
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled synthesis of trimetallic nitrogen-incorporated CoNiFe layered double hydroxide electrocatalysts for boosting the oxygen evolution reaction†
Anh Thi Nguyet Nguyen ,
Minji Kim and
Jun Ho Shim*
,
Minji Kim and
Jun Ho Shim*
Department of Chemistry and Institute of Basic Science, Daegu University, Gyeongsan 38453, Republic of Korea. E-mail: junhoshim@daegu.ac.kr
First published on 27th April 2022
Abstract
The development of non-precious trimetallic electrocatalysts exhibiting high activity and stability is a promising strategy for fabricating efficient electrocatalysts for the oxygen evolution reaction (OER). In this study, trimetallic nitrogen-incorporated CoNiFe (N–CoNiFe) was produced to solve the low OER efficiency using a facile co-precipitation method in the presence of ethanolamine (EA) ligands. A series of CoNiFe catalysts at different EA concentrations were also investigated to determine the effects of the ligand in the co-precipitation of a trimetallic system. The introduction of an optimized EA concentration (20 mM) improved the electrocatalytic performance of N–CoNiFe dramatically, with an overpotential of 318 mV at 10 mA cm−2 in 1.0 M KOH and a Tafel slope of 72.2 mV dec−1. In addition, N–CoNiFe shows high durability in the OER process with little change in the overpotential (ca. 16.0 mV) at 10 mA cm−2 after 2000 cycles, which was smaller than that for commercial Ir/C (38.0 mV).
Introduction
Hydrogen is a promising clean fuel and an important part of the future of renewable energy in addressing the global energy demand and environmental issues.1 Despite this, one of the major problems in the hydrogen economy is its clean production because most of the hydrogen currently used is produced from fossil fuel via the steam reforming process, with carbon dioxide as a byproduct.2 Therefore, considerable effort has been made to produce hydrogen from water electrolysis, which is an environmentally clean, potentially cost-effective, and renewable source of hydrogen.3 Water electrolysis consists of two half-cell reactions, the hydrogen evolution reaction (HER) at the cathode and the oxygen evolution reaction (OER) at the anode. For the reaction to proceed, electrochemical water splitting theoretically requires a high potential of 1.23 V between the anode and cathode because of the four-electron transfer in the OER mechanism.4 The sluggish kinetics leads to lower efficiencies for water splitting. Accordingly, a highly efficient electrocatalyst is needed to decrease the overpotential as much as possible.Currently, noble metals and noble metal-based oxides (especially Ru, Ir, and their oxides)5,6 are considered the most effective electrocatalysts for water splitting, but they are unavailable for large-scale application owing to their high cost and low natural abundance.4 Over the past decades, tremendous progress has been made in transition metal-based electrocatalysts, such as metal oxide,7,8 metal phosphide,9 and metal chalcogenides.10 These catalysts can be promising alternative materials with good OER performances for electrochemical water splitting. Moreover, the interactions between the various compositions in a multi-metallic structure can produce additional synergistic effects, optimize the adsorption energies, or increase the electrical conductivity compared to monometallic derivatives.11 For example, Kuai et al. prepared a broader range of amorphous Fe–Ni–O(x) using an aerosol-spray-assisted approach.12 Fe–Ni mixed oxides exhibited better OER activity than pure Fe or Ni oxides because of the synergistic effect between different metal sites. In particular, the Fe6Ni10Ox composition showed an overpotential of as low as 0.286 V (vs. RHE) at 10 mA cm−2 and a Tafel slope of 48 mV dec−1. Yang et al. reported that increasing the Fe and Ni concentrations in Co-MOF produced different catalytic efficiencies.13 The bimetallic CoFe-MOF prepared hydrothermally showed better OER performance with an overpotential of 0.355 V (vs. RHE) at 10 mA cm−2 in 0.1 M KOH and a decent Tafel slope of 49 mV dec−1, which is better than the monometallic Co-MOF. On the other hand, we demonstrated that CoFe nanocubes with tunable porosity could significantly increase the OER and ORR activity in a previous investigation.14 The CoFe composition in microporous catalysts (average pore diameter, d ≤ 2 nm) had a larger effect on the OER than the ORR, whereas microporous/mesoporous structures (d ≈ 35.6 nm) favored the ORR catalytic activity.14 Smith et al. extended the combination of trimetallic Ni–Co–Fe system, in which the Fe concentration was maintained at 20–40% and the onset potential and Tafel slope were the lowest, regardless of whether the other metal component was Ni or Co.15
Recently, transition metal-based layered double hydroxides (LDHs) have attracted considerable attention in water oxidation because of many advantages, such as rich active site comparison with the 0D and 1D materials, large surface area, excellent structural and compositional tunability, strong electrostatic interactions between layers and interlayers anions, hierarchical porosity accelerating the diffusion of water molecules and release of gaseous products, and their promising electrocatalytic performance.16 Among various LDHs systems, such as NiFe,17 NiMn,8 NiCo,18 and CoFe,19 NiFe LDH or NiFe (oxy)hydroxides are the most active OER catalyst compared to other bimetallic LDHs under alkaline conditions.17 On the other hand, the effect of promoting cooperation between different metal ions in this OER catalyst may not be limited to Ni and Fe metals. A recent study reported that the addition of a third metal (e.g., Co, Cr, Mn, or Mo) could further improve the catalytic activity.20–22 Qu et al. synthesized CoNiFe-LDH nanocage with cation exchange reaction using a metal–organic framework as a template, which exhibited a low overpotential of 299 mV (10 mA cm−2, 1 M KOH) with a Tafel slope of 50 mV dec−1.21 Jin et al. reported that the trimetallic properties of Ni, Fe, and Cr provide opportunities to generate strong electron interactions within the metal hydroxide matrix and enhance the synergy between metals toward the OER.22 The introduction of multivalent Cr ion into NiFe can readily adopt various oxidation states during the OER reactions, indicating a positive effect on active species for water oxidation.22 Although the identity of the catalytic active sites remains unclear, a third metal introduction and its positive influence on the local electronic structure produces a favorable OER process.
Incorporating a heteroatom into a metallic compound was a practical approach to modify the electronic structure to boost the intrinsic activity of electrocatalysts.23–28 For example, sulfur-doped cobalt,25 nitrogen-doped cobalt oxide,26 sulfur-doped NiCoFe LDH nanosheet,27 and phosphorus-doped Ni3S2/CoFe2O4 arrays on nickel foam28 exhibited significantly enhanced electrocatalytic performance toward OER. Lu et al. revealed a facile and practical pathway for hierarchical fabrication of 3D porous S-incorporated NiCoFe LDH nanosheets on carbon cloth.27 S doping increases the electrical conductivity of LDH, and the high catalytic efficiency for the OER was attributed to the enhancement of substantial active sites on the surface of S–NiCoFe LDH.27 Joo et al. suggested that the addition of gaseous boronization into NiFe-LDH enhances the OER electrocatalytic activity.29 This strategy increases the oxyhydroxides ratio of LDHs, which has a positive effect on the OER performance.29 On the other hand, because of the poor electrical conductivity of the LDHs, carbon materials, including graphene oxide (GO) and carbon nanotubes, doped with heteroatoms, such as B, N, S, and P have been demonstrated to be electrochemically active. Sun et al. prepared vertically aligned ternary NiCoFe-LDHs on defect-rich and N-doped crumpled GO by in situ routes.30 Owing to the synergistic effect between NiCoFe-LDH and N–GO, the prepared composite contained a larger number of active sites and fast electron transfer as an efficient bifunctional catalyst for OER/ORR.30
This paper reports a trimetallic nitrogen-incorporated CoNiFe LDHs (N–CoNiFe) fabricated using a rapid, inexpensive, and facile co-precipitation method in the presence of ethanolamine (EA). The enhanced OER performance was attributed to the following reasons: (1) synergistic effect between the different metal sites; (2) fully and uniformly exposed active sites in the two-dimensional sheets; (3) LDHs structure allows the intercalation of water molecules and anions, provide better bulk redox activity; (4) successful incorporation of the nitrogen element into the CoNiFe boosts the OER activity of N–CoNiFe considerably by enhancing its electrical conductivity and tuning the energy barrier for the adsorption of OER intermediates on the catalyst surface.
Experimental
Chemicals
Cobalt(II) acetate tetrahydrate (Co(Ac)2·4H2O), iron(III) chloride (FeCl3), and ethanolamine (EA) were purchased from Sigma-Aldrich. Nickel(II) chloride hexahydrate (NiCl2·6H2O) was obtained from Duksan Co. (Korea). E-TEK supplied the commercial Ir/C catalysts (20 wt% loading) and Vulcan XC-71 as the supporting material. The aqueous solutions were prepared using deionized water. All other chemicals were used as received.Synthesis of trimetallic nitrogen-incorporated CoNiFe LDHs
Briefly, Co(Ac)2·4H2O, NiCl2·6H2O, and FeCl3 ([Co2+]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Ni2+]:[Fe3+] = 1:2:1 molar ratio) were dissolved in 15 mL of H2O in a 100 mL three-neck flask. Subsequently, 2 mL of EA was added dropwise with magnetic stirring and heated to 100 °C for 2 h. The catalyst suspension was heated on a heating mantle equipped with a reflux condenser to maintain the temperature and solvent volume during the reaction. After cooling to room temperature, the precipitated gel was collected by centrifugation, washed once with deionized water, and freeze-dried to obtain trimetallic N–CoNiFe LDHs. The resulting powder product was calcined at 200 °C with a heating rate of 5 °C min−1 and maintained for 1 h under a N2 atmosphere. Combinations of monometallic and bimetallic catalysts were prepared using the same method and denoted as N–Co, N–Ni, N–Fe, N–NiFe, N–CoNi, and N–CoFe catalysts, respectively. N-free CoNiFe synthesized without EA was also prepared to compare the effect of nitrogen incorporation on the electrocatalytic activity.
:[Ni2+]:[Fe3+] = 1:2:1 molar ratio) were dissolved in 15 mL of H2O in a 100 mL three-neck flask. Subsequently, 2 mL of EA was added dropwise with magnetic stirring and heated to 100 °C for 2 h. The catalyst suspension was heated on a heating mantle equipped with a reflux condenser to maintain the temperature and solvent volume during the reaction. After cooling to room temperature, the precipitated gel was collected by centrifugation, washed once with deionized water, and freeze-dried to obtain trimetallic N–CoNiFe LDHs. The resulting powder product was calcined at 200 °C with a heating rate of 5 °C min−1 and maintained for 1 h under a N2 atmosphere. Combinations of monometallic and bimetallic catalysts were prepared using the same method and denoted as N–Co, N–Ni, N–Fe, N–NiFe, N–CoNi, and N–CoFe catalysts, respectively. N-free CoNiFe synthesized without EA was also prepared to compare the effect of nitrogen incorporation on the electrocatalytic activity.
Characterization
The morphologies of all samples were observed by high-resolution transmission electron microscopy (HR-TEM, Philip, CM-200) and scanning electron microscopy (SEM, S-4800) equipped with EDS (energy-dispersive X-ray spectroscopy). Powder X-ray diffraction (XRD, Rigaku D/max-2500) was performed using Cu Kα radiation (λ = 1.54 Å). The surface properties of the catalysts were examined by X-ray photoelectron spectroscopy (XPS, AXIS Nova) using Al Kα monochromatized radiation. The Brunauer–Emmett–Teller (BET) surface area and pore size distributions of catalysts were analyzed using a BELSORP-mini II (Mictrotrac-BEL, Japan).Electrochemical measurements
All electrochemical experiments were carried out in a three-electrode system using mercury–mercury oxide (Hg/HgO), Pt wire, and glassy carbon (GC) as the reference, counter, and working electrodes, respectively, on a rotating disk electrode (RDE, BAS Bioanalytical System). Typically, each prepared catalyst was mixed with the supporting carbon material (1:1 weight ratio) and dispersed ultrasonically in water for 30 min (2.0 mg mL−1). Subsequently, 15 μL of catalyst ink was dropped onto a GC electrode (3 mm diameter, 0.42 mg cm−2) and dried at room temperature under vacuum. The electrode was covered with 5 μL of 0.5 wt% Nafion. For comparison, Ir/C was prepared using the same method. All electrochemical measurements were performed at a scan rate of 10 mV s−1 in aqueous O2-saturated 1.0 M KOH solutions. All potentials presented in this study are referenced to the reversible hydrogen electrode (RHE) according to the Nernst equation. To determine the electrochemical surface area (ECSA), cyclic voltammetry (CV) was conducted over the potential range of 0.92 to 1.02 V (vs. RHE), which is located at the non-faradaic region to explore the electrochemical double-layer capacitance (Cdl). Electrochemical impedance spectroscopy (EIS) was performed at η = 337.4 mV (vs. RHE) in a frequency range over 0.1 Hz to 100 kHz by applying an AC voltage of 5 mV amplitude. For the OER measurements, linear sweep voltammetry (LSV) was conducted by sweeping the potential from 1.22 V to 1.82 V (vs. RHE) at a scan rate of 10 mV s−1 and a fixed rotation rate of 1600 rpm.
Results and discussion
Morphological and structural characterization
Fig. 1 presents the facile and one-step synthesis of trimetallic N–CoNiFe LDHs electrocatalysts by precipitation. The as-prepared complexes were formed between the transition metals (Co, Ni, and Fe) and EA at high temperatures via dative covalent bonds, in which both electrons originate from the amino (–NH2) and hydroxyl (–OH) groups. EA plays a crucial role as a ligand for fabricating metal hydroxide M(OH)x, the nucleophilicity of the donor atoms, stabilizing agent, and a nitrogen/oxygen source that can tune the electronic, solubility, and steric properties of the catalytic systems.31–33 Finally, calcination at 200 °C for 1 h under a N2 atmosphere helps to stabilize the structure of the metal-based LDHs.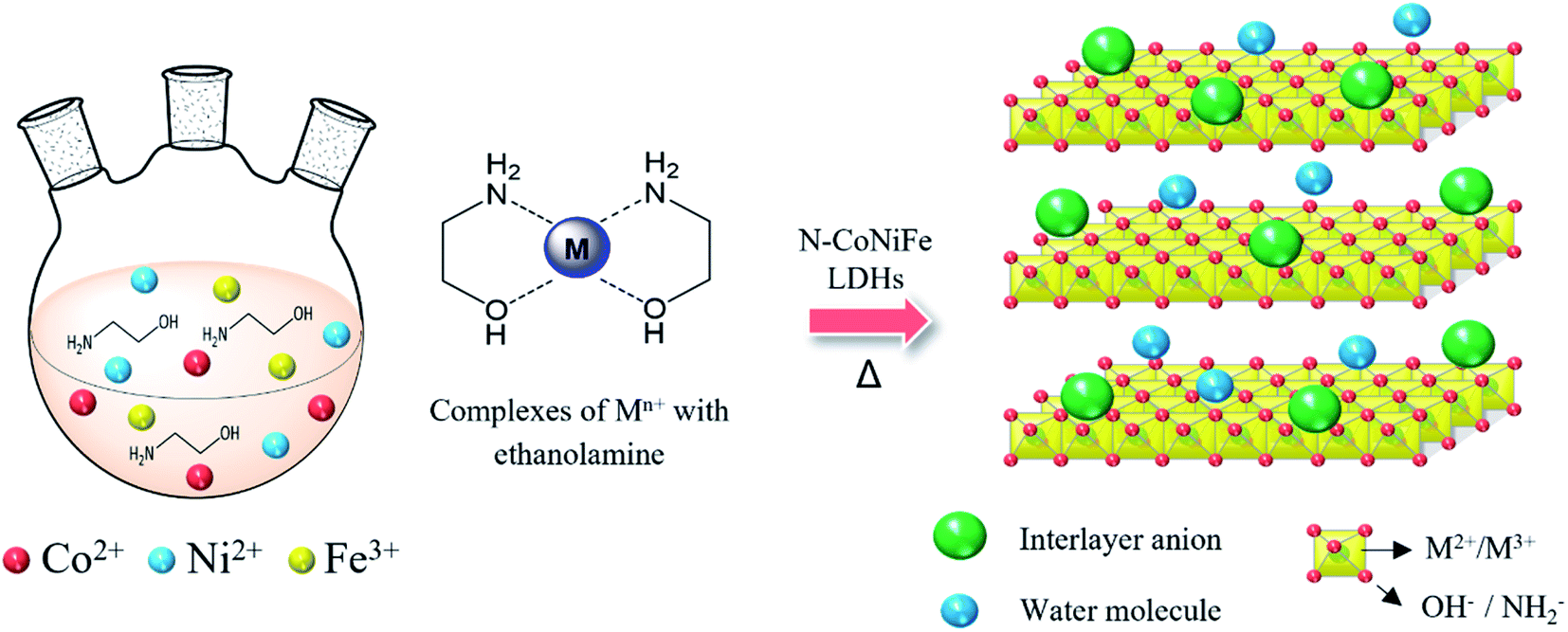 | ||
| Fig. 1 Schematic diagram of the preparation of N-incorporated CoNiFe LDHs. | ||
The morphology of the prepared catalysts was examined by SEM. Flower-shaped particles were observed in N–Co (Fig. S1a†) with a particle diameter of 1–3 μm. Nanoflake-like structures formed in the main framework were identified in Ni-containing N–Ni (Fig. S1b†) and N–CoNi (Fig. 2b). The N-free trimetallic CoNiFe (Fig. 2d) catalyst synthesized in an aqueous solution exhibited a uniform particle morphology in which particle linkages can be observed. On the other hand, the catalysts synthesized in the presence of FeCl3 and EA showed a highly assembled plate-like structure (Fig. S1c†). In contrast to N–Fe, N–NiFe, and N–CoFe, relatively thin nanosheets were formed throughout the trimetallic N–CoNiFe (Fig. 2e and f). The structure of LDHs with a thin plate-like structure increases the specific surface area of the material and the active catalyst sites improves electrolyte diffusion, and accelerates charge transfer to remove the gas produced rapidly during the catalytic reaction, ultimately leading to high OER catalytic activity.
 | ||
| Fig. 2 SEM images of (a) N–NiFe, (b) N–CoNi, (c) N–CoFe, (d) CoNiFe, and (e and f) N–CoNiFe LDHs. | ||
In Fig. 3, the XRD patterns of N–NiFe, N–CoNi, N–CoFe, CoNiFe, and N–CoNiFe were analyzed to identify the phase constitution of as-prepared catalysts. The XRD peaks at 11.1°, 22.4°, 33.5°, and 59.8° 2θ were assigned to the (003), (006), (012), and (110) planes of NiFe LDH (Fig. 3b), respectively. The relative peak position and intensity are well-known characteristics for correctly identifying N–CoNiFe LDHs with a hydrotalcite-like structure (Fig. 3a).34 Fig. 3d showed sharp XRD peaks of N–CoNi, revealing the formation of crystalline Co(OH)2 and Ni(OH)2 structures. On the other hand, the XRD patterns of N-free CoNiFe (Fig. 3c) and N–CoFe (Fig. 3e) showed relatively poor signal-to-noise ratios and very broad peaks due to the low crystallinity. These results indicate that EA plays an important role as a ligand in forming metallic LDH structures. Moreover, the XRD peaks of N–CoFe and CoNiFe showed the same 2θ values as that of N–Fe (Fig. S2a†), suggesting poor precipitation of Co2+ and Ni2+ without EA compared to Fe3+ species.
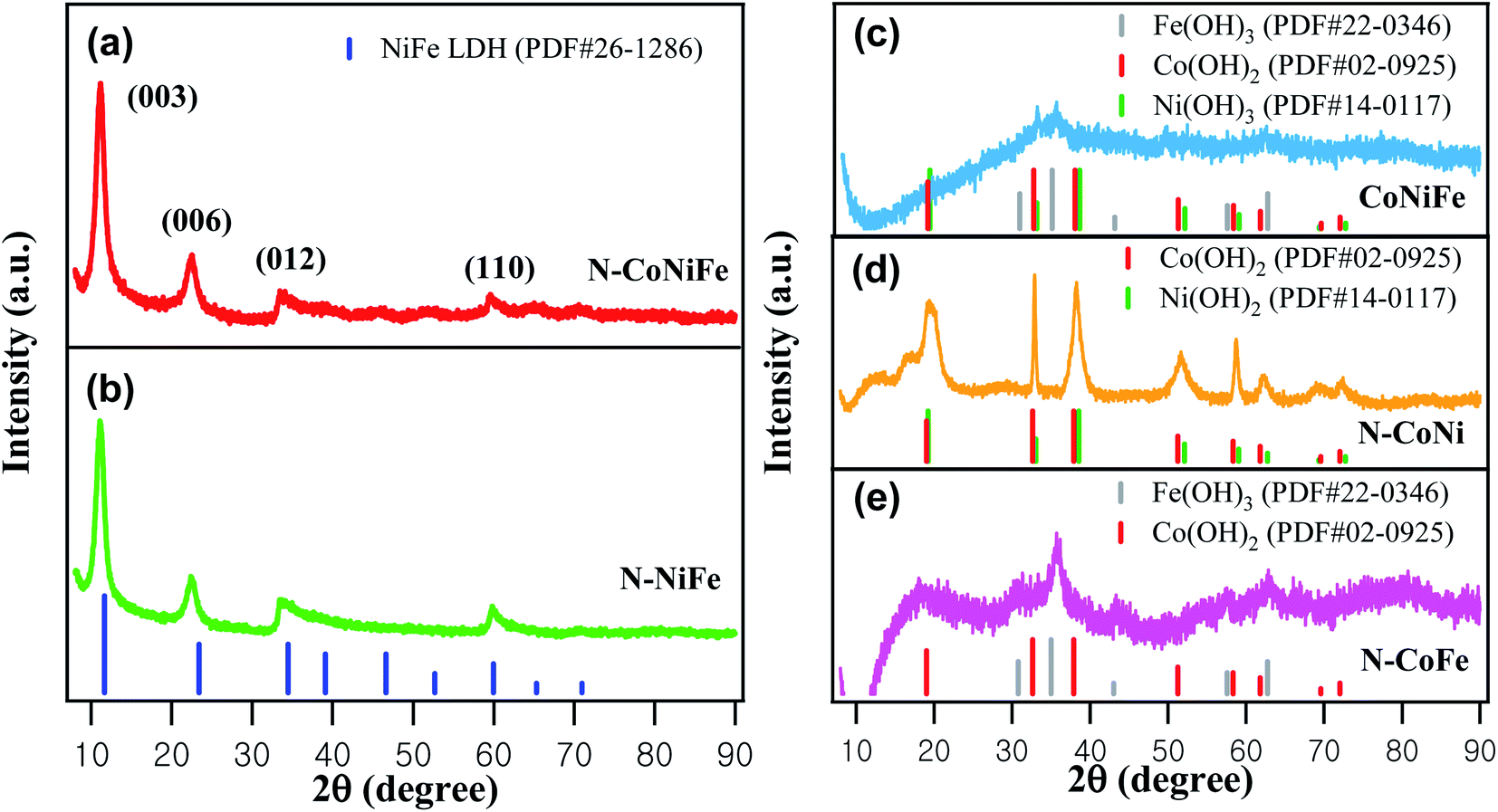 | ||
| Fig. 3 XRD patterns of (a) N–CoNiFe LDH, (b) N–NiFe, (c) CoNiFe, (d) N–CoNi, and (e) N–CoFe. | ||
TEM (Fig. 4) revealed the more detailed morphology and structure of the as-synthesized N–CoNiFe LDHs. The porous crystalline nanosheet displays lattice fringes with the interplanar spacing of 0.24 and 0.23 nm (inset in Fig. 4a), which are related to the interplanar spacing of the (012) crystal plane of N–CoNiFe LDHs phase (Fig. 3). The energy-dispersive spectroscopy (EDS) mapping of N–CoNiFe LDHs (Fig. 4b) revealed the presence and uniform distribution of Co, Ni, Fe, and N, suggesting that nitrogen had been incorporated successfully into the CoNiFe LDHs nanostructure. SEM-EDS (Fig. S3a†) confirmed that N–CoNiFe consists of the above elements with a Co, Ni, Fe, N, and O atomic% of 9.81%, 15.99%, 7.50%, 4.63%, and 62.06%, respectively.
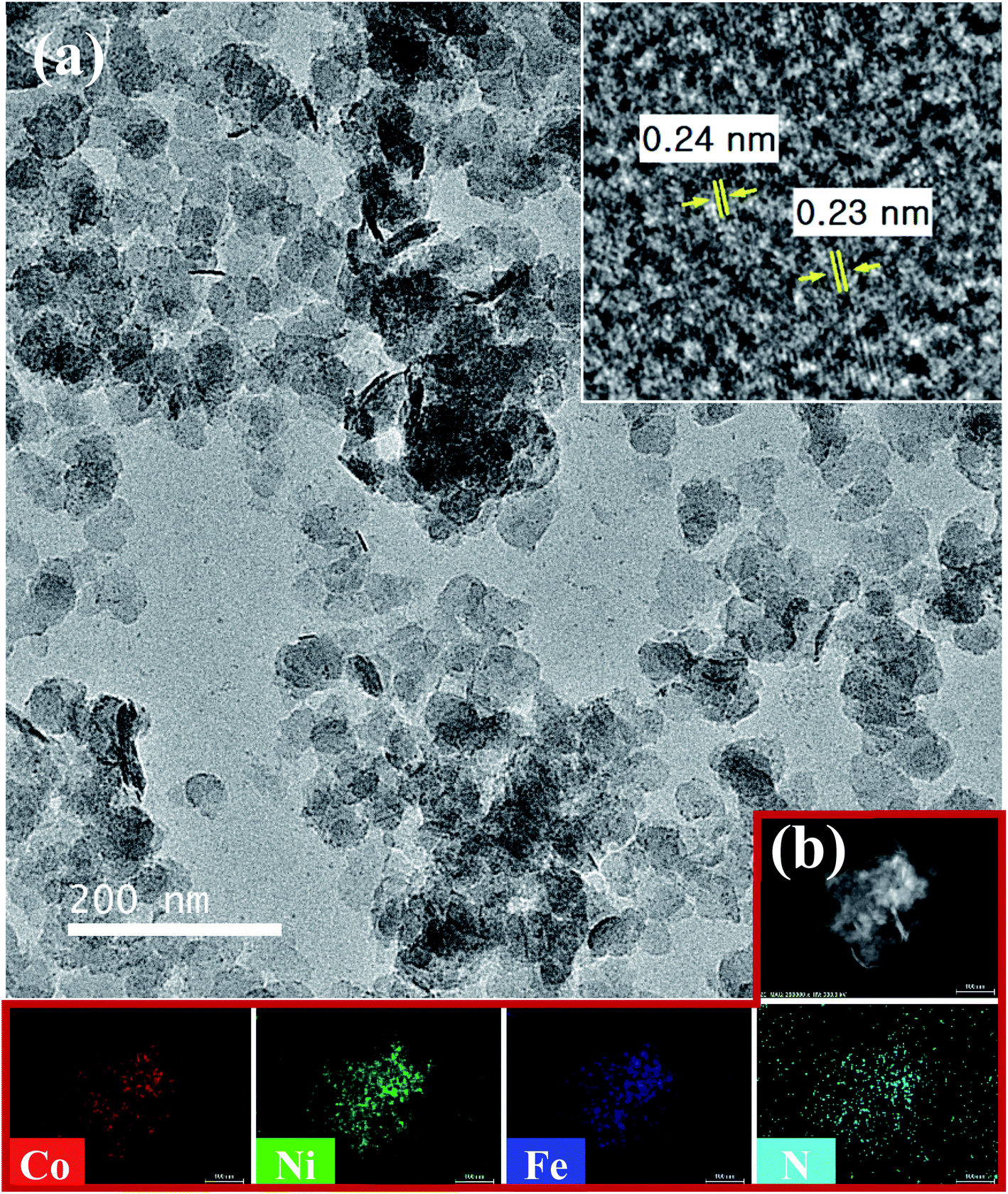 | ||
| Fig. 4 (a) TEM and (b) TEM-EDS elemental mapping (scale bar: 100 nm) images of N–CoNiFe. Inset: HRTEM image of N–CoNiFe LDHs. | ||
XPS revealed the surface elemental composition, chemical nature, and valence state of the as-prepared trimetallic N–CoNiFe. XPS of monometallic catalysts (N–Co, N–Ni, and N–Fe) were also performed to determine the relationship between structural features. Fig. S4† shows typical XPS survey scans of N–CoNiFe LDHs, which indicate the presence of C 1s, N 1s, O 1s, Co 2p, Ni 2p, and Fe 2p, showing that nitrogen had been incorporated successfully into the trimetallic CoNiFe system. The high-resolution of C 1s spectrum of N–CoNiFe shown in Fig. 5a could be divided into four different peaks with binding energies of 284.2 eV, 285.0 eV, 286.4 eV, and 288.1 eV corresponding to sp2 hybridized C–C, sp2 bonded with nitrogen C–N, hydroxy C–O,35 and carboxyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O,36 respectively. Fig. 5b presents the high-resolution XPS spectrum of N 1s, showing the formation of four peaks centered at 397.8 eV for cyanide CN,37 398.5 eV for pyridinic N, 399.0 eV for metal–nitrogen (M–Nx) bonding, and 400.0 eV for pyrrolic N.38,39 The presence of M–Nx species with a peak area of 34.6% indicates the bonding between the metal atoms and amino group via a coordination interaction. The O 1s peak of N–CoNiFe, located at approximately 530.9 eV was fitted to metal–oxygen (M–Ox) bonding, C–O–C, and CO/M–OH40 (Fig. 5c).
O,36 respectively. Fig. 5b presents the high-resolution XPS spectrum of N 1s, showing the formation of four peaks centered at 397.8 eV for cyanide CN,37 398.5 eV for pyridinic N, 399.0 eV for metal–nitrogen (M–Nx) bonding, and 400.0 eV for pyrrolic N.38,39 The presence of M–Nx species with a peak area of 34.6% indicates the bonding between the metal atoms and amino group via a coordination interaction. The O 1s peak of N–CoNiFe, located at approximately 530.9 eV was fitted to metal–oxygen (M–Ox) bonding, C–O–C, and CO/M–OH40 (Fig. 5c).
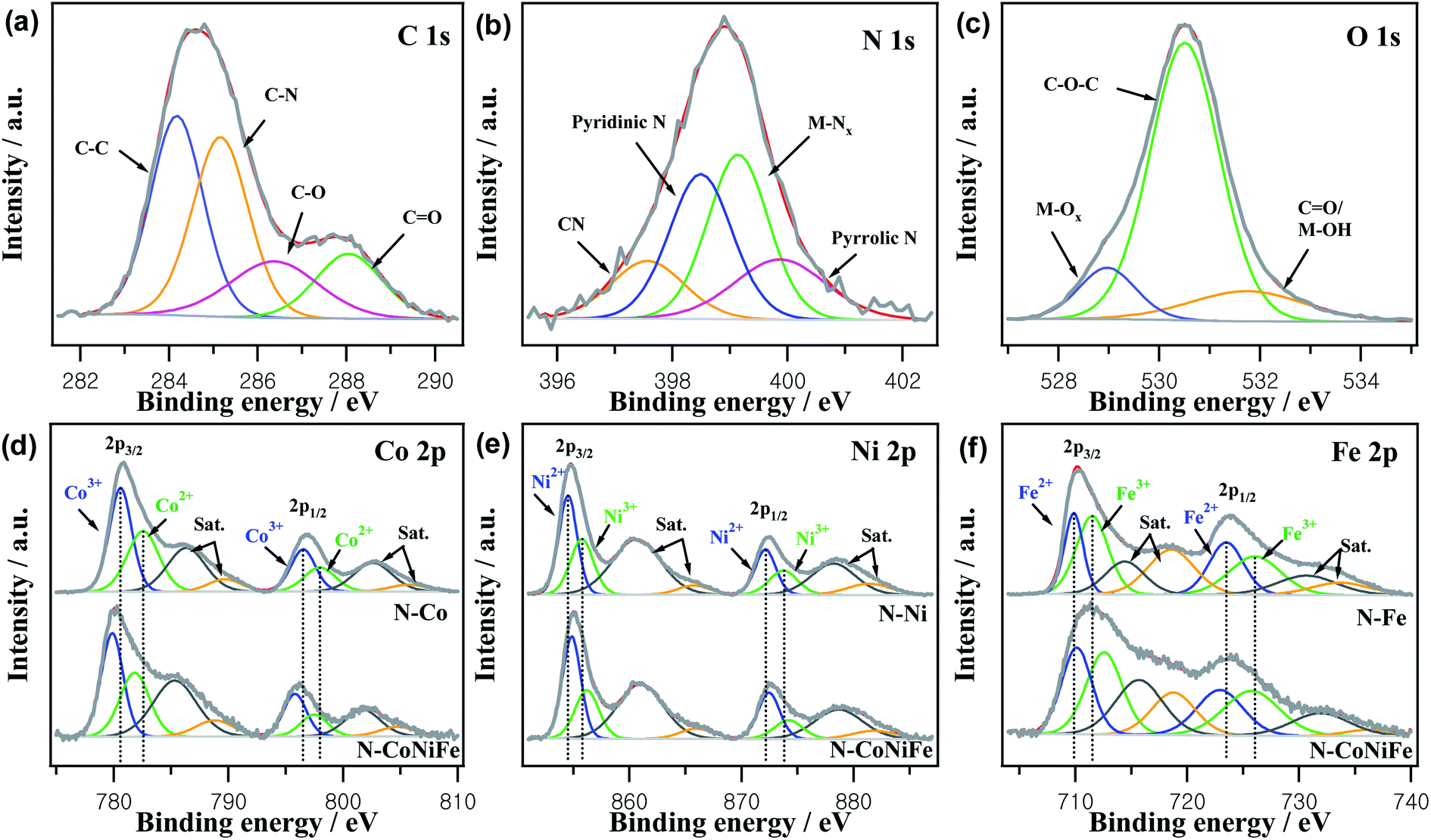 | ||
| Fig. 5 XPS analysis of (a) C 1s, (b) N 1s, and (c) O 1s of N–CoNiFe LDHs. Deconvoluted XPS spectra of (d) Co 2p, (e) Ni 2p, and (f) Fe 2p of N–Co, N–Ni, N–Fe, and N–CoNiFe LDHs catalysts, respectively. | ||
The XPS spectra of Co 2p (Fig. 5d) could be deconvoluted into two spin–orbit doublets of Co3+ and Co2+. The major peaks of N–CoNiFe at 780.1 eV for Co 2p3/2 and 796.0 eV for Co 2p1/2 could be split into two distinct peaks of Co3+ (assigned to the 779.9 eV peak in Co 2p3/2 and 795.8 eV peak in Co 2p1/2) and Co2+ (corresponding to the 781.8 eV peak in Co 2p3/2 and 797.5 eV peak in Co 2p1/2). The shake-up satellite peaks were observed at 785.1 eV and 801.9 eV, which is typical for cobalt oxide.21,26 Regarding N–CoNiFe, the deconvolution of Co 2p3/2 and Co 2p1/2 exhibited no significant shift compared to N–Co, showing that the other metals (Ni and Fe) did not influence the oxidation state of the Co atom. Fig. 5e represents the high-resolution spectra of the Ni 2p, comprising two spin–orbit doublets characteristic of two satellite peaks and Ni2+ and Ni3+. Two main peaks of N–CoNiFe observed at 855.0 eV (Ni2+ at 854.9 eV and Ni3+ at 856.2 eV) and 872.6 eV (Ni2+ at 872.5 eV and Ni3+ at 874.2 eV) were attached to Ni 2p3/2 and Ni 2p1/2 orbits, and two satellite peaks at 861.0 eV and 879.0 eV.21,41 For the Fe 2p XPS spectra in Fig. 5f, two peaks at 711.7 eV and 723.7 eV were assigned to Fe 2p3/2 and Fe 2p1/2, respectively. The Fe2+ peaks at 710.1 eV and Fe3+ at 712.6 eV were observed in the Fe 2p3/2 region, along with Fe2+ and Fe3+ peaks at 722.9 eV and 725.7 eV, respectively, in the Fe 2p1/2 region.42 Compared to the deconvoluted Ni 2p and Fe 2p of monometallic N–Ni and N–Fe, the peaks of N–CoNiFe were shifted ∼0.4 eV to a higher binding energy for Ni 2p and ∼1.2 eV for Fe 2p, showing a change in the chemical state of Ni and Fe due to the synergistic effect between the different metal sites after the co-precipitation process. XPS deconvolution indicated the successful fabrication of N–CoNiFe LDH in this work. Compared to Co 2p, the Ni 2p and Fe 2p binding energy of N–CoNiFe shifts positively to 0.4 eV and 1.1 eV, respectively, indicating an intimate interaction between Ni and Fe and modification of the electronic structure by Ni, Fe, or Fe/Ni incorporation.43,44 The high-valence species enrichment can enhance the electro-oxidation reactions,43 which helps improve the OER performance.
Electrocatalytic performance
The electrochemical performance of the as-prepared electrocatalysts toward the OER was studied in a three-electrode system using a 1 M KOH electrolyte. A series of CoNiFe catalysts prepared with different EA concentrations were investigated to examine the effects of EA on its OER activity in the co-precipitation of the trimetallic system. As shown in Fig. S5a,† the LSV curves showed inferior OER activity of CoNiFe without using EA as a ligand and a stabilizing agent. N–CoNiFe was co-precipitated with a large amount of N/O doping compared to the CoNiFe counterpart because of the strong complexing with EA. The ligand acts as both an activator and deactivator of the catalytic system, depending on its concentration.45,46 Thus, the ligand concentration was kept at a minimum to control the formation of metal complexes. The best OER catalyst was determined using 2 mL of EA at the lowest overpotential and highest current density compared to the other concentrations. The metal concentration was optimized systematically to achieve the best synergistic effect between the metal sites (Fig. S5b†). An appropriate ratio of [Co2+]:[Ni2+]:[Fe3+] = 1:2:1 was established, which corresponds to 15 mM, 30 mM, and 15 mM of the Co2+, Ni2+, and Fe3+ solutions, respectively. An irreversible anodic peak was observed at 1.4 V, which was assigned to the oxidation of Ni from the low valence states (Ni0 or Ni2+) to high valence states (Ni3+ or Ni4+).47,48 The Ni oxidation peak was not found in the CoNiFe catalyst because of poor precipitation in the absence of EA during the synthesis process. In particular, while the oxidation peak at 1.30–1.45 V (vs. RHE) was shifted negatively as the Co2+ concentration increased, the opposite phenomenon was observed with a change in the Fe3+ concentration. Interestingly, the overpotential of N–CoNiFe was influenced relatively weakly by the amount of Co2+, as shown in Fig. S5b,† indicating that the ligand acetate (–OOCCH3) in the cobalt precursor shows stronger binding to the center metal than EA because of the delocalized electrons between the two oxygen atoms, which is stable towards EA. XPS also demonstrated that Co is in a stable oxidation state after co-precipitation.
Fig. 6 shows the OER LSV curves for as-prepared catalysts and commercial Ir/C obtained in 1.0 M KOH at a scan rate of 10 mV s−1. The overpotential required to drive 10 mA cm−2 corresponds to the 10% efficiency solar water-splitting units that are generally presented as a vital parameter to evaluate the OER performance of a catalyst. The trimetallic N–CoNiFe LDHs displayed an overpotential (η) of 318 mV at 10 mA cm−2, which is lower than those of bimetallic N–NiFe (346 mV), N–CoNi (421 mV), N–CoFe (391 mV), CoNiFe (492 mV), and commercial Ir/C (358 mV) (Fig. 6a and c). The current density of N–CoNiFe at η = 400 mV was 61.3 mA cm−2, which was 2.5-, 9.3-, 5.1-, 41- and 3.3-fold higher than those of N–NiFe, N–CoNi, N–CoFe, CoNiFe and Ir/C, respectively (Fig. 6d). The Tafel plots extracted from the polarization curves were also analyzed to obtain more information on the OER kinetics of the catalyst. A relatively low Tafel slope is preferable because it reveals more advantageous kinetics and higher catalytic activity. As shown in Fig. 6b, the Tafel slope of N–CoNiFe was 72.2 mV dec−1, which was lower than their catalyst counterparts: N–NiFe (84.9 mV dec−1); N–CoNi (115.6 mV dec−1); N–CoFe (83.5 mV dec−1); CoNiFe (105.2 mV dec−1); and Ir/C (74.1 mV dec−1).
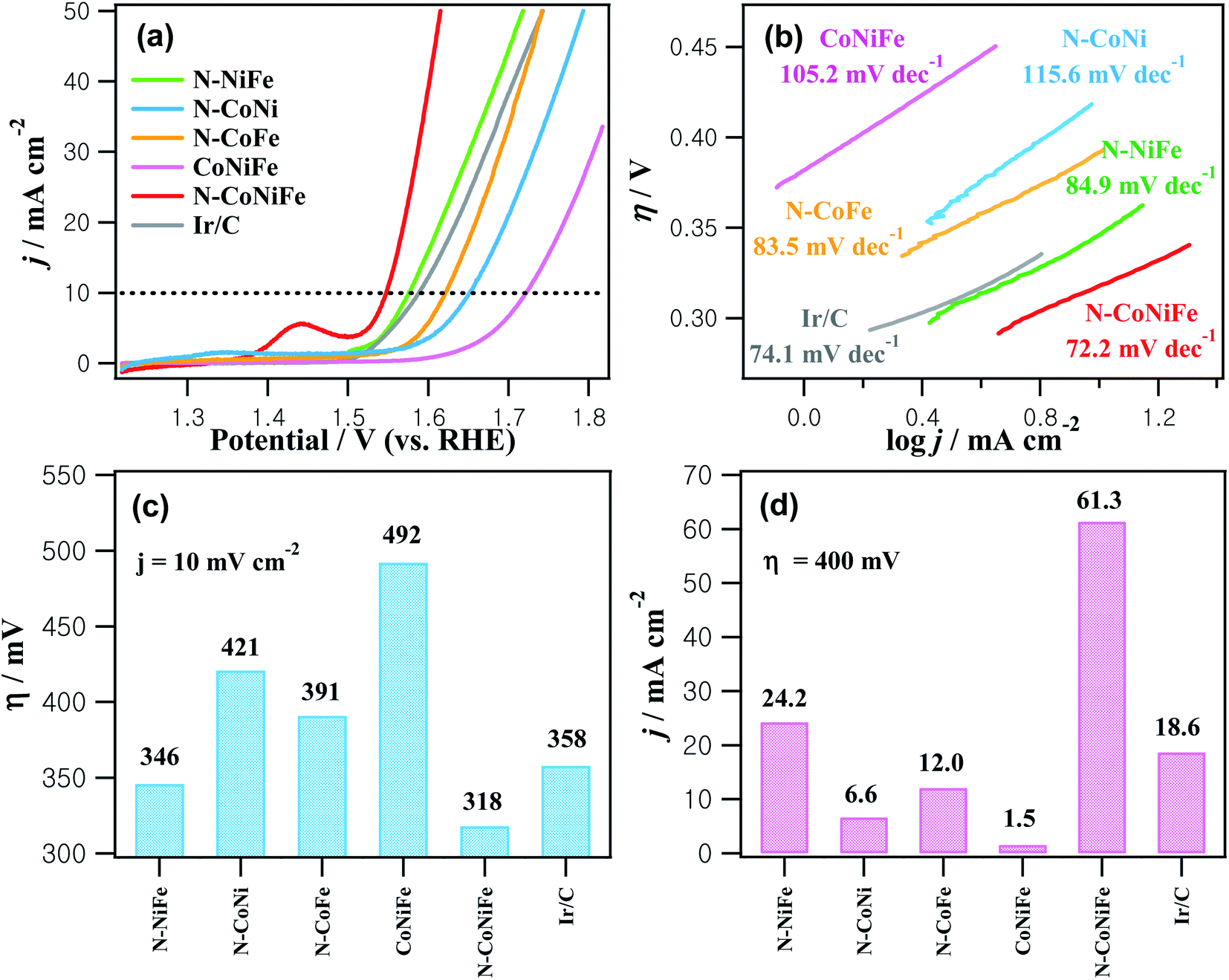 | ||
| Fig. 6 (a) LSV curves recorded in an O2-saturated 1.0 M KOH at a scan rate of 10 mV s−1 (rotational speed of 1600 rpm). (b) Tafel plots. (c) Comparative overpotential at 10 mV cm−2. (d) Current densities at η = 400 mV. The electrodes used in the electrocatalytic OER: N–NiFe; N–CoNi; N–CoFe; CoNiFe; N–CoNiFe; and Ir/C. | ||
The high electrocatalytic performance of N–CoNiFe for the OER was investigated by measuring the double-layer capacitance (Cdl) to estimate the electrochemically active surface area (ECSA). The Cdl value was determined by cyclic voltammetry at various scan rates (Fig. S6†) and calculated by plotting Δj = janodic − jcathodic at 1.0 V (vs. RHE) as a function of the scan rate. The ECSA (cm2) value was determined using the following equation:49
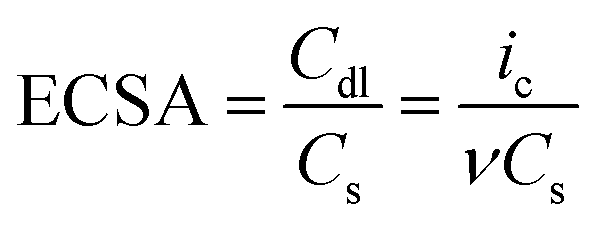
Fig. 7a and Table 1 show that the trimetallic N–CoNiFe LDHs catalyst had the highest Cdl value (3.44 mF cm−2) and ECSA (127.4 cm2) compared to bimetallic N–NiFe, N–CoNi, N–CoFe, and CoNiFe. These results show that the LDH-formed N–CoNiFe catalyst can absorb and desorb water molecules and gas products, suggesting that the N–CoNiFe LDHs sample can provide more active sites than the other samples, resulting in superior OER activity. The BET specific surface area and pore distribution were determined to characterize the surface area of CoNiFe and N–CoNiFe. In the case of N–CoNiFe, the BET specific surface area in Fig. S7† was 161.4 m2 g−1, which was significantly larger than that of the N-free CoNiFe catalyst. The pore size distribution of N–CoNiFe (0.665 cm3 g−1) was calculated by Barrett–Joyner–Halenda method revealing a relatively wider distribution compared with CoNiFe. The LDH structure of N–CoNiFe exhibiting mesoporosity (average pore diameter, d ≈ 12.8 nm) is beneficial in terms of O2 generation because of the improved active site densities. EIS was carried out at η = 337.4 mV to determine the different charge transfer rates during the OER of the catalysts. As shown in Fig. 7b, a relatively higher charge transfer resistance (Rct) was observed in N–CoNi and N-free CoNiFe, increasing the potential barrier and reaction kinetics of the catalyst. The low crystallinity of N-free CoNiFe synthesized in the absence of EA leads to higher Rct curves with increased resistance to charge transfer (lower rate constant). On the other hand, the overall resistance of N–CoNiFe LDHs had the lowest Rct value among the catalysts, suggesting that the N–CoNiFe LDHs have favorable charge transfer kinetics and mass transfer in the OER process. Indeed, the observed Rct values were similar to the trend of an OER performance resulting in the reduced Rct value and more active OER.
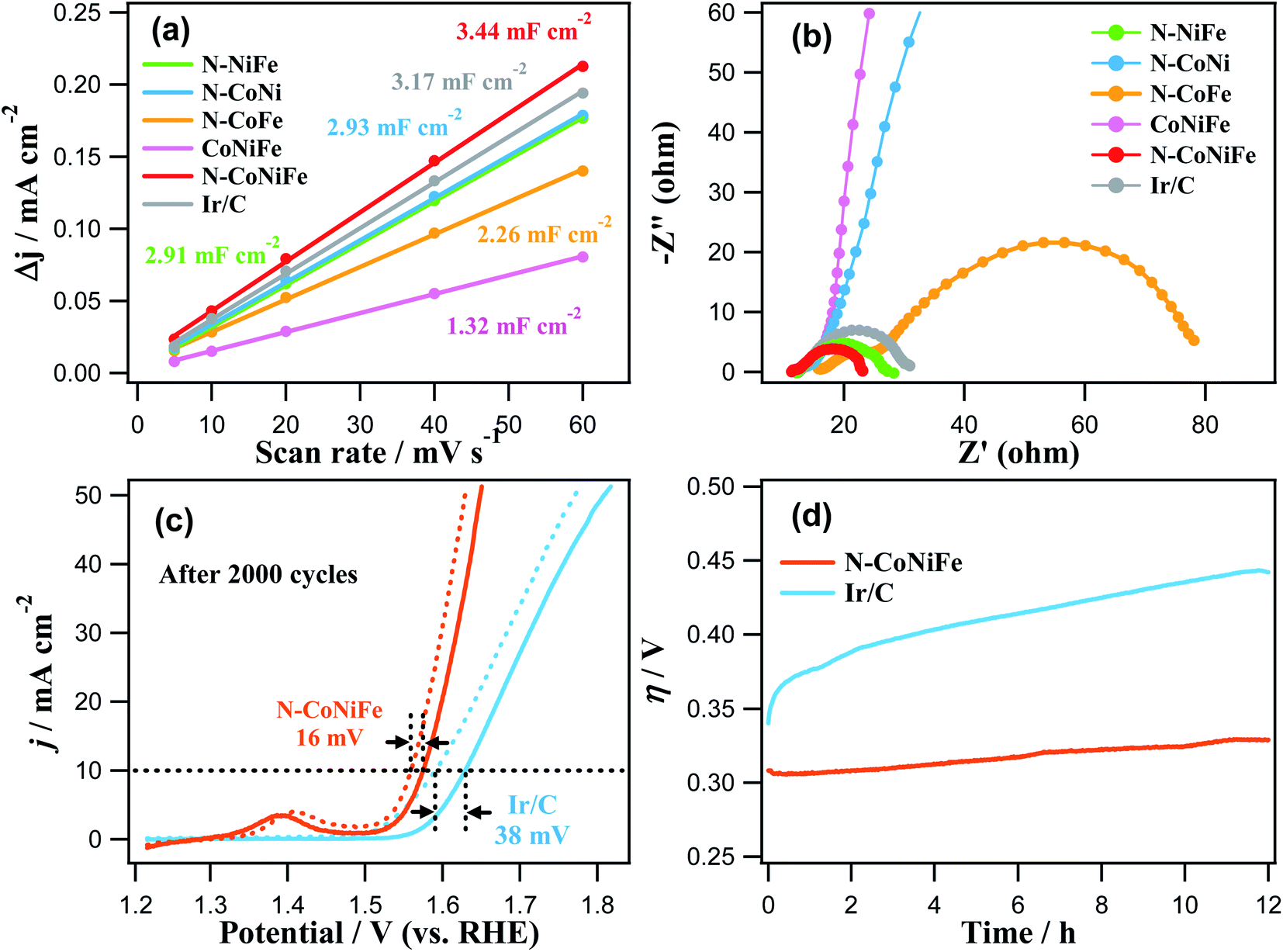 | ||
| Fig. 7 (a) Plots of the capacitive current density (Δj in 1.0 V) as a function of the scan rate shown in Fig. S6.† (b) EIS plots of N–NiFe, N–CoNi, N–CoFe, CoNiFe, N–CoNiFe, and Ir/C. (c) Stability test of the N–CoNiFe and Ir/C for the OER before and after 2000 cycles on the GC electrode in a 1.0 M KOH solution at a scan rate of 10 mV s−1. (d) Chronopotentiometric response of N–CoNiFe and Ir/C at the current density of 10 mA cm−2. | ||
| Samples | η (mV) (at 10 mA cm−2) | j (mA cm−2) (at η = 400 mV) | Tafel slope (mV dec−1) | Cdl (mF cm−2) | ECSA (cm2) |
|---|---|---|---|---|---|
| N–NiFe | 346 | 24.2 | 84.9 | 2.91 | 107.8 |
| N–CoNi | 421 | 6.6 | 115.6 | 2.93 | 108.5 |
| N–CoFe | 391 | 12.0 | 83.5 | 2.26 | 83.7 |
| CoNiFe | 492 | 1.5 | 105.2 | 1.32 | 48.9 |
| N–CoNiFe | 318 | 61.3 | 72.2 | 3.44 | 127.4 |
| Ir/C | 358 | 18.6 | 74.1 | 3.17 | 117.4 |
In addition to the catalytic performance, N–CoNiFe also showed high stability and durability in the OER process. As shown in Fig. 7c, the overpotential of N–CoNiFe at 10 mA cm−2 was positively shifted 16 mV after 2000 CV cycles, which is smaller than commercial Ir/C (38 mV). These results suggest that the N–CoNiFe catalyst has high catalytic activity and good stability in the OER after a long-term cycle. The SEM and SEM-EDS results showed that the surface morphology and composition of the N–CoNiFe catalyst remained almost unchanged before and after the stability cycling (Fig. S3a and S8†). Moreover, the N–CoNiFe LDHs remained stable over the 12 h OER process at the current density of 10 mA cm−2, whereas the η values of Ir/C increased rapidly in just a few hours (Fig. 7d). Table S1† shows a list of several reported CoNiFe catalysts with various structural features. The intrinsically high activity of N–CoNiFe was attributed to the following aspects. First, the synergistic effects of metal species play a vital role in enhancing the electronic conductivity and altering the adsorption energies for the reaction intermediate. Second, the improving specific surface area and active sites in the porous structure of nanosheets provided by nitrogen doping allow rapid electrolyte penetration and charge/ion transfer, as well the more accessible release of gases produced during water electrolysis.
Conclusions
Nitrogen-incorporated trimetallic CoNiFe LDHs catalysts were prepared using a rapid, facile, controllable, and economical method. The N–CoNiFe LDHs showed good electrocatalytic efficiency and high stability for the OER, with a low overpotential of 318 mV at 10 mA cm−2, and a low Tafel slope of 72.2 mV dec−1 in a 1.0 M KOH solution. In addition, the catalyst shows high durability in the OER process with little change in the overpotential (ca. 16.0 mV) at 10 mA cm−2 after 2000 cycles, which was smaller than commercial Ir/C (38.0 mV). Thus, the results show that the N–CoNiFe LDH is a promising nanomaterial in terms of high-performance and cost-effective catalyst for the OER. Moreover, the method for fabricating N-incorporated trimetallic LDHs catalyst is a practical approach to achieve large-scale production.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Dongil Culture and Scholarship Foundation (2021), and the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MSIT) (No. 2019R1F1A1049614 and No. 2021R1F1A1063825).References
- S. E. Hosseini and M. A. Wahid, Hydrogen production from renewable and sustainable energy resources: promising green energy carrier for clean development, Renewable Sustainable Energy Rev., 2016, 57, 850–866 CrossRef CAS.
- F. Dawood, M. Anda and G. M. Shafiullah, Hydrogen production for energy: an overview, Int. J. Hydrogen Energy, 2020, 45, 3847–3869 CrossRef CAS.
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Future cost and performance of water electrolysis: an expert elicitation study, Int. J. Hydrogen Energy, 2017, 42, 30470–30492 CrossRef CAS.
- X. Li, X. Hao, A. Abudula and G. Guan, Nanostructured catalysts for electrochemical water splitting: current state and prospects, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC.
- A. T. N. Nguyen and J. H. Shim, Facile one-step synthesis of Ir-Pd bimetallic alloy networks as efficient bifunctional catalysts for oxygen reduction and oxygen evolution reactions, J. Electroanal. Chem., 2018, 827, 120–127 CrossRef CAS.
- Y. Kweon, S. Noh and J. H. Shim, Low content Ru-incorporated Pd nanowires for bifunctional electrocatalysis, RSC Adv., 2021, 11, 28775–28784 RSC.
- J.-W. Zhang, H. Zhang, T.-Z. Ren, Z.-Y. Yuan and T. J. Bandosz, FeNi doped porous carbon as an efficient catalyst for oxygen evolution reaction, Front. Chem. Sci. Eng., 2021, 15, 279–287 CrossRef.
- L. Yang, L. Chen, D. Yang, X. Yu, H. Xue and L. Feng, NiMn layered double hydroxide nanosheets/NiCo2O4 nanowires with surface rich high valence state metal oxide as an efficient electrocatalyst for oxygen evolution reaction, J. Power Sources, 2018, 392, 23–32 CrossRef CAS.
- W. Li, D. Xiong, X. Gao and L. Liu, The oxygen evolution reaction enabled by transition metal phosphide and chalcogenide pre-catalysts with dynamic changes, Chem. Commun., 2019, 55, 8744–8763 RSC.
- Y. Du, S. Khan, X. Zhang, G. Yu, R. Liu, B. Zheng, R. Nadimicherla, D. Wu and R. Fu, In situ preparation of porous carbon nanosheets loaded with metal chalcogenides for a superior oxygen evolution reaction, Carbon, 2019, 149, 144–151 CrossRef CAS.
- J.-Y. Xue, C. Li, F.-L. Li, H.-W. Gu, P. Braunstein and J.-P. Lang, Recent advances in pristine tri-metallic metal–organic frameworks toward the oxygen evolution reaction, Nanoscale, 2020, 12, 4816–4825 RSC.
- L. Kuai, J. Geng, C. Chen, E. Kan, Y. Liu, Q. Wang and B. Geng, A reliable aerosol-spray-assisted approach to produce and optimize amorphous metal oxide catalysts for electrochemical water splitting, Angew. Chem., Int. Ed. Engl., 2014, 53, 7547–7551 CrossRef CAS PubMed.
- C. Yang, W.-J. Cai, B.-B. Yu, H. Qiu, M.-L. Li, L.-W. Zhu, Z. Yan, L. Hou and Y.-Y. Wang, Performance enhancement of oxygen evolution reaction through incorporating bimetallic electrocatalysts in two-dimensional metal–organic frameworks, Catal. Sci. Technol., 2020, 10, 3897–3903 RSC.
- S. Jo, S. Noh, K.-R. Wee and J. H. Shim, Structural Features of Porous CoFe Nanocubes and Their Performance for Oxygen-involving Energy Electrocatalysis, ChemElectroChem, 2020, 7, 3725–3732 CrossRef CAS.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, Water oxidation catalysis: electrocatalytic response to metal stoichiometry in amorphous metal oxide films containing iron, cobalt, and nickel, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed.
- Z. Cai, X. Bu, P. Wang, J. C. Ho, J. Yang and X. Wang, Recent advances in layered double hydroxide electrocatalysts for the oxygen evolution reaction, J. Mater. Chem. A, 2019, 7, 5069–5089 RSC.
- R. Chen, S.-F. Hung, D. Zhou, J. Gao, C. Yang, H. Tao, H. B. Yang, L. Zhang, L. Zhang, Q. Xiong, H. M. Chen and B. Liu, Layered structure causes bulk NiFe layered double hydroxide unstable in alkaline oxygen evolution reaction, Adv. Mater., 2019, 31, 1903909 CrossRef CAS PubMed.
- J. Jiang, A. Zhang, L. Li and L. Ai, Nickel–cobalt layered double hydroxide nanosheets as high-performance electrocatalyst for oxygen evolution reaction, J. Power Sources, 2015, 278, 445–451 CrossRef CAS.
- F. Yang, K. Sliozberg, I. Sinev, H. Antoni, A. Bähr, K. Ollegott, W. Xia, J. Masa, W. Grünert, B. R. Cuenya, W. Schuhmann and M. Muhler, Synergistic effect of cobalt and iron in layered double hydroxide catalysts for the oxygen evolution reaction, ChemSusChem, 2017, 10, 156–165 CrossRef CAS PubMed.
- K. Yue, J. Liu, Y. Zhu, C. Xia, P. Wang, J. Zhang, Y. Kong, X. Wang, Y. Yan and B. Y. Xia, In situ ion-exchange preparation and topological transformation of trimetal–organic frameworks for efficient electrocatalytic water oxidation, Energy Environ. Sci., 2021, 14, 6546–6553 RSC.
- F. Li, Z. Sun, H. Jiang, Z. Ma, Q. Wang and F. Qu, Ion-exchange synthesis of ternary FeCoNi-layered double hydroxide nanocage toward enhanced oxygen evolution reaction and supercapacitor, Energy Fuels, 2020, 34, 11628–11636 CrossRef CAS.
- Y. Yang, L. Dang, M. J. Shearer, H. Sheng, W. Li, J. Chen, P. Xiao, Y. Zhang, R. J. Hamers and S. Jin, Highly active trimetallic NiFeCr layered double hydroxide electrocatalysts for oxygen evolution reaction, Adv. Energy Mater., 2018, 8, 1703189 CrossRef.
- J.-T. Ren, Y.-S. Wang, L. Chen, L.-J. Gao, W.-W. Tian and Z.-Y. Yuan, Binary FeNi phosphides dispersed on N,P-doped carbon nanosheets for highly efficient overall water splitting and rechargeable Zn-air batteries, Chem. Eng. J., 2020, 389, 124408 CrossRef CAS.
- X.-W. Lv, W.-S. Xu, W.-W. Tian, H.-Y. Wang and Z.-Y. Yuan, Activity promotion of core and shell in multifunctional core–shell Co2P@NC electrocatalyst by secondary metal doping for water electrolysis and Zn-air batteries, Small, 2021, 17, 2101856 CrossRef CAS PubMed.
- M. Al-Mamun, Z. Zhu, H. Yin, X. Su, H. Zhang, P. Liu, H. Yang, D. Wang, Z. Tang, Y. Wang and H. Zhao, The surface sulfur doping induced enhanced performance of cobalt catalysts in oxygen evolution reactions, Chem. Commun., 2016, 52, 9450–9453 RSC.
- S. V. S. Mers, V. Maruthapandian and V. Ganesh, Highly efficient bifunctional electrocatalyst using structurally architectured N-doped cobalt oxide, ChemistrySelect, 2018, 3, 8752–8762 CrossRef CAS.
- L.-M. Cao, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Template-directed synthesis of sulphur doped NiCoFe layered double hydroxide porous nanosheets with enhanced electrocatalytic activity for the oxygen evolution reaction, J. Mater. Chem. A, 2018, 6, 3224–3230 RSC.
- J.-J. Duan, R.-L. Zhang, J.-J. Feng, L. Zhang, Q.-L. Zhang and A.-J. Wang, Facile synthesis of nanoflower-like phosphorus-doped Ni3S2/CoFe2O4 arrays on nickel foam as a superior electrocatalyst for efficient oxygen evolution reaction, J. Colloid Interface Sci., 2021, 581, 774–782 CrossRef CAS PubMed.
- I.-K. Ahn, S.-Y. Lee, H. G. Kim, G.-B. Lee, J.-H. Lee, M. Kim and Y.-C. Joo, Electrochemical oxidation of boron-doped nickel–iron layered double hydroxide for facile charge transfer in oxygen evolution electrocatalysts, RSC Adv., 2021, 11, 8198–8206 RSC.
- D. Zhou, Z. Cai, X. Lei, W. Tian, Y. Bi, Y. Ji, N. Han, T. Gao, Q. Zhang, Y. Kuang, J. Pan, X. Sun and X. Duan, NiCoFe-layered double hydroxides/N-doped graphene oxide array colloid composite as an efficient bifunctional catalyst for oxygen electrocatalytic reactions, Adv. Energy Mater., 2017, 1701905 Search PubMed.
- M. Amjad, S. H. Sumrra, M. S. Akram and Z. H. Chohan, Metal-based ethanolamine-derived compounds: a note on their synthesis, characterization and bioactivity, J. Enzyme Inhib. Med. Chem., 2016, 31, 88–97 CrossRef CAS PubMed.
- S. Sahu, U. K. Sahu and R. K. Patel, Synthesis of thorium–ethanolamine nanocomposite by the co-precipitation method and its application for Cr(vi) removal, New J. Chem., 2018, 42, 5556–5569 RSC.
- M. S. Masoud, A. E. Ali, H. M. Ahmed and E. A. Mohamed, Spectral studies and thermal analysis of new vanadium complexes of ethanolamine and related compounds, J. Mol. Struct., 2013, 1050, 43–52 CrossRef CAS.
- C. Dong, L. Han, C. Zhang and Z. Zhang, Scalable dealloying route to mesoporous ternary CoNiFe layered double hydroxides for efficient oxygen evolution, ACS Sustainable Chem. Eng., 2018, 6, 16096–16104 CrossRef CAS.
- T. Feng, Q. Zeng, S. Lu, M. Yang, S. Tao, Y. Chen, Y. Zhao and B. Yang, Morphological and interfacial engineering of cobalt-based electrocatalysts by carbon dots for enhanced water splitting, ACS Sustainable Chem. Eng., 2019, 7, 7047–7057 CrossRef CAS.
- C. L. Chiang and J. M. Yang, in Novel Fire Retardant Polymers and Composite Materials, ed. D.-Y. Wang, Woodhead Publishing, 2017, pp. 295–312 Search PubMed.
- A. Cano, J. Rodríguez-Hernández, A. Shchukarev and E. Reguera, Intercalation of pyrazine in layered copper nitroprusside: synthesis, crystal structure and XPS study, J. Solid State Chem., 2019, 273, 1–10 CrossRef CAS.
- T.-N. Tran, H.-Y. Lee, J.-D. Park, T.-H. Kang, B.-J. Lee and J.-S. Yu, Synergistic CoN-decorated Pt catalyst on two-dimensional porous Co–N-doped carbon nanosheet for enhanced oxygen reduction activity and durability, ACS Appl. Energy Mater., 2020, 3, 6310–6322 CrossRef CAS.
- H. Peng, Z. Mo, S. Liao, H. Liang, L. Yang, F. Luo, H. Song, Y. Zhong and B. Zhang, High performance Fe- and N-doped carbon catalyst with graphene structure for oxygen reduction, Sci. Rep., 2013, 3, 1765 CrossRef.
- J.-C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Systematic XPS studies of metal oxides, hydroxides and peroxides, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
- Y. Xue, Y. Wang, H. Liu, X. Yu, H. Xue and L. Feng, Electrochemical oxygen evolution reaction catalyzed by a novel nickel–cobalt-fluoride catalyst, Chem. Commun., 2018, 54, 6204–6207 RSC.
- H. Liu, Y. Li, M. Yuan, G. Sun, Q. Liao and Y. Zhang, Solid and macroporous Fe3C/N-C nanofibers with enhanced electromagnetic wave absorbability, Sci. Rep., 2018, 8, 16832 CrossRef PubMed.
- W. Liu, J. Xie, Y. Guo, S. Lou, L. Gao and B. Tang, Sulfurization-induced edge amorphization in copper–nickel–cobalt layered double hydroxide nanosheets promoting hydrazine electrooxidation, J. Mater. Chem. A, 2019, 7, 24437–24444 RSC.
- H. Wang and S. Tao, Fabrication of a porous NiFeP/Ni electrode for highly efficient hydrazine oxidation boosted H2 evolution, Nanoscale Adv., 2021, 3, 2280–2286 RSC.
- K. Matyjaszewski, in Encyclopedia of Materials: Science and Technology, ed. K. H. J. Buschow, R. W. Cahn, M. C. Flemings, B. Ilschner, E. J. Kramer, S. Mahajan and P. Veyssière, Elsevier, Oxford, 2001, pp. 356–365 Search PubMed.
- Y. I. Yermakov, B. N. Kuznetsov and V. A. Zakharov, in Studies in Surface Science and Catalysis, ed. Elsevier, 1981, vol. 8, pp. 1–58 Search PubMed.
- L.-A. Stern and X. Hu, Enhanced oxygen evolution activity by NiOx and Ni(OH)2 nanoparticles, Faraday Discuss., 2014, 176, 363–379 RSC.
- B. H. R. Suryanto, Y. Wang, R. K. Hocking, W. Adamson and C. Zhao, Overall electrochemical splitting of water at the heterogeneous interface of nickel and iron oxide, Nat. Commun., 2019, 10, 5599 CrossRef CAS PubMed.
- H. Lei, M. Chen, Z. Liang, C. Liu, W. Zhang and R. Cao, Ni2P hollow microspheres for electrocatalytic oxygen evolution and reduction reactions, Catal. Sci. Technol., 2018, 8, 2289–2293 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra00919f |
| This journal is © The Royal Society of Chemistry 2022 |