
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel 2D-AuSe nanostructures as effective platinum replacement counter electrodes in dye-sensitized solar cells
Esmie Mposa a,
Rudo K. Sitholea,
Zakhele Ndalaa,
Grace N. Ngubenia,
Kalenga P. Mubiayia,
Poslet M. Shumbulab,
Lerato F. E. Machogo-Phao*ac and
Nosipho Moloto*a
a,
Rudo K. Sitholea,
Zakhele Ndalaa,
Grace N. Ngubenia,
Kalenga P. Mubiayia,
Poslet M. Shumbulab,
Lerato F. E. Machogo-Phao*ac and
Nosipho Moloto*a
aMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Private Bag 3, Wits, 2050, South Africa. E-mail: leratoma@mintek.co.za; Nosipho.Moloto@wits.ac.za; Tel: +27 11 709 4111 Tel: +27 11 717 6774
bDepartment of Chemistry, University of Limpopo, Private Bag X1106 Sovenga, 0727, South Africa
cAnalytical Services Division, Mintek, 200 Malibongwe Drive, Randburg, South Africa
First published on 28th April 2022
Abstract
Studies to improve the efficiency of dye-sensitized solar cells (DSSCs) include, but are not limited to, finding alternatives such as 2D layered materials as replacement counter electrodes (CEs) to the commonly used Pt. Herein, we report for the first time, the use of AuSe as a counter electrode for the reduction of triiodide ions (I3−) to iodide ions (I−). The colloidal synthesis of gold selenide nanostructures produced α-AuSe and β-AuSe dominated products as determined by XRD. Electron microscopy showed α-AuSe having belt-like structures while β-AuSe had a plate-like morphology. EDS mapping confirmed the elemental composition and homogeneity of the AuSe CEs. Cyclic voltammetry curves of the AuSe CEs displayed the double set of reduction–oxidation peaks associated with the reactions in the I3−/I− electrolyte and therefore were comparable to the Pt CV curve. The α-AuSe CE showed better electrocatalytic activity with a reduction current of 6.1 mA than that of β-AuSe and Pt CEs, which were 4.2 mA and 4.8 mA, respectively. The peak-to-peak separation (ΔEpp) for the α-AuSe CE was also more favourable with a value of 532 mV over that of the β-AuSe CE of 739 mV however, both values were larger than that of the Pt CE, which was found to be 468 mV. The EIS and Tafel plot data showed that α-AuSe had the best catalytic activity compared to β-AuSe and was comparable to Pt. The DSSC using α-AuSe as a CE had the highest PCE (6.94%) as compared to Pt (4.89%) and β-AuSe (3.47%). The lower efficiency for Pt was attributed to the poorer fill factor. With these novel results, α-AuSe is an excellent candidate to be used as an alternative CE to Pt in DSSCs.
1. Introduction
Dye-sensitized solar cells (DSSCs) have emerged as a powerful photovoltaic technology alternative to conventional silicon solar cells because of their advantages such as low cost, use of environmentally friendly materials, simple fabrication techniques and acceptable photoelectric conversion efficiencies.1,2 To date, the highest reported efficiency of lab-scale DSSCs is 14.3%, an increase from 7.1–7.9% first reported in 1991.3–5 A typical DSSC has four main components; a photoanode made of a mesoporous oxide layer (typically, TiO2) deposited on a transparent conductive glass substrate; a monolayer of dye sensitizer covalently bonded to the surface of the TiO2, an electrolyte containing redox couple (typically, I−/I3−), and a counter electrode made of a platinum coated conductive glass substrate.6,7 The functioning of DSSCs relies on the interconnection of a multicomponent system; therefore improving the device efficiency considers a multifaceted approach. Research on the modification of some of the cell components has been explored. This includes alternative synthesis methods for nanostructured semiconductor materials such as the photoanode for high dye loading and fast electron transport, the search for dyes/sensitizers with strong visible light-harvesting ability, redox electrolytes for fast and efficient hole transport, and as substitutes for the electron-rich platinum (Pt) counter electrode (CE).8–11The counter electrode is a key constituent which assists in collecting electrons from the external circuit passing them to the electrolyte which in turn donates these electrons to the previously oxidized dye molecules.12 The key properties of a good CE material are; (i) high conductivity for charge transport, (ii) good electrocatalytic activity for reducing the electrolyte and (iii) excellent stability.13 To date, Pt has been the preferred CE choice due to its metallic nature and willingness to donate electrons, however, it is susceptible to corrosion by the commonly used liquid iodide–triiodide (I−/I3−) electrolyte due to the formation of PtI4.14,15 Variations such as different carbonaceous materials, carbon–metal composites, polymers and inorganic materials such as metal chalcogenides and metal oxides have been explored as replacement CEs for platinum.1,8,16–18 In addition to the properties listed above, a high surface area is an added advantage for a counter electrode as it allows for faster catalytic redox activity. Transition metal chalcogenides with a two-dimensional layered structure have this feature and have therefore drawn much attention as potential alternative CEs in DSSCs.19 Some of the reported power conversion efficiencies (PCEs) when using layered materials as CEs include; 6.23% for graphene,20 6.6% for MoS2 (ref. 21) and 7.01% for MoSe2.22 Another layered transition metal chalcogenide is gold selenide (AuSe), a relatively unexplored material which is electron rich and therefore has potential as a counter electrode material in DSSCs. AuSe crystallizes in monoclinic space group C2/m with two polymorphs, α-AuSe and β-AuSe with lattice parameters, a = 12.202(2) and 8.355(2) Å, b = 3.690(7) and 3.663(1) Å and c = 8.433(2) and 6.262(1) Å, respectively. Both crystal phases of AuSe contain Au1+ and Au3+ where the Au1+ is coordinated linearly to two Se atoms whilst Au3+ is surrounded by four Se atoms in a square planar geometry.23,24 Herein, we report for the first time, the electrocatalytic properties of α- and β-AuSe in the redox reaction of the iodide–triiodide system.
2. Experimental section
2.1 Chemicals
Gold(III) chloride hydrate (99%), elemental selenium powder (99%), oleylamine (OLA, 70%), toluene (anhydrous, 95%), 1-methyl-2-pyrrolidinone (anhydrous NMP, 99.5%), lithium iodide (99.9%), iodine (≥99.8%), lithium perchlorate (≥95%), 4-tert-butylpyridine (98%), sodium iodide (anhydrous, ≥99.9%), acetonitrile (≥99.9%), N-719 dye (95%), white titania paste, reflector (TiO2, 20.0 wt%), Whatman™ glass microfiber filter paper, fluorine doped tin oxide (FTO) coated glass slide (surface resistivity ∼ 7 Ω sq−1), were obtained from Sigma-Aldrich and used without any further processing. Absolute ethanol 99% was obtained from MK Chemical.2.2 Synthesis of gold selenide
(A) For the synthesis of the predominantly α-AuSe sample, in a three-neck round bottom flask, OLA was heated to 100 °C followed by the addition of 0.28 g selenium dissolved in OLA. The temperature was further raised to 200 °C and at this point 0.17 g gold chloride dissolved in OLA was subsequently added. The reaction was carried out at 200 °C and aged for 1 h.(B) In the synthesis of the predominantly β-AuSe sample, OLA, selenium powder and gold chloride were all placed in a three-neck flask and the temperature was raised to 200 °C over a period of 20 min. Once the required temperature was reached the reaction was aged for 1 h.
In both experiments, the mixtures were subjected to strong magnetic stirring and inert conditions during the heating as well as aging processes. Inert conditions were achieved by passing of nitrogen gas. Upon aging, ethanol was used to flocculate the particles as well as to wash off any excess OLA. The nanomaterials were then collected by centrifugation.
X-ray diffraction. A Bruker D2 phaser (D2-205530) diffractometer using secondary graphite monochromated Cu Kα radiation (λ 1.5418 Å) at 30 kV and 10 mA was used to measure the powder XRD patterns on the as-synthesized materials. Measurements were taken using a glancing angle of incidence detector at an angle of 2°, for 2θ values over 5–90° in steps of 0.036° with a step time of 0.5 s and at a temperature of 294 K.
Transmission electron microscopy. Transmission electron microscopy (TEM) was carried out on a FEI Technai T12 TEM microscope operated at an acceleration voltage of 120 kV with a beam spot size of 3 in TEM mode. The samples were initially suspended in toluene followed by placing a drop of the suspended nanomaterials on a lacey-carbon copper grid. The grid was allowed to dry at room temperature before analysis.
Counter electrode fabrication. The counter electrode ink was prepared by dispersing 40 mg of the as-synthesized AuSe in 1 mL NMP followed by sonication for 30 min. The ink was drop-casted onto pre-cleaned FTO substrates (area ∼ 1.56 cm2) followed by baking on a hotplate at 80 °C for 10 min to allow for evaporation of the solvent and a subsequent layer was deposited and allowed to bake as previous. For reference and comparative studies, two layers of platinum were sputter-coated onto a pre-cleaned FTO substrate.
Photo-anode fabrication. The titania (TiO2) paste was applied onto the conductive side of the pre-cleaned FTO substrate using the doctor blade method. The substrates were then annealed at 350 °C for 30 min to remove any residual organic compounds and to make sure the TiO2 is bonded to the plate to enable better contact with the N-719 dye. The photo-sensitizing dye was made by dissolving the N-719 dye in methanol (3.0 × 10−4 M). A drop of the dye mixture was then added onto the annealed TiO2 substrate and left overnight to dry at ambient conditions in the dark.
Device assembly. The photo-anode electrode with the sensitized TiO2 side facing up was placed on the bench as the bottom plate of the cell and the counter electrode, active side facing down was placed on top of the photo-anode. The two electrodes were offset from each other and the Whatman filter paper was placed in between to act as a sponge for the redox electrolyte solution. The redox electrolyte solution was composed of 0.05 M iodine, 0.1 M lithium iodide, 0.1 M potassium iodide, 0.1 M sodium iodide and 0.5 M 4-tert-butylpyridine. The assembled device was secured with binder clips before doing the J–V measurements. The FTO used for the fabricated DSSCs of all three CEs had the same active area (∼1.56 cm2) for comparisons.
2.3 Characterization of the counter electrode (CE) thin films
Scanning electron microscopy (SEM) and energy dispersive spectroscopy (EDS) mapping were carried out on a VEGA3 TESCAN SEM microscope. The AuSe-CEs were mounted on aluminium stubs with carbon tape and analysed accordingly. Cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), and Tafel polarization measurements were done using Biologic: SP-300. A three-electrode system was used to conduct CV measurements at a scan rate of 100 mV s−1 where Ag/AgCl electrode acted as the reference electrode, Pt wire as the auxiliary electrode and the prepared AuSe-CEs acted as the working electrode. The electrolyte was made of 0.1 M LiClO4, 0.001 M I2 and 0.01 M LiI dissolved in anhydrous acetonitrile. The electrolyte was purged with nitrogen gas before each scan. EIS measurements were obtained using a symmetrical cell with two identical electrodes in the redox electrolyte used for DSSCs. The electrodes were analysed between 100 kHz and 100 mHz at varying open circuit potentials for each sample. The Tafel polarization measurements were conducted at a potential window of −1.0 to 1.0 V with a scan rate of 100 mV s−1. The photocurrent–voltage (J–V) characteristic curves of the DSCCs were measured in ambient conditions using the HP 4141B source measure unit (SMU) under controlled illumination of 100 mW cm−2 (AM 1.5 G).3. Results and discussion
Materials used in this study were previously synthesized and discussed at great length in previous publications.23,24 X-ray diffraction was used to determine the structure of the as-synthesized powders. Fig. 1 shows the obtained diffraction patterns together with the matching standard patterns. For the two synthetic methods, mixed phased gold selenide samples were obtained with sample A having more peaks belonging to α-AuSe (PDF 00-020-0457) and sample B having more of the β-AuSe (PDF 00-020-0458) peaks. Both materials showed impurities of elemental Au (PDF 00-004-0784) and additionally sample B showed those of elemental Se (PDF 00-006-0362). | ||
| Fig. 1 Powder X-ray diffraction patterns of as-synthesized AuSe matched to standard powder patterns of α-AuSe (A), β-AuSe (B), and elemental Au & elemental Se (key: α = α-AuSe, β = β-AuSe, $ = elemental Au and * = elemental Se). | ||
The morphology of the samples was analysed by TEM and the results are depicted in Fig. 2. Sample A revealed long belt-like structures of varying lengths and widths, on the other hand, sample B showed short plate-like structures. Owing to the sample distribution displayed on the copper grids (sample A having more belts and sample B showing more plates) it was concluded that the belts were of the α-phase while the plates were of β-phase. The mixed morphologies of the synthesized α-AuSe and β-AuSe were discussed in previous publications.23,24
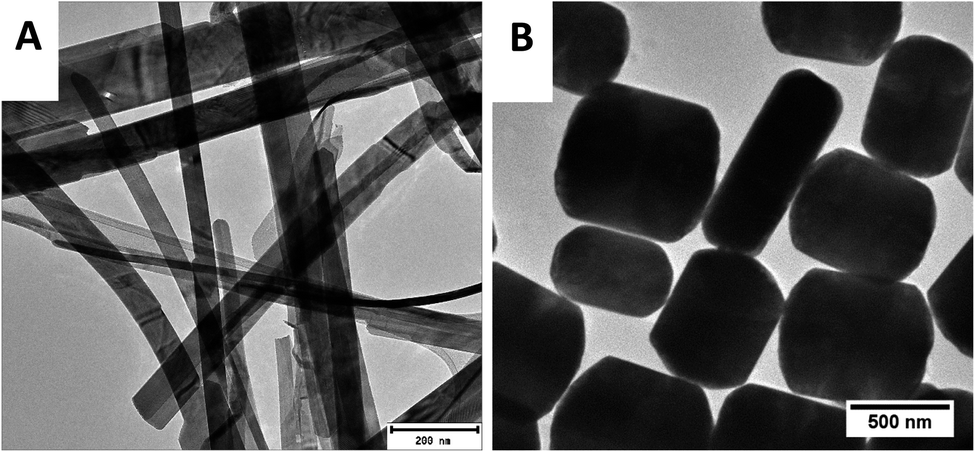 | ||
| Fig. 2 TEM images of the as-obtained gold selenide materials. (A) Showing the α-AuSe nanobelts and (B) the β-AuSe nanoplates. | ||
Upon depositing the materials onto the FTO glass substrates, SEM was performed on the AuSe-CE films to confirm that no morphological changes occurred. Fig. 3(i) shows the images obtained during the SEM analysis where image A displays the aggregated nanobelts (α-AuSe CE) and image B shows the aggregated nanoplates (β-AuSe CE) unaltered. SEM-EDS mapping micrographs in columns (ii) and (iii) displayed even distributions of Au and Se throughout the films, respectively for both CEs. The summed mappings in column (iv) illustrates the uniformity in elemental distribution of both Au & Se giving complete homogenous AuSe-CEs.
 | ||
| Fig. 3 (i) SEM images of (A) α-AuSe and (B) β-AuSe counter electrodes. Elemental mapping of (ii) Au, (iii) Se and (iv) summed mapping of Au and Se. | ||
Elemental composition was further confirmed by EDS analysis as illustrated in Fig. 4. Both CEs contained Au, Se and traces of C attributed to the capping agent. In addition, the α-AuSe-CE (Fig. 4A) also showed Sn, Si and O which comes from the FTO glass substrate used.
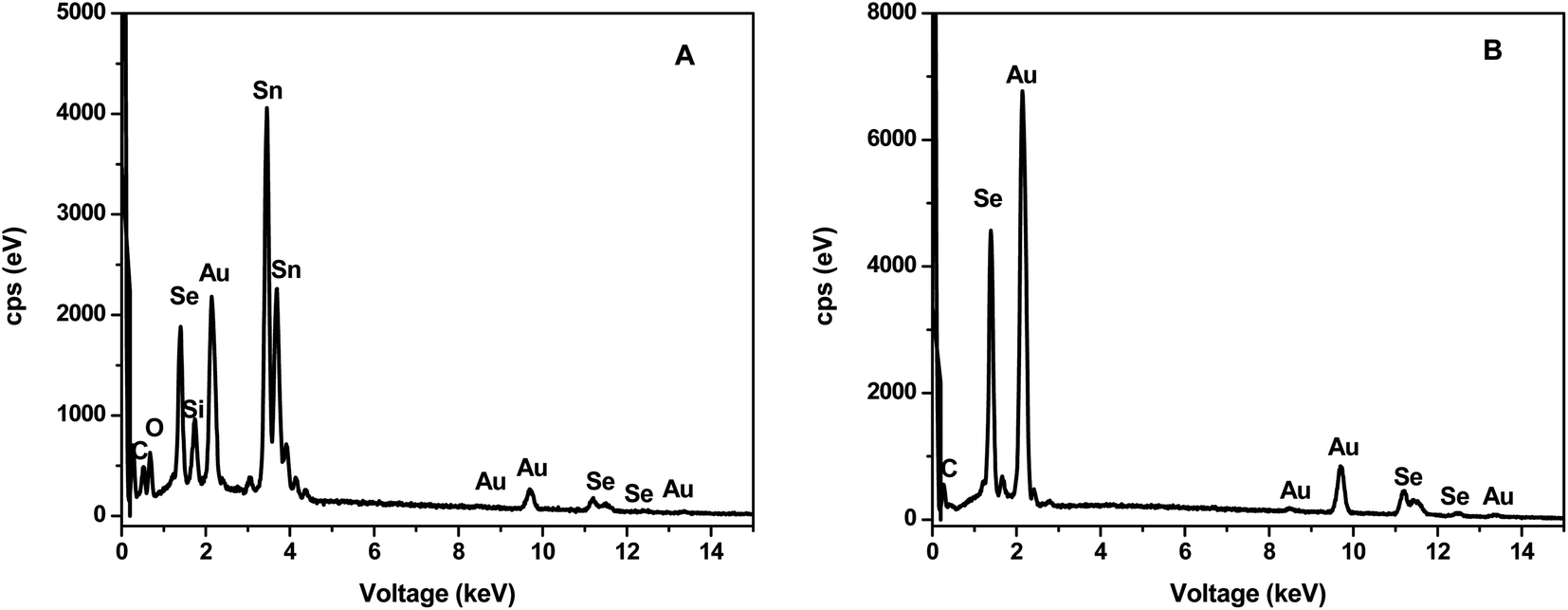 | ||
| Fig. 4 SEM-EDS of (A) α-AuSe-CE and (B) β-AuSe-CE showing the elemental composition of the materials on the FTO glass substrates. | ||
The electrocatalytic activity of the CEs for the I−/I3− redox couple were evaluated using cyclic voltammetry (CV) at a scan rate of 100 mV s−1. Fig. 5 shows CV curves of the α-AuSe and β-AuSe CEs in comparison to that of the Pt CE. The curves of both AuSe CEs display similar duck shapes to that of Pt and therefore show compelling evidence that they can be used as alternative counter electrodes in DSSCs. The CV curves are composed of two pairs of reduction–oxidation peaks: the first pair on the left (Red1/Ox1) and the second pair on the right, (Red2/Ox2). The two pairs of redox peaks on the CV curves have similar shapes as the Pt CE suggesting that the AuSe CEs have similar catalytic activity during the redox process.17,25 The negative (left side) and positive (right side) are represented by eqn (1) and (2), respectively.26,27
| I3− + 2e− ↔ 3I− | (1) |
| 3I2 + 2e− ↔ 3I3− | (2) |
 | ||
| Fig. 5 Cyclic voltammograms of the Pt, α-AuSe and β-AuSe counter electrodes at a scan rate of 100 mV s−1 for the I−/I3− redox couple. | ||
Two parameters that speak to the quality and performance of the CE are deduced from the resulting CV curves. These are the cathodic peak current density |Current Red1| and the change in peak-to-peak separation (ΔEpp) between Red1 and Ox1. A larger peak current density and smaller ΔEpp indicates good electrocatalytic activity of a CE.26,28 Table 1 compares the discussed parameters for each CE, showing α-AuSe having a higher |Current Red1| of 6.1 mA than that of Pt (4.8 mA) and β-AuSe (4.2 mA). The nanobelt structure of α-AuSe gives them a much larger apparent surface area and active catalytic sites, leading to a higher current density than the Pt electrode. These findings suggest that the α-AuSe CE is the better of the two AuSe CEs. On the other hand, the α-AuSe and the β-AuSe have higher ΔEpp as compared to the Pt electrode which is attributed to the higher overpotential losses in these electrodes than in the Pt electrode which can be caused by the thickness and adhesion of the electrode films thus affecting the test results.28,29 The ΔEpp values are in the order of Pt < α-AuSe < β-AuSe. Despite the larger ΔEpp value for the α-AuSe, it could still serve as an alternative to the Pt electrode.
| CE | |Current Red1 (mA)| | ΔEpp (mV) |
|---|---|---|
| Pt | 4.8 | 468 |
| α-AuSe | 6.1 | 532 |
| β-AuSe | 4.2 | 739 |
The behaviour of the CEs over different scan rate conditions was investigated and is shown in Fig. 6(a) and (b) for α-AuSe and β-AuSe, respectively. For both CEs there is an increase in |Current Red1| and ΔEpp with an increase in scan rate. Fig. 6(c) and (d) show the anodic and cathodic current as a function of the square root of scan rate for the two samples. The current density and square root of the scan rate have a relationship, according to eqn (3). As the scan rate increases, the diffusion layer becomes thinner, and the electrochemical polarization increases, resulting in a high overpotential and limited reversibility.30 Because of the linear relationship, diffusion limits in cathodic and anodic processes may affect iodide species movement on the counter electrode surface and of evidence is the disappearance of the Red1 peak at 10 mV s−1 suggesting this condition is not favourable for the reduction of triiodide ions using the AuSe CEs.31,32 This linear relationship reveals that the reaction of the I3−/I− redox couple at both CEs is dominated by ionic diffusion-controlled transport, and there is no specific interaction between the I3−/I− redox couple and the CEs.33,34
| ip = 2.69 × 105n3/2AD1/2V1/2C | (3) |
 | ||
| Fig. 6 Cyclic voltammograms for (a) α-AuSe and (b) β-AuSe CEs at varying scan rates from 10 mV s−1 to 100 mV s−1. Dependence of anodic and cathodic peak currents on square root of scan rates for (c) α-AuSe and (d) β-AuSe CEs. | ||
The stability of a counter electrode can be evaluated using successive cycle voltammetry scanning and by studying the dark current–voltage.35–37 The reproducibility and stability of the AuSe CEs were investigated at a scan rate of 100 mV s−1 for 60 consecutive redox cycles and the resultant curves are presented in Fig. 7(a) and (b) for α-AuSe and β-AuSe, respectively. There were minimal changes in the shapes of the CV curves. Inserts in the respective graphs show the first and sixtieth cycle and only slight changes were observed. By comparison, of the two CEs, the α-AuSe has excellent reproducibility and stability as compared to the β-AuSe, which shows some difference between the first and the sixtieth cycle with the anodic and cathodic peak current densities decreasing after the sixtieth cycle. This shows compelling evidence that the α-AuSe CE possess excellent reversible redox activity.
 | ||
| Fig. 7 Cyclic voltammograms for, (a) α-AuSe and (b) β-AuSe CEs for 60 consecutive cycles, with inserts showing the 1st and the 60th cycle for both electrodes. | ||
The electrical impedance spectroscopy (EIS) technique was used to investigate the electrocatalytic processes of the DSSCs, specifically the ability of the CEs to transfer charge to the electrolyte. For a robust counter electrode, high electrical conductivity is expected to easily facilitate electron transfers from the external circuit to reduce the I3− ions.38 Fig. 8(a) shows the Nyquist plot of Pt, α-AuSe and β-AuSe which showed typically one semicircle located in the higher frequency region which can be attributed to a charge transfer resistance and (c) the electrochemical equivalent circuit whose components show four important parameters, Rs, Rct, Cdl and Zw. Where the Rs is the total ohmic series resistance, Rct the charge transfer resistance at an interface between the CE and the electrolyte, Cdl which is the double layer capacitance which denotes the charge storage capacity of the CEs and Zw which represents the Nernst diffusion impedance in the electrolyte often employed when a line is 45° to the semi-circle at lower frequency region and explains if the interaction between the CE and the electrolyte is diffusion-controlled.33,34 The two main parameters, Rs and Rct were obtained using the Z-fit in EC-Chem software from Biologic and are listed in Table 2 for each CE. The smaller the Rs value, the higher the conductivity of the CE material and the following trend was observed Pt < α-AuSe < β-AuSe. This suggests that in terms of the two phases, the α-AuSe is the most conductive as compared to the β-AuSe. The Rs values of the AuSe electrodes are slightly higher than Pt which could be due to a lack of adhesive strength between the AuSe CE layer and the FTO glass, resulting in a larger contact resistance that decreases the conductivity of the electrode.28 The charge transfer process represented by the Rct values also suggest that the α-AuSe has better charge transfer kinetics with lower Rct values as compared to the β-AuSe, represented in the order Pt < α-AuSe < β-AuSe. This can be attributed to the morphology of α-AuSe where elongated belt-like structures form charge transfer channels as compared to the less anisotropic plate-like β-AuSe.
 | ||
| Fig. 8 (a) Nyquist plots, (b) Tafel polarization curves for the symmetric cells fabricated with Pt, α-AuSe and β-AuSe CEs and (c) the equivalent circuit. | ||
| CE | Rs (ohm) | Rct (ohm) | Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) J0 (mA cm−2) J0 (mA cm−2) |
LogJlim (mA cm−2) |
|---|---|---|---|---|
| Pt | 40.91 | 2587 | −5.30 | −3.25 |
| α-AuSe | 41.18 | 11499 |
−5.95 | −3.04 |
| β-AuSe | 41.43 | 14439 |
−6.05 | −3.37 |
To further investigate the catalytic activity of the CEs, Tafel polarization measurements were conducted on the symmetric cells. Fig. 8(b) shows plots of logarithmic current density (logJ0) versus potential (V) at room temperature. From the Tafel plots, we can obtain the exchange current density (J0) and the limiting diffusion current density (Jlim) which are characteristic to the catalytic activity of the CEs. The parameters are influenced by both anodic or cathodic contributions of the CE and are better explained using the following equations:
| J0 = RT/nFRct | (4) |
| Jlim = 2nFDC/l | (5) |
Fig. 9 shows the photocurrent density–voltage (J–V) curves of the DSSCs assembled from the Pt and AuSe CEs, both in dark and upon illumination. The dark current for the three devices did not show much difference hence signifying relatively good stability similar to what is observed in the successive CV scans. The respective parameters including the open circuit voltage (Voc), short circuit current density (Jsc), fill factor (FF) and power conversion efficiency (PCE) are summarized in Table 3.
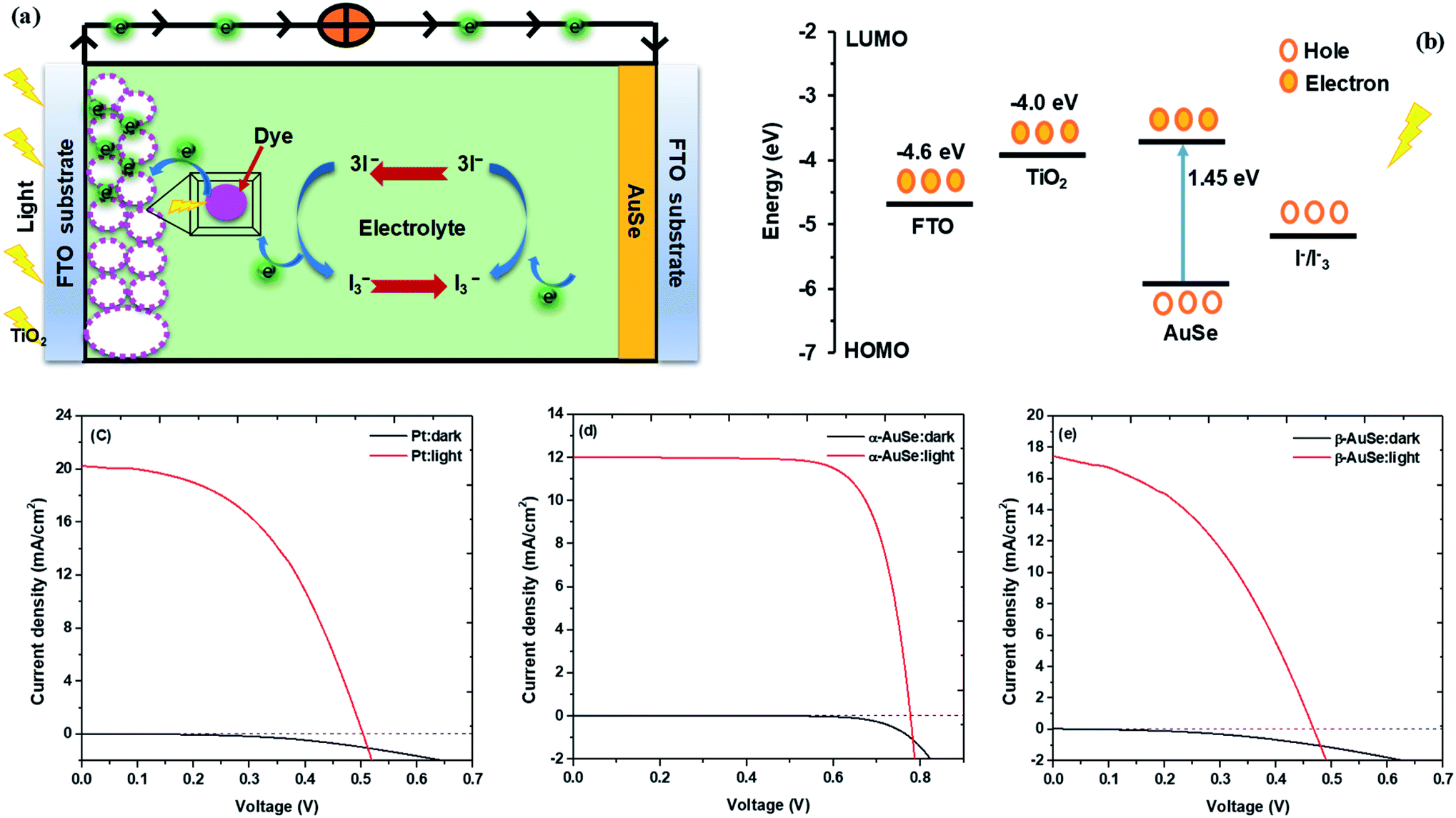 | ||
| Fig. 9 (a) Schematic of a DSSC, (b) energy diagram of the DSCC, (c) J–V curves of DSSCs with Pt, (d) α-AuSe and (e) β-AuSe CEs in the dark and under 100 mW cm−2 illumination. | ||
| Counter electrode | Jsc (mA cm2) | Voc (V) | FF (%) | PCE (%) |
|---|---|---|---|---|
| Pt | 20.26 ± 0.08 | 0.52 ± 0.02 | 46 ± 0.2 | 4.89 ± 0.23 |
| α-AuSe | 12.00 ± 0.03 | 0.79 ± 0.01 | 73 ± 0.4 | 6.94 ± 0.09 |
| β-AuSe | 17.43 ± 0.10 | 0.49 ± 0.02 | 41 ± 1.0 | 3.47 ± 0.18 |
From the results shown in Table 3, of the AuSe phases, it can be observed that the α-AuSe has the highest efficiency of 6.94% as compared to the β-AuSe, which has an efficiency of 3.47%, however α-AuSe has a higher efficiency than Pt due to a better Voc. The Voc usually increases with an increasing bandgap, α-AuSe has been shown to have a bandgap of 1.45 eV while the β-AuSe bandgap is narrower.27,39 Notably, the α-AuSe has the highest FF. A high voltage generally results in a high FF. The low FF values are due to the high Rs values and low shunt resistance (Rsh) values which are caused by increasing recombination at interfaces of the DSSCs.40 The lower efficiency of the Pt CE is due to recombination as indicated by the low Voc and FF; this can be attributed to the roughness of the thin film which creates recombination sites.41 The PCEs of the α-AuSe CE is comparable to some of the reported PCEs of different layered materials as CEs. Examples include; 6.23% for graphene,20 6.6% for MoS2,21 7.01% for MoSe2,22 6.30% for WS2,42 7.25% for MoTe2,43 7.99% for WS2/MoTe2,44 7.66% for TiS2 and 8.80% for TiS2 nanosheets assembled and decorated on graphene (TiS2–G)16 as shown in Fig. 10. The lower values are attributed to the lower FF. This could be due to the presence of the mixed phases of AuSe as observed in the XRD. Nevertheless, the Jsc are comparable.
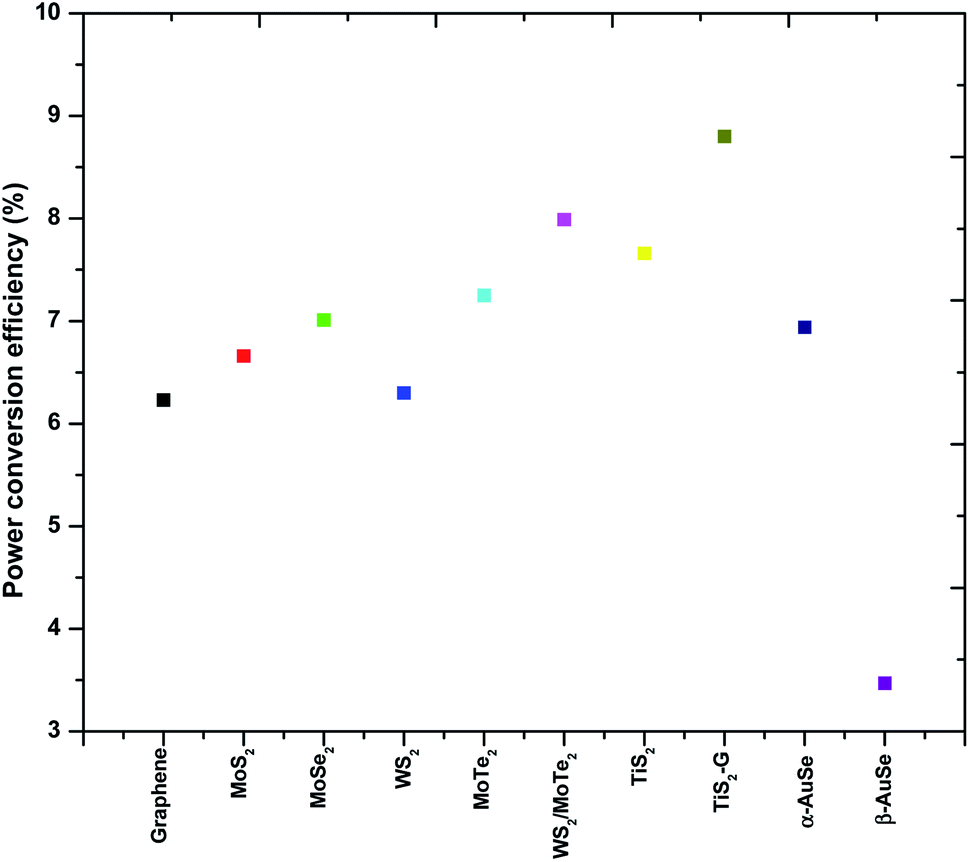 | ||
| Fig. 10 A comparison plot showing the trend in power conversion efficiencies (PCEs) of a few 2-dimensional layered transition metal chalcogenides. | ||
4. Conclusion
In summary, colloidal gold selenide was successfully synthesized and the as synthesized materials were effectively incorporated onto FTO glass to form homogenized counter electrode films. Powder XRD showed biphasic materials where both α and β-AuSe phases present in each sample, however, a level of dominance was observed in each. TEM depicted long belt-like structures for the α-AuSe dominated sample and short plate-like structures for the β-AuSe dominated sample. SEM analysis confirmed the morphology of the nanostructures on the CE films, it further validated the elemental composition with EDS mapping. Cyclic voltammetry showed compelling evidence that both α-AuSe and β-AuSe have potential use in dye sensitized solar cells as counter electrodes with good electrocatalytic activity towards reduction of I3− ions in the iodide–triiodide redox couple. The higher cathodic peak current (|Current Red1|) of 6.1 mA and lower ΔEpp value of 532 mV for the α-AuSe CE, makes it the more preferred of the two CEs, compared to 4.2 mA and 739 mV for the β-AuSe CE. The two AuSe CEs gave larger ΔEpp values than the Pt CE (468 mV). The EIS and Tafel plot data followed the same trend hence indicating the catalytic activities of β-AuSe < α-AuSe < Pt. The results from complete DSSCs were extracted from the J–V curves, α-AuSe had the highest PCE (6.94%) as compared to Pt (4.89%) and β-AuSe (3.47%). The lower efficiency for Pt was attributed to the fabrication process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the University of the Witwatersrand, School of Chemistry, Wits Microscopy and Microanalysis Unit (MMU) for their facilities and Mr Siyasanga Mpelane at the University of Johannesburg for the worked performed on SEM analyses. Gratitude also goes to Mintek, Tiso Foundation and the NRF for their financial support.References
- S. Yun, Y. Liu, T. Zhang and S. Ahmad, Nanoscale, 2015, 7, 11877–11893 RSC.
- S. P. Mohanty, V. More and P. Bhargava, RSC Adv., 2015, 5, 18647–18654 RSC.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J. I. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894–15897 RSC.
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef.
- S. Yun, Y. Qin, A. R. Uhl, N. Vlachopoulos, M. Yin, D. Li, X. Han and A. Hagfeldt, Energy Environ. Sci., 2018, 11, 476–526 RSC.
- M. Kokkonen, P. Talebi, J. Zhou, S. Asgari, S. A. Soomro, F. Elsehrawy, J. Halme, S. Ahmad, A. Hagfeldt and S. G. Hashmi, J. Mater. Chem. A, 2021, 9, 10527–10545 RSC.
- S. Yun, A. Hagfeldt and T. Ma, Adv. Mater., 2014, 26, 6210–6237 CrossRef CAS PubMed.
- M. Ye, X. Wen, M. Wang, J. Iocozzia, N. Zhang, C. Lin and Z. Lin, Mater. Today, 2015, 18, 155–162 CrossRef CAS.
- J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan and G. Luo, Chem. Rev., 2015, 115, 2136–2173 CrossRef CAS PubMed.
- M. R. Narayan, Renewable Sustainable Energy Rev., 2012, 16, 208–215 CAS.
- S. Thomas, T. G. Deepak, G. S. Anjusree, T. A. Arun, S. V. Nair and A. S. Nair, J. Mater. Chem. A, 2014, 2, 4474–4490 RSC.
- D. Nechiyil, B. P. Vinayan and S. Ramaprabhu, J. Colloid Interface Sci., 2017, 488, 309–316 CrossRef CAS PubMed.
- Y. S. Yen, H. H. Chou, Y. C. Chen, C. Y. Hsu and J. T. Lin, J. Mater. Chem., 2012, 22, 8734–8747 RSC.
- S. S. Nemala, P. Kartikay, R. K. Agrawal, P. Bhargava, S. Mallick and S. Bohm, Sol. Energy, 2018, 169, 67–74 CrossRef CAS.
- E. Olsen, G. Hagen and S. E. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 63, 267–273 CrossRef CAS.
- X. Meng, C. Yu, B. Lu, J. Yang and J. Qiu, Nano Energy, 2016, 22, 59–69 CrossRef CAS.
- X. Li, J. Bai, B. Zhou, X. Yuan, X. Zhang and L. Liu, Chem.–Eur. J., 2018, 24, 11444–11450 CrossRef CAS PubMed.
- U. Ahmed, M. Alizadeh, N. A. Rahim, S. Shahabuddin, M. S. Ahmed and A. K. Pandey, Sol. Energy, 2018, 174, 1097–1125 CrossRef CAS.
- E. Singh, K. S. Kim, G. Y. Yeom and H. S. Nalwa, RSC Adv., 2017, 7, 28234–28290 RSC.
- S. S. Nemala, P. Kartikay, S. Prathapani, H. L. M. Bohm, P. Bhargava, S. Bohm and S. Mallick, J. Colloid Interface Sci., 2017, 499, 9–16 CrossRef CAS PubMed.
- S. Vijaya, G. Landi, J. J. Wu and S. Anandan, Electrochim. Acta, 2019, 294, 134–141 CrossRef CAS.
- X. Yuan, B. Zhou, X. Zhang, Y. Li and L. Liu, Electrochim. Acta, 2018, 283, 1163–1169 CrossRef CAS.
- L. F. E. Machogo, P. Tetyana, R. Sithole, S. S. Gqoba, N. Phao, M. Airo, P. M. Shumbula, M. J. Moloto and N. Moloto, Appl. Surf. Sci., 2018, 456, 973–979 CrossRef CAS.
- L. F. E. Machogo, M. Mthimunye, R. K. Sithole, P. Tetyana, N. Phao, G. N. Ngubeni, M. Mlambo, P. S. Mduli, P. M. Shumbula and N. Moloto, New J. Chem., 2019, 43, 5773–5782 RSC.
- X. Wu, J. Duan, Y. Zhao, X. Yang, H. Chen, B. He and Q. Tang, Sol. Energy, 2019, 180, 85–91 CrossRef CAS.
- J. Wu, Q. Li, L. Fan, Z. Lan, P. Li, J. Lin and S. Hao, J. Power Sources, 2008, 181, 172–176 CrossRef CAS.
- X. Qian, H. Li, L. Shao, X. Jiang and L. Hou, ACS Appl. Mater. Interfaces, 2016, 8, 29486–29495 CrossRef CAS PubMed.
- Y. Liu, S. Yun, X. Zhou, Y. Hou, T. Zhang, J. Li and A. Hagfeldt, Electrochim. Acta, 2017, 242, 390–399 CrossRef CAS.
- S. Yun, J. Shi, Y. Si, M. Sun, Y. Zhang, A. Arshad and C. Yang, J. Colloid Interface Sci., 2021, 601, 12–29 CrossRef CAS PubMed.
- M. Wu, Y. Wang, X. Lin, N. Yu, L. Wang, L. Wang, A. Hagfeldt and T. Ma, Phys. Chem. Chem. Phys., 2011, 13, 19298–19301 RSC.
- S. Biallozor and A. Kupniewska, Electrochem. Commun., 2000, 2, 480–486 CrossRef CAS.
- M. Ikegami, K. Miyoshi and T. Miyasaka, Appl. Phys. Lett., 2007, 90, 153122 CrossRef.
- G. Yue, W. Wu, X. Liu and H. Zheng, Sol. Energy, 2018, 167, 137–146 CrossRef CAS.
- T. Zhang, S. Yun, X. Li, X. Huang, Y. Hou, Y. Liu and J. Li, J. Power Sources, 2017, 340, 325–336 CrossRef CAS.
- C. Wang, S. Yun, Q. Fan, Z. Wang, Y. Zhang, F. Han, Y. Si and A. Hagfeldt, J. Mater. Chem. A, 2019, 7, 14864–14875 RSC.
- S. Yun, H. Pu, J. Chen, A. Hagfeldt and T. Ma, ChemSusChem, 2014, 7, 442–450 CrossRef CAS PubMed.
- S. Yun, A. Hagfeldt and T. Ma, ChemCatChem, 2014, 6, 1584–1588 CrossRef CAS.
- Q. S. Jiang, R. Chen, X. Guo, Y. Zhu, W. Cheng, W. Li, X. Yang and H. C. Chen, Sol. Energy, 2019, 179, 59–66 CrossRef CAS.
- C. Tang, L. Zhang, C. Zhang, J. Macleod, K. Ostrikov and A. Du, Nanoscale Horiz., 2020, 5, 366–371 RSC.
- T. Kolokoto, V. Mashindi, L. F. E. Machogo-phao, Z. B. Ndala, N. P. Shumbula, S. S. Nkabinde, G. N. Ngubeni, S. S. Gqoba, K. P. Mubiayi and N. Moloto, RSC Adv., 2021, 11, 31159–31173 RSC.
- Z. Huang, G. Natu, Z. Ji, M. He, M. Yu and Y. Wu, J. Phys. Chem., 2012, 116, 26239–26246 CAS.
- S. Hussain, S. Shaikh, D. Vikraman, R. S. Mane, O. S. Joo, M. Naushad and J. Jung, RSC Adv., 2015, 5, 103567–103572 RSC.
- S. Hussain, S. A. Patil, D. Vikraman and N. Mengal, Sci. Rep., 2018, 6, 4–11 Search PubMed.
- S. Hussain, S. A. Patil, A. A. Memon, D. Vikraman, H. G. Abbas, S. H. Jeong, H. S. Kim, H. S. Kim and J. Jung, Inorg. Chem. Front., 2018, 5, 3178–3183 RSC.
| This journal is © The Royal Society of Chemistry 2022 |