
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilver-catalyzed three-component reaction of uracils, arylboronic acids, and selenium: synthesis of 5-arylselanyluracils†
Yuki Murata ,
Saori Tsuchida,
Rena Nezaki,
Yuki Kitamura,
Mio Matsumura* and
Shuji Yasuike*
,
Saori Tsuchida,
Rena Nezaki,
Yuki Kitamura,
Mio Matsumura* and
Shuji Yasuike*
School of Pharmaceutical Sciences, Aichi Gakuin University, 1-100 Kusumoto-cho, Chikusa-ku, Nagoya 464-8650, Japan. E-mail: m-matsu@dpc.agu.ac.jp; s-yasuik@dpc.agu.ac.jp
First published on 13th May 2022
Abstract
Herein, we describe a simple and general multi-component synthesis of 5-arylselanyluracils by the regioselective C–H selenation of uracils. Reactions of uracils with arylboronic acid and Se powder in the presence of AgNO3 (10 mol%) at 120 °C under aerobic conditions afforded various 5-arylselanyluracils. The source of the introduced selanyl group was prepared from a commercially available arylboronic acid and Se powder in the reaction system, thereby ensuring a simple and efficient protocol. This reaction represents the first example of the synthesis of a 5-arylselanyluracil in a multi-component system.
Introduction
5-Substituted uracils have played very important roles in a wide range of fields, including organic synthesis, medicinal chemistry, and drug discovery.1 Among these, 5-selanyluracils have attracted significant interest due to their potential as biological and pharmaceutical therapeutic agents (Fig. 1). For instance, 5-phenylselanyluracil I is a potent inhibitor of DHUDase and UedPase,2 selenoimidazole nucleoside II potently inhibits human and malarial orotate phosphoribosyltransferase,3 and selenocyanic acid III acts as a hypoxic radiosensitizer and genomic DNA molecular probe.4 Owing to the importance of 5-selanyluracils, as mentioned above, a number of synthetic strategies have been developed to prepare 5-arylselanyluracils from uracils. Conversions from 5-halouracil include nucleophilic substitution reaction with phenylselenol promoted by microwave irradiation5 and Cu-mediated cross-coupling reaction with a diaryl diselenide.6 A more versatile method involves the reaction of 5-unsubstituted uracils with phenylselanyl chloride or a diaryl diselenide. Anzai reported the first synthesis of 5-phenylselanyluracil by reacting uracil with phenylselanyl chloride,7 while Kim et al. developed the reaction of a 5-unsubstituted uracil with phenylselanyl chloride in the presence of a silver reagent, such as Ag2O, AgBF4, or AgOCOCF3.8 These authors also reported the reactions of uracils with diphenyl diselenide using Mn(OAc)2 and hypervalent iodine reagents, such as (diacetoxyiodo)benzene.9,10 On the other hand, Yotphan et al. reported the Cu-catalyzed C–H selenation of a uracil with diphenyl diselenide, and iodine-persulfate-promoted selenation,11,12 while Cheng et al. developed the oxidative coupling of a uracil with a diaryl diselenide using a NaI–H2O2 system.13 Ma et al. and He et al. performed reactions of uracils with diaryl diselenides in electrolytic syntheses in the presence of NH4I14 and KI,15 respectively. Furthermore, Choudhury et al. recently reported photoreaction under irradiation of visible light using Rose Bengal as a photocatalyst.16 However, selenium source, such as the diaryl diselenides, arylselanyl chlorides, and arylselenols used in these reactions, is poorly commercially available and/or require complicated synthetic procedures. Moreover, these reactions are limited to two-components, and the efficient syntheses of 5-selanyluracils using multi-component reactions and readily available selenium sources have, to the best of our knowledge, not been reported to date.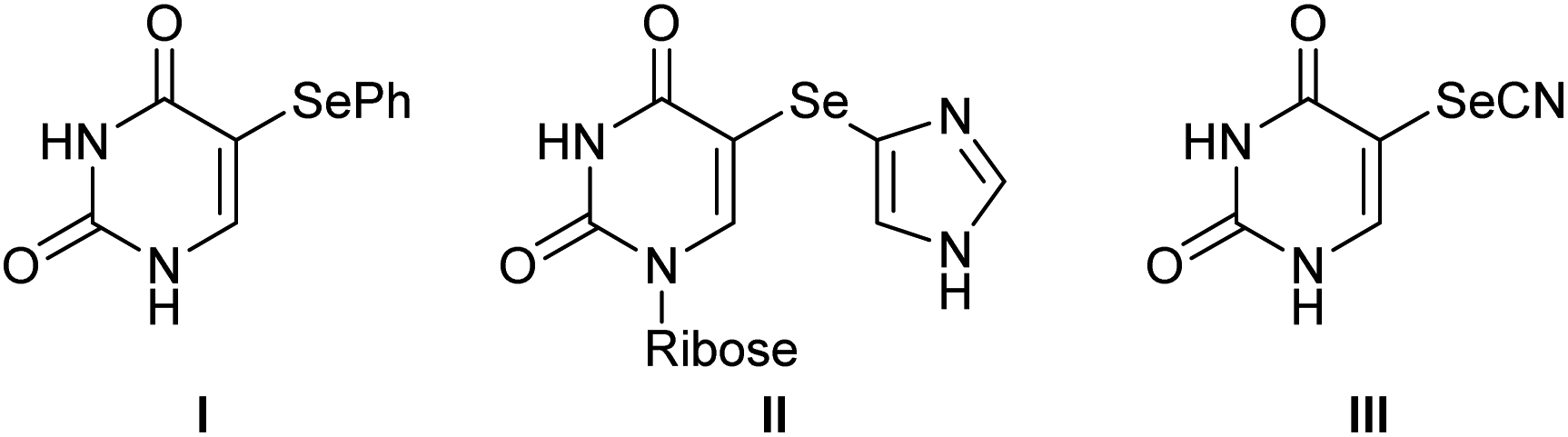 | ||
| Fig. 1 Biologically active 5-selanyluracils. | ||
Ag-catalyzed reactions are powerful tools for the formation of carbon–carbon and carbon–heteroatom bonds and are attracting increasing attention in modern organic chemistry.17 Among these, Liu and co-workers developed Ag-catalyzed three-component reactions for C–Se bond formation using an arylboronic acid, Se powder, and an epoxide or acetylene derivative;18–21 the reactions with epoxides catalyzed by AgNO3 afford β-hydroxyselenides through epoxide ring-opening and selenation (Scheme 1a).18 Reactions with 2-alkynyl-anisoles, -thioanisoles, -aryl esters, and alkynone O-methyloximes catalyzed by AgNO3 and AgNO2 gave furan and thiophene derivatives, isochromenones, and isoxazoles through the tandem cyclization and selenation (Scheme 1b and c).19–21 Clearly, the use of Se powder, which is commercially available, stable, and easy-to-handle, as a reagent for the construction of C–Se bonds provids a more straightforward and attractive alternative. However, the previous methods used either for ring-opening reactions of epoxides or for electrophilic annulation reactions to form heterocycles. Inspired by the aforementioned reports, in this paper we present the simple and efficient C–H selenation of uracils (Scheme 1d). The developed protocol involves the Ag-catalyzed regioselective three-component reaction of uracils, selenium, and arylboronic acids under aerobic conditions, and are first examples of using for C–H selenation.
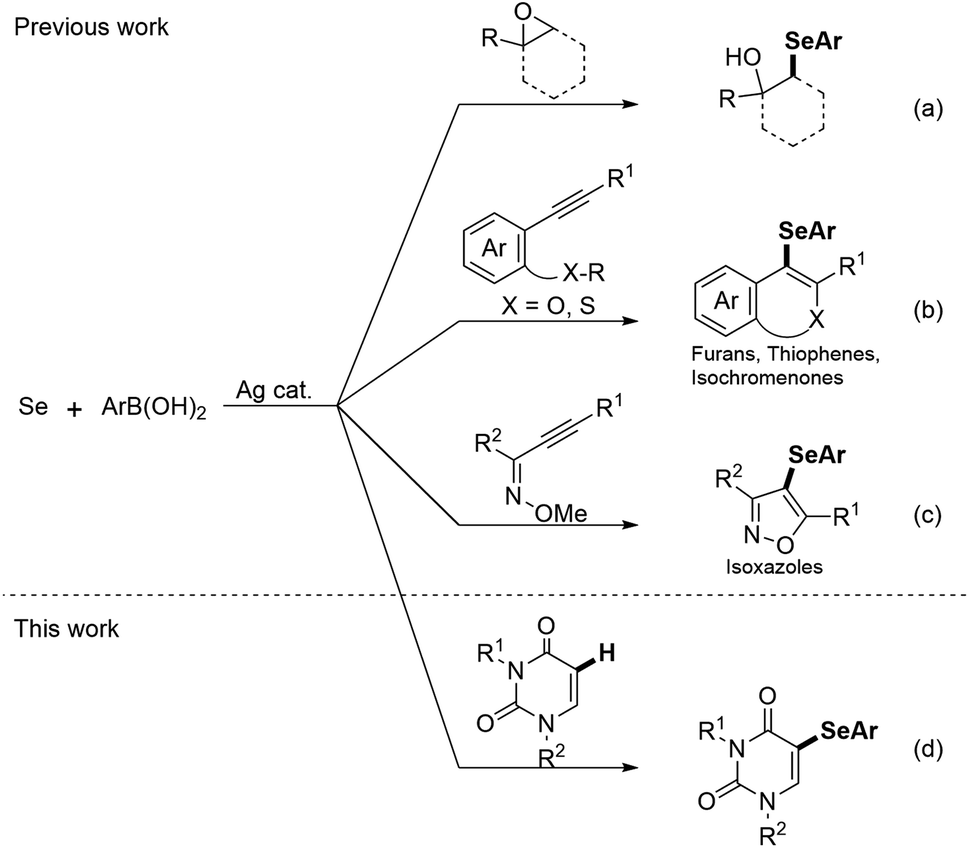 | ||
| Scheme 1 Diverse Ag catalyzed reaction using selenium powder and boronic acid. | ||
Results and discussion
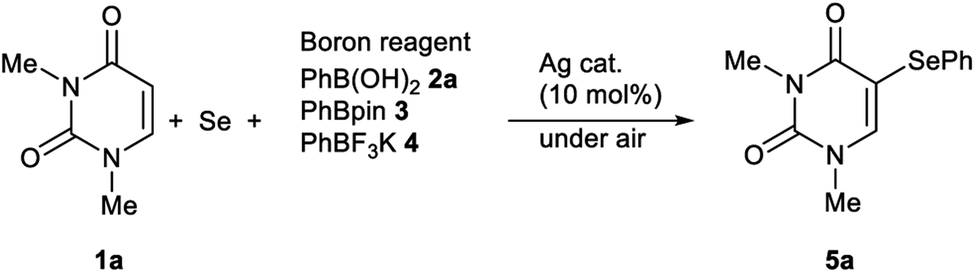
Initially, we focused on identifying the optimal experimental conditions for the synthesis of 5-phenylselanyluracil 5a using a Ag-catalyzed three-component reaction involving N,N-dimethyluracil 1a, Se powder and phenylboron reagents 2a–4, the results of which, including screening for suitable Ag catalysts, solvents, and aryl donors, are summarized in Table 1. We first reacted 1a (0.5 mmol) with Se powder (0.5 mmol) and phenylboronic acid 2a (0.5 mmol) using a variety of available Ag catalysts (0.05 mmol) under aerobic conditions in DMSO at 120 °C (entries 1–8). AgNO3 and AgNO2 gave coupling product 5a in high yields (97% and 75%, respectively); however, the other silver catalysts examined did not produce 5a. Therefore, AgNO3 was identified to be the best catalyst for this reaction in terms of the yield of product 5a (entry 1). The reaction also did not proceed in the absence of the Ag catalyst (entry 9). Solvent screening revealed that the reaction proceeded effectively only in DMSO, whereas NMP, DMF, MeCN, 1,4-dioxane, toluene, and 1,2-DCE were inefficient reaction solvents (entries 1, and 10–15). The reaction under oxygen produced 5a in a high yield (89%) that was almost the same as that obtained under aerobic conditions, while the reaction yield was suppressed (8%) under argon (entries 16 and 17). This reaction gives the best result under aerobic conditions, which are operationally superior reaction conditions (entry 1). A lower yield was obtained at 100 °C compared to 120 °C (entry 18), and decreasing the AgNO3 loading from 10 to 5 or 1 mol% significantly decreased the yield of 5a and extended the reaction time (entries 1, 19 and 20). The reaction of 1a with 2-phenyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane 3 gave coupling product 5a in 64% yield, while no reaction was observed with potassium phenyltrifluoroborate 4 (entries 21 and 22). These results show that boronic acid 2a is a superior aryl-group donor. The regiochemistry of 5-selanyluracil 5a was elucidated by 1H NMR spectroscopy and single-crystal X-ray analysis (Fig. 2); the 1H NMR spectrum of 5a is consistent with that of a standard sample.13
|
||||||
|---|---|---|---|---|---|---|
| Entry | Boron reagent | Ag cat. | Temp. (°C) | Solvent | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a–4 (0.5 mmol), Se powder (0.5 mmol), Solvent (3 mL).b GC yield using biphenyl as internal standard.c Isolated yield.d Under O2 (1 atm).e Under Ar.f Ag cat. (5 mol%).g Ag cat. (1 mol%). | ||||||
| 1 | 2a | AgNO3 | 120 | DMSO | 4 | 97 (96)c |
| 2 | 2a | AgNO2 | 120 | DMSO | 4 | 75 |
| 3 | 2a | AgOAc | 120 | DMSO | 24 | — |
| 4 | 2a | AgOTf | 120 | DMSO | 24 | — |
| 5 | 2a | AgBF4 | 120 | DMSO | 24 | — |
| 6 | 2a | AgSbF6 | 120 | DMSO | 24 | — |
| 7 | 2a | Ag2CO3 | 120 | DMSO | 24 | — |
| 8 | 2a | Ag2O | 120 | DMSO | 24 | — |
| 9 | 2a | — | 120 | DMSO | 24 | — |
| 10 | 2a | AgNO3 | 100 | 1,4-Dioxane | 24 | 22 |
| 11 | 2a | AgNO3 | 80 | MeCN | 24 | 18 |
| 12 | 2a | AgNO3 | 120 | NMP | 24 | 7 |
| 13 | 2a | AgNO3 | 100 | Toluene | 24 | 7 |
| 14 | 2a | AgNO3 | 80 | 1,2-DCE | 24 | 3 |
| 15 | 2a | AgNO3 | 120 | DMF | 24 | — |
| 16d | 2a | AgNO3 | 120 | DMSO | 4 | 89 |
| 17e | 2a | AgNO3 | 120 | DMSO | 24 | 8 |
| 18 | 2a | AgNO3 | 100 | DMSO | 24 | 52 |
| 19f | 2a | AgNO3 | 120 | DMSO | 24 | 87 |
| 20g | 2a | AgNO3 | 120 | DMSO | 24 | 11 |
| 21 | 3 | AgNO3 | 120 | DMSO | 6 | 64 |
| 22 | 4 | AgNO3 | 120 | DMSO | 24 | — |
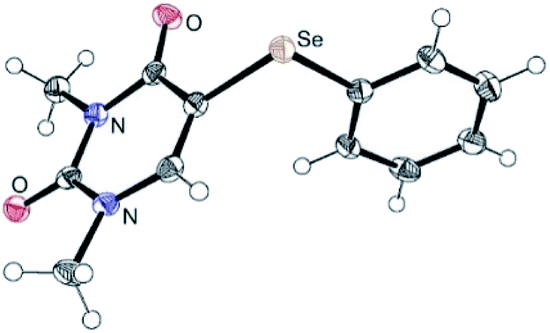 | ||
| Fig. 2 ORTEP drawing 5a with 50% probability (CCDC 2093049). | ||
To demonstrate the efficiency and generality of this three component system, the reactions of various uracils 1 (0.5 mmol), Se powder (0.5 mmol), and boronic acids 2 (0.5 mmol) were investigated under the optimized conditions, the results of which are summarized in Table 2. Reactions of N,N-dimethyluracil 1a with Se powder and arylboron reagents 2b–f bearing various electron-donating and -withdrawing groups at the 4-position of the benzene ring afforded the corresponding products 5c–f in good yields; however 5b, which bears an electron-donating methoxy group, was obtained in poor yield (19%). Di(4-methoxyphenyl)selenide was unexpectedly isolated as the main product in 66% yield when 4-methoxyphenylboronic acid 2b was used. The reaction of sterically hindered ortho-substituted boronic acids gave the corresponding 5-selanyluracils 5g and 5h without any difficulties. Unfortunately, the reaction of arylboronic acids containing heterocyclic rings, such as benzothiophene and pyridine, afforded 5i in low yield (16%), and while 5j was not produced. Moreover, reactions using vinyl and alkylboranoic acids did not yield the corresponding product 5k and 5l, and uracil 1a were recovered.
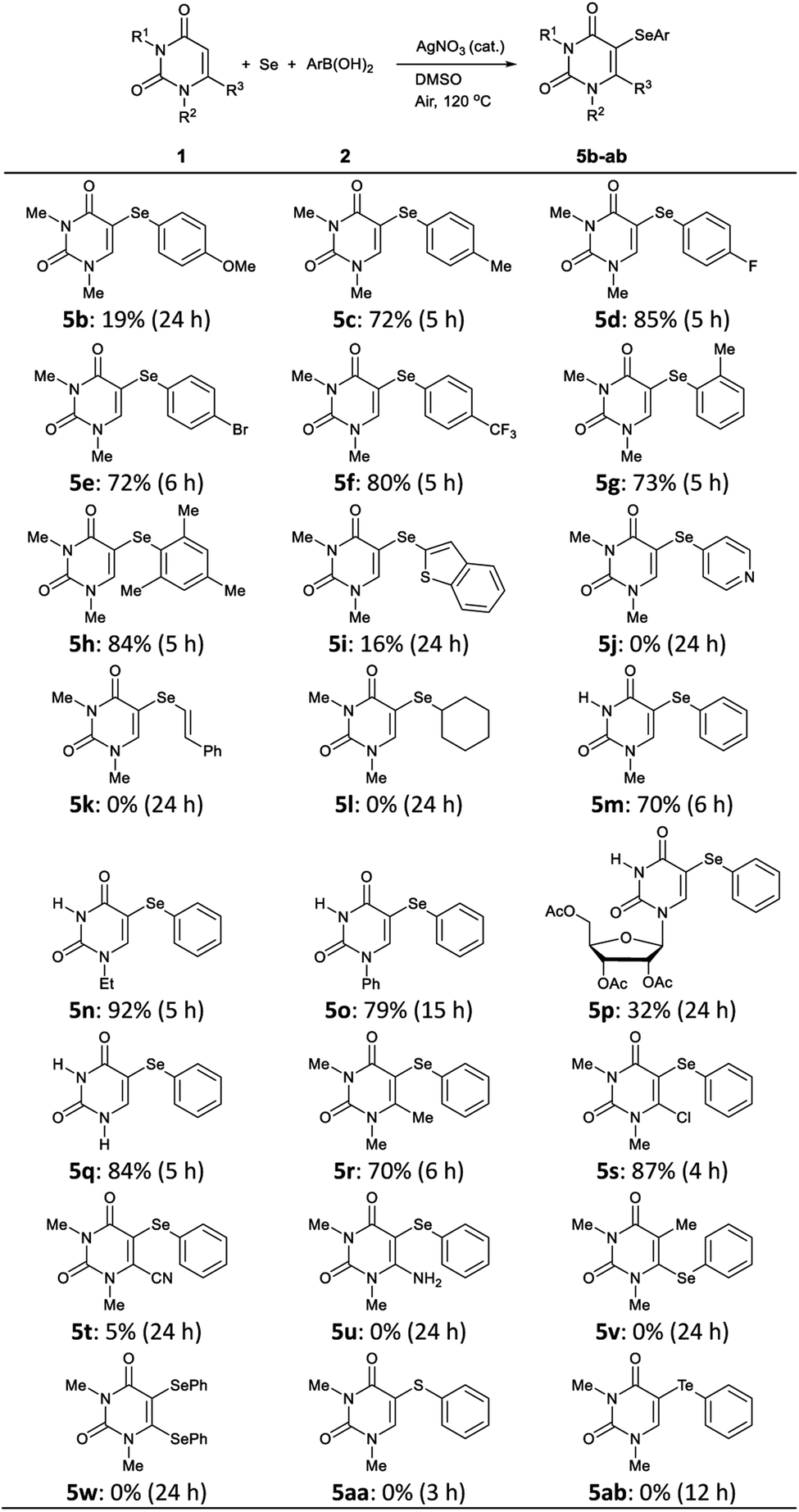
Various uracils 1 were then treated with Se powder and phenylboronic acid 2a under the same reaction conditions. Mono-substituted uracils 1m–o bearing methyl, ethyl, and phenyl groups at their 1-position gave products 5m–o, respectively, in satisfactory yields. Uracil 1p bearing a triacetyl-D-ribosyl group also gave the desired product 5p, albeit in low yield (32%). The reaction proceeded smoothly even with unsubstituted uracil 1q to give the 5-selanyluracil 5q in 84% yield. 6-Methyl and 6-chloro substituted uracils gave coupling products 5r and 5s in good yields, whereas nitrile 1t reacted to produce 5t in extremely low yield (5%), while amine 5u was not obtained. In the reaction of 5-methyluracil 1v, selenation at 6-position did not proceed and 6-selanyluracil 5v was not obtained. We also attempted to double C–H selenation of 1a using 2 equiv. of Se powder and phenylboronic acid 2a, but the corresponding diselanyluracil 5w was not obtained; rather 5-selanyluracil 5a was isolated in 95% yield. These results suggest that this reaction proceeds only at the 5-position of uracil. Finally, other chalcogen sources, such as sulfur and tellurium powder, were subjected to the reaction conditions with 1a and 2a; however, neither chalcogen reacted and the corresponding products 5aa and 5ab were not produced.
Diphenyl diselenide 6 and 5-selanyluracil 5a were observed when the reaction of 1a, 2a, and Se powder was monitored by gas chromatography (Fig. 3). Therefore, we carried out a control experiment to investigate the reaction pathway and mechanism. The reaction of phenylboronic acid 2a with Se powder under the standard conditions gave diphenyl diselenide 6 in 78% yield [Scheme 2, eqn (1)]. The two-component reaction of uracil 1a with 6 gave the corresponding 5-selanyluracil 5a in high yield [eqn (2)]; this reaction was completely inhibited when a radical scavenger, such as TEMPO [(2,2,6,6-tetramethyl-piperidin-1-yl)oxyl] or diphenylethylene, was included. In addition, the three-component reaction of 1a, 2a, and Se powder did not proceed in the presence of a radical scavenger [eqn (3)]. These results suggest that these reactions proceed by a radical mechanism that involves the formation of diphenyl diselenide. Selenide 7 was detected by GC-MS when phenylboronic acid 2a was reacted with Se powder and diphenylethylene under the standard conditions [eqn (4)]; 7 was also formed when 6 was reacted with diphenylethylene [eqn (5)]. Moreover, the reaction of phenylboronic acid with diphenylethylene gave triphenylethylene 8 in 30% yield [eqn (6)]. These results suggest that phenylselanyl and phenyl radicals are generated in this reaction system.
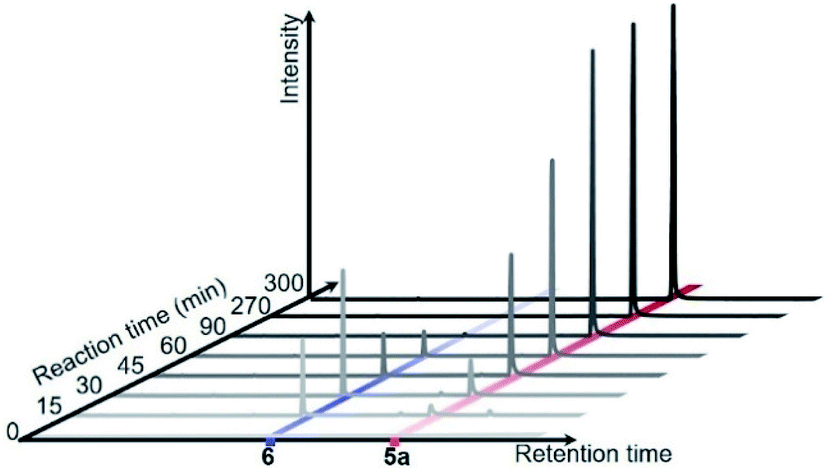 | ||
| Fig. 3 Monitor of the reaction using 1a, 2a and Se powder by GC. | ||
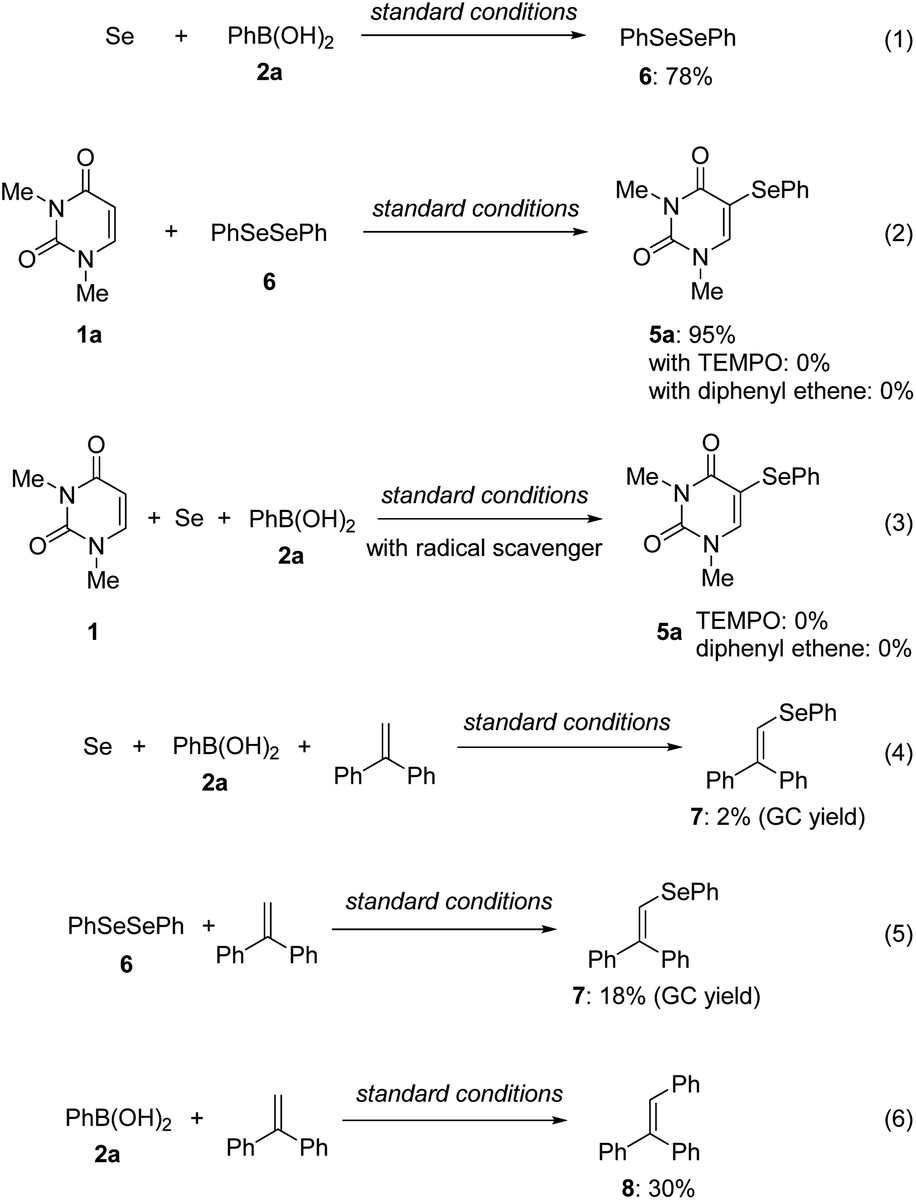 | ||
| Scheme 2 Control experiments. | ||
Based on the above control experiments, a possible mechanism for the three-component reaction is proposed in Scheme 3. Initially, the aryl radical is generated from arylboronic acid 2 by the action of Ag2+, which is formed by the aerobic or DMSO oxidation of AgNO3. The aryl radical then reacts with selenium powder to form arylselanyl radical A, which immediately undergoes radical coupling to form diaryl diselenide B.18,21 Diselenide B then receives one electron from Ag+ to generate the aryl selenide anion C and A. Radical A then reacts with uracil 1 to produce intermediate D, which is then converted into 3-selenyluracil 5 via E and F. On the other hand, the generated selenide anion C is oxidized to the stable diaryl diselenide B and reused in our reaction system, which improves the reaction efficiency. As a more direct route, it is also possible that uracil 1 reacts with radical species A that was initially generated from boronic acid 2 and selenium, to produce the 3-selanyluracil 5.
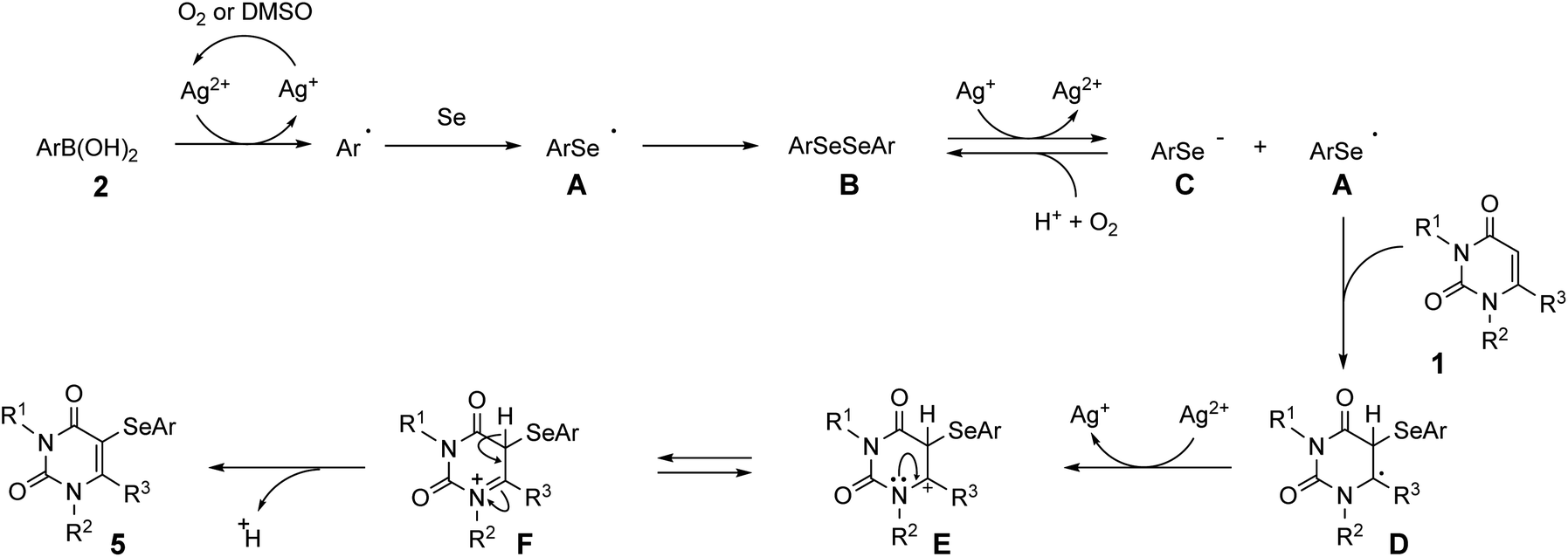 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In conclusion, we developed a Ag-catalyzed three-component reaction for the synthesis of a 5-selanyluracils from a uracil, Se powder, and an arylboronic acid. This protocol enables various functionalized uracils and arylboronic acids to be converted into desired products. The reaction proceeds with high regioselectivity, the required reagents, including the arylboronic acid and Se powder, are inexpensive and easy to handle, and the protocol can easily be performed under aerobic conditions. Detailed mechanistic studies into this three-component reaction and its application to medicinal chemistry are currently in progress and will be reported in the future.Experimental
General information
All chromatographic separations were accomplished with Silica Gel 60N (Kanto Chemical Co., Inc.). Thin-layer chromatography (TLC) was performed with Macherey-Nagel Sil G25 UV254 pre-coated TLC plates. Reagents were used without further purification unless otherwise specified. Melting points were recorded on a Yanagimoto micro melting point hot-stage apparatus (MP-S3) and are not corrected. IR spectra were recorded on a SHIMADZU FTIR-8400S spectrophotometer and are reported in frequency of absorption (cm−1). Only selected IR peaks are reported. 1H NMR (TMS: δ = 0.00 ppm as an internal standard), 13C NMR (CDCl3: δ = 77.00 ppm as an internal standard), 19F NMR (trifluoromethylbenzene: δ = −64.0 ppm as an external standard) and 77Se NMR (Ph2Se2: δ = 436.15 ppm as an internal standard) spectra were recorded on a JEOL ECZ-400S (400 MHz, 100 MHz, 376 MHz and 76 MHz) spectrometer in CDCl3 unless otherwise stated. GC-MS (EI) spectra were recorded on Agilent 5977E Diff-SST MSD-230V spectrometer. HRMS (ESI) were recorded on Agilent 6230 Accurate-Mass TOF LC/MS system. The X-ray diffraction measurements carried out using a Rigaku XtaLAB Synergy, single source at home/near, HyPix3000 diffractometer. Spectroscopic data of 5-arylselanyluracils 5a, p, q, and 5s are in accordance with the literature.8,11,13Synthesis of 5-arylselanyluracil
Arylboronic acid (2) (0.5 mmol, 1.0 eq.), Se powder (40 mg, 0.5 mmol, 1.0 eq.), AgNO3 (8.5 mg, 0.05 mmol, 10 mol%), and N,N-dimethyluracil derivative (1) (0.5 mmol) were added to dimethylsulfoxide (3 mL) in around-bottom flask. After stirring at 120 °C for 5–24 h, the mixture was cooled to room temperature and evaporated to dryness under reduced pressure. The crude product was purified on a silica gel column chromatography to give the desired product 5a–i and 5m–t.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Mp 119–121 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.56 (dt, J = 9.1, 2.5 Hz, 2H, Ar-H), 7.14 (s, 1H, H-6), 6.86 (dt, J = 9.1, 2.5 Hz, 2H, Ar-H), 3.81 (s, 3H, OCH3), 3.37 (s, 3H, N–CH3), 3.34 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ = 161.9 (s, C), 160.0 (s, C), 151.5 (s, C), 143.7 (s, CH), 136.3 (s, CH), 118.1 (s, C), 115.2 (s, CH), 105.2 (s, C), 55.3 (s, CH3), 37.1 (s, CH3), 28.6 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 321.5 (s). IR (KBr): νmax/cm−1 1711 vs. (C
:1). Mp 119–121 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.56 (dt, J = 9.1, 2.5 Hz, 2H, Ar-H), 7.14 (s, 1H, H-6), 6.86 (dt, J = 9.1, 2.5 Hz, 2H, Ar-H), 3.81 (s, 3H, OCH3), 3.37 (s, 3H, N–CH3), 3.34 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ = 161.9 (s, C), 160.0 (s, C), 151.5 (s, C), 143.7 (s, CH), 136.3 (s, CH), 118.1 (s, C), 115.2 (s, CH), 105.2 (s, C), 55.3 (s, CH3), 37.1 (s, CH3), 28.6 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 321.5 (s). IR (KBr): νmax/cm−1 1711 vs. (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1670 vs. (CO). HRMS (ESI): m/z calcd for C13H14N2O3Se + H+; 327.0248 [M + H]+: found: 327.0251.:1). Mp 113–114 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.46 (d, J = 8.2 Hz, 2H, Ar-H), 7.30 (s, 1H, H-6), 7.11 (d, J = 7.8 Hz, 2H, Ar-H), 3.38 (s, 3H, N–CH3), 3.36 (s, 3H, N–CH3), 2.33 (s, 3H, 4′-CH3). 13C NMR (100 MHz, CDCl3): δ = 161.9 (s, C), 151.6 (s, C), 145.0 (s, CH), 138.3 (s, C), 133.6 (s, CH), 130.3 (s, CH), 125.0 (s, C), 104.2 (s, C), 37.1 (s, CH3), 28.6 (s, CH3), 21.2 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 324.9 (s). IR (KBr): νmax/cm−1 1705 vs. (CO), 1651 vs. (CO). HRMS (ESI): m/z calcd for C13H14N2O2Se + H+; 311.0299 [M + H]+: found: 311.0301.:1). Mp 85–87 °C (ether). 1H NMR (400 MHz, CDCl3): δ = 7.56 (dd, J = 8.7, 5.5 Hz, 2H, Ar-H), 7.41 (s, 1H, H-6), 7.00 (t, J = 8.9 Hz, 2H, Ar-H), 3.40 (s, 3H, N–CH3), 3.38 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ = 162.8 (d, 1JC,F = 249 Hz, CF), 161.9 (s, C), 151.5 (s, C), 145.9 (s, CH), 135.5 (d, 3JC,F = 8.7 Hz, CH), 123.6 (d, 4JC,F = 3.9 Hz, C), 116.6 (d, 2JC,F = 21.2 Hz, CH), 103.6 (s, C), 37.2 (s, CH3), 28.7 (s, CH3). 19F NMR (376 MHz, CDCl3): δ = −114.3 (s). 77Se NMR (76 MHz, CDCl3): δ = 326.0 (s). IR (KBr): νmax/cm−1 1715 vs. (CO), 1655 vs. (CO). HRMS (ESI): m/z calcd for C12H11FN2O2Se + H+; 315.0048 [M + H]+: found: 315.0048.:1). Mp 115–117 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.53 (s, 1H, H-6), 7.42–7.37 (m, 4H, Ar-H), 3.42 (s, 3H, N–CH3), 3.39 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ = 161.8 (s, C), 151.6 (s, C), 147.1 (s, CH), 133.9 (s, CH), 132.4 (s, CH), 128.5 (s, C), 122.1 (s, C), 102.4 (s, C), 37.2 (s, CH3), 28.8 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 328.2 (s). IR (KBr): νmax/cm−1 1707 vs. (CO), 1647 vs. (CO). HRMS (ESI): m/z calcd for C12H11BrN2O2Se + H+; 374.9247 [M + H]+: found: 374.7246.:1). Mp 139–141 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.71 (s, 1H, H-6), 7.54–7.48 (m, 4H, Ar-H), 3.46 (s, 3H, N–CH3), 3.41 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ = 161.8 (s, C), 151.6 (s, C), 148.8 (s, CH), 135.6 (s, C), 130.7 (s, CH), 129.3 (q, 2JC,F = 32.8 Hz, C), 126.0 (q, 3JC,F = 3.9 Hz, CH), 123.9 (q, 1JC,F = 272 Hz, CF3), 100.9 (s, C), 37.3 (s, CH3), 28.9 (s, CH3). 19F NMR (376 MHz, CDCl3): δ = −63.9 (s). 77Se NMR (76 MHz, CDCl3): δ = 335.0 (s). IR (KBr): νmax/cm−1 1711 vs. (CO), 1655 vs. (CO). HRMS (ESI): m/z calcd for C13H11F3N2O2Se + H+; 365.0016 [M + H]+: found: 365.0017.:1). Mp 147–149 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.33 (dd, J = 8.0, 1.4 Hz, 1H, Ar-H), 7.25 (s, 1H, H-6), 7.23–7.18 (m, 2H, Ar-H), 7.10 (td, J = 7.2, 2.1 Hz, 1H, Ar-H), 3.40 (s, 3H, N–CH3), 3.38 (s, 3H, N–CH3), 2.46 (s, 3H, 2′-CH3). 13C NMR (100 MHz, CDCl3): δ = 161.9 (s, C), 151.6 (s, C), 145.6 (s, CH), 139.2 (s, C), 132.4 (s, CH), 130.4 (s, CH), 129.9 (s, C), 127.9 (s, CH), 126.9 (s, CH), 102.5 (s, C), 37.2 (s, CH3), 28.7 (s, CH3), 22.0 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 290.5 (s). IR (KBr): νmax/cm−1 1701 s (CO), 1651 vs. (CO). HRMS (ESI): m/z calcd for C13H14N2O2Se + H+; 311.0299 [M + H]+: found: 311.0298.:1). Mp 142–144 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.01 (s, 2H, 3′,5′-H), 6.27 (s, 1H, H-6), 3.38 (s, 3H, N–CH3), 3.25 (s, 3H, N–CH3), 2.49 (s, 6H, 2′,6′-CH3), 2.31 (s, 3H, 4′-CH3). 13C NMR (100 MHz, CDCl3): δ = 161.9 (s, C), 151.4 (s, C), 143.8 (s, C), 139.7 (s, C), 137.3 (s, CH), 129.2 (s, CH), 123.7 (s, C), 105.7 (s, C), 37.2 (s, CH3), 28.4 (s, CH3), 24.0 (s, CH3), 21.1 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 221.7 (s). IR (KBr): νmax/cm−1 1701 vs. (CO), 1684 vs. (CO). HRMS (ESI): m/z calcd for C15H18N2O2Se + H+; 339.0612 [M + H]+: found: 339.0612.:1). Mp 148–150 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.71–7.67 (m, 2H, Ar-H), 7.51 (d, J = 0.9 Hz, 1H, benzothiophene-3′H), 7.30–7.23 (m, 3H, H-6, Ar-H), 3.31 (s, 3H, N–CH3), 3.28 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ = 161.6 (s, C), 151.4 (s, C), 144.6 (s, CH), 143.6 (s, C), 140.0 (s, C), 132.8 (s, CH), 125.0 (s, CH), 124.7 (s, C), 124.6 (s, CH), 123.5 (s, CH), 121.8 (s, CH), 104.2 (s, C), 37.3 (s, CH3), 28.7 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 263.6 (s). IR (KBr): νmax/cm−1 1709 s (CO), 1655 vs. (CO). HRMS (ESI): m/z calcd for C14H12N2O2SSe + H+; 352.9863 [M + H]+: found: 352.9869.:1). Mp 190–193 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 8.79 (s, 1H, N–H), 7.53–7.50 (m, 2H, Ar-H), 7.47 (s, 1H, H-6), 7.30–7.27 (m, 3H, Ar-H), 3.37 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ = 161.9 (s, C), 150.8 (s, C), 149.0 (s, CH), 132.5 (s, CH), 129.5 (s, CH), 129.3 (s, C), 127.9 (s, CH), 103.9 (s, C), 36.1 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 319.6 (s). IR (KBr): νmax/cm−1 1717 vs. (CO), 1697 vs. (CO). HRMS (ESI): m/z calcd for C11H10N2O2Se + H+; 282.9986 [M + H]+: found: 282.9981.:1). Mp 173–175 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 9.22 (brs, 1H, N–H), 7.54–7.50 (m, 2H, Ar-H), 7.49 (s, 1H, H-6), 7.30–7.27 (m, 3H, Ar-H), 3.78 (q, J = 7.3 Hz, 2H, CH2), 1.29 (t, J = 7.3 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ = 162.0 (s, C), 150.5 (s, C), 148.0 (s, CH), 132.4 (s, CH), 129.4 (s, CH), 127.8 (s, CH), 104.0 (s, C), 44.2 (s, CH2), 14.4 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 319.4 (s). IR (KBr): νmax/cm−1 1697 vs. (CO), 1668 vs. (CO). HRMS (ESI): m/z calcd for C12H12N2O2Se + H+; 297.0142 [M + H]+: found: 297.0143.:3). Mp 198–201 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 8.87 (s, 1H, N–H), 7.64 (s, 1H, H-6), 7.57 (dd, J = 6.2, 3.0 Hz, 2H, Ar-H), 7.49–7.41 (m, 3H, Ar-H), 7.32–7.26 (m, 5H, Ar-H). 13C NMR (100 MHz, CDCl3): δ = 161.7 (s, C), 150.0 (s, C), 147.9 (s, CH), 137.9 (s, CH), 132.8 (s, C), 129.6 (s, CH), 129.5 (s, CH), 129.1 (s, CH), 129.0 (s, C), 128.0 (s, CH), 126.1 (s, CH), 105.0 (s, CH). 77Se NMR (76 MHz, CDCl3): δ = 322.7 (s). IR (KBr): νmax/cm−1 1726 s (CO), 1663 vs. (CO). HRMS (ESI): m/z calcd for C16H12N2O2Se + H+; 345.0142 [M + H]+: found: 345.0142.:1). 1H NMR (400 MHz, CDCl3): δ = 7.38–7.35 (m, 2H, Ar-H), 7.25–7.20 (m, 3H, Ar-H), 3.52 (s, 3H, N–CH3), 3.40 (m, 3H, N–CH3), 2.71 (s, 3H, 6-CH3). 13C NMR (100 MHz, CDCl3): δ = 161.5 (s, C), 157.1 (s, C), 151.9 (s, C), 131.5 (s, C), 130.2 (s, CH), 129.2 (s, CH), 126.7 (s, CH), 103.3 (s, C), 33.6 (s, CH3), 29.2 (s, CH3), 21.9 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 302.8 (s). IR (KBr): νmax/cm−1 1701 s (CO), 1647 vs. (CO). HRMS (ESI): m/z calcd for C13H14N2O2Se + H+; 333.0113 [M + H]+: found: 333.0115.:3). Mp 127–129 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.68 (dd, J = 7.8, 1.8 Hz, 2H, Ar-H), 7.35–7.30 (m, 3H, Ar-H), 3.65 (s, 3H, N–CH3), 3.38 (m, 3H, N–CH3), 2.71 (s, 3H, 6-CH3). 13C NMR (100 MHz, CDCl3): δ = 160.1 (s, C), 150.5 (s, C), 134.2 (s, CH), 131.6 (s, C), 129.5 (s, CH), 128.9 (s, CH), 128.3 (s, C), 113.9 (s, C), 111.1 (s, C), 35.8 (s, CH3), 29.4 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 365.0 (s). IR (KBr): νmax/cm−1 2234 w (C
O), 1670 vs. (CO). HRMS (ESI): m/z calcd for C13H14N2O3Se + H+; 327.0248 [M + H]+: found: 327.0251.:1). Mp 113–114 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.46 (d, J = 8.2 Hz, 2H, Ar-H), 7.30 (s, 1H, H-6), 7.11 (d, J = 7.8 Hz, 2H, Ar-H), 3.38 (s, 3H, N–CH3), 3.36 (s, 3H, N–CH3), 2.33 (s, 3H, 4′-CH3). 13C NMR (100 MHz, CDCl3): δ = 161.9 (s, C), 151.6 (s, C), 145.0 (s, CH), 138.3 (s, C), 133.6 (s, CH), 130.3 (s, CH), 125.0 (s, C), 104.2 (s, C), 37.1 (s, CH3), 28.6 (s, CH3), 21.2 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 324.9 (s). IR (KBr): νmax/cm−1 1705 vs. (CO), 1651 vs. (CO). HRMS (ESI): m/z calcd for C13H14N2O2Se + H+; 311.0299 [M + H]+: found: 311.0301.:1). Mp 85–87 °C (ether). 1H NMR (400 MHz, CDCl3): δ = 7.56 (dd, J = 8.7, 5.5 Hz, 2H, Ar-H), 7.41 (s, 1H, H-6), 7.00 (t, J = 8.9 Hz, 2H, Ar-H), 3.40 (s, 3H, N–CH3), 3.38 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ = 162.8 (d, 1JC,F = 249 Hz, CF), 161.9 (s, C), 151.5 (s, C), 145.9 (s, CH), 135.5 (d, 3JC,F = 8.7 Hz, CH), 123.6 (d, 4JC,F = 3.9 Hz, C), 116.6 (d, 2JC,F = 21.2 Hz, CH), 103.6 (s, C), 37.2 (s, CH3), 28.7 (s, CH3). 19F NMR (376 MHz, CDCl3): δ = −114.3 (s). 77Se NMR (76 MHz, CDCl3): δ = 326.0 (s). IR (KBr): νmax/cm−1 1715 vs. (CO), 1655 vs. (CO). HRMS (ESI): m/z calcd for C12H11FN2O2Se + H+; 315.0048 [M + H]+: found: 315.0048.:1). Mp 115–117 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.53 (s, 1H, H-6), 7.42–7.37 (m, 4H, Ar-H), 3.42 (s, 3H, N–CH3), 3.39 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ = 161.8 (s, C), 151.6 (s, C), 147.1 (s, CH), 133.9 (s, CH), 132.4 (s, CH), 128.5 (s, C), 122.1 (s, C), 102.4 (s, C), 37.2 (s, CH3), 28.8 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 328.2 (s). IR (KBr): νmax/cm−1 1707 vs. (CO), 1647 vs. (CO). HRMS (ESI): m/z calcd for C12H11BrN2O2Se + H+; 374.9247 [M + H]+: found: 374.7246.:1). Mp 139–141 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.71 (s, 1H, H-6), 7.54–7.48 (m, 4H, Ar-H), 3.46 (s, 3H, N–CH3), 3.41 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ = 161.8 (s, C), 151.6 (s, C), 148.8 (s, CH), 135.6 (s, C), 130.7 (s, CH), 129.3 (q, 2JC,F = 32.8 Hz, C), 126.0 (q, 3JC,F = 3.9 Hz, CH), 123.9 (q, 1JC,F = 272 Hz, CF3), 100.9 (s, C), 37.3 (s, CH3), 28.9 (s, CH3). 19F NMR (376 MHz, CDCl3): δ = −63.9 (s). 77Se NMR (76 MHz, CDCl3): δ = 335.0 (s). IR (KBr): νmax/cm−1 1711 vs. (CO), 1655 vs. (CO). HRMS (ESI): m/z calcd for C13H11F3N2O2Se + H+; 365.0016 [M + H]+: found: 365.0017.:1). Mp 147–149 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.33 (dd, J = 8.0, 1.4 Hz, 1H, Ar-H), 7.25 (s, 1H, H-6), 7.23–7.18 (m, 2H, Ar-H), 7.10 (td, J = 7.2, 2.1 Hz, 1H, Ar-H), 3.40 (s, 3H, N–CH3), 3.38 (s, 3H, N–CH3), 2.46 (s, 3H, 2′-CH3). 13C NMR (100 MHz, CDCl3): δ = 161.9 (s, C), 151.6 (s, C), 145.6 (s, CH), 139.2 (s, C), 132.4 (s, CH), 130.4 (s, CH), 129.9 (s, C), 127.9 (s, CH), 126.9 (s, CH), 102.5 (s, C), 37.2 (s, CH3), 28.7 (s, CH3), 22.0 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 290.5 (s). IR (KBr): νmax/cm−1 1701 s (CO), 1651 vs. (CO). HRMS (ESI): m/z calcd for C13H14N2O2Se + H+; 311.0299 [M + H]+: found: 311.0298.:1). Mp 142–144 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.01 (s, 2H, 3′,5′-H), 6.27 (s, 1H, H-6), 3.38 (s, 3H, N–CH3), 3.25 (s, 3H, N–CH3), 2.49 (s, 6H, 2′,6′-CH3), 2.31 (s, 3H, 4′-CH3). 13C NMR (100 MHz, CDCl3): δ = 161.9 (s, C), 151.4 (s, C), 143.8 (s, C), 139.7 (s, C), 137.3 (s, CH), 129.2 (s, CH), 123.7 (s, C), 105.7 (s, C), 37.2 (s, CH3), 28.4 (s, CH3), 24.0 (s, CH3), 21.1 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 221.7 (s). IR (KBr): νmax/cm−1 1701 vs. (CO), 1684 vs. (CO). HRMS (ESI): m/z calcd for C15H18N2O2Se + H+; 339.0612 [M + H]+: found: 339.0612.:1). Mp 148–150 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.71–7.67 (m, 2H, Ar-H), 7.51 (d, J = 0.9 Hz, 1H, benzothiophene-3′H), 7.30–7.23 (m, 3H, H-6, Ar-H), 3.31 (s, 3H, N–CH3), 3.28 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ = 161.6 (s, C), 151.4 (s, C), 144.6 (s, CH), 143.6 (s, C), 140.0 (s, C), 132.8 (s, CH), 125.0 (s, CH), 124.7 (s, C), 124.6 (s, CH), 123.5 (s, CH), 121.8 (s, CH), 104.2 (s, C), 37.3 (s, CH3), 28.7 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 263.6 (s). IR (KBr): νmax/cm−1 1709 s (CO), 1655 vs. (CO). HRMS (ESI): m/z calcd for C14H12N2O2SSe + H+; 352.9863 [M + H]+: found: 352.9869.:1). Mp 190–193 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 8.79 (s, 1H, N–H), 7.53–7.50 (m, 2H, Ar-H), 7.47 (s, 1H, H-6), 7.30–7.27 (m, 3H, Ar-H), 3.37 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ = 161.9 (s, C), 150.8 (s, C), 149.0 (s, CH), 132.5 (s, CH), 129.5 (s, CH), 129.3 (s, C), 127.9 (s, CH), 103.9 (s, C), 36.1 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 319.6 (s). IR (KBr): νmax/cm−1 1717 vs. (CO), 1697 vs. (CO). HRMS (ESI): m/z calcd for C11H10N2O2Se + H+; 282.9986 [M + H]+: found: 282.9981.:1). Mp 173–175 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 9.22 (brs, 1H, N–H), 7.54–7.50 (m, 2H, Ar-H), 7.49 (s, 1H, H-6), 7.30–7.27 (m, 3H, Ar-H), 3.78 (q, J = 7.3 Hz, 2H, CH2), 1.29 (t, J = 7.3 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ = 162.0 (s, C), 150.5 (s, C), 148.0 (s, CH), 132.4 (s, CH), 129.4 (s, CH), 127.8 (s, CH), 104.0 (s, C), 44.2 (s, CH2), 14.4 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 319.4 (s). IR (KBr): νmax/cm−1 1697 vs. (CO), 1668 vs. (CO). HRMS (ESI): m/z calcd for C12H12N2O2Se + H+; 297.0142 [M + H]+: found: 297.0143.:3). Mp 198–201 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 8.87 (s, 1H, N–H), 7.64 (s, 1H, H-6), 7.57 (dd, J = 6.2, 3.0 Hz, 2H, Ar-H), 7.49–7.41 (m, 3H, Ar-H), 7.32–7.26 (m, 5H, Ar-H). 13C NMR (100 MHz, CDCl3): δ = 161.7 (s, C), 150.0 (s, C), 147.9 (s, CH), 137.9 (s, CH), 132.8 (s, C), 129.6 (s, CH), 129.5 (s, CH), 129.1 (s, CH), 129.0 (s, C), 128.0 (s, CH), 126.1 (s, CH), 105.0 (s, CH). 77Se NMR (76 MHz, CDCl3): δ = 322.7 (s). IR (KBr): νmax/cm−1 1726 s (CO), 1663 vs. (CO). HRMS (ESI): m/z calcd for C16H12N2O2Se + H+; 345.0142 [M + H]+: found: 345.0142.:1). 1H NMR (400 MHz, CDCl3): δ = 7.38–7.35 (m, 2H, Ar-H), 7.25–7.20 (m, 3H, Ar-H), 3.52 (s, 3H, N–CH3), 3.40 (m, 3H, N–CH3), 2.71 (s, 3H, 6-CH3). 13C NMR (100 MHz, CDCl3): δ = 161.5 (s, C), 157.1 (s, C), 151.9 (s, C), 131.5 (s, C), 130.2 (s, CH), 129.2 (s, CH), 126.7 (s, CH), 103.3 (s, C), 33.6 (s, CH3), 29.2 (s, CH3), 21.9 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 302.8 (s). IR (KBr): νmax/cm−1 1701 s (CO), 1647 vs. (CO). HRMS (ESI): m/z calcd for C13H14N2O2Se + H+; 333.0113 [M + H]+: found: 333.0115.:3). Mp 127–129 °C (CH2Cl2–hexane). 1H NMR (400 MHz, CDCl3): δ = 7.68 (dd, J = 7.8, 1.8 Hz, 2H, Ar-H), 7.35–7.30 (m, 3H, Ar-H), 3.65 (s, 3H, N–CH3), 3.38 (m, 3H, N–CH3), 2.71 (s, 3H, 6-CH3). 13C NMR (100 MHz, CDCl3): δ = 160.1 (s, C), 150.5 (s, C), 134.2 (s, CH), 131.6 (s, C), 129.5 (s, CH), 128.9 (s, CH), 128.3 (s, C), 113.9 (s, C), 111.1 (s, C), 35.8 (s, CH3), 29.4 (s, CH3). 77Se NMR (76 MHz, CDCl3): δ = 365.0 (s). IR (KBr): νmax/cm−1 2234 w (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1711 s (CO), 1663 vs. (CO). HRMS (ESI): m/z calcd for C13H11N3O2Se + H+; 322.0095 [M + H]+: found: 322.0099.
N), 1711 s (CO), 1663 vs. (CO). HRMS (ESI): m/z calcd for C13H11N3O2Se + H+; 322.0095 [M + H]+: found: 322.0099.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a Nagai Memorial Research Scholarship from the Pharmaceutical Society of Japan (Y. K.), and JSPS KAKENHI (Grant Number JP19K07005) (S. Y.). The authors also thank Aichi Gakuin University for generous financial support.Notes and references
- M. Ahmadian and D. E. Bergstrom, in Modified Nucleosides: in Biochemistry, Biotechnology and Medicine, ed. P. Herdewijn, Wiley-VCH, Weinheim, 2008, pp. 251–276 Search PubMed.
- N. M. Goudgaon, F. N. M. Naguib, M. H. el Kouni and R. F. Schinazi, J. Med. Chem., 1993, 36, 4250 CrossRef CAS PubMed.
- M. Abdo, Y. Zhang, V. L. Schramm and S. Knapp, Org. Lett., 2010, 12, 2981 CrossRef PubMed.
- M. Sosnowska, S. Makurat, M. Zdrowowicz and J. Rak, J. Phys. Chem. B, 2017, 121, 6139 CrossRef CAS PubMed.
- W.-P. Fang, Y.-T. Cheng, Y.-R. Cheng and Y.-J. Cherng, Tetrahedron, 2005, 61, 3107 CrossRef CAS.
- F. Botha, M. Slaíčková, R. Pohl and M. Hocek, Org. Biomol. Chem., 2016, 14, 10018 RSC.
- K. Anzai, J. Heterocycl. Chem., 1979, 16, 567 CrossRef CAS.
- C. H. Lee and Y. H. Kim, Tetrahedron Lett., 1991, 32, 2401 CrossRef CAS.
- D. H. Lee and Y. H. Kim, Synlett, 1995, 349 CrossRef.
- K. R. Roh, H. K. Chang and Y. H. Kim, Heterocycles, 1998, 48, 437 CrossRef CAS.
- M. Noikham and S. Yotphan, Eur. J. Org. Chem., 2019, 2759 CrossRef CAS.
- D. Beukeaw, M. Noikham and S. Yotphan, Tetrahedron, 2019, 75, 130537 CrossRef.
- X.-D. Li, Y.-T. Gao, Y.-J. Sun, X.-Y. Jin, D. Wang, L. Liu and L. Cheng, Org. Lett., 2019, 21, 6643 CrossRef CAS PubMed.
- Q. Wang, X.-L. Ma, Y.-Y. Chen, C.-N. Jiang and Y.-L. Xu, Eur. J. Org. Chem., 2020, 4384 CrossRef CAS.
- J.-Y. Chen, C.-T. Zhong, Q.-W. Gui, Y.-M. Zhou, Y.-Y. Fang, K.-J. Liu, Y.-W. Lin, Z. Cao and W.-M. He, Chin. Chem. Lett., 2021, 32, 475 CrossRef CAS.
- D. Ali, T. Parvin and L. H. Choudhury, J. Org. Chem., 2022, 87, 1230 CrossRef CAS PubMed.
- For selected reviews: (a) Q.-Z. Zheng and N. Jiao, Chem. Soc. Rev., 2016, 45, 4590 RSC; (b) G. Fang, X. Cong, G. Zanoni, Q. Liu and X. Bi, Adv. Synth. Catal., 2017, 359, 1422 CrossRef CAS; (c) M. Neetha, T. Aneeja, C. M. A. Afsina and G. Anilkumar, ChemCatChem, 2020, 12, 5330 CrossRef CAS.
- T. Leng, G. Wu, Y.-B. Zhou, W. Gao, J. Ding, X. Huang, M. Liu and H. Wu, Adv. Synth. Catal., 2018, 360, 4336 CrossRef CAS.
- C. An, C.-Y. Li, X.-B. Huang, W.-X. Gao, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Org. Lett., 2019, 21, 6710 CrossRef CAS PubMed.
- G.-Q. Jin, W.-X. Gao, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, RSC Adv., 2020, 10, 30439 RSC.
- J. Wu, Y.-F. Yang, X.-B. Huang, W.-X. Gao, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, ACS Omega, 2020, 5, 23358 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2093049. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra01685k |
| This journal is © The Royal Society of Chemistry 2022 |