.5 Hz, 1H), 5.18 (s, 2H), 3.25 (s, 3H), 1.87 (s, 3H). 13C NMR (125 MHz, CDCl3) δ: 170.4, 161.8, 144.6, 136.9 (q, J = 5.0 Hz), 135.2 (d, J = 2.5 Hz), 134.9, 129.4, 127.7, 124.2, 122.1, 121.8, 52.3, 37.1, 22.4. HRMS (ESI) m/z: calculated for C16H15F3N2O2 [M + Na]+ 347.0978; found 347.0977.
N-Methyl-N-(4-((5-methyl-2-oxopyridin-1(2H)-yl)methyl)phenyl)acetamide (11b)
Replacing 9a with 9c, compound 11b was obtained by the same method as 11a (0.21 g, 77% yield). 1H NMR (500 MHz, CDCl3) δ: 7.35 (d, J = 7.9 Hz, 2H), 7.23 (d, J = 9.2 Hz, 1H), 7.16 (d, J = 7.8 Hz, 2H), 7.11 (s, 1H), 6.58 (d, J = 9.2 Hz, 1H), 5.13 (s, 2H), 3.23 (s, 3H), 2.08 (s, 3H), 1.86 (s, 3H). 13C NMR (125 MHz, CDCl3) δ: 170.4, 161.9, 144.1, 142.4, 136.3, 134.7, 129.1, 127.4, 120.9, 115.6, 51.6, 37.1, 22.4, 17.1. HRMS (ESI) m/z: C16H18N2O2 [M + Na]+ 293.1260; found 293.1262.
Biological assay
Cell lines and culture. The human embryonic lung fibroblast cell line (HFL1) and the mouse embryonic lung fibroblast cell line (NHI3T3) used in this study were purchased from Bokang Biotechnology Co. LTD (Qingdao, China). Cell lines were cultivated in F12K/DMEM containing 10% (v/v) heat-inactivated fetal bovine serum (FBS), 100 units per mL penicillin, and 100 μg mL−1 streptomycin.
CCK8 assay
For cytotoxicity assay, the cells grown in the logarithmic phase were seeded into 96-well plates (5 × 103 cells per well) for 24 h, and then exposed to different concentrations of the test compounds for 72 h. The attached cells were incubated with CCK8 (Sigma, USA) for another 1–4 h. Then, the absorbance at 450 nm was measured using a multifunction microplate reader (Molecular Devices, Flex Station 3), and each experiment was performed at least in triplicate. The cytotoxic effects of each compound were expressed as IC50 values, which represent the drug concentrations required to cause 50% tumour cell growth inhibition, and were calculated with GraphPad Prism Software version 5.02 (GraphPad Inc., La Jolla, CA, USA).
Anti-cell-migration study
NHI3T3 cells were plated in a 6-well culture dish at 5 × 105 cells per dish and grown for 24 h, and the non-migrated cells were scraped off the upper surface of the membrane with a 10 μL pipette. The medium was then replaced by 10% serum 1640 medium and treated with compound 9d at the indicated concentrations for another 24 h. After washing with phosphate buffer solution (PBS), the cell images were immediately detected using a Zeiss LSM 570 laser scanning confocal microscope (Carl Zeiss, Germany). The cell migration assay was performed using Transwell inserts with an 8.0 μM pore size. The cells (5 × 104 cells in 0.2 mL of serum-free medium) were added to the upper chamber. Various concentrations of chemicals in 0.5 L of medium with 10% FBS were applied to the lower chamber. After 9d-48 h, the migrated cells attached to the lower surface were stained with crystal violet (500 μL of 5 mg mL−1 crystal violet dissolved in 20% methanol) (Sigma-Aldrich) and allowed to incubate for 10 min. The membrane was then washed several times with PBS, and the cells that penetrated the filter were counted under a microscope.
Immunofluorescence microscopy
The NHI3T3 cells were fixed in 4% paraformaldehyde, stained with specific primary antibodies, including those against E-cadherin and α-SMA, at 4 °C overnight and then incubated with FITC-conjugated anti-rabbit IgG (Solarbio). The nuclei were stained with DAPI. Representative micrographs were observed using a confocal laser scanning microscope (Leica, Wetzlar, Germany).
In vivo biosafety evaluation
Kunming mice (5–6 weeks old, 18–24 g) in pathogen-free condition, obtained from the Vital River of Beijing University, were maintained at constant room temperature and fed a standard rodent chow and water. The mice were randomly divided into experimental group and control group (n = 10, five male and five female). The mice were caudal vein injected with 9d at a dose of 100/150/300 mg kg−1, and the control group was caudal vein injected with NaCl solution. The mice died after neck dissection, and the tissues of the liver, spleen and kidney were taken for histopathological examination. The major organs (liver, kidney, heart, and spleen) were excised, fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 6 μm sections for hematoxylin and eosin (H&E) staining by standard procedures. Images were captured using a microscope.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Foundation of Guangdong Innovation Team Project (2016ZT06Y337), Shandong Provincial Natural Science Foundation (ZR2021QB172), and BZMC Scientific Research Foundation (BY2020KYQD08) for financial support of this study.
Notes and references
- K. K. Brown, F. J. Martinez, S. L. Walsh, V. J. Thannickal, A. Prasse, R. Schlenker-Herceg, R.-G. Goeldner, E. Clerisme-Beaty, K. Tetzlaff, V. Cottin and A. U. Wells, Eur. Respir. J., 2020, 55, 2000085 CrossRef CAS PubMed.
- V. Navaratnam, K. M. Fleming, J. West, C. J. Smith, R. G. Jenkins and A. Fogarty, Thorax, 2011, 66, 462–467 CrossRef CAS PubMed.
- M. Wijsenbeek and V. Cottin, N. Engl. J. Med., 2020, 383, 958–968 CrossRef CAS PubMed.
- G. Sgalla, A. Comes, M. Lerede and L. Richeldi, Expert Opin. Invest. Drugs, 2021, 30, 1183–1195 CrossRef CAS PubMed.
- O. Hilberg, U. Simonsen, R. du Bois and E. Bendstrup, Clin. Respir. J., 2012, 6, 131–143 CrossRef CAS PubMed.
- S. T. Lehtonen, A. Veijola, H. Karvonen, E. Lappi-Blanco, R. Sormunen and S. Korpela, Respir. Res., 2016, 17, 14 CrossRef PubMed.
- L. Knüppel, Y. Ishikawa, M. Aichler, K. Heinzelmann, R. Hatz and J. Behr, Am. J. Respir. Cell Mol. Biol., 2017, 57, 77–90 CrossRef PubMed.
- T. E. King, W. Z. Bradford and S. Castro-Bernardini, et al., N. Engl. J. Med., 2014, 370, 2083 CrossRef PubMed.
- P. W. Noble, C. Albera and W. Z. Bradford, et al., Lancet, 2011, 377, 1760 CrossRef CAS.
- I. M. Westra, D. Oosterhuis, G. M. Groothuis and P. Olinga, PLoS One, 2014, 9, e95462 CrossRef PubMed.
- K. Yamagami, T. Oka, Q. Wang, T. Ishizu, J. K. Lee, K. Miwa, H. Akazawa, A. T. Naito, Y. Sakata and I. Komuro, Am. J. Physiol.: Heart Circ. Physiol., 2015, 309, H512–H522 CrossRef CAS PubMed.
- D. Tampe and M. Zeisberg, Nat. Rev. Nephrol., 2014, 10, 226–237 CrossRef CAS PubMed.
- Y. Xi, Y. Li, P. Xu, S. Li, Z. Liu, H. Tung, X. Cai, J. Wang, H. Huang, M. Wang, M. Xu, S. Ren, S. Li, M. Zhang, Y. J. Lee, L. Huang, D. Yang, J. He, Z. Huang and W. Xie, Sci. Adv., 2021, 7, eabg9241 CrossRef CAS PubMed.
- J. G. F. Cleland, P. Pellicori and A. Gonzálezand, Nat. Med., 2021, 27, 1338–1344 CrossRef PubMed.
- J. C. Dussaul and C. Chatziantoniou, Cell Death Differ., 2007, 14, 1343 CrossRef PubMed.
- X. Bai, P. Nie, Y. Lou, Y. Zhu, S. Jiang, B. Li and P. Luo, Eur. J. Pharmacol., 2021, 911, 174503 CrossRef CAS PubMed.
- R. M. Hazem, S. A. Antar, Y. K. Nafea, A. A. Al-Karmalawy, M. A. Saleh and M. F. El-Azab, Life Sci., 2022, 288, 120185 CrossRef CAS PubMed.
- H. Taniguchi, M. Ebina and Y. Kondoh, Eur. Respir. J., 2010, 35, 821 CrossRef CAS PubMed.
- C. Wang, Y. He, W. Sun, C. Wu, Z. Li and L. Sun, J. Clin. Pharm. Ther., 2021, 1 Search PubMed.
- C. Zang, Y. Zheng, Y. Wang and L. Li, Eur. J. Med. Res., 2021, 26, 129 CrossRef CAS PubMed.
- J. Chen, M. Lu, B. Liu, Z. Chen, Q. Li, L. Tao and G. Hu, Bioorg. Med. Chem. Lett., 2012, 22, 2300–2302 CrossRef CAS PubMed.
- J. Chen, Z. Pen, M. Lu, X. Xiong, Z. Chen, Q. Li, Z. Cheng, D. Jiang, L. Tao and G. Hu, Bioorg. Med. Chem. Lett., 2018, 28, 222 CrossRef CAS PubMed.
- Y. Y. Wan, G. Y. Tian and H. S. Guo, et al., Respir. Res., 2013, 14, 56 CrossRef PubMed.
- Z. Guo, P. Li, G. Wang, Q. Kang, C. Tu, B. Jiang, J. Zhang, W. Wang and T. Wang, Front. Pharmacol., 2021, 12, 682823 CrossRef CAS PubMed.
- Q. Chun, K. Zhu, Y. Bai, H. Shang, D. Zhang, M. Zhao, P. Zheng and X. Jin, Front. Hum. Neurosci., 2021, 15, 735569 CrossRef PubMed.
- Y. Fan, Y. Fu, Y. Zhou, Y. Liu, B. Hao and R. Shang, BMC Pharmacol. Toxicol., 2022, 23, 2 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a
a