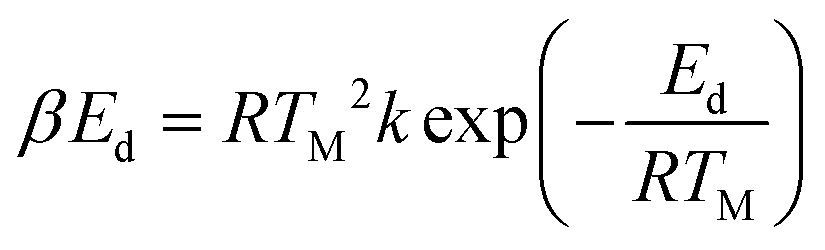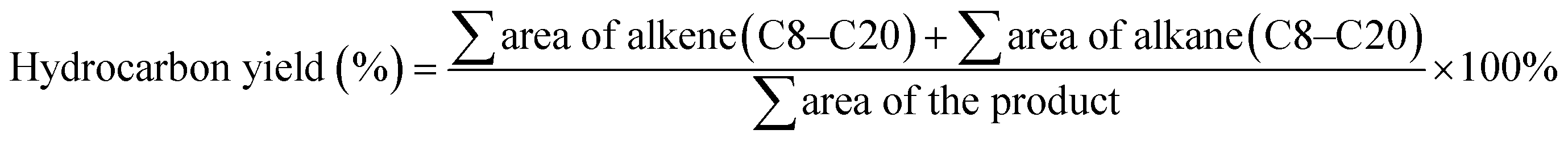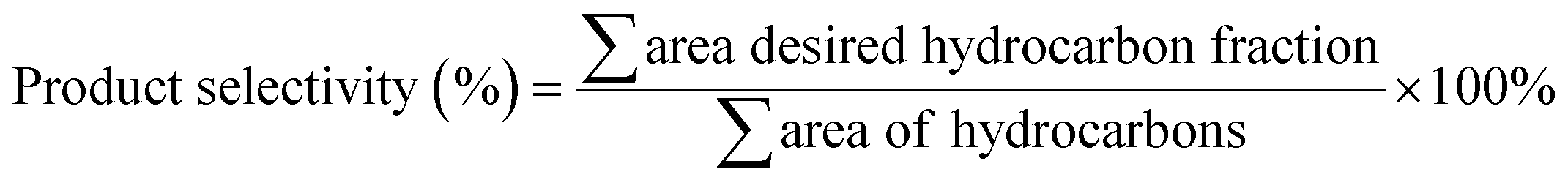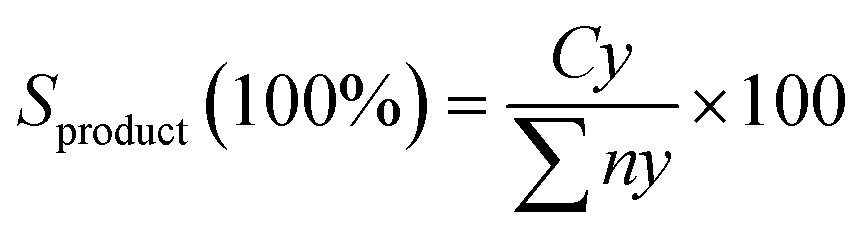DOI:
10.1039/D2RA00853J
(Paper)
RSC Adv., 2022,
12, 16903-16917
Chemoselective decarboxylation of ceiba oil to diesel-range alkanes over a red mud based catalyst under H2-free conditions†
Received
9th February 2022
, Accepted 20th May 2022
First published on 8th June 2022
Abstract
Concerns over global greenhouse gas emissions such as COx and NOx as well as the depletion of petroleum fossil resources have motivated humankind to seek an alternative energy source known as green diesel. In this study, green diesel was produced via a deoxygenation (DO) reaction of ceiba oil under a H2-free atmosphere over Ni modified red mud-based catalysts, which have been synthesized via a precipitation – deep-deposition assisted autoclave method. The obtained catalyst was further characterized by XRF, XRD, BET, FTIR, TPD-NH3, FESEM, and TGA. Based on the catalytic activity test, all Ni/RMOx catalysts facilitated greater DO activity by yielding 83–86% hydrocarbon yield and 70–85% saturated diesel n-(C15 + C17) selectivity. Ni/RMO3 was the best catalyst for deoxygenizing the ceiba oil owing to the existence of a high acidic strength (12717.3 μmol g−1) and synergistic interaction between Fe–O and Ni–O species, thereby producing the highest hydrocarbon yield (86%) and n-(C15 + C17) selectivity (85%). According to the reusability study, the Ni/RMO3 could be reused for up to six consecutive runs with hydrocarbon yields ranging from 53% to 83% and n-(C15 + C17) selectivity ranging from 62% to 83%.
1. Introduction
The global warming issue and depletion of fossil fuels are the two most threatening problems of our present day civilization. According to NASA,1 in 2021, CO2 levels have indeed reached 416 ppm. This recent inexorable rise in CO2 levels reveals a highly consistent link with the use of fossil fuels. The resulting rise in CO2 levels will continue to melt the ice in Antarctica, resulting in a ‘doomsday glacier’, causing global sea levels to rise and affecting people all over the world. Aside from that, the global energy crises are the outcome of the potential future depletion of fossil fuel. Renewable sources of energy such as green diesel are generally considered a replaceable alternative to traditional fossil fuels due to their similar physicochemical properties criteria, feedstock availability, and environmental friendliness.2–4 In general, green diesel can be produced via catalytic hydrodeoxygenation (HDO) and deoxygenation (DO) of oxygenated compounds (triglycerides and fatty acid devatives). In HDO processes, oxygenated species are removed under high pressure conditions with the presence of H2, whereas in DO processes, oxygenated species are removed in the form of CO, CO2, and H2O via decarbonylation/decarboxylation (deCOx) pathways under H2-free conditions.5,6 Due to the fact that no H2 is needed in the DO process, DO pathways are usually preferred.
Ceiba pentandra, often known as kapok or silk-cotton tree, is a tropical tree of the Malvaceae family that grows quickly (up to 13 feet per year).7 Pentandra is a drought-tolerant tree and grown in waste land thus readily and abundantly available.8 The pods of these trees are leathery, ellipsoid, pendulous capsules that are 10–25 cm long and 3–6 cm in diameter. The capsules split open into five valves, revealing a mass of woolly, yellowish grey, and glossy fibre in which 120–175 seeds are lodged.9 Ceiba seeds are blackish in colour and have a composition of 13% water, 5% ash, 20% crude fibre, 6% fat, 29% protein, and 20% carbohydrates. Apparently, ceiba seeds consist of a yellow and pleasant oil that is within the range of 20–28% by weight with high amount of free fatty acid (FFA) (FFA = 6%).3 As a result, ceiba oil is an inedible feedstock that has been used successfully for oil sorption and biodiesel generation utilising a variety of catalysts.10–12 Ceiba oil has not been used for green diesel generation, hence it was employed as a prospective feedstock source in this study. Apparently, ceiba seeds consist of a yellow and pleasant oil that is within the range of 20–28% by weight with high amount of free fatty acid (FFA) (FFA = 6%).3 As a result, ceiba oil is an inedible feedstock that has been used successfully for oil sorption and biodiesel generation utilising a variety of catalysts.10–12 At present, the transformation of ceiba oil to diesel is rarely reported in DO studies. Thus, in the present study, special focus will be focused on the DO of ceiba oil to green diesel.
A catalyst is a substance that increases the rate of a chemical reaction without being consumed in the process. Many studies in the literature have reported on the DO process using noble metal catalysts such as Pd, Pt, and Ru.13–16 However, noble metal catalysts are expensive, which limits it applicability for commercialization. As a result, the utilisation of industrial waste or naturally occurring solids containing catalytically active metals such as Fe, Ni, V, and others as a substitute for commercial catalysts can assist in minimizing the cost of catalyst utilization.17,18 Bauxite residue (BR), often known as red mud, is made during alkali leaching of bauxite and is considered a low-grade Fe ore containing 30% to 60% Fe and Al (10–20%).17,19,20 Because of its high alkalinity, currently, its storage sites pose a significant safety and environmental risk.21 It is noteworthy to mention that red mud waste disposal is an expensive operation that accounts for 5% of the production cost.22 Indeed, red mud is mainly composed of Fe (30–60%), which can be considered an ideal precursor for synthesizing an effective DO catalyst.23 Fe has recently been discovered to have strong oxophilic effects and favours a robust redox reaction, making it easier to break C–C and C–O bonds.24,25 Likewise, a red mud catalyst has demonstrated an excellent plastic pyrolytic activity, producing ∼67% liquid fuel yield.26,27 Previous reports also affirmed that the presence of Fe species in red mud resulted in a good organic liquid yield (17–74%) but noted that those findings were limited to bio-oil production.28–30 Several studies on gasification production processes have shown that a Fe-catalyzed system can prevent coking activity and prolong the life span of the catalyst. Apparently, strong basic sites of Fe will transform the coke by assisting the Boudouard reaction, in which carbon combines with CO2 to produce CO gas.31 Up to now, the Fe species is also a good metal support, as it is capable of improving the surface area of the catalyst and permits homogenous dispersion of the active metal, thus effectively rendering excellent deoxygenation activity.32,33 Indeed, the use of a modified red mud catalyst for green diesel production via DO has not yet been investigated. As a result, the current research will focus on the creation of a red mud-based catalyst modified with Ni for the production of green diesel from DO of ceiba oil.
2. Experimental
2.1 Materials and chemicals
Red mud was collected from three different red mud waste areas; Semabok, Bachang, and Durian Tunggal from Melaka. Ceiba Pentandra L. seeds were purchased from West Java, Indonesia. The ceiba oils were extracted from the seeds by using a cold-pressing method.34 These feedstocks were used for the DO reaction without further treatment and purification. The physicochemical properties of the feedstock were determined using the American Oil Chemists' Society (AOCS) method, and the results are tabulated in Table 1.35 The majority of the ceiba oil consists of palmitic acid (C16:0) at 19.2%, oleic acid (C18:1) at 17.4%, linoleic acid (C18:2) at 39.6%, and malvaloyl*18 CE (18:CE) at 18.5%. Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, purity >99%) was purchased from R&M, Malaysia. Concentrated hydrochloric acid (HCl, 85–87% purity) was purchased from J. T. Baker (USA). Ammonia solution (NH3, 30% purity) was purchased from R & M Chemical (UK). Solvents such as ethanol, hexane, and acetone were acquired by Merck & Co., USA. Analytical grade n-hexane (purity >98%, Merck, Malaysia) and absolute ethanol (purity >98%) were used as solvents. A standard solution that consists of alkanes and alkenes (C8–C20) and an internal standard 1-bromohexane (CHBR, purity >98% (GC grade)) for gas chromatography (GC) analysis were purchased from Sigma Aldrich, Malaysia and used without further purification. n-Hexane (GC grade) with a purity >98% from Merck (Germany) was used for dilution (2.1%).
Table 1 The physiochemical properties of ceiba oila
| Oil properties |
Ceiba oil |
Method |
| Traces% of lauric acid and myristic acid (0.1%), palmitoleic acid ‘(0.3%), stearic acid (2.6%), linolenic acid (1.5%), arachinic acid (0.56%) and others (0.34%). |
| Acid value (mgKOH g−1) |
11.9 |
AOCS Ca 5a-40 |
| FFA value (%) |
5.9 |
AOCS Ca 5a-40 |
| Fatty acid composition of oil (%) |
|
AOCS Cel-62 & Cel-661 |
| Palmitic (C16:0) |
19.2 |
|
| Oleic (C18:1) |
17.4 |
|
| Linoleic (C18:2) |
39.6 |
|
| Malvaloyl (18:CE) |
18.5 |
|
2.2 Preparation of the Ni supported red mud catalyst
Approximately 30 g of three different types of red muds (RMx, x = 1, 2, 3) were separately ground and dissolved in mixtures of 150 mL of 37% HCl and 100 mL of distilled water. The mixtures were then stirred for 24 h at 30 rpm and centrifuged. Subsequently, the obtained liquid was further titrated with 30% ammonia that had been diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with distilled water until the liquid reached a pH of 11. The precipitant was then filtered, washed with distilled water, and dried in the oven for 24 h. The obtained solid was then calcined at 550 °C for 3 h under atmospheric pressure, and all the catalysts were denoted as RMOx: RMO1, RMO2, and RMO3. The deep-deposition technique was used in order to dope the Ni promoter on the RMO1, RMO2, and RMO3. Ni was successfully been deposited by slowly adding a 0.6 M nickel salt solution into RMO1. The Ni deposition was performed by adding 1 M ammonia dropwise until the pH reached 10 under vigorous stirring. The slurry was then put into an autoclave machine at 220 °C for 24 h. The obtained solid was then filtered and washed using distilled water. The product was dried in the oven overnight and calcined at 550 °C for 3 h. The catalyst was denoted as Ni/RMO1. Similar steps were repeated for RMO2 and RMO3. The catalysts were denoted as Ni/RMO2 and Ni/RMO3. These catalysts were further reduced under a H2 atmosphere at 550 °C for 3 h, and the catalysts were denoted as Ni/rRM1, Ni/rRM2, and Ni/rRM3.
:1 with distilled water until the liquid reached a pH of 11. The precipitant was then filtered, washed with distilled water, and dried in the oven for 24 h. The obtained solid was then calcined at 550 °C for 3 h under atmospheric pressure, and all the catalysts were denoted as RMOx: RMO1, RMO2, and RMO3. The deep-deposition technique was used in order to dope the Ni promoter on the RMO1, RMO2, and RMO3. Ni was successfully been deposited by slowly adding a 0.6 M nickel salt solution into RMO1. The Ni deposition was performed by adding 1 M ammonia dropwise until the pH reached 10 under vigorous stirring. The slurry was then put into an autoclave machine at 220 °C for 24 h. The obtained solid was then filtered and washed using distilled water. The product was dried in the oven overnight and calcined at 550 °C for 3 h. The catalyst was denoted as Ni/RMO1. Similar steps were repeated for RMO2 and RMO3. The catalysts were denoted as Ni/RMO2 and Ni/RMO3. These catalysts were further reduced under a H2 atmosphere at 550 °C for 3 h, and the catalysts were denoted as Ni/rRM1, Ni/rRM2, and Ni/rRM3.
2.3 Catalyst characterization
The identification of the crystallography and structural properties of all catalysts was carried out using powder X-ray diffraction (XRD). The XRD analyses were performed using a Shimadzu diffractometer, model XRD-6000, with a scan speed of 4 °C min−1 with a 2θ range within 5° to 40°. The diffractograms that were produced were matched with the published International Centre for Diffraction Data (ICDD) files in order to determine the crystallinity phases of the synthesized materials. The Thermo-Finnigan Shopmatic 1990 series N2 sorption analyser was used to analyse the specific surface area and pore distribution using N2-adsorption and desorption techniques. The catalysts were degassed for 12 h at 150 °C to eliminate contaminations and moisture on the catalyst surface. The adsorption and desorption processes of N2 were evaluated in a vacuum chamber at −196 °C. The acidity of the catalysts were investigated using temperature-programmed desorption with NH3 as the probe molecules (TPD-NH3). The analysis was performed using a Thermo-Finnigan TPDRO analyzer model 1100 equipped with a corresponding thermal conductivity detector (TCD). Approximately 0.05 g of sample was added to a quartz tube, and the sample was initially pre-treated at 150 °C under N2 conditions to remove excess moisture. The pre-treated samples were exposed to NH3 for 1 h at room temperature for NH3 adsorption. Subsequently, the excess NH3 was flushed out using N2 (20 mL min−1) at room temperature for 35 min. The treated catalyst was heated from 50 °C to 900 °C at a heating rate of 10 °C min−1 in a flow of He (30 mL min−1). The desorbed NH3 was detected by the TCD. The acidic sites was calculated by using eqn (1):
Acidic sites,
| |
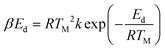 | (1) |
where
β is the strength of the binding energy,
Ed is the activation desorption energy,
TM is the maximum temperature of TPD spectrum,
R is the ideal gas constant (8.314 kJ per mol per kelvin), and
T is absolute temperature. The surface morphology and elemental composition of the catalysts were observed using a field emission scanning electron microscope and energy dispersive X-ray (FESEM/EDX) (Hitachi S-3400N). The elemental analysis was also estimated using X-ray fluorescence spectroscopy (XRF) (Rigaku, RIX 3100). The XRF was operated at 50 kV and 70 mA using a wavelength dispersive spectrometer that was equipped with a rhodium tube, LiF 200 crystal, and scintillation counter. The chemical functional group of the comprised catalyst was determined using a PerkinElmer (PC) Spectrum 100 FTIR with a resolution of 4 cm
−1 within the range of 300–4000 cm
−1. A thermogravimetric analysis, TGA instrument (TGA 1000i, Instrument Specialists Inc., USA) was used to determine the thermal stability of the catalyst under an inert environment. The powder sample was first placed in an alumina crucible and then heated from 25 °C to 900 °C at a heating rate of 30 °C min
−1 under nitrogen gas flow rate of 40 mL min
−1.
2.4 DO reaction of ceiba oil
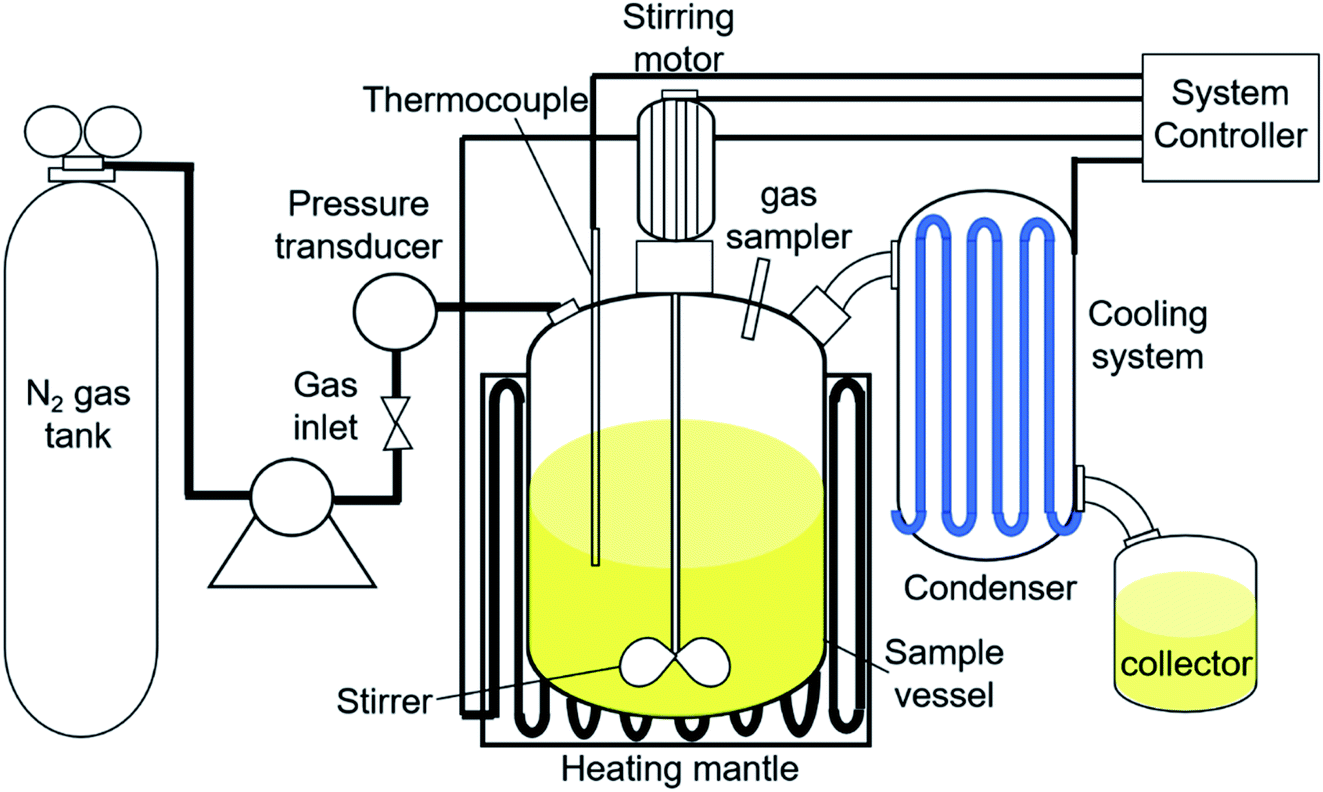
The catalytic DO reaction of ceiba oil was performed in a 250 mL mechanically stirred semi-batch reactor under inert N2 flow (Fig. 1). Basically, the DO reaction of ceiba oil was carried out at 350 °C under the flow of inert gases (50 cc min−1) for 2 h using 10 g of ceiba oil and 3 wt% catalyst loading. Prior to the reaction, the oxygen in the reactor was removed by purging with N2 gas at a flow rate of 20 cc min−1. The inert N2 gas was then continuously flowed at this rate. Vapor or volatile species generated during the DO process was condensed into the liquid product using an external water cooling circulator at 22 °C and collected using a vessel collector. The liquid product was then evaluated by using the gas chromatography-flame ionisation detector (GC-FID) and gas chromatography-mass spectrometer (GC-MS); meanwhile, the gaseous product was collected using a sampling gas bag and further analysed by using gas chromatography-thermal conductivity detector (GC-TCD). A thermogravimetric analysis, TGA instrument (TGA 1000i, Instrument Specialists Inc, USA) was used to determine the extent of coke formation on the spent catalyst in an oxidative environment. The powder sample was first placed in an alumina crucible and then heated from 25 °C to 900 °C at a heating rate of 30 °C min−1 under oxygen flow rate of 40 mL min−1.
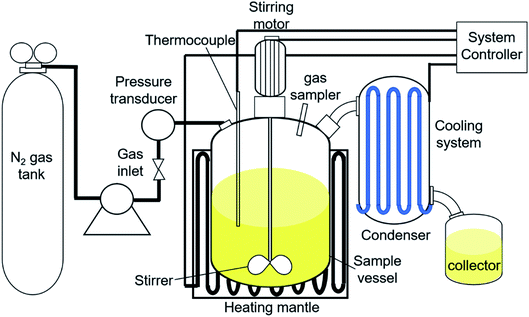 |
| | Fig. 1 Reactor set-up for DO of ceiba oil. | |
2.5 Product characterization
The liquid products were identified using alkane and alkene standards (C8–C20) procured from Sigma-Aldrich (USA). The liquid product was analyzed qualitatively using GC-FID (Shimadzu GC-14B) equipped with an HP-5 capillary column (length of 30 m; inner diameter of 0.32 mm; film at 300 °C) based on the previous study by Baharudin et al.36 First, the liquid product was diluted with GC grade n-hexane. 1-Bromohexane was used as an internal standard for quantitative analysis. A 1 mL aliquot of a sample was injected into the GC column. The injection temperature was 250 °C, and nitrogen gas served as the carrier gas. The initial temperature of the oven was set at 40 °C, and it was held there for 6 min; then, it was ramped up to 270 °C at a heating rate of 7 °C min−1.37 Literature data also supported the GC-FID oven programme method.38–40 The green diesel conversion, product selectivity, and hydrocarbon yield were defined as in eqn (2) and (3).41| |
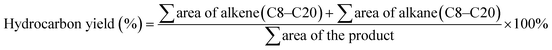 | (2) |
| |
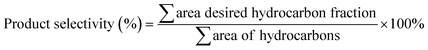 | (3) |
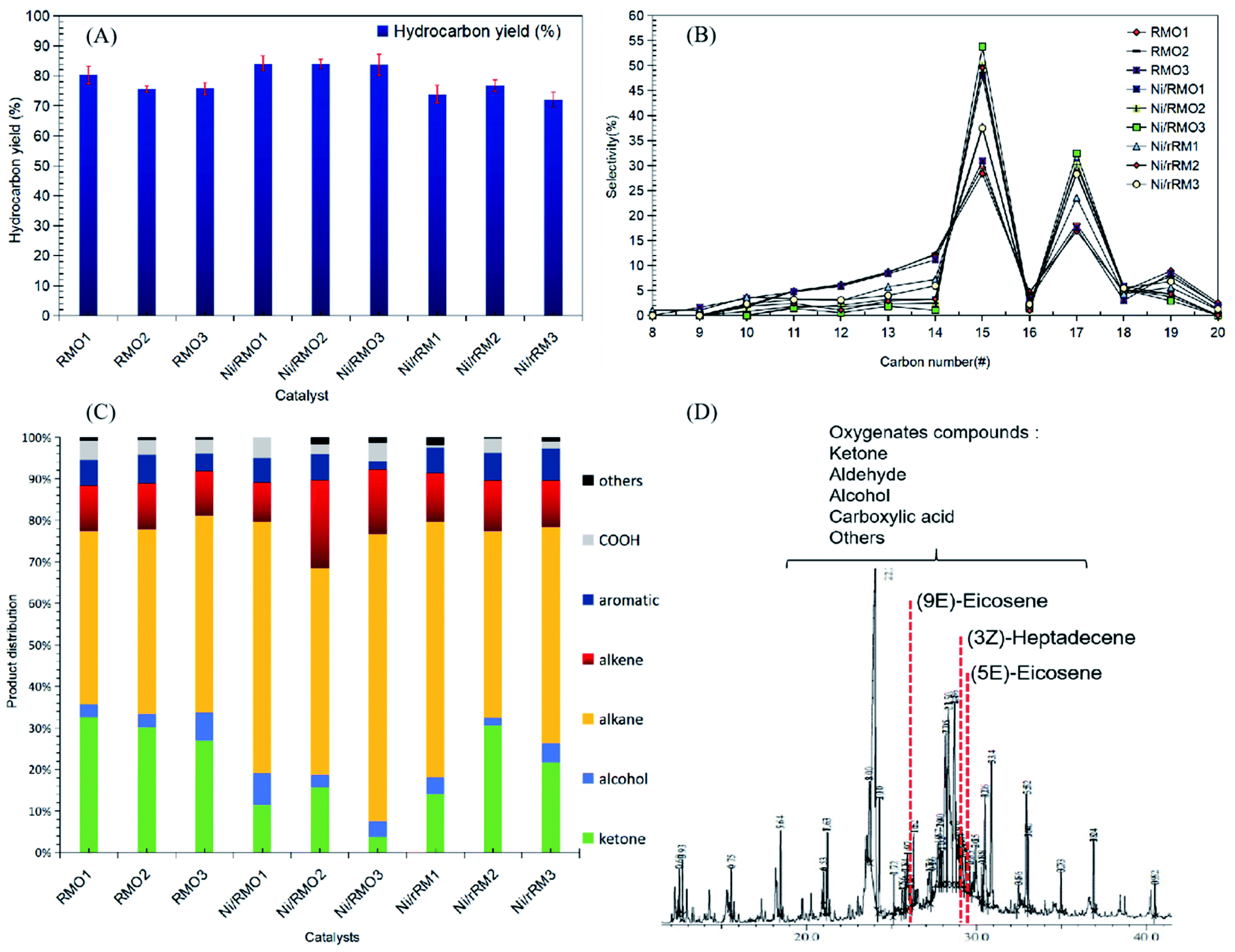
The organic compounds in the liquid product were further investigated by a GC–MS (model Shimadzu QP2010 Plus) fitted with a Zebron ZB-5 MS column (30 m × 0.25 mm × 0.25 μm) using a splitless inlet. The liquid deoxygenated products were diluted to 100 ppm using GC-grade hexane (purity >98%). The component peaks in the GC-MS spectrum were identified from the National Institute of Standards and Testing (NIST) library based on a probability of agreement of ≥95%.42 The selectivity of the deoxygenated products was determined by using eqn (4):
| |
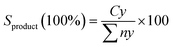 | (4) |
where
Sproduct is the yield of the organic compound (%),
Cy is the area of the desired organic compound, and ∑
ny is the total area of the organic compounds. The chemical functional group of a comprised liquid product was determined using a PerkinElmer (PC) Spectrum 100 FTIR with a resolution of 4 cm
−1 in the range of 300–4000 cm
−1. To quantify the concentration of the gases (CO
2, CO)
43 obtained from the DO reaction, 1 μL of gas was injected into an off-line gas chromatograph (GC) (Agilent G1540N, USA) connected to a thermal conductivity detector (TCD).
3. Results and discussion
3.1 Characterization of fresh red mud
TGA analysis was performed on fresh red mud 1–3 (RMx), and the results are displayed in Fig. S1.† Based on the TGA analysis, minor weight loss (3–8%) was observed in the temperatures range of ∼50–200 °C owing to the elimination of physically absorbed water, Al2O3·3H2O (Al2O3·3H2O → Al2O3 + 3H2O) and chemically bounded water, 2 (Al(OH)3 → Al2O3 + 3H2O) on the RMx particles.44,45 This chemically bound water might come from the dehydration of the hydrate molecule in the minerals and the decomposition of gibbsite phases.46 Notably, detection of rich Fe species in all fresh red mud was confirmed by FESEM-EDX analysis. The Fe content in RM2 was the highest (58%), while others were 35–46% (Fig. 2A–C). The Fe% in RMx was in accordance with previous findings (30–60%).47–49 It is noteworthy to mention that all the RMx were red in colour, which is due to the rich Fe(III) oxide species that comprise approximately 20–60% of its mass.50,51 The morphology structure of RMx is displayed in Fig. 2D–F. There are obvious variations in the surface morphology. Fig. 2D–F shows the morphology structure of the red mud. It is clear that RM1 exhibits a spongy-like structure. Meanwhile, RM2 shows an assembly of small aggregates, and RM3 exhibits agglomerated particles. Indeed, when compared to RM1 and RM3, the Fe rich red mud (RM2) exhibits a greater homogeneity of the aggregate size; this reveals that the high Fe content in red mud contributes to the homogeneity of the aggregate size of the material.52–54 Fig. S2† shows the XRD patterns for RMx. The majority of the RMx displayed peaks belonging to hematite (Fe2O3, ICDD card no. 00-006-0502), aluminium oxide hydrate (Al2O3·3H2O, ICDD card no. 00-001-0307), quartz (SiO2, ICDD card no. 00-001-0649), and anatase (TiO2, ICDD card no. 00-001-1292).55,56 Similarly, insignificant peaks were also discovered for other elements such as goethite (FeO(OH)) and gibbsite (Al(OH)3).57 This finding was found to be in accordance with the weight loss of chemically bounded water molecule in the findings of the TGA analysis.
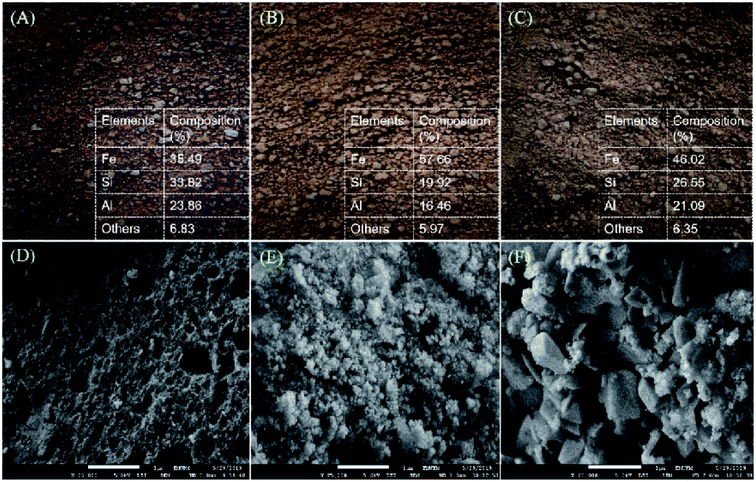 |
| | Fig. 2 Picture and EDX result for (A) RM1, (B) RM2 (C) RM3 and FESEM images of (D) RM1, (E) RM2 (F) RM3. *others: * = TiO2, FeO(OH), Al(OH)3. | |
3.2 Characterization of the red mud based nickel catalyst
As explained in Section 2.2, the RMx was further calcined at 550 °C for 3 h to fully transform the RMx to the RMOx species-based catalyst, which was further characterized by XRF analysis. The Fe content (referred to as Fe2O3 species) in all RMOx species was found to be within the range of 97–99% (Table 2). Because RMOx is primarily composed of Fe species, it has a great potential to be used as a catalyst. Further Ni inclusion (20 wt%) resulted in a significant reduction in the Fe content (53–74%), whereas the Ni species (referred to as NiO2 and NiO) were discovered to be in the range of 26–38%. The Ni species were prominent in Ni/RMO1 (38%), suggesting that the rich porous structure of RMO1 (Fig. 2D) has the ability to trap more Ni species into the pores during the catalyst synthesis.58 Notably, the Ni and Fe contents were reduced slightly (∼2–5%), indicating that a few of the oxide species (Fe2O3, NiO2, NiO) in the reduced catalysts successfully converted to metallic Fe and Ni.
Table 2 Crystallite size and elemental composition of the RMOx, Ni/RMOx and Ni/rRMx catalysts
| Catalyst |
Crystallite size (Fe2O3) 2θ: 33.4° |
Elemental composition (%) |
| Fe2O3 |
NiO2/NiO |
Othersa |
| Other = SiO2, TiO2, Al(OH)3. |
| RMO1 |
172.4 |
99.1 |
— |
0.9 |
| RMO2 |
129.4 |
97.0 |
— |
3.0 |
| RMO3 |
129.4 |
97.1 |
— |
2.9 |
| Ni/RMO1 |
64.7 |
61.2 |
37.2 |
1.7 |
| Ni/RMO2 |
64.7 |
64.8 |
34.6 |
0.6 |
| Ni/RMO3 |
64.7 |
73.0 |
26.3 |
0.7 |
| Ni/rRM1 |
96.3 |
60.2 |
38.0 |
1.8 |
| Ni/rRM2 |
64.7 |
67.2 |
32.5 |
0.2 |
| Ni/rRM3 |
96.3 |
70.5 |
28.7 |
0.8 |
The XRD patterns of the RMOx, Ni/RMOx and Ni/rRMx catalysts are shown in Fig. 3. The XRD of RMOx exhibited a hematite structure of Fe2O3 at 2θ = 24.3°, 33.4°, 35.8°, 38.4°, 49.7°, 54.3°, and 62.7° (ICDD card no. 00-001-1053) on the RMOx, Ni/RMOx, and Ni/rRMx. The NiO2 (ICDD card no. 01-085-1977) and NiO (ICDD card no. 03-065-2901) species were detected on all Ni/RMOx catalysts. As for Ni/rRMx, typical peaks corresponding to NiO, metallic Ni, Fe2O3, Fe3O4, FeO, and metallic Fe were observed. The formation of metallic Ni and Fe indicated the successful H2 gas reduction of Ni2+ to Ni0 and Fe3+ to Fe0. The existence of the oxide species after the H2 reduction method suggested that at low temperatures, the H2 method (550 °C for 3 h) does not completely reduce the nickel oxide and iron oxides. Indeed, previous studies concurred that reducing nickel oxide to metallic nickel requires temperatures ranging from 600 to 900 °C for 1–3 h,59–61 whereas reducing hematite to metallic iron takes 4–10 h at temperatures ranging from 650 to 1000 °C. Altogether, all red mud catalysts dominantly owing to alkaline oxide species (Fe2O3), indicating that all these catalysts are very resistant to coking and particularly boost the catalyst lifespan.
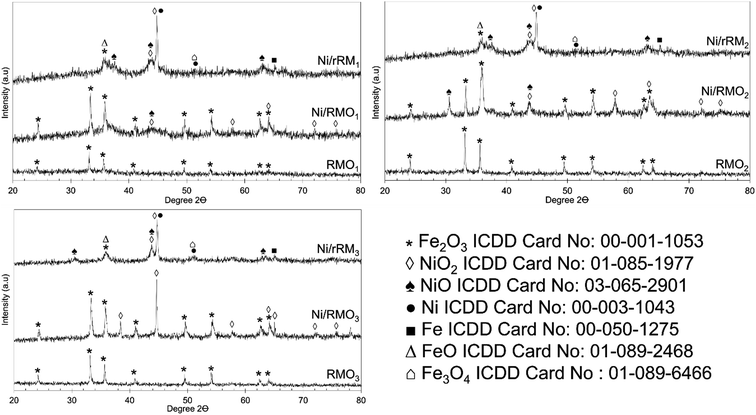 |
| | Fig. 3 XRD diffractogram of RMOx, Ni/RMOx and Ni/rRMx catalysts. | |
The surface area, total pore volume, and mean pore diameter of the synthesized catalysts were determined using the BET and BJH methods, and the results are tabulated in Table 3. Among the RMOx catalysts, RMO1 had the highest surface area (135 m2 g−1) compared to RMO2 and RMO3. Upon the addition of Ni, the surface area of the Ni/RMOx catalysts gradually decreased, implying the successful deposition of the Ni species on the RMOx surface.62 In contrast, for Ni/RMO3, the surface area increased remarkably after the addition of the Ni species. The reduction of the surface area is ascribed to the coverage of surfaces and the blockage of pores by the Ni species. Fig. S3B† affirmed this finding, whereby Ni/RMO3 displayed a type IV H2 isotherm with a large hysteresis gap, while Ni/RMO1 and Ni/RMO2 catalysts demonstrated a Type III H3 isotherm with a narrow hysteresis gap, illustrating a narrow range of pore necks due to pore blockage (Fig. S3A–C†).63 The surface area of the reduced catalysts was lower than that of the RMOx and Ni/RMOx counterparts. This discovery, which is also consistent with prior research, demonstrates that reducing red mud catalysts to temperatures >400 °C reduces the surface area of the metal oxide-based catalysts.64 Notably, there is no link between pore size and pore volume trends with the lowering of the surface area on Ni/rRMx. As demonstrated in Fig. S4A,† RMOx catalysts had mesopores with pore diameters varying from 2 to 50 nm. Meanwhile, in Ni/RMOx and Ni/rRMx catalysts, multiscale pores (meso- and macro-pores) with pore sizes ranging from 2 to 50 nm and 50 to 130 nm were discovered, with Ni/rRM2 showing the highest pore volume and diameter (Fig. 4B and C).43
Table 3 Textural properties of RMOx, Ni/RMOx and Ni/rRMx catalysts
| Catalysts |
N2 adsorption–desorption analysis |
| Surface areaa (m2 g−1) |
Pore size diameterb (nm) |
Pore volumeb (cm3 g−1) |
| Measured by BET analysis. Measured by BJH analysis. |
| RMO1 |
135 |
7.8 |
0.27 |
| RMO2 |
77 |
9.0 |
0.18 |
| RMO3 |
73 |
7.4 |
0.14 |
| Ni/RMO1 |
108 |
15.0 |
0.27 |
| Ni/RMO2 |
58 |
13.3 |
0.19 |
| Ni/RMO3 |
83 |
9.5 |
0.20 |
| Ni/rRM1 |
97 |
9.3 |
0.23 |
| Ni/rRM2 |
50 |
53.8 |
0.68 |
| Ni/rRM3 |
56 |
15.4 |
0.21 |
 |
| | Fig. 4 FESEM images of (A) RMO1, (B) RMO3, (C) Ni/RMO3 and (D) Ni/rRM3 catalysts. | |
The acid density and strength of acidity for the RMOx and modified RMOx catalysts were measured using TPD-NH3. According to Abdulkareem et al. (2016),65 the acid site plays a critical role in controlling the C–O cleavage activity of the feedstock toward the desired hydrocarbon product, and this study claimed that the DO activity preferred a catalyst with rich weak and medium acidic sites, as it enables the oxygenated species to be removed from the fatty acid derivatives via deCOx pathways. The results showed that all the catalysts exhibited weak (Tmax <250 °C), medium (Tmax >250 °C < 500 °C), and strong (Tmax >500 °C) (Table 4 and Fig. S5A–C†) acidic sites, whereby strong acidic sites predominated.66 Despite this fact, the strong acidity of the catalyst was desirable to provide the catalytic C–C cracking, leading to the formation of light hydrocarbons (alkanes). The trend in the acidity profiles of catalysts is as follows: Ni/rRM3 > Ni/rRM2 > Ni/rRM1 > Ni/RMO3 > Ni/RMO1 > Ni/RMO2 > RMO1 > RMO2 > RMO3. Ni/rRM3 has the strongest acidic sites, while RMO3 has the weakest acidic sites. The result also showed that only RMO3 has weak and medium sites. This result confirms the feasibility of distributing the Ni species for tuning and maximizing the acidity, hence simultaneously shifting the weak and medium acid sites to strong acid sites. A similar finding was observed in a previous study;67 hence, it can be concluded that Ni species play an important role in designing a strong acidic catalyst. Further thermal reduction treatment results in the majority of the Ni/rRMx catalysts exhibiting a remarkable increase in the strong acid sites. It is believed that removal of the oxide species on Ni/RMOx effectively exposes the Ni0 species and Fe0, which in turn enhances its acidity. The finding is consistent with those of prior works showing the significant role of Ni0 acidic compounds at the SBA-15 and Al2O3 support surfaces.68 A similar observation was found on Fe0-rich catalysts.64 Because Ni/rRM3 exhibited a significant amount of acidic sites (40799.6 μmol g−1), the morphological analysis for RMO3, Ni/RMO3, and Ni/rRM3 was performed. The result was also compared with the highest surface area catalysts (RMO1). It is worth noting that the largest RMO1 particles (particle size: 0.0083–0.6 μm, 1.2–2.4 μm) (Fig. 4A) had no correlation with the formation of the large surface area (Table 3). RMO3 has larger particles that were coated with small aggregates (particle size: 0.0077–0.2 μm, 1.0–1.7 μm) (Fig. 4B). Yet, these aggregates went unnoticed with the addition of Ni (Fig. 4C), implying that the increase in the surface area of the Ni/RMO3 catalyst is related to the homogeneous particle RMO3 distribution by Ni. In this study, indeed, the addition of the Ni species and the H2 reduction approach resulted in significant changes in the morphology of the RMO3 support. The FESEM micrograph shows that Ni/RMO3 has plate-like structures (0.037–0.16 μm thick), while the FESEM micrograph of Ni/rRM3 shows that it forms uniform nanosheets (0.008–0.048 μm thick) decorated by Ni foam (Fig. 4D). This implies that the iron and nickel oxides (see XRD) must have been largely consumed during the H2 reduction approach. The resulting uniform nanosheets of Ni/rRM3 are possibly interesting for applications in DO reactions because of the presence of both meso- and macropores.
Table 4 Acidity profiles of RMOx, Ni/RMOx and Ni/rRMx catalysts
| Catalysts |
Acidity strengtha (μmol g−1) |
| Weak <200 °C |
Medium >200 °C < 500 °C |
Strong >500 °C |
| Determined by TPD-NH3. |
| RMO1 |
— |
2955.5 |
2955.5 |
| RMO2 |
— |
2955.5 |
2621.4 |
| RMO3 |
205.9 |
503.3 |
877.8 |
| Ni/RMO1 |
— |
— |
10445.9 |
| Ni/RMO2 |
— |
— |
5975.9 |
| Ni/RMO3 |
— |
— |
12717.3 |
| Ni/rRM1 |
— |
25274.7 |
19042.2 |
| Ni/rRM2 |
— |
— |
25381.7 |
| Ni/rRM3 |
— |
— |
40799.6 |
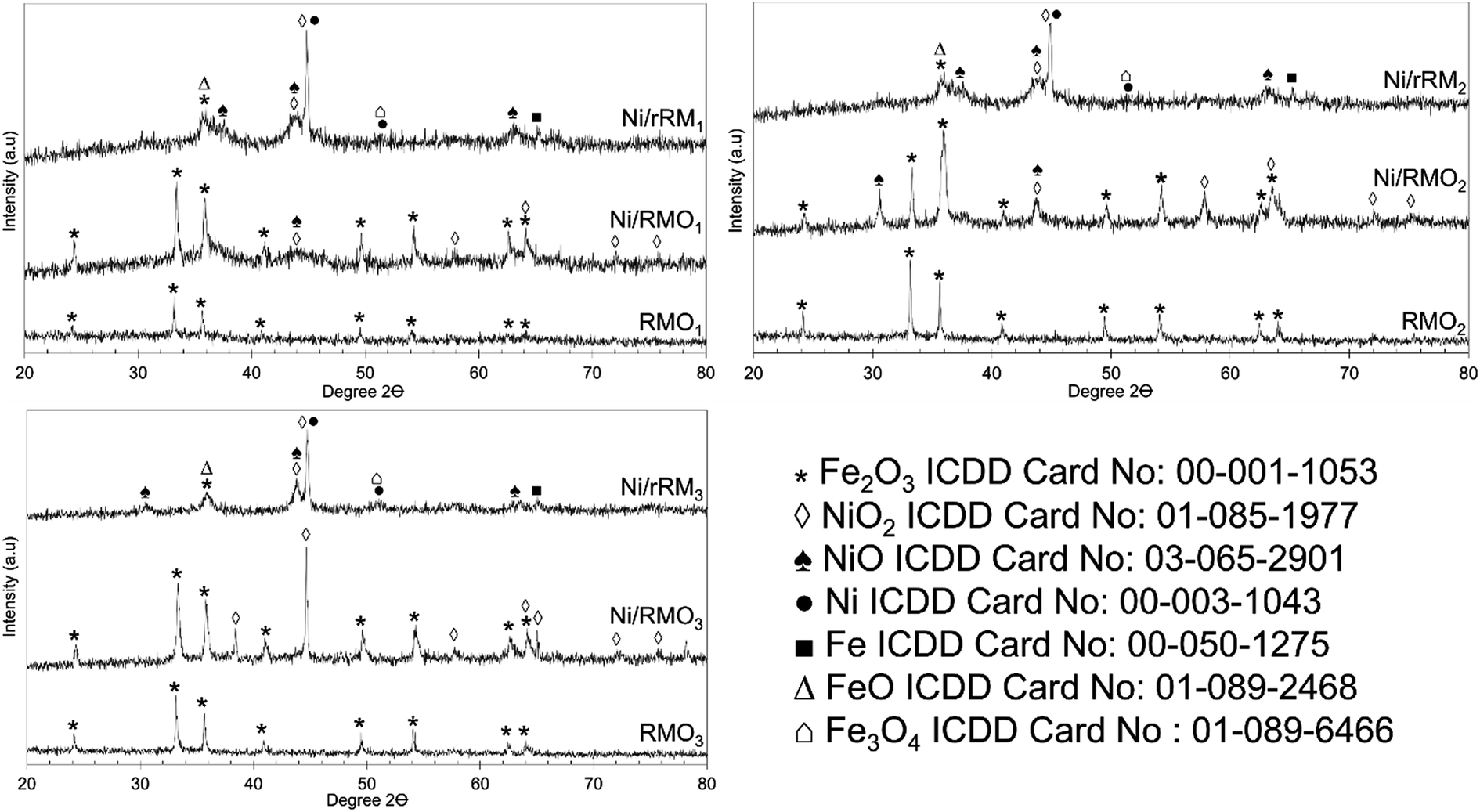
TGA analysis was performed to investigate the stability of the catalysts, and the result is shown in Fig. 5. All of the catalysts had insignificant weight loss during thermal treatment up to 850 °C, suggesting that all catalysts have good stability. The addition of Ni and the H2 reduction approach barely changed the stability of the red-mud based catalyst, implying that the red-mud based support catalyst itself preserves its stability. When compared with the findings of the acidity trend (see Table 3), the increase in the acid sites does not play a critical role in improving the stability of the modified red mud-based catalyst. Our result is unlike Kaya's finding,69 who demonstrated that acid modification of red mud followed by calcination apparently increased the thermal stability of the red-mud based material. To summarize, the correlation between stability and acidity should be further investigated. Based on the TGA result, all the catalysts exhibited two stages of weight gain (∼1–4%) at temperatures from 50–200 °C and 450–850 °C (3–5%) due to the adsorption of the water and N2 adsorption.70 The weight gained at 450–850 °C was dominated by the Ni/rRM3 catalyst, which was attributed to the formation of multi-scale (meso- and macro-) porosity structures (Fig. S4C†) that increased the tendency of N2 adsorption, generating Ni3N, Fe2N, and ε-Fe3N 71–74. This finding is aligned with previous studies showing that N2 can form a bond with the metal to form a nitride compound at high temperatures (>400 °C).71–75
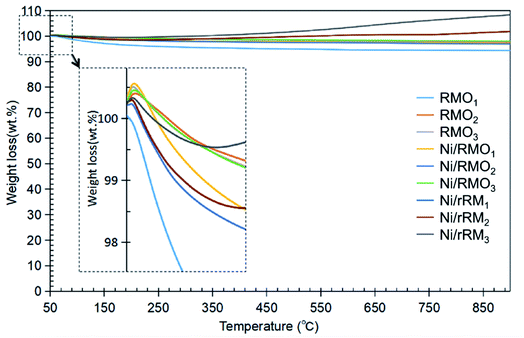 |
| | Fig. 5 TGA thermogram for RMOx, Ni/RMOx and Ni/rRMx catalysts. | |
3.3 Catalytic DO profile of ceiba oil
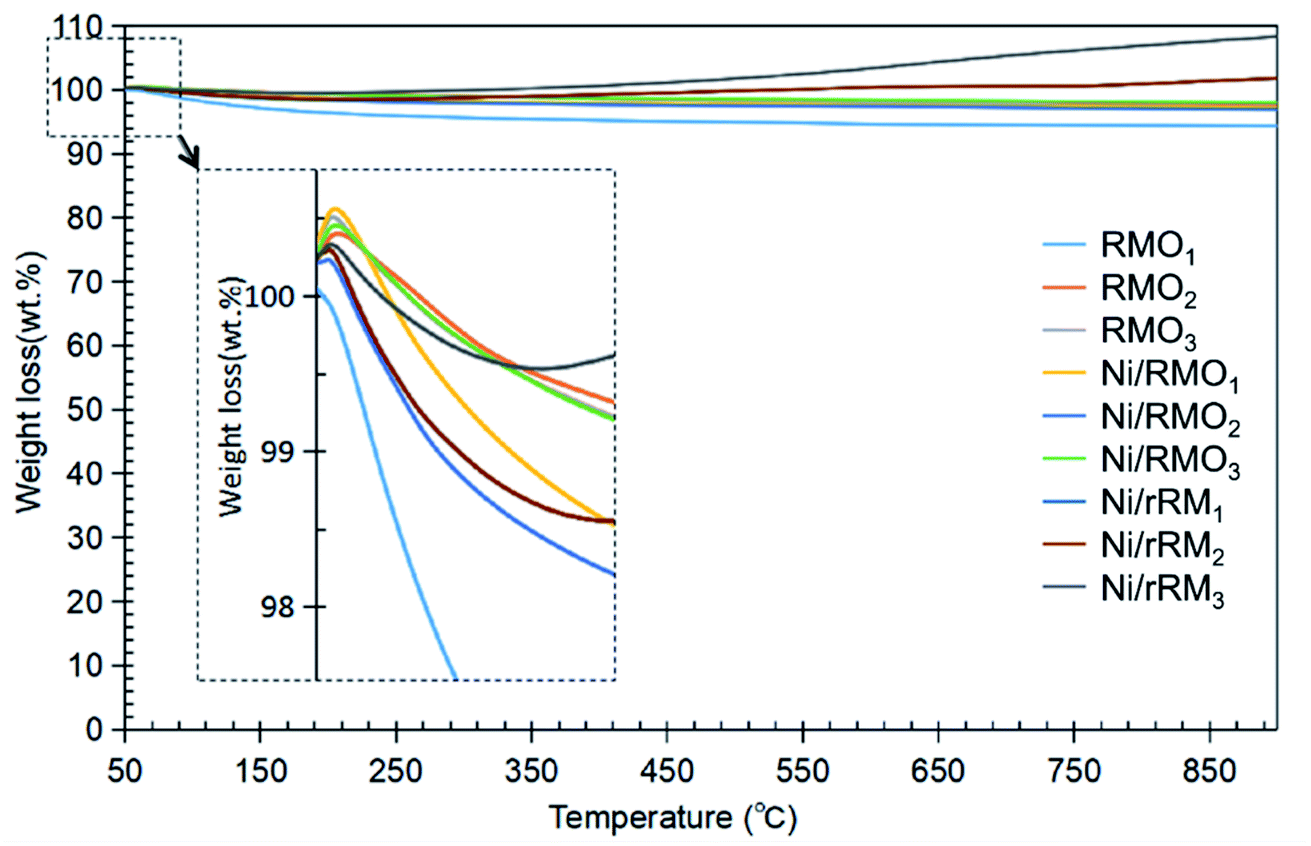
From Table 1, the fatty acid composition of ceiba oil is mainly composed of 39.6% linoleic acid (C18:2), 17.4% oleic acid (C18:1), 18.5% malvaloyl*18 CE (C18:CE), and 19.2% palmitic acid (C16:0). The C16 and C18 fatty acid derivatives will undergo the deCOx reaction to form n-heptadecenes (n-C17) and n-pentadecenes (n-C15).76 A blank experiment was performed under the same condition to determine the product distribution during the DO of ceiba oil without the presence of the catalyst. The blank reaction exhibited a low n-(C8–C20) hydrocarbon yield (<20%) and low n-C15 + C17 selectivity, implying that a catalyst was required to produce a higher yield and selectivity of the deoxygenated product (Fig. 6A and B). In the case of the catalysed reaction, the majority of the reaction shows an excellent catalytic DO reaction performance. Ni/RMO3 yielded the highest hydrocarbon fraction (∼86%), while Ni/rRM3 showed the poorest DO activity with a hydrocarbon yield of ∼73%. This could be due to the presence of a high proportion of strong acidic sites in Ni/rRM3 (40799.6 μmol g−1, see Table 4), which prone toward C–C cracking than C–O cleavage activity. Furthermore, the large pore size of Ni/rRMx catalysts (see Table 3) also allowed molecules to diffuse across acid active areas, resulting in further cracking and the formation of volatile species.77,78 In the case of Ni/RMO3, the preferential amount of strong acidic sites (12717.3 μmol g−1) enables them to have a high DO activity, which is likely to undergo C–O bond cleavage. Indeed, the Ni/RMOx catalysed process promotes outstanding DO activity and n-C15 + C17 selectivity compared to RMOx and Ni/rRMx. This finding strongly affirmed that the interaction between the Ni–O species and Fe–O phase motivated the deCOx activity. The deterioration of the DO activity by Ni/rRMx is due to the generation of rich O-vacancies (Ni0, Fe0) that can be confirmed through XRD results showing existence of metallic Ni and Fe peaks in all Ni/rRMx catalysts (see Fig. 3). These rich-O vacancies species that act as acid sites (see TPD-NH3, the acidity of Ni/rRMx > Ni/RMOx) will invoke the occurrence of extensive undesired C–C cracking and result in coking, which reduces the DO activity.64,68 This finding is aligned with prior studies showing that rich metallic Ni (Ni0) supported on Al2O3 and SBA-15 catalysts is easily deactivated during the DO reaction due to the high coking activity (∼17–60%).68 Fig. S6† shows the number of saturated n-C15 + C17 is significantly higher than the number of unsaturated n-C15 + C17, suggesting that metal sites were the reason for the greater hydrogenation activity.79,80 The hydrogenation reaction may occur as a result of the H2 produced in situ through the cracking (C–C cleavage) reaction of the deoxygenated product.81 In addition, saturation of the deCOx product can be achieved via the occurence of decarboxylation. It is noteworthy to mention that the high strength acidic sites (Table 4) favoured the formation of a high degree of saturated hydrocarbon species via the decarboxylation reaction. Overall, the occurrence of the decarboxylation and hydrogenation reactions indicates the existence of a rich saturated hydrocarbon species.
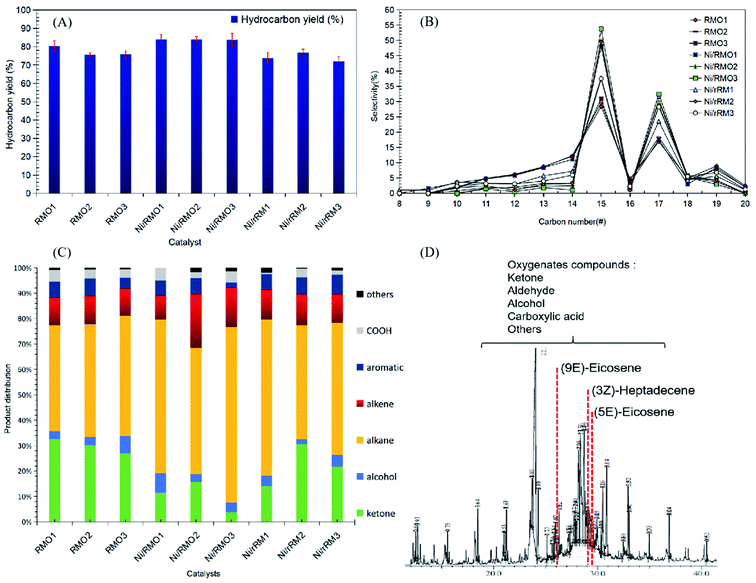 |
| | Fig. 6 Data collected from catalysed DO liquid product (A) hydrocarbon yield and (B) n-(C15 + C17) selectivity, (C) product distribution and (D) GCMS of deoxygenated compound. Reaction condition: T = 350 °C, 2 h reaction time, 3 wt% of catalyst loading. | |
The DO activity of all catalysed DO reactions was monitored by FTIR analysis, and the result is shown in Fig. S7.† The FTIR spectra of ceiba oil showed the main absorption bands at 2917 cm−1 and 2850 cm−1 (–CH stretching), 1740 cm−1 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester stretching), and 1120 cm−1 (C–O–C stretching). Based on an earlier work,82 Putra et al. proposed that the DO activity can be evaluated by comparing the decrease in the CO and C–O–C peak intensities. This is confirmed, as there is a slight shift of the CO peak at 1740 cm−1 in ceiba oil to 1702 cm−1 in the liquid products, indicating transformation of carboxylic acid from ester via triglyceride cracking. The following results are also in agreement with the GC-MS results, whereby the oxygenated species were discovered to be less in all DO products, while aliphatic alkanes and alkenes were the principal components that were detected (Fig. 6C).83 Notably, the DO reaction of ceiba oil catalyzed by Ni/RMO3 showed the highest amount of alkane hydrocarbon fractions within the range n-(C8–C20) with product distribution 70%; meanwhile, the lowest alkane hydrocarbon fraction is 42% for RMO1. Again, this shows that the Ni/RMO3 catalyst favors the formation of saturated hydrocarbon-like fuel.
O ester stretching), and 1120 cm−1 (C–O–C stretching). Based on an earlier work,82 Putra et al. proposed that the DO activity can be evaluated by comparing the decrease in the CO and C–O–C peak intensities. This is confirmed, as there is a slight shift of the CO peak at 1740 cm−1 in ceiba oil to 1702 cm−1 in the liquid products, indicating transformation of carboxylic acid from ester via triglyceride cracking. The following results are also in agreement with the GC-MS results, whereby the oxygenated species were discovered to be less in all DO products, while aliphatic alkanes and alkenes were the principal components that were detected (Fig. 6C).83 Notably, the DO reaction of ceiba oil catalyzed by Ni/RMO3 showed the highest amount of alkane hydrocarbon fractions within the range n-(C8–C20) with product distribution 70%; meanwhile, the lowest alkane hydrocarbon fraction is 42% for RMO1. Again, this shows that the Ni/RMO3 catalyst favors the formation of saturated hydrocarbon-like fuel.
According to the results, the amount of hydrocarbon detected by GC-FID and GC–MS did not complement very well. This is primarily due to the ability of Fe-promoted catalysts to facilitate isomerization reactions that yield hydrocarbon isomer compounds (Z) and (E), such as 5,9-eicosene (E) and 3-heptadecene (Z) that have been detected through the GC–MS analysis (Fig. 6D).84 Despite the fact that both GC-FID and GC–MS analysis yielded slightly different hydrocarbon yields in the n-(C8–C20) range, it is obvious that straight chain hydrocarbons (saturated and unsaturated) are the major compound in the deoxygenated liquid product. Based on the GC-MS finding, the deoxygenated liquid product exhibited the presence of oxygenated species (ketones, alcohols, carboxylic acids), and the oxygenated species was the highest (47%) for the RMO1 catalysed DO reaction due to the high alkalinity of red mud.85
3.4 Mass balance
A mass balance profile for the DO reaction of ceiba oil into the liquid hydrocarbon product using RMx, Ni/RMOx, and Ni/rRMx is tabulated in Table 5. According to eqn (4), theoretically, the DO reaction of ceiba oil will produce the hydrocarbon liquid product via deCOx by releasing CO2, CO, and H2O as by-products. Hence, the mass fractions including the ceiba oil feedstock, liquid product, and by-products (CO2 gas, CO gas, water) will be recorded. Based on the results, a low mass fraction of the liquid product (<12 wt%) was obtained experimentally compared to the theoretical value (68 wt%) with a deviation of <55 wt%. These contradictory results were most readily explained by the formation of undesirable by-products (char + residue), which amounted to a total of 41–46 wt% remaining in the semi-batch reactor after the reaction. Vitolo et al. reported that the formation of the by-products (char + residue) was caused by a low degree of volatilization of ceiba during DO at a high temperature (350 °C).86 The majority of the Ni-containing catalysts resulted in a high mass fraction of the liquid product (∼11 wt%), especially Ni/RMO3. This might be due to the preferential amount of strong acidic sites (12717.3 μmol g−1), contributing to a good balance of the occurrence of C–O cleavage and limiting secondary cracking that leads to the formation of volatile species.3 In contrast, Ni/rRM3 has the lowest mass fraction of the liquid product (4.73 wt%), which might be due to the high acidity site (see TPD-NH3) (40799.6 μmol g−1), that prone to have an aggressive catalytic cracking. This also could be confirmed, as Ni/rRM3 produces the highest fraction of gaseous (48.37 wt%) as the by-product compared to other catalysts. Besides, it is worth noting that all the catalysed DO reactions produced a small quantity of water (3 wt%), which may be produced by fatty acid hydrolysis or the decarbonylation process, but this amount was insignificant because it readily evaporated into gas during the high temperature of the DO reaction. Overall, Ni/RMO3 has proven to be effective in deoxygenizing the ceiba oil due to the formation of more condensable liquid-fuel products.
Table 5 Mass balance profile for catalytic DO of ceiba oil
| Theoretical deCOx |
Ceiba → liquid (oil) + 3 mol CO2/CO (g) + 3 mol H2O (aq) + by product (4) |
| Reactiona |
Feedstock |
Liq-productb |
Gasc |
Waterd |
Char + residuee |
| (g) |
(g) |
(wt%) |
(g) |
(wt%) |
(g) |
(wt%) |
(g) |
(wt%) |
| Deoxygenation condition: reaction temperature of 350 °C, 60 min reaction time, 3 wt% of catalyst, under inert condition with 400 rpm stirring rate. Mass fraction for Liq-product = [(mass of liq-product/mass of feedstock) × 100]. Material fraction for gas = [(mass of feedstock − mass of liq-product − mass of (char + residue) − mass of water)/mass of feedstock × 100]. Material fraction for water = [(mass of water/mass of feedstock) × 100]. Material fraction for (char + residue) (Y) = [(mass of (char + residue)/mass of feedstock) × 100]. |
| Theoritical data (deCOx) |
10.00 |
6.89 |
68.90 |
2.49 |
24.90 |
0.62 |
6.20 |
— |
— |
| RMO1 |
10.12 |
0.64 |
6.32 |
5.10 |
50.00 |
0.04 |
0.40 |
4.38 |
43.28 |
| RMO2 |
10.08 |
0.71 |
7.04 |
5.21 |
51.39 |
0.04 |
0.40 |
4.15 |
41.17 |
| RMO3 |
10.04 |
0.94 |
9.36 |
4.79 |
47.70 |
0.09 |
0.90 |
4.22 |
42.03 |
| Ni/RMO1 |
10.07 |
1.08 |
10.64 |
4.50 |
44.69 |
0.10 |
1.00 |
4.39 |
43.59 |
| Ni/RMO2 |
10.01 |
1.02 |
10.19 |
4.48 |
43.86 |
0.08 |
0.80 |
4.52 |
45.15 |
| Ni/RMO3 |
10.02 |
1.09 |
10.88 |
4.67 |
46.61 |
0.04 |
0.39 |
4.22 |
42.12 |
| Ni/rRM1 |
10.05 |
1.02 |
10.15 |
4.30 |
43.48 |
0.12 |
1.19 |
4.54 |
45.17 |
| Ni/rRM2 |
10.05 |
1.02 |
10.15 |
4.30 |
42.69 |
0.20 |
2.00 |
4.54 |
45.17 |
| Ni/rRM3 |
10.15 |
0.48 |
4.73 |
5.74 |
48.37 |
0.20 |
1.97 |
4.56 |
44.93 |
3.5 Optimization studies
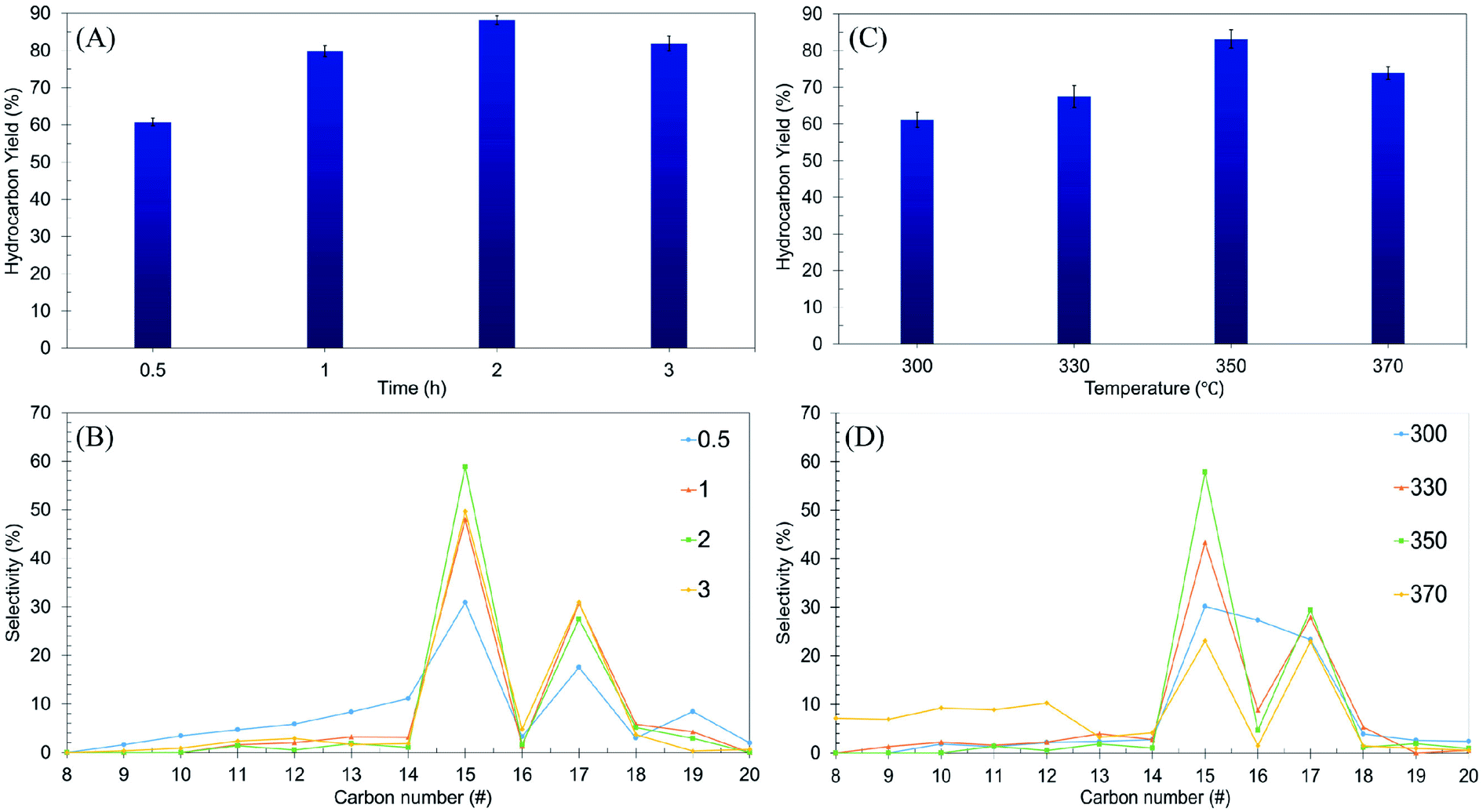
Using the one-variable-at-a-time technique, the effects of reaction time (0.5–3 h) and reaction temperature (300–370 °C) were examined using the Ni/RMO3 catalyst. The impact of reaction time on hydrocarbon yield and n-(C15 + C17) selectivity was investigated and reported in a volcano shape graph (Fig. 7A and B). The lowest hydrocarbon yield (60%) and n-(C15 + C17) selectivity (42%) were achieved when the reaction time was within 0.5 h. This demonstrated that for 0.5 h, there is a low degree of DO activity due to an insufficient amount of energy for the catalyst to initiate the reaction.5 Also, when the time was prolonged up to 2 h, the hydrocarbon yield and n-(C15 + C17) selectivity increased remarkably, suggesting that a longer residence time was required for a high degree of interaction between the reactant molecule and the catalyst surface.87 Beyond 3 h of reaction time, the DO activity decreased. The hydrocarbon yield and n-(C15 + C17) selectivity decreased from 89% to 81% and 86% to 79%, respectively. Notably, the light hydrocarbon fractions increased from 2% to 7% due to the cracking reaction at longer reaction times.88 According to the findings, the most efficient period for the DO reaction is 2 h, which results in a hydrocarbon yield of 89% and n-(C15 + C17) selectivity of 86%.
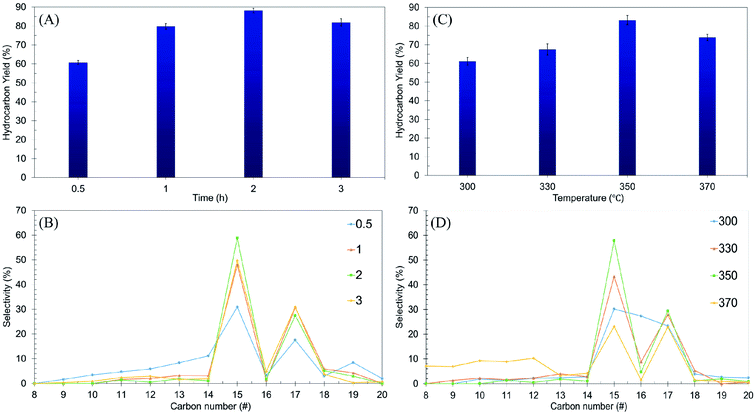 |
| | Fig. 7 Optimization studies of ceiba: (A and B) the effects of reaction time on hydrocarbon yield and n-(C15 + C17) selectivity at reaction parameters: 350 °C and 3 wt% catalyst loading, (C and D) the effects of reaction temperature on hydrocarbon yield and n-(C15 + C17) selectivity at reaction parameters: 3 wt% catalyst loading and 2 h reaction time with a stirring rate of 300 rpm and under an inert atmosphere. | |
The effect of reaction temperature was further studied, and the result is shown in Fig. 7C and D. The results showed that increasing the temperature from 300 °C to 350 °C resulted in a significant increase in the hydrocarbon yield (from 61% to 83%) and n-(C15 + C17) selectivity (from 53% to 87%). Further increase of the reaction temperature to >350 °C resulted in a decrease in the DO activity. The n-(C15 + C17) selectivity was significantly reduced, indicating that thermal cracking is favored at high temperatures, resulting in the formation of volatile species and light fractions.89 This finding is in agreement with an increase in the n-(C8–C12) selectivity from 3% to 42%. Overall, it can be inferred that reaction time and reaction temperature have a substantial influence on the DO of ceiba oil. Therefore, the optimum conditions for the DO reaction are the use of a 3 wt% catalyst loading for a 2 h reaction time at 350 °C under N2 flow.
The composition of the gases collected from the Ni/RMO3 catalysed DO reaction process at optimum reaction conditions was further analysed by GC-TCD, and the result is shown in Fig. S8.† Theoretically, the DO of ceiba oil in the absence of H2 will favour the decarboxylation and decarbonylation reactions and produce CO2 and CO.90 However, only CO2 was detected by GC-TCD, confirming that the oxygenate species were removed via decarboxylation pathways. Aside from that, H2 gas was detected, which could be prompted by the cracking process and the water–gas-shift reaction (WGS). The WGS reaction is reversible, and the equation is as follows: CO + H2O → CO2 + H2. The CO gas remained undetected. The absence of the CO gas in the TCD analysis is due to the effective WGS reaction.91
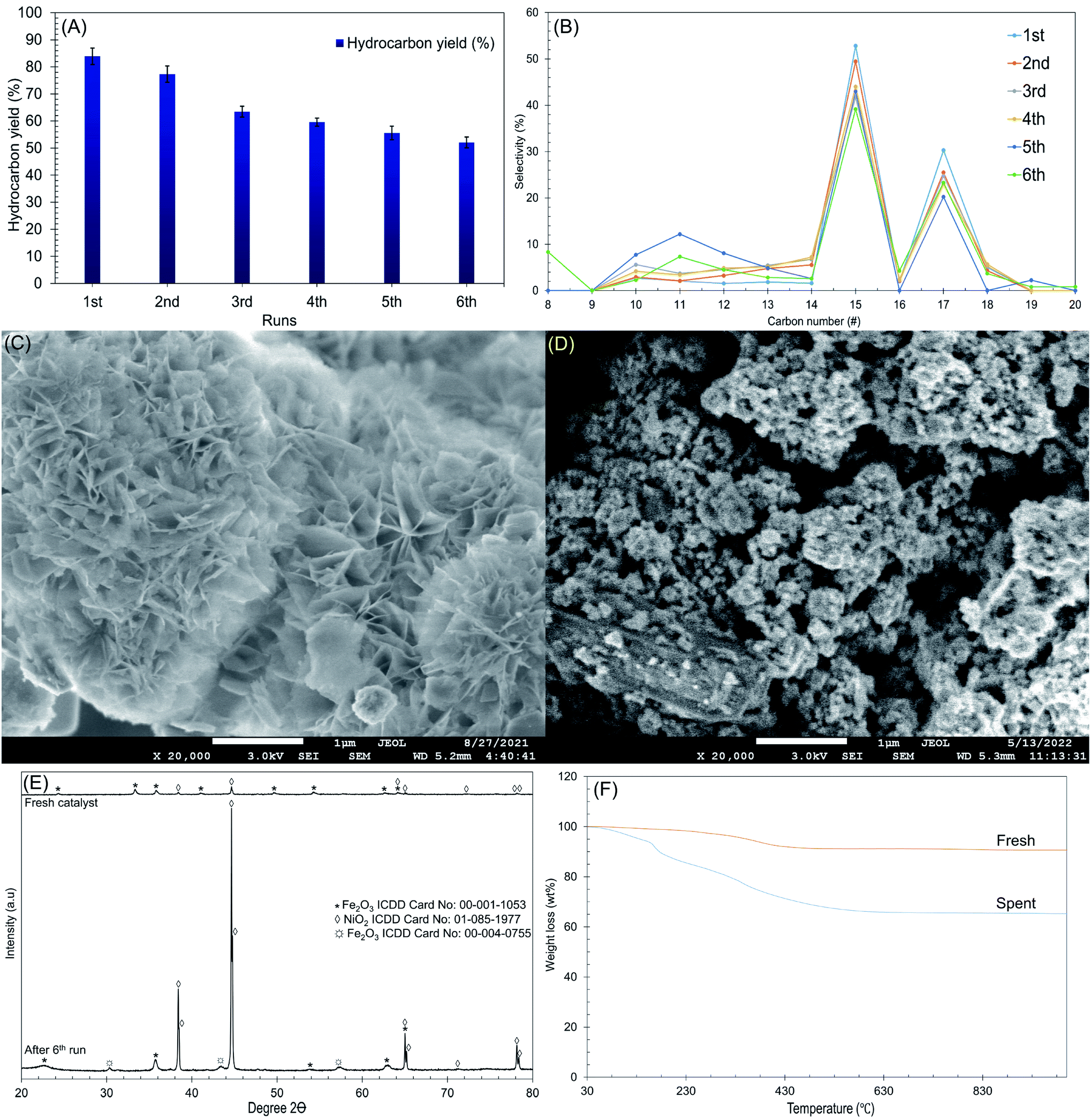
3.6 Reusability and stability of the Ni/RMO3 catalysts
The reusability and stability of Ni/RMO3 were further investigated at the following reaction condition: 2 h reaction, 350 °C reaction temperature using 3 wt% catalyst loading under N2 flow. The result is shown in Fig. 8A and F. To remove adsorbed organics, the catalyst from each cycle was washed numerous times with hexane. The reusability was studied for six cycles, and the gradual reduction in the hydrocarbon yield and n-C15 + C17 was observed (Fig. 8A and B). After the 6th run, the structure of the catalyst transformed from plate-like to agglomeration, indicating that the metal oxides has been sintered (Fig. 8C and D). Apparently, sintering of metal oxides largely occurs by high temperature reaction processes and results in the loss of active sites, thereby reducing the DO activity.92 Furthermore, XRD results revealed that the intensity of the NiO2 peak at 2θ: 38.4°, 65.1° and 78.2° (ICDD card no. 01-085-1977) were dramatically increased on the spent catalyst. This is because the NiO2 in the fresh catalyst exhibited as an amorphous form has been oxidised throughout the reaction and transitioned to crystalline form, resulting into distinct intensity in the NiO2 peak of the spent catalyst. Noted, high DO reaction also lead to structural transformation, which was proven by the new formation of syn-maghemite cubic Fe2O3 which appeared at 2θ: 30.3°, 44.7° and 57.4° (ICDD card no. 00-004-0755) on spent catalyst (Fig. 8E).93 Considering the existence of rhombohedral hematite Fe2O3 in a fresh catalyst, high thermal reaction temperatures apparently lead to structure distortion and alteration.94 Apart from that, TGA analysis also confirmed that the employed catalysts suffered from coke coverage (Fig. 8F). According to the TGA results, the oxidation of the deposits from these reactions occurred at a comparatively higher decomposition temperature (550 °C), with coke deposits accounting for 23 wt%, as suggested by the presence of hard coke. Soft coke (decomposition temperature = 160 °C) was also found but in smaller quantities (7 wt%). Based on this finding, it can be suggested that the coke deposits on Ni/RMO3 are mainly polynuclear aromatic coke. Overall, Ni/RMO3 after DO suffer with loss of actives sites by sintering and coking but the coke content was lower (46–60 wt%) than prior study.37,68 It should be noted that Ni/RMO3 is made from waste bauxite, which has been catastrophic in taking lives and devastating surrounding areas. As a result, this research is critical for developing an alternative strategy on how waste management can be used to provide long-term value, such as alternative sources of metal recovery with low cost.95
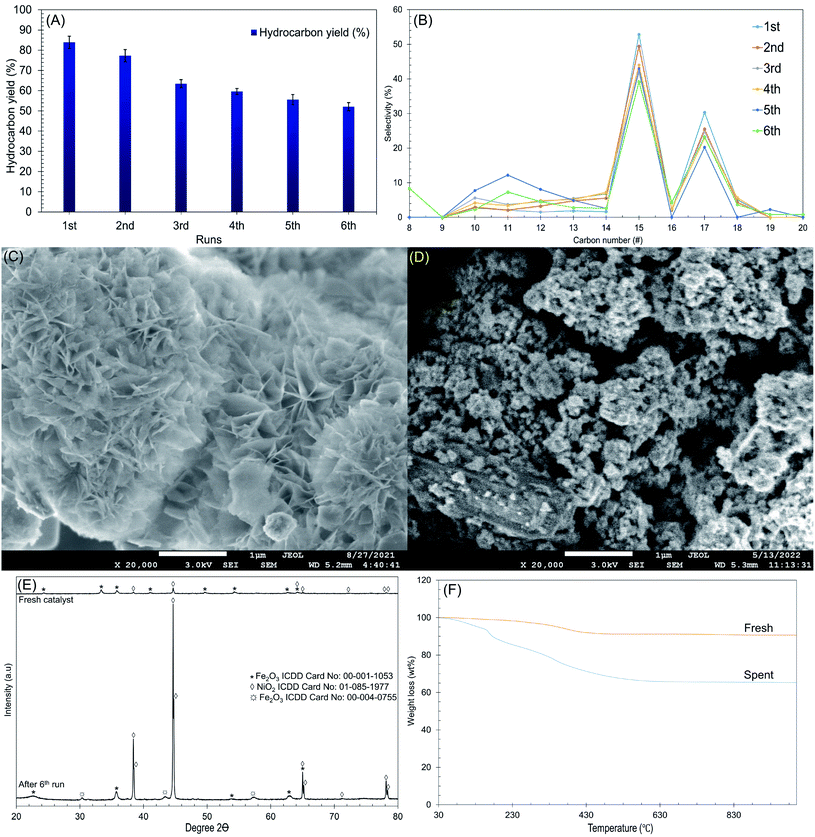 |
| | Fig. 8 Data collected from the Ni/RMO3 catalyst's deoxygenated liquid product after the 6th run at T = 350 °C, 2 h reaction time, 3 wt% of catalyst loading (A) hydrocarbon yield, (B) n-(C15 + C17) selectivity with number of DO cycles, (C) FESEM images for the fresh catalyst, (D) FESEM images for the spent catalyst, (E) TGA of Ni/RMO3 catalyst, and (F) XRD diffractogram of the catalyst. | |
4. Conclusion
The potential of red-mud derived catalysts (RMOx, Ni/RMOx, and Ni/rRMx) for the chemoselective reaction on the DO of ceiba oil was successfully investigated. The results show that the removal of oxygenated species in the production of diesel-range alkanes n-(C15 + C17). Based on the DO profile, the liquid hydrocarbon yield increased in the order of Ni/RMO3 > Ni/RMO1 > Ni/RMO2 > RMO1 > Ni/rRM2 > RMO3 > RMO2 > Ni/rRM1 > Ni/rRM3. The Ni/RMO3 was found to have an effective DO activity with a hydrocarbon yield of ∼86% and n-(C15 + C17) selectivity of up to 85%. This was attributed to a synergistic impact between Ni–O and Fe–O species in the catalyst as well as high-strength acidic sites, which enhance the C–O cleavage, thereby promoting the DO activity. It was also shown that Ni/RMOx catalysts enhance the formation of saturated alkanes with selectivity ranging from 49% to 70%, owing to the high strong acidic active sites that promote the saturation process, resulting in the production of more alkanes. Based on the optimization study, reaction time and reaction temperature play a critical role in improving the DO activity. A high hydrocarbon yield (83%) and n-(C15 + C17) selectivity (87%) were observed under the optimum conditions: 3 wt% of catalyst loading, 2 h of reaction time at 350 °C reaction temperature. Despite the fact that the Ni/RMO3 catalyst showed tremendous potential for converting ceiba oil to diesel fuel, it was obscured by coke, particularly hard coke, which produces aromatic species (ketones, alcohols, and carboxylic acids) that deactivate the catalyst. To summarise, transformation of waste bauxite to an effective DO catalyst demonstrated an efficient management of waste bauxite pollution in the future, which is in accordance with the worldwide mission of transforming “Waste to Wealth”.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the financial support from Ministry of Higher Education Malaysia for Fundamental Research Grant Scheme (FRGS-MRSA/1/2019/STG01/UPM/01/30), Galakan Penyelidik Muda (GGPM) (GGPM-2020-015) and GP-2021-K023310 for funding this research work.
References
- NASA: Climate Change and Global Warming, accessed 14 July 2021, https://climate.nasa.gov/.
- M. H. M. Ashnani, A. Johari, H. Hashim and E. Hasani, Renewable Sustainable Energy Rev., 2014, 35, 244–257 CrossRef.
- N. Asikin-mijan, H. V. Lee, J. C. Juan, A. R. Noorsaadah, H. Chyuan and S. M. Razali, Appl. Catal., A, 2018, 552, 38–48 CrossRef CAS.
- M. S. Gamal, N. Asikin-Mijan, W. N. A. W. Khalit, M. Arumugam, S. M. Izham and Y. H. Taufiq-Yap, Fuel Process. Technol., 2020, 208, 106519 CrossRef CAS.
- N. Asikin-Mijan, H. V. Lee, T. S. Marliza and Y. H. Taufiq-Yap, J. Anal. Appl. Pyrolysis, 2018, 129, 221–230 CrossRef CAS.
- H. Zhang, H. Lin and Y. Zheng, Appl. Catal., B, 2014, 160–161, 415–422 CrossRef CAS.
- M. A. Abdullah, A. U. Rahmah and Z. Man, J. Hazard. Mater., 2010, 177, 683–691 CrossRef CAS PubMed.
- A. Demirbas, Energy Convers. Manage., 2009, 50, 14–34 CrossRef CAS.
- P. Sivakumar, S. Sindhanaiselvan, N. N. Gandhi, S. S. Devi and S. Renganathan, Fuel, 2013, 103, 693–698 CrossRef CAS.
- M. A. Abdullah, M. Afzaal, Z. Ismail, A. Ahmad, M. S. Nazir and A. H. Bhat, Desalin. Water Treat., 2015, 54, 3044–3053 CrossRef CAS.
- F. A. Santoso, S. Soe’eib, A. Suryandari and N. Asri, Proc. Int. Conf. Green Technol., 2017, 8, 209–215 Search PubMed.
- M. Balajii and S. Niju, Renewable Energy, 2020, 146, 2255–2269 CrossRef CAS.
- K. Alharbi, W. Alharbi, E. F. Kozhevnikova and I. V. Kozhevnikov, ACS Catal., 2016, 6, 2067–2075 CrossRef CAS.
- L. Chen, Y. Zhu, H. Zheng, C. Zhang and Y. Li, Appl. Catal., A, 2012, 411–412, 95–104 CrossRef CAS.
- I.-H. Choi, J.-S. Lee, C.-U. Kim, T.-W. Kim, K.-Y. Lee and K.-R. Hwang, Fuel, 2018, 215, 675–685 CrossRef CAS.
- S. Janampelli and S. Darbha, Catal. Commun., 2019, 125, 70–76 CrossRef CAS.
- S. Sushil and V. S. Batra, Appl. Catal., B, 2008, 81, 64–77 CrossRef CAS.
- M. Abou Rjeily, C. Gennequin, H. Pron, E. Abi-Aad and J. H. Randrianalisoa, Pyrolysis-catalytic upgrading of bio-oil and pyrolysis-catalytic steam reforming of biogas: a review, Springer International Publishing, 2021 Search PubMed.
- K. Jayasankar, P. K. Ray, A. K. Chaubey, A. Padhi, B. K. Satapathy and P. S. Mukherjee, Int. J. Miner., Metall. Mater., 2012, 19, 679–684 CrossRef CAS.
- G. Ning, B. Zhang, C. Liu, S. Li, Y. Ye and M. Jiang, Minerals, 2018, 8(102), 1–16 Search PubMed.
- S. Wang, H. M. Ang and M. O. Tadé, Chemosphere, 2008, 72, 1621–1635 CrossRef CAS PubMed.
- S. Kumar, R. Kumar and A. Bandopadhyay, Resour., Conserv. Recycl., 2006, 48, 301–314 CrossRef.
- H. Sutar, S. C. Mishra, S. Sahoo and A. P. Chakraverty, Am. Chem. Sci. J., 2014, 4, 255–279 CrossRef.
- T. Nordgreen, T. Liliedahl and K. Sjöström, Fuel, 2006, 85, 689–694 CrossRef CAS.
- A. M. Robinson, J. E. Hensley and J. Will Medlin, ACS Catal., 2016, 6, 5026–5043 CrossRef CAS.
- P. A. Bozkurt, O. Tosun and M. Canel, J. Energy Inst., 2017, 90, 355–362 CrossRef CAS.
- V. Patil, S. Adhikari and P. Cross, Bioresour. Technol., 2018, 270, 311–319 CrossRef CAS PubMed.
- X. Lim, A. Sanna and J. M. Andrésen, Fuel, 2014, 119, 259–265 CrossRef CAS.
- D. Wang, D. Wang, J. Yu, Z. Chen, Y. Li and S. Gao, Fuel Process. Technol., 2019, 186, 81–87 CrossRef CAS.
- H. Jahromi and F. A. Agblevor, Appl. Catal., B, 2018, 236, 1–12 CrossRef CAS.
- S. K. Kim, M. T. Mehran, U. Mushtaq, T. H. Lim, J. W. Lee, S. B. Lee, S. J. Park and R. H. Song, Energy Convers. Manage., 2016, 130, 119–129 CrossRef CAS.
- J. Sun, A. M. Karim, H. Zhang, L. Kovarik, X. S. Li, A. J. Hensley, J. S. McEwen and Y. Wang, J. Catal., 2013, 306, 47–57 CrossRef CAS.
- Y. M. Sani, W. M. A. W. Daud and A. R. Abdul Aziz, Appl. Catal., A, 2014, 470, 140–161 CrossRef CAS.
- H. C. Ong, A. S. Silitonga, H. H. Masjuki, T. M. I. Mahlia, W. T. Chong and M. H. Boosroh, Energy Convers. Manage., 2013, 73, 245–255 CrossRef CAS.
- N. Asikin-Mijan, H. V. Lee, J. C. Juan, A. R. Noorsaadah, H. C. Ong, S. M. Razali and Y. H. Taufiq-Yap, Appl. Catal., A, 2018, 552, 38–48 CrossRef CAS.
- K. B. Baharudin, Y. H. Taufiq-Yap, J. Hunns, M. Isaacs, K. Wilson and D. Derawi, Microporous Mesoporous Mater., 2019, 276, 13–22 CrossRef CAS.
- N. Asikin-Mijan, H. V. Lee, J. C. Juan, A. R. Noorsaadah and Y. H. Taufiq-Yap, RSC Adv., 2017, 7, 46445–46460 RSC.
- R. Fréty, J. G. A. Pacheco, M. R. Santos, J. F. Padilha, A. F. Azevedo, S. T. Brandão and L. A. M. Pontes, J. Anal. Appl. Pyrolysis, 2014, 109, 56–64 CrossRef.
- J. Asomaning, P. Mussone and D. C. Bressler, J. Anal. Appl. Pyrolysis, 2014, 105, 1–7 CrossRef CAS.
- S. S. Lam, A. D. Russell, C. L. Lee and H. A. Chase, Fuel, 2012, 92, 327–339 CrossRef CAS.
- G. Abdulkareem-Alsultan, N. Asikin-Mijan, N. Mansir, H. V. Lee, Z. Zainal, A. Islam and Y. H. Taufiq-Yap, J. Anal. Appl. Pyrolysis, 2019, 137, 171–184 CrossRef CAS.
- G. Baskar and S. Soumiya, Renewable Energy, 2016, 98, 101–107 CrossRef CAS.
- W. N. A. W. Khalit, N. Asikin-Mijan, T. S. Marliza, M. S. Gamal, M. R. Shamsuddin, M. I. Saiman and Y. H. Taufiq-Yap, Biomass Bioenergy, 2021, 154, 106248 CrossRef CAS.
- V. M. Sglavo, R. Campostrini, S. Maurina, G. Carturan, M. Monagheddu, G. Budroni and G. Cocco, J. Eur. Ceram. Soc., 2000, 20, 235–244 CrossRef CAS.
- H. Nath, P. Sahoo and A. Sahoo, Powder Technol., 2015, 269, 233–239 CrossRef CAS.
- C. Wu and D. Liu, J. Nanomater., 2012, 1–6 Search PubMed.
- L. Wang, G. Hu, F. Lyu, T. Yue, H. Tang, H. Han, Y. Yang, R. Liu and W. Sun, Minerals, 2019, 9, 281 CrossRef CAS.
- Z. Chen, D. Wang, H. Yang, Y. Zhang, Y. Li, C. Li, J. Yu and S. Gao, Carbon Resour. Convers., 2021, 4, 10–18 CrossRef CAS.
- J. K. Sadangi, S. P. Das, A. Tripathy and S. K. Biswal, Sep. Sci. Technol., 2018, 53, 2186–2191 CrossRef CAS.
- D. Mombelli, S. Barella, A. Gruttadauria and C. Mapelli, Appl. Sci., 2019, 9, 4902 CrossRef CAS.
- L. Wang, N. Sun, H. Tang and W. Sun, Minerals, 2019, 9(362), 1–19 Search PubMed.
- S. Alam, S. K. Das and B. H. Rao, J. Cleaner Prod., 2017, 168, 679–691 CrossRef CAS.
- G. Alkan, C. Schier, L. Gronen, S. Stopic and B. Friedrich, Metals, 2017, 7, 458 CrossRef.
- S. Agatzini-Leonardou, P. Oustadakis, P. E. Tsakiridis and C. Markopoulos, J. Hazard. Mater., 2008, 157, 579–586 CrossRef CAS PubMed.
- M. S. S. Lima, L. P. Thives, V. Haritonovs and K. Bajars, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 251, 1–10 Search PubMed.
- M. A. Khairul, J. Zanganeh and B. Moghtaderi, Resour., Conserv. Recycl., 2019, 141, 483–498 CrossRef.
- Mineral Processing and Extractive Metallurgy Review : An International Journal TRENDS IN RED MUD UTILIZATION – A REVIEW.
- S. S. Kushwaha, D. Kishan, M. S. Chauhan and S. Khetawath, Mater. Today: Proc., 2018, 5, 20500–20512 CAS.
- R. Y. Chen, Oxid. Met., 2017, 88, 687–717 CrossRef CAS.
- J. T. Richardson, M. Lei, B. Turk, K. Forster and M. V. Twigg, Appl. Catal., A, 1994, 110, 217–237 CrossRef CAS.
- Q. Jeangros, T. W. Hansen, J. B. Wagner, C. D. Damsgaard, R. E. Dunin-Borkowski, C. Hebert and A. Hessler-Wyser, J. Mater. Sci., 2013, 48, 2893–2907 CrossRef CAS.
- Y. Zheng, F. Wang, X. Yang, Y. Huang, C. Liu, Z. Zheng and J. Gu, J. Anal. Appl. Pyrolysis, 2017, 126, 169–179 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- J. Weber, A. Thompson, J. Wilmoth, V. S. Batra, N. Janulaitis and J. R. Kastner, Appl. Catal., B, 2019, 241, 430–441 CrossRef CAS.
- G. Abdulkareem-Alsultan, N. Asikin-Mijan, H. V. Lee and Y. H. Taufiq-Yap, Chem. Eng. J., 2016, 304, 61–71 CrossRef CAS.
- S. H. Shuit, E. P. Ng and S. H. Tan, J. Taiwan Inst. Chem. Eng., 2015, 52, 100–108 CrossRef CAS.
- Z. P. Hu, C. C. Weng, C. Chen and Z. Y. Yuan, Appl. Catal., A, 2018, 562, 49–57 CrossRef CAS.
- Z. Zhang, X. Hu, L. Zhang, Y. Yang, Q. Li, H. Fan, Q. Liu, T. Wei and C.-Z. Li, Fuel Process. Technol., 2019, 191, 138–151 CrossRef CAS.
- K. Kaya, S. F. Kurtoǧlu, A. Uzun and S. Soyer-Uzun, Ind. Eng. Chem. Res., 2018, 57, 7156–7168 CrossRef CAS.
- M. L. Guzman-Castillo, X. Bokhimi, A. Toledo-Antonio, J. Salmones-Blasquez and F. Hernandez-Beltran, J. Phys. Chem. B, 2001, 105, 2099–2106 CrossRef CAS.
- N. Zhao, W. Wang, X. Lei, Z. Ye, X. Chen, H. Ding and H. Yang, J. Mater. Sci.: Mater. Electron., 2017, 28, 15701–15707 CrossRef CAS.
- F. Tessier, A. Navrotsky, R. Niewa, A. Leineweber, H. Jacobs, S. Kikkawa, M. Takahashi, F. Kanamaru and F. J. DiSalvo, Solid State Sci., 2000, 2, 457–462 CrossRef CAS.
- D. L. Kuznetsov, G. G. Ugodnikov and I. E. Filatov, Tech. Phys. Lett., 2008, 34, 87–89 CrossRef CAS.
- S. H. Gage, D. A. Ruddy, S. Pylypenko and R. M. Richards, Catal. Today, 2018, 306, 9–15 CrossRef CAS.
- B. Ouyang, Y. Zhang, Z. Zhang, H. J. Fan and R. S. Rawat, Small, 2017, 13, 1604265 CrossRef PubMed.
- N. Asikin-Mijan, H. V. Lee, J. C. Juan, A. R. Noorsaadah, G. Abdulkareem-Alsultan, M. Arumugam and Y. H. Taufiq-Yap, J. Anal. Appl. Pyrolysis, 2016, 120, 110–120 CrossRef CAS.
- S. R. Naqvi and M. Naqvi, Int. J. Energy Res., 2018, 42, 1352–1362 CrossRef CAS.
- P. Maneechakr and S. Karnjanakom, ACS Omega, 2021, 6, 20006–20014 CrossRef CAS PubMed.
- L. Xu, R. Nie, X. Lyu, J. Wang and X. Lu, Fuel Process. Technol., 2020, 197, 106205 CrossRef CAS.
- A. Kurimoto, R. P. Jansonius, A. Huang, A. M. Marelli, D. J. Dvorak, C. Hunt and C. P. Berlinguette, Angew. Chem., Int. Ed., 2021, 60, 11937–11942 CrossRef CAS PubMed.
- I. Shimada, S. Kato, N. Hirazawa, Y. Nakamura, H. Ohta, K. Suzuki and T. Takatsuka, Ind. Eng. Chem. Res., 2017, 56, 75–86 CrossRef CAS.
- R. Putra, W. W. Lestari, F. R. Wibowo and B. H. Susanto, Bull. Chem. React. Eng. Catal., 2018, 13, 245–255 CrossRef CAS.
- N. Asikin-Mijan, H. V. Lee, Y. H. Taufiq-Yap, G. Abdulkrem-Alsultan, M. S. Mastuli and H. C. Ong, Energy Convers. Manage., 2017, 141, 325–338 CrossRef CAS.
- E. W. Qian, N. Chen and S. Gong, J. Mol. Catal. A: Chem., 2014, 387, 76–85 CrossRef CAS.
- M. Renz, Eur. J. Org. Chem., 2005, 2005, 979–988 CrossRef.
- S. Vitolo, M. Seggiani, P. Frediani, G. Ambrosini and L. Politi, Fuel, 1999, 78, 1147–1159 CrossRef CAS.
- N. Asikin-mijan, H. V. V. Lee, G. Abdulkareem-alsultan, A. Afandi, C. Production, N. Asikin-mijan, H. V. V. Lee, G. Abdulkareem-alsultan, A. Afandi, Y. H. H. Taufiq-Yap, C. Production, N. Asikin-mijan, H. V. V. Lee, G. Abdulkareem-alsultan, A. Afandi and Y. H. H. Taufiq-Yap, J. Cleaner Prod., 2017, 167, 1048–1059 CrossRef CAS.
- G. A. Alsultan, N. Asikin-Mijan, H. V. Lee, A. S. Albazzaz and Y. H. Taufiq-Yap, Energy Convers. Manage., 2017, 151, 311–323 CrossRef CAS.
- S. Bezergianni, S. Voutetakis and A. Kalogianni, Ind. Eng. Chem. Res., 2009, 48, 8402–8406 CrossRef CAS.
- M. S. Gamal, N. Asikin-Mijan, M. Arumugam, U. Rashid and Y. H. Taufiq-Yap, J. Anal. Appl. Pyrolysis, 2019, 144, 104690 CrossRef.
- N. Aliana-Nasharuddin, N. Asikin-Mijan, G. Abdulkareem-Alsultan, M. I. Saiman, F. A. Alharthi, A. A. Alghamdi and Y. H. Taufiq-Yap, RSC Adv., 2019, 10, 626–642 RSC.
- G. Victor, A. Martinez-Hernandez and M. A. Gracia-Pinilla, Appl. Catal., A, 2020, 594, 117455 CrossRef.
- C.-H. Wu, J.-S. Ma and C.-H. Lu, Curr. Appl. Phys., 2012, 12, 1190–1194 CrossRef.
- P. Guionneau, C. Brigouleix, Y. Barrans, A. E. Goeta, J.-F. Létard, J. A. K. Howard, J. Gaultier and D. Chasseau, C. R. Acad. Sci., Ser. IIc: Chim., 2001, 4, 161–171 CrossRef CAS.
- W. Elekwachi, C. Wizor and H. Nwankwoala, Int. J. Emerg. Eng. Res. Technol., 2019, 7, 1–11 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *c,
Mohd Izham Saiman*ab,
G. Abdulkareem Alsultan
*c,
Mohd Izham Saiman*ab,
G. Abdulkareem Alsultan