
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An in situ fluorine and ex situ titanium two-step co-doping strategy for efficient solar water splitting by hematite photoanodes†
Kyoungwoong
Kang‡
a,
Hemin
Zhang‡
 *b,
Jeong Hun
Kim
a,
Woo Jin
Byun
a and
Jae Sung
Lee
*a
*b,
Jeong Hun
Kim
a,
Woo Jin
Byun
a and
Jae Sung
Lee
*a
aSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 44919, Republic of Korea. E-mail: jlee1234@unist.ac.kr
bCollege of Materials Science and Engineering, Sichuan University, Chengdu 610065, China. E-mail: hmzhang@scu.edu.cn
First published on 12th February 2022
Abstract
A unique two-step co-doping strategy of in situ fluorine doping followed by ex situ titanium doping enhances the performance of the hematite photoanode in photoelectrochemical water splitting much more effectively than single-step co-doping strategies that are either all in situ or all ex situ. The optimized fluorine, titanium co-doped Fe2O3 photoanode without any cocatalyst achieves 1.61 mA cm−2 at 1.23 VRHE under 100 mW cm−2 solar irradiation, which is ∼2 and 3 times those of titanium or fluorine singly-doped Fe2O3 photoanodes, respectively. The promotional effect is attributed to the synergy of the two dopants, in which the doped fluorine anion substitutes oxygen of Fe2O3 to increase the positive charges of iron sites, while the doped titanium cation substitutes iron to increase free electrons. Moreover, excess titanium on the surface suppresses the drain of in situ doped fluorine and agglomeration of hematite during the high-temperature annealing process, and passivates the surface trap states to further promote the synergy effects of the two dopants.
1. Introduction
Water splitting with sunlight has attracted great research interest in recent decades as an ideal route to renewable and storable hydrogen (“green hydrogen”).1–3 Among several options for solar hydrogen production, the photoelectrochemical (PEC) cell has been regarded as a promising technology for practical applications because it is more efficient than the particulate photocatalytic cell, and cheaper than the photovoltaic–electrolyzer cell.4 The performance of a PEC cell is determined by its photoelectrode material, which requires a narrow band gap, appropriate conduction and valence band positions, low cost, and high operational stability. Despite intensive research and many technical advances in the last few decades, no photoelectrode material has met all of these requirements.5Hematite (α-Fe2O3) is one of the most extensively studied photoanode materials because of its highly desirable properties including ample visible-light absorption, good stability in aqueous solutions, non-toxicity, earth-abundance, and low cost. With its band gap of 2.1 eV, an ideal hematite photoanode can generate a photocurrent density as high as 12.6 mA cm−2 under solar irradiation of 100 mW cm−2 (AM 1.5G) or a solar to hydrogen (STH) conversion efficiency of 15.5%.6 However, the state-of-the-art hematite photoanodes reported so far have only achieved less than half of the theoretical limit because of its intrinsically poor optoelectronic properties of short diffusion length of the photogenerated holes (2–4 nm), low electrical conductivity (10−2 cm2 V−1 s−1), and slow water oxidation kinetics.7,8
A number of modification strategies have been developed to alleviate the shortcomings of hematite such as doping, nanostructuring, hetero-/homo-junctions, surface modifications and co-catalysts.9–13 In particular, doping and nanostructuring are the most effective ways to reduce the recombination of photogenerated electrons and holes in the bulk of hematite. Foreign atom doping can increase the charge carrier density to enhance the electrical conductivity of hematite, while nanostructuring can help overcome the problem of its extremely short hole diffusion length.14 Hence, doping and nanostructuring in combination could exert a synergistic effect in reduction of bulk recombination.
The most common dopants for hematite are tetravalent metal cations (M4+ = Ti4+, Sn4+, Zr4+, Si4+, or Pt4+), which provide an additional free electron when these M4+ ions substitute for Fe3+ in the hematite lattice. Recently, fluorine anions were used as an effective dopant by replacing oxygen atoms in metal oxide semiconductors (TiO2, WO3, and ZnO) including hematite owing to its ionic radius (119 pm) being similar to that of oxygen (126 pm), which resulted in multiple desirable effects – promotion of electron transfer from the localized states to the conduction band, enhanced light harvesting, and decreased band gap.15–17 In light of these previous studies, we conjectured that a hematite photoanode co-doped with metal cations and non-metal anions might provide a new path to improved PEC performance. There have been several reports on co-doped Fe2O3 with different cations,18–20 but this type of cation (M4+) and anion (F−) co-doping of hematite has never been investigated previously.
In this work, we explore the synergistic co-doping effects of the fluorine anion (F−) and titanium cation (Ti4+) into the hematite lattice according to a unique two-step doping process – in situ F−-doping during the hydrothermal synthesis of FeOOH nanorods followed by ex situ Ti4+-doping during FeOOH-to-Fe2O3 conversion under high-temperature annealing. It is demonstrated that the two-step co-doping process promotes the PEC performance of the hematite photoanode much more effectively than single-step co-doping strategies that are either all in situ or all ex situ. In the finally obtained F−, Ti4+-codoped Fe2O3 nanorods (F,Ti:Fe2O3), the F− anion substitutes oxygen of Fe2O3 to enhance the positive charges of the iron sites, while Ti4+ substitutes iron to generate more free electrons, thereby both contributing to electrical conductivity and n-type character. In addition, excess titanium remaining on the surface suppressed the drain of in situ doped fluorine and agglomeration of hematite during the high-temperature annealing process and passivated the surface trap states to further promote the synergy effects of the two dopants. This beneficial side effect is only possible for our two-step co-doping strategy. As a result, the F,Ti:Fe2O3 photoanode without any cocatalyst generates a photocurrent density of 1.61 mA cm−2 at 1.23 VRHE under 1-sun irradiation, far outperforming those of pristine and singly-doped hematite photoanodes.
2. Experimental section
2.1. Fabrication of F,Ti:Fe2O3 nanorods on F:SnO2 (FTO) coated glass
Several pieces of FTO (PECTM 8, 6–9 Ω, Pilkington) were ultrasonically cleaned by using a chemical agent (deconex® 11 Universal) solution, ethanol and acetone, respectively, which made the surface sufficiently hydrophilic. The FeOOH nanorods were hydrothermally synthesized on the FTO glass. Briefly, into a 10 mL aqueous solution of 0.15 M FeCl3·6H2O, and 1 M NaNO3 at pH of ∼1.5, FTO glass was immersed and kept in an electric oven at 100 °C for 4 h to obtain a thin yellow film of FeOOH nanorods on FTO. For F-doping, NH4F was added in the precursor solution to obtain F:FeOOH nanorods. For Ti-doping, a diluted titanium(IV) chloride solution using 2-methoxyethanol was deposited on FeOOH or F:FeOOH nanorods by spin-coating at 2000 rpm for 20 s. For single-step F and Ti in situ co-doping, NH4F and titanium chloride were added in the precursor solution to obtain F,Ti:FeOOH nanorods. For single-step ex situ co-doping, NH4F and titanium butoxide were diluted in 2-methoxyethanol and deposited on the surface of FeOOH nanorods by spin-coating. The FeOOH or F:FeOOH nanorods with/without titanium were heated at 550 °C for 2 h to obtain crystalline hematite nanorod films followed by a second annealing at 700 °C for 10 min to achieve higher crystallinity.2.2. Characterization
X-ray diffraction (XRD) patterns were collected by using a PW3040/60 X'per PRO, PANalytical, using Cu-Kα (λ = 1.54056 Å) radiation, an accelerating voltage of 40 kV, and a current of 30 mA. Raman spectra were obtained using a 0.2 mW 532 nm laser (AFM-Raman, WITec, alpha300R). Ultraviolet-visible absorbance was measured by using a UV-Vis spectrometer (UV-2401 PC, Shimadzu). Surface morphology and structure were investigated using a scanning electron microscope (SEM-S4800, HITACHI) and transmission electron microscope (TEM, JEOL, JEM-2100F, 200 kV). High-angle annular dark-field (HAADF) and energy dispersive X-ray spectroscopy (EDS) analyses were carried out using a FEI Titan3 G2 60-300 microscope equipped with a double-sided Cs corrector. The surface atomic composition and atomic depth profiling were performed by X-ray photoelectron spectroscopy (XPS, Thermo-Fisher, Kα). Time-of-flight secondary ion mass spectrometry (TOF-SIMS) data were obtained on an instrument (ION-TOF GmbH, Germany) using a 10 keV Bi+ primary ion beam source.2.3. Photoelectrochemical performance measurements
Solar water oxidation was performed in a three-electrode cell with the photoanode as a working electrode, a platinum mesh as a counter electrode and Ag/AgCl as a reference electrode in NaOH (1 M, pH = 13.6) electrolyte. Photocurrent (J)–potential (V) curves and electrochemical impedance spectra (EIS) were recorded under simulated solar light generated by a solar simulator (91170, Oriel) with an air mass 1.5 G filter. Light intensity of the solar simulator was calibrated to 1 sun (100 mW cm−2) using a reference cell certified by the National Renewable Energy Laboratories (USA). All electrochemical measurements were performed on a potentiostat (IviumStat, Ivium Technologies). The Mott–Schottky plots were measured by sweeping the 0.4–1.0 VRHE range with an alternative current (AC) frequency of 1000 Hz under dark conditions. EIS spectra were recorded at 1.23 VRHE with an AC potential frequency range of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–0.1 Hz. Z-View software (Scribner Associates) was used for fitting the experimental EIS data to an equivalent circuit model. The incident photon-to-electron conversion efficiency (IPCE) was measured using a Xe lamp (300 W, Oriel) and a monochromator with a bandwidth of 10 nm at 1.23 VRHE in the same electrolyte. The on-line gas evolution analysis of H2 and O2 was conducted using a gas chromatograph (Agilent, GC 7890) equipped with a packed column (Supelco, Carboxen 1000) and a thermal conductivity detector (TCD).
000–0.1 Hz. Z-View software (Scribner Associates) was used for fitting the experimental EIS data to an equivalent circuit model. The incident photon-to-electron conversion efficiency (IPCE) was measured using a Xe lamp (300 W, Oriel) and a monochromator with a bandwidth of 10 nm at 1.23 VRHE in the same electrolyte. The on-line gas evolution analysis of H2 and O2 was conducted using a gas chromatograph (Agilent, GC 7890) equipped with a packed column (Supelco, Carboxen 1000) and a thermal conductivity detector (TCD).
The electrochemically active surface area (EASA) was determined by measuring the capacitive current associated with double-layer charging from the scan-rate dependence of cyclic voltammograms (100–200 mV s−1). The double layer capacitance (Cdl) was evaluated by plotting ΔJ = (Ja − Jc) at 1.1 VRHE against the scan rate, in which the linear slope is equivalent to twice Cdl. The charge carrier density (ND) is inversely proportional to the slope and can be extracted using the following equation:
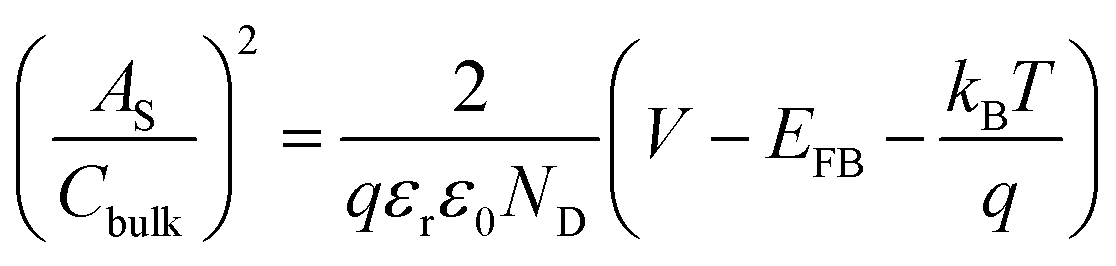
 is the surface area-corrected space charge capacitance, V is the applied potential, EFB is the flat band potential of the electrode, εr = 32, ε0 = 8.85 × 10−12 C2 J−1 m−1, q = 1.602 × 10−19 C, T = 298 K, and kB = 1.38 × 10−23 J K−1 for hematite.
is the surface area-corrected space charge capacitance, V is the applied potential, EFB is the flat band potential of the electrode, εr = 32, ε0 = 8.85 × 10−12 C2 J−1 m−1, q = 1.602 × 10−19 C, T = 298 K, and kB = 1.38 × 10−23 J K−1 for hematite.
3. Results and discussion
3.1. Fabrication of the F,Ti:Fe2O3 nanorod photoanode
A schematic synthesis procedure of F,Ti:Fe2O3 nanorods grown on FTO glass is shown in Fig. 1. First, F-doped β-FeOOH nanorods on FTO glass were synthesized by a hydrothermal reaction in NH4F-containing solution to achieve in situ F-doping (Fig. S1 in the ESI†). Then, a dilute titanium butoxide solution was uniformly deposited on the surface of F:FeOOH nanorods by spin-coating at a speed of 2000 rpm for 20 s. Finally, titanium-deposited F:FeOOH nanorods were annealed at 550 °C for 2 h and then 700 °C for 10 min to execute ex situ Ti-doping and complete the two-step co-doping process that gave the F,Ti:Fe2O3/FTO photoanode. Annealing of all the photoanodes was carried out under the air conditions. Moderate conditions (instead of 800 °C or longer time) were selected to compromise between the loss of doped fluorine and the high crystallinity obtained during the transformation of FeOOH into Fe2O3.21 The pristine Fe2O3, single-doped F:Fe2O3, and Ti:Fe2O3 photoanodes were also synthesized according to the same procedure except for the doping steps. Single-step co-doping was also tried for comparison through either all in situ or all ex situ doping.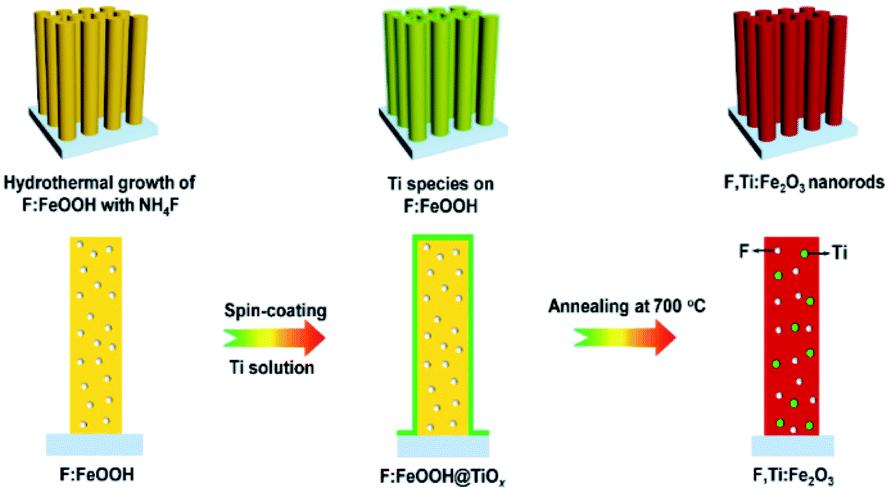 | ||
| Fig. 1 Schematic synthesis procedure of F,Ti:Fe2O3 nanorods via the two-step co-doping process of in situ F-doping and ex situ external Ti-doping. | ||
3.2. Morphology of the F,Ti:Fe2O3 nanorod photoanode
SEM images of the F,Ti:Fe2O3 photoanode in Fig. 2a and b show the morphology of the annealed nanorods with diameters of 80–120 nm and lengths of 350–500 nm. All three photoanodes (F:Fe2O3, Ti:Fe2O3, and F,Ti:Fe2O3) exhibit an identical nanorod morphology, but Ti:Fe2O3 and F,Ti:Fe2O3 photoanodes show relatively smaller diameters compared with Fe2O3 and F:Fe2O3 (Fig. S2†). This suggests that the excess titanium remaining on the surface suppresses the agglomeration of Fe2O3 nanorods in spite of the rather high annealing temperature (700 °C).22 The TEM image of a single nanorod in Fig. 2c shows a diameter of ∼78 nm and a length of ∼515 nm, and the high-resolution TEM (HRTEM) image in Fig. 2d displays a single crystalline nanorod with a d-spacing of 0.255 nm corresponding to the (110) crystal plane of hematite. Some nanorods exhibit a porous structure (Fig. 2e), probably because NH4F evolves NH3 gas during the hydrothermal synthesis. Elemental mapping images of Fe, O, Ti, and F (Fig. 2f–i) show the spatially uniform distribution, indicating the homogeneity of co-doping via the two-step process; in situ F anion doping followed by ex situ Ti cation doping. There is no significant Sn signal that might have diffused from FTO (Fig. 2j), suggesting that the thermal damage of FTO could be negligible. | ||
| Fig. 2 SEM (top view (a) and cross-sectional (b)), TEM (c), HRTEM (d), HAADF (e), and elemental mapping images of Fe (f), O (g), Ti (h), F (i), and Sn (j) of F,Ti:Fe2O3 nanorods. | ||
3.3. Physical characterization of F,Ti:Fe2O3/FTO photoanodes
All of the hematite photoanodes show similar XRD patterns of the rhombohedral α-Fe2O3 phase (JCPDS no. 33-0664) in Fig. 3a, which demonstrates that doping F and/or Ti does not change the crystal structure of hematite. They all show a strong (110) peak at 2θ = 35.61°, indicating a highly orientated growth. Compared with Ti:Fe2O3, F:Fe2O3 shows a very weak peak of (104) at 2θ = 33.15°, indicating that fluorine doping further enhances the orientated crystal growth and gives a higher intensity ratio of (110)/(104) peaks (Fig. S3†). It is known that the electron transport along the hematite (110) plane is four orders-of-magnitude higher than that of the (104) plane,23 and such a directional charge flow is quite favourable for high PEC performance. While in situ doped fluorine is involved in the crystallization process of hematite, titanium doping does not show this phenomenon since titanium is introduced ex situ from the external surface and has no way to exert an influence on the already-constructed crystal structure.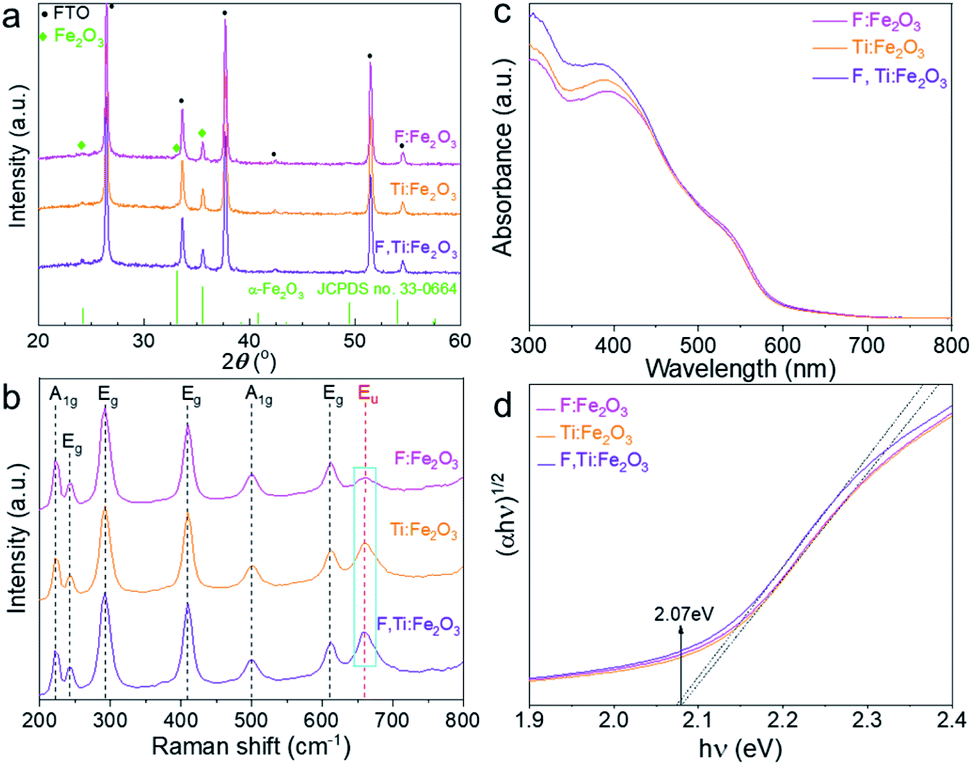 | ||
| Fig. 3 XRD patterns (a), Raman spectra (b), light absorption spectra (c) and Tauc plots (d) of Fe2O3, F:Fe2O3, Ti:Fe2O3, and F,Ti:Fe2O3 photoanodes. | ||
All Raman spectra in Fig. 3b match well with those of hematite with no impurity phase like maghemite (γ-Fe2O3) or magnetite (Fe3O4). The peaks at 229 cm−1 and 500 cm−1 are assigned to the A1g mode, and the other four peaks at 249, 295, 414, and 615 cm−1 are assigned to Eg modes.24 In general, the A1g mode is related to the symmetric stretching of oxygen atoms, while the Eg mode is related to symmetric and asymmetric bending of oxygen with respect to iron in the tetrahedral voids. In particular, the forbidden vibration mode (Eu) at ∼660 cm−1 (marked rectangle in Fig. 3b) represents structural disorders.25,26 Thus, Ti:Fe2O3 shows an increase of the Eu peak compared with F:Fe2O3, while F,Ti:Fe2O3 displays the highest peak intensity, demonstrating that in situ F− doping generates little disorder in the structure, whereas ex situ Ti4+ doping produces much lattice stress or local lattice disorder. The variation of the Eu mode caused by F− and/or Ti4+ doping demonstrates successful introduction of the dopants into the hematite lattice.
Both Ti,F:Fe2O3 and Ti:Fe2O3 show slightly improved light absorption as shown in Fig. 3c compared with F:Fe2O3. Interestingly, light absorption by F:Fe2O3 is lower at 420 nm but becomes higher at 550 nm than that by Ti:Fe2O3, suggesting different mechanisms of absorption enhancement induced by F− or Ti4+ doping. This is probably related to their different doping mechanisms. The in situ doped fluorine anions prefer to substitute oxygen atoms of Fe2O3, while the titanium cations substitute iron atoms. Xie et al. reported that F− doped hematite generated a defect level to reduce apparent band gap, which led to a significant enhancement of light absorption.27 Ti4+ doping might induce some intra-band gap states as well, leading to a narrow band gap.28 Consequently, F,Ti:Fe2O3 shows a highest absorption at 420 nm but a lower one at 550 nm than that of F:Fe2O3. Light harvesting efficiency (LHE, defined as 1–10−A where A is the measured absorbance) is used to evaluate the light absorption capability of electrodes (Fig. S4†). Compared with single-doped hematite, the co-doped hematite shows slightly higher LHE. Specifically, F:Fe2O3 shows the highest LHE above the wavelength of 500 nm. From the Tauc plots (Fig. 3d), however, all the derived band gaps are almost the same (∼2.07 eV), indicating that F or Ti doping does not introduce significant intra-band levels in the band gap of hematite, which is in accordance with the literature reports.29
3.4. The states and compositions of dopants in F,Ti:Fe2O3
XPS in Fig. 4 shows Fe 2p1/2, Fe 2p3/2 and Fe3+ satellite peaks at 724.5, 711.1 and 718.8 eV, respectively, indicative of α-Fe2O3.30 The F:Fe2O3 and F,Ti:Fe2O3 photoanodes show a little larger areas of Fe3+ satellite peaks relative to Ti:Fe2O3, suggesting that the positive charges of Fe sites are enhanced in hematite lattices by the strong electronegativity of F dopants. On the other hand, Ti:Fe2O3 shows a smaller area of Fe3+ satellite peaks relative to that of F:Fe2O3, indicating that the positive charges of Fe sites were weakened or Fe2+ ions were generated in hematite lattices by electronic contribution of Ti dopants. The F dopant in F:FeOOH, F:Fe2O3 and F,Ti:Fe2O3 can be verified by the F 1s XPS peak at ∼684.1 eV (Fig. S5†), which is consistent with fluorine in metal fluorides and demonstrates the presence of Fe–F bonds in F-doped hematite.31 Both F:Fe2O3 and F,Ti:Fe2O3 photoanodes show a lower intensity than that of F:FeOOH, indicating some loss of F dopants during the annealing process. However, F,Ti:Fe2O3 shows a slightly higher intensity of F dopants than that of F:Fe2O3, suggesting that ex situ Ti doping can suppress the loss of F dopants to some extent. This could be a source of the synergistic promotion effect of F and Ti co-doping. This result was further demonstrated by TOF-SIMS in Fig. S6.† The fluorine signals from F:Fe2O3 and F,Ti:Fe2O3 are almost an order-of-magnitude higher than Ti:Fe2O3, thereby confirming the success of in situ F doping. In addition, F,Ti:Fe2O3 shows slightly stronger fluorine signals relative to that of F:Fe2O3, which verifies again fluorine preservation by the excess Ti remaining on the external surface of Fe2O3 nanorods.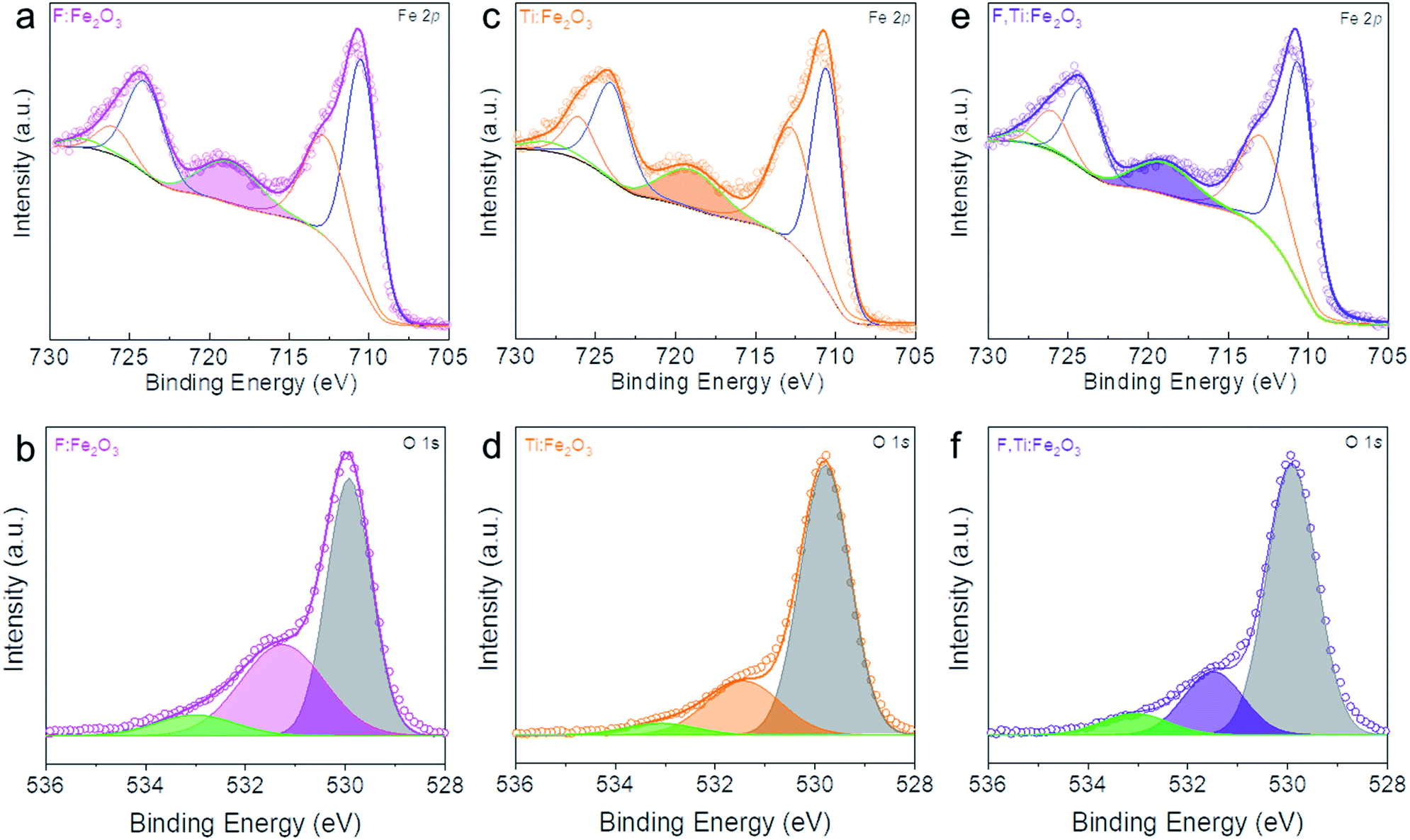 | ||
| Fig. 4 Fe 2p and O 1s XPS spectra of F:Fe2O3 (a) and (b), Ti:Fe2O3 (c) and (d) and F,Ti:Fe2O3 (e) and (f). The coloured peaks in (a), (c), and (e) denote the Fe3+ satellite peak. Core-level O 1s XPS spectra in (b), (d), and (f) show the Fe–O bond, oxygen vacancy, and surface hydroxyl group peaks, respectively, from low to high binding energies. | ||
The XPS peaks at ∼464 and 458 eV in Fig. S7† are assigned to Ti 2p1/2 and Ti 2p3/2 of Ti4+ states, respectively.32 Importantly, the interaction between F and Ti dopants in F,Ti:Fe2O3 is manifested in Ti 2p1/2 and Ti 2p3/2 peaks of F,Ti:Fe2O3, which show a shift of 0.15 eV towards higher binding energies relative to those of Ti:Fe2O3, indicating that F dopants also promote the positive charges of Ti dopants because of their strong electronegativity. During annealing, the doped fluorine would tend to escape from the hematite lattice, while the external surface-coated titanium would attempt to go into the lattices. Consequently, the already-doped fluorine is retained more in the lattice by the ex situ Ti doping from the external surface. The F and Ti atomic concentrations in the F,Ti:Fe2O3 photoanode determined by TOF-SIMS are 2% and 3%, respectively (Fig. S8†), but 1.5% F and 4.7% Ti are determined on the surface. This more significant concentration gradient of Ti is reasonable considering the doping procedure – in situ F-doping followed by ex situ external Ti-doping.
In addition, Sn diffusion from FTO is usually inevitable during the high-temperature annealing process,33 although the elemental mapping image in Fig. 2j did not show a significant Sn signal. Thus, the Sn signal was monitored by more sensitive TOF-SIMS for F,Ti:Fe2O3 photoanodes treated at different annealing temperatures (Fig. S9†). The Sn signal appears from the sample annealed at 700 °C, and gets stronger for the samples annealed at 800 and 900 °C. Therefore, F,Ti:Fe2O3 actually contains a small amount of Sn dopant diffused from FTO in addition to intentional dopants of F and Ti. The lattice oxygen peak of F,Ti:Fe2O3 shows a little shift (only ∼0.1 eV) towards higher binding energies from that of Fe2O3, probably due to strong electron-withdrawing capability of F dopants. Deconvoluted O 1s spectra in Fig. 4b, d and f show the Fe–O bond, oxygen vacancy, and surface hydroxyl group peaks, respectively, from low to high binding energies. The F:Fe2O3 photoanode shows the largest area of oxygen vacancies (middle peak) due to the substitution of oxygen and surface trap states, while Ti:Fe2O3 shows a relatively small area due to the passivation of surface trap states by ex situ Ti doping. Importantly, the co-doped F,Ti:Fe2O3 photoanode contains an intermediate amount of oxygen vacancies. An appropriate amount of oxygen vacancies could significantly contribute to the enhanced PEC performance by improving the electrical conductivity of the photoanode.11,34
3.5. Photoelectrochemical water oxidation performance
The PEC water oxidation performance of the optimized F,Ti:Fe2O3 photoanode was studied under simulated 1 sun irradiation (100 mW cm−2) in 1 M NaOH electrolyte in a three-electrode cell with the photoanode, Ag/AgCl (3 M NaCl), and Pt mesh as the working, reference, and counter electrodes, respectively. The F:Fe2O3 photoanode with the optimized F dopants (by addition of 30 mg NH4F in the precursor solution, Fig. S10†) shows a photocurrent density of 0.35 mA cm−2 at 1.23 VRHE. The F:Fe2O3 photoanode with optimized Ti4+ dopants (by spin-coating of 10 mM TiCl4 solution, Fig. S11†) generates a photocurrent of 0.90 mA cm−2 at 1.23 VRHE, which is ∼3 times that of F:Fe2O3.In the study of the performance of PEC water oxidation over these modified hematite photoanodes, first of all, we would like to verify the efficacy of our two-step co-doping strategy relative to a single step co-doping that is either in situ only or ex situ only. As shown in the photocurrent density (J)–applied voltage (V) curves in Fig. 5a, the photocurrent generation at 1.23 VRHE under 1 sun irradiation over the F,Ti:Fe2O3 photoanode co-doped by the two-step process is much higher (1.6 mA cm−2 at 1.23 VRHE) than that over the photoanode co-doped in an ex situ single step (0.96 mA cm−2) or in situ single step (0.42 mA cm−2). The J–V curves in Fig. 5b represent the performance of the photoanodes for photo-oxidation of a hole scavenger (0.5 M H2O2) in the same electrolyte. Since the surface charge recombination of the highly reactive H2O2 is negligible, this performance represents charge transfer characteristics in the bulk of the photoanode. In this case as well, the F,Ti:Fe2O3 photoanode shows a similar trend of the performance gaps depending on the co-doping method. Thus, our two-step co-doping strategy produces the F,Ti:Fe2O3 photoanode that demonstrates the synergy effect of co-doping in the most prominent manner. As mentioned, the second step of external Ti doping can suppress the drain of already-doped F and agglomeration of hematite nanorods during the high temperature annealing process. These beneficial side effects cannot be expected for in situ or ex situ single-step doping methods.
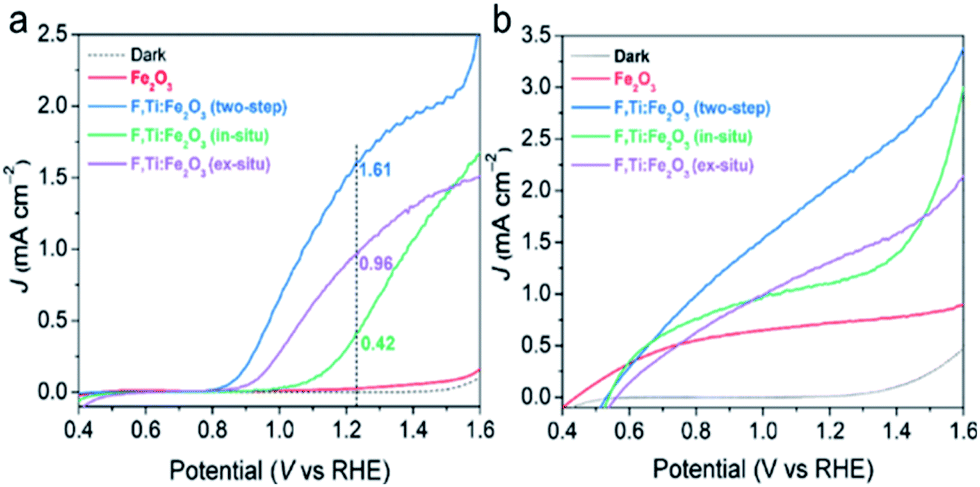 | ||
| Fig. 5 Comparison of single-step (in situ or ex situ) and two-step (in situ followed by ex situ) co-doping of F and Ti into hematite to fabricate the F,Ti:Fe2O3 photoanode. (a) J–V curves of water oxidation (without H2O2). (b) J–V curves of H2O2 oxidation. The PEC water oxidation was performed under 1 sun irradiation (100 mW cm−2) in 1 M NaOH electrolyte. | ||
The J–V curves of different photoanodes with (dotted lines) and without H2O2 (solid lines) are summarized in Fig. 6a. Compared with single F- or Ti-doped Fe2O3 photoanodes, F and Ti co-doped hematite shows synergistically enhanced PEC performance for oxidation of water as well as H2O2. The photocurrent onset potential (Von) is another important kinetic parameter, which is usually related to Fermi-level pinning due to surface trap states. It can be determined from the first-order derivative of the J–V curve (Fig. S12†).35
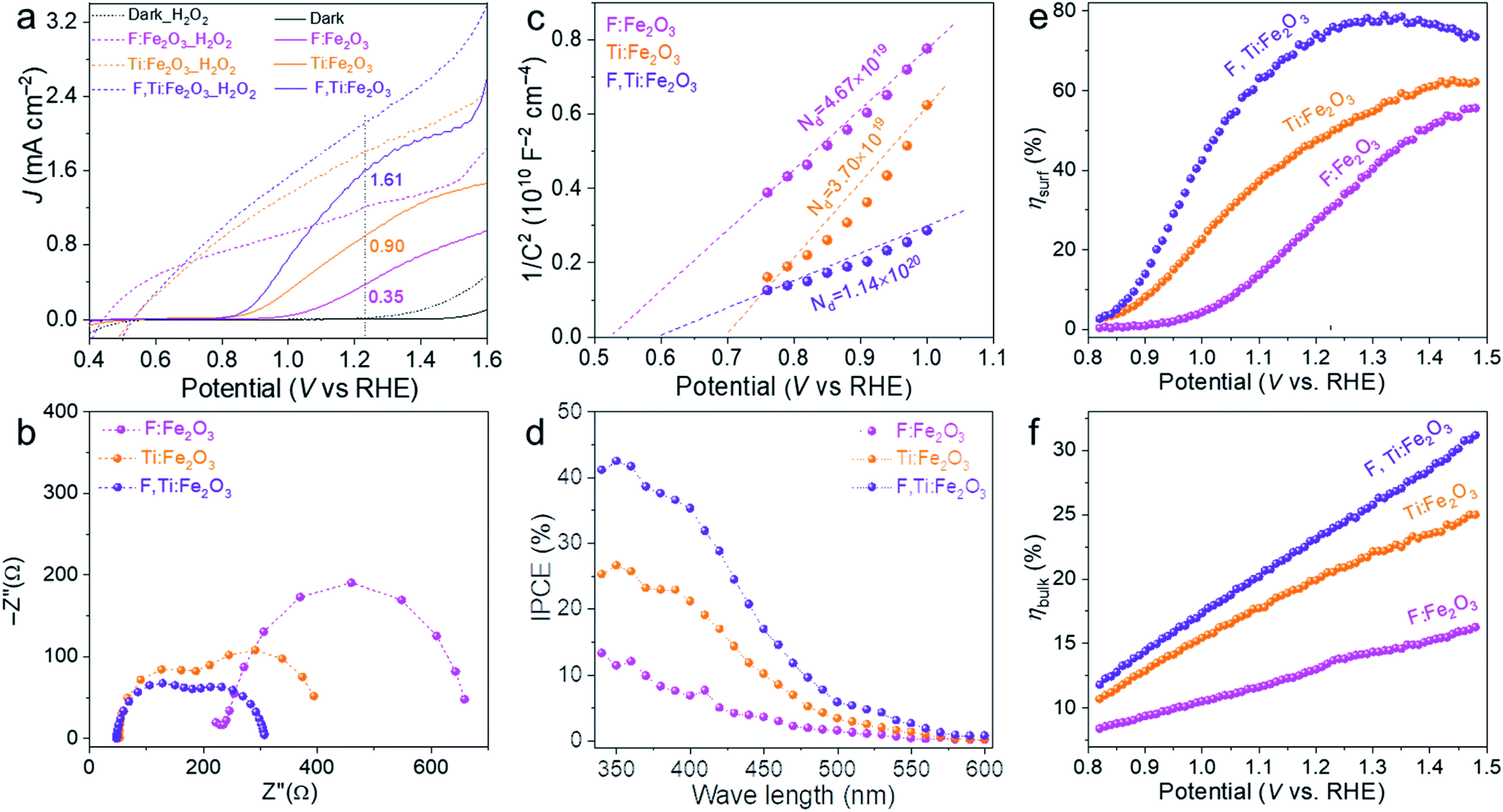 | ||
| Fig. 6 J–V curves with (dashed) and without (solid) the H2O2 hole scavenger in electrolyte (a), Nyquist plots (b), Mott–Schottky plots (c), IPCE (d), ηsurf (e) and ηbulk (f) of F:Fe2O3, Ti:Fe2O3, and F,Ti:Fe2O3. The PEC water oxidation was performed under 1 sun irradiation (100 mW cm−2) in 1 M NaOH electrolyte. | ||
The result shows that F and Ti dopants exert significant influences on Von. F-Doping (F:Fe2O3) causes a negative shift of Von by 210 mV induced by its highly polarized surface and facile surface reaction, while Ti-doping (Ti:Fe2O3) shifts Von by 340 mV towards the negative direction by the passivation of surface trapping states. Note that F-doping cannot eliminate most of the surface trapping states while Ti-doping can passivate them more effectively. The F,Ti:Fe2O3 photoanode shows a cathodic shift of nearly 400 mV from that of bare Fe2O3, recording the lowest Von of 0.74 VRHE, which demonstrates the synergetic effect of F- and Ti-doping. Thus, F-doping promotes a facile surface reaction by surface polarization and increases charge carrier density as a typical impurity doping, which leads to a negative shift of Von and improved electrical conductivity. The ex situ external Ti-doping from spin-coated titanium also improves the electrical conductivity, but in addition, some Ti remaining on the external surface of Fe2O3 forms a surface passivation layer that passivates the surface trap states to cause a cathodic shift of Von.36 Indeed, TiO2 is the most frequently used material as a passivation layer for many photoanode materials. This inadvertent formation of the Ti passivation layer also provides an additional advantage of the two-step co-doping strategy using ex situ Ti doping.
The gases evolved from the F,Ti:Fe2O3 photoanode and the Pt counter electrode were quantified by gas chromatography (Fig. S13†), which was carried out at a constant potential of 1.30 VRHE for 60 min. The O2 and H2 gases evolved with their ratio close to the stoichiometry (O2/H2 = 1/2) as shown in Fig. S14.† The faradaic efficiencies (ratio of gas evolution/photocurrent generation) of O2 and H2 evolution reactions are 93.5% and 98.5%, respectively (Fig. S15†). The results demonstrate that the measured photocurrents do come mostly from O2 and H2 evolution reactions during the PEC water splitting without any significant parasitic process.
Another informative indicator of catalytic activity is the EASA, which can be determined by measuring the capacitive current associated with double-layer charging from the scan-rate dependence of cyclic voltammograms (Fig. S16†). Apparently, the Ti-doped photoanode shows a larger EASA than that of the F-doped one, indicating that the Ti dopants not only passivate surface trapping states but also provide more catalytically active sites by suppressing agglomeration of hematite nanorods during the annealing step (Fig. 2 and S2†). Consequently, the F and Ti co-doped photoanode shows more than 2 times higher EASA than that of bare Fe2O3.
The charge transfer characteristics were investigated by EIS analysis at 1.23 VRHE under 1 sun irradiation (Fig. S17†). The Nyquist plots in Fig. 6b were fitted to a typical two-RC-unit equivalent circuit, where Rs is the sheet resistance involving the electrolyte, FTO resistance and external contact; Rtrap/Cbulk denotes the electron pathway within the bulk of the electrode (the semicircle at the high frequency region); Rct/Css represents the interface between the electrode and electrolyte (the semicircle at the low frequency region).37 Overall, both semicircles at high and low frequency regions gradually become smaller along with the order of F:Fe2O3 > Ti:Fe2O3 > F,Ti:Fe2O3, indicating the decrease of the charge transfer resistance both in bulk and at the interface, which is consistent with the corresponding J–V curves. Compared with F:Fe2O3, Rs values of Ti:Fe2O3 and F,Ti:Fe2O3 decrease by 70% and 84%, respectively. In the case of Rtrap, F,Ti:Fe2O3 shows 76% decrease, while Rtrap of Ti:Fe2O3 decreases by 51% relative to that of F:Fe2O3. Moreover, Rct values of Ti:Fe2O3 and F,Ti:Fe2O3 decrease by 50% and 72% relative to that of F:Fe2O3, respectively. These results indicate that the Ti dopants could make more contribution to the PEC performance of the hematite photoanode than the F dopants. The greatest decrease of all resistances for F,Ti:Fe2O3 verifies the effectiveness of the synergetic co-doping effect. It should be noted that the concentration of Ti dopants is over 3 times higher than that of F dopants according to TOF-SIMS (Fig. S8†), which indicates that F dopants on the surface promote charge transfer very efficiently. This might arise from the highly polarized surface induced by its strong electronegativity and the facile surface reaction with a low oxygen-evolving overpotential.24 In all cases, F and Ti co-doped F,Ti:Fe2O3 shows a significant synergy effect, resulting in much smaller values of Rs, Rtrap and Rct.
Mott–Schottky plots (Fig. 6c) were obtained from the bulk capacitance data of EIS spectra, which give the flat band potential (EFB) from the x-intercept and the donor density (ND) from the slope. All samples have positive slopes, indicating that they are n-type semiconductors.38 Compared with Ti:Fe2O3, F:Fe2O3 shows 26% increase in ND and a negative shift of EFB from 0.70 to 0.53 VRHE. Although F-doping gives a larger ND, the increased overpotential (difference between Von and EFB) by the negative shift of EFB leads to the sluggish kinetics of water oxidation. The overpotential would cause hole accumulation at the surface and subsequent surface recombination until sufficiently positive potentials are applied for appreciable charge transfer across the interface.39 However, Ti doping lowers the overpotential by the positive shift of EFB even without a comparable increase of ND, leading to significantly improved photocurrents. In particular, F,Ti:Fe2O3 displays a synergistic effect of the two dopants showing the largest amount of ND (over 3 times that of Ti:Fe2O3) and substantially improving the poor electrical conductivity of hematite.
The IPCE represents a quantitative measure of the photoactivity; IPCE = (1240 × Jlight)/(λ × Plight), where Plight is the measured irradiance at a specific wavelength (λ) of incident light and Jlight is the measured photocurrent density. Single doping (F or Ti) and co-doping improve the IPCE in the entire wavelength range of 340–600 nm (Fig. 6d), showing the trend of F,Ti:Fe2O3 > Ti:Fe2O3 > F:Fe2O3 which is consistent with their corresponding J–V curves and EIS results. Besides, the independent IPCE can be integrated with the standard AM 1.5G solar spectrum to calculate the photocurrent density using the following equation:
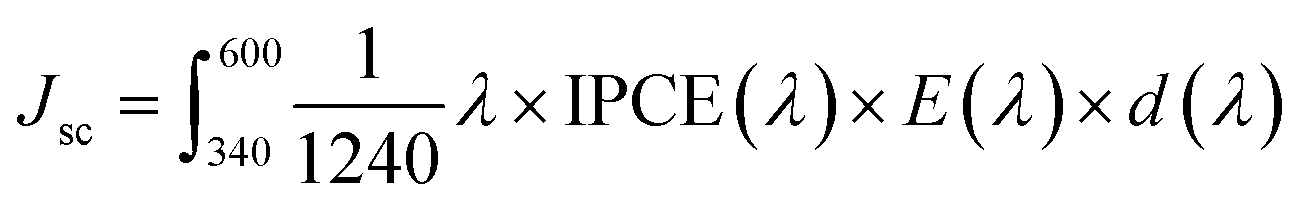
The bulk (ηbulk) and surface (ηsurf) charge separation efficiencies were obtained by comparing oxidation photocurrents of water and a hole scavenger (0.5 M H2O2) in Fig. 6a according to the procedure described in Fig. S18.† The ηbulk represents the fraction of holes that reach the electrode|electrolyte interface without recombination in the bulk, while ηsurf is the fraction of those holes at the interface that is injected successfully into the electrolyte to oxidize water.40 As shown in Fig. 6e, ηsurf of F:Fe2O3 and Ti:Fe2O3 reaches the maximum of 55% and 62% at 1.45 VRHE, respectively, indicating that F or Ti doping has comparable contribution to ηsurf, while F,Ti:Fe2O3 achieves a higher ηsurf of 78% at a lower potential of 1.3 VRHE, indicating a prominent synergy effect of co-doping. In case of ηbulk (Fig. 6f), Ti-doping (Ti:Fe2O3 and F,Ti:Fe2O3) is much more effective than F-doping (F:Fe2O3). The results of both ηsurf and ηbulk are consistent with the EIS results.
4. Conclusions
For the first time in this work, we studied substitutional co-doping of both Fe3+ cations and O2− anions of hematite by Ti4+ and F−, respectively, to improve the performance of hematite photoanodes for PEC water splitting. In particular, we developed a novel two-step co-doping strategy of in situ F− anion doping, followed by ex situ Ti4+ cation doping. The promotional effects were much more pronounced when our unique two-step doping strategy was employed – in situ F-doping followed by ex situ external Ti-doping, rather than single-step co-doping strategies that were all in situ or all ex situ. The in situ F-doping induced a large ratio of (110)/(104) crystal planes and a highly polarized surface. Ex situ Ti-doping plays multiple roles – improving the electrical conductivity of hematite, passivating surface trapping states, suppressing the agglomeration of hematite nanorods, and protecting already-doped fluorine. As a result, the optimized F,Ti:Fe2O3 photoanode without any cocatalyst generates a photocurrent density of 1.61 mA cm−2 at 1.23 VRHE under 1 sun irradiation. Besides, its Von exhibited a cathodic shift of ∼200 mV relative to F:Fe2O3. This two-step co-doping strategy of in situ anions and ex situ cations could be applied to design efficient photoelectrodes in general for solar energy conversion.Author contributions
Kyoungwoong Kang: methodology, synthesis and characterization of materials, formal analysis, writing original draft. Hemin Zhang: conceptualization, resources, data analysis, writing & editing. Jeong Hun Kim: data discussion, validation. Woo Jin Byun: gas evolution measurements. Jae Sung Lee: supervision, conceptualization, review & editing, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Climate Change Response Project (NRF-2019M1A2A2065612), the Basic Science Grant (NRF-2018R1A2A1A05077909), and the Korea-China Key Joint Research Program (2017K2A9A2A11070341) funded by the Ministry of Science and ICT and by the 2019 Research Fund (1.190013.01) of UNIST. H. Zhang acknowledges the financial support from the Talent Introduction Program of Sichuan University (Grant No. YJ202180).References
- Y. Kuang, Q. Jia, G. Ma, T. Hisatomi, T. Minegishi, H. Nishiyama, M. Nakabayashi, N. Shibata, T. Yamada, A. Kudo and K. Domen, Nat. Energy, 2017, 2, 16191 CrossRef CAS.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827–832 CrossRef CAS.
- J. H. Kim, D. Hansora, P. Sharma, J.-W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- H. Dotan, O. Kfir, E. Sharlin, O. Blank, M. Gross, I. Dumchin, G. Ankonina and A. Rothschild, Nat. Mater., 2013, 12, 158 CrossRef CAS PubMed.
- S. C. Warren, K. Voïtchovsky, H. Dotan, C. M. Leroy, M. Cornuz, F. Stellacci, C. Hébert, A. Rothschild and M. Grätzel, Nat. Mater., 2013, 12, 842 CrossRef CAS.
- T. H. Jeon, G.-h. Moon, H. Park and W. Choi, Nano Energy, 2017, 39, 211–218 CrossRef CAS.
- A. J. E. Rettie, W. D. Chemelewski, D. Emin and C. B. Mullins, J. Phys. Chem. Lett., 2016, 7, 471–479 CrossRef CAS PubMed.
- H. Zhang, Y. K. Kim, H. Y. Jeong and J. S. Lee, ACS Catal., 2019, 9, 1289–1297 CrossRef CAS.
- H. Zhang, W. Y. Noh, F. Li, J. H. Kim, H. Y. Jeong and J. S. Lee, Adv. Funct. Mater., 2019, 29, 1805737 CrossRef.
- H. Zhang, J. H. Park, W. J. Byun, M. H. Song and J. S. Lee, Chem. Sci., 2019, 10, 10436–10444 RSC.
- H. Zhang, D. Li, W. J. Byun, X. Wang, T. J. Shin, H. Y. Jeong, H. Han, C. Li and J. S. Lee, Nat. Commun., 2020, 11, 4622 CrossRef PubMed.
- J. Y. Kim, J.-W. Jang, D. H. Youn, G. Magesh and J. S. Lee, Adv. Energy Mater., 2014, 4, 1400476 CrossRef.
- C. Lohaus, A. Klein and W. Jaegermann, Nat. Commun., 2018, 9, 4309 CrossRef PubMed.
- L. Luo, W. Tao, X. Hu, T. Xiao, B. Heng, W. Huang, H. Wang, H. Han, Q. Jiang, J. Wang and Y. Tang, J. Power Sources, 2011, 196, 10518–10525 CrossRef CAS.
- X. Wang, L.-L. Wang, D. Guo, L.-L. Ma, B.-L. Zhu, P. Wang, G.-C. Wang, S.-M. Zhang and W.-P. Huang, Catal. Today, 2019, 327, 182–189 CrossRef CAS.
- G. Yang, Z. Jiang, H. Shi, M. O. Jones, T. Xiao, P. P. Edwards and Z. Yan, Appl. Catal., B, 2010, 96, 458–465 CrossRef CAS.
- A. Annamalai, H. Lee and S. Choi, Sci. Rep., 2016, 6, 23183 CrossRef CAS.
- A. G. Tamirat, W.-N. Su, A. A. Dubale, H.-M. Chen and B.-J. Hwang, J. Mater. Chem. A, 2015, 3, 5949–5961 RSC.
- J. Wang, C. Du, Q. Peng, J. Yang, Y. Wen, B. Shan and R. Chen, Int. J. Hydrogen Energy, 2017, 42, 29140–29149 CrossRef CAS.
- J. Cai, J. Liu, Z. Gao, A. Navrotsky and S. L. Suib, Chem. Mater., 2001, 13, 4595–4602 CrossRef CAS.
- J. Y. Kim, G. Magesh, D. H. Youn, J.-W. Jang, J. Kubota, K. Domen and J. S. Lee, Sci. Rep., 2013, 3, 2681 CrossRef PubMed.
- S. Kment, P. Schmuki, Z. Hubicka, L. Machala, R. Kirchgeorg, N. Liu, L. Wang, K. Lee, J. Olejnicek, M. Cada, I. Gregora and R. Zboril, ACS Nano, 2015, 9, 7113–7123 CrossRef CAS PubMed.
- J. Xiao, F. Zhao, J. Zhong, Z. Huang, L. Fan, L. Peng, S.-F. Zhou and G. Zhan, Chem. Eng. J., 2020, 402, 126163 CrossRef CAS.
- Y.-S. Hu, A. Kleiman-Shwarsctein, A. J. Forman, D. Hazen, J.-N. Park and E. W. McFarland, Chem. Mater., 2008, 20, 3803–3805 CrossRef CAS.
- A. M. Jubb and H. C. Allen, ACS Appl. Mater. Interfaces, 2010, 2, 2804–2812 CrossRef CAS.
- J. Xie, W. Liu, J. Xin, F. Lei, L. Gao, H. Qu, X. Zhang and Y. Xie, ChemSusChem, 2017, 10, 4465–4471 CrossRef CAS PubMed.
- R. Mazzaro, S. Boscolo Bibi, M. Natali, G. Bergamini, V. Morandi, P. Ceroni and A. Vomiero, Nano Energy, 2019, 61, 36–46 CrossRef CAS.
- A. Srivastav, A. Verma, A. Banerjee, S. A. Khan, M. Gupta, V. R. Satsangi, R. Shrivastav and S. Dass, Phys. Chem. Chem. Phys., 2016, 18, 32735–32743 RSC.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy, Perkin Elmer Co., Eden Prairie, Minnesota, USA, 1992 Search PubMed.
- P. Zhang, T. Tachikawa, M. Fujitsuka and T. Majima, ChemSusChem, 2016, 9, 617–623 CrossRef CAS PubMed.
- A. Pu, J. Deng, M. Li, J. Gao, H. Zhang, Y. Hao, J. Zhong and X. Sun, J. Mater. Chem. A, 2014, 2, 2491–2497 RSC.
- M. Li, Y. Yang, Y. Ling, W. Qiu, F. Wang, T. Liu, Y. Song, X. Liu, P. Fang, Y. Tong and Y. Li, Nano Lett., 2017, 17, 2490–2495 CrossRef CAS PubMed.
- J. H. Kim, Y. J. Jang, J. H. Kim, J.-W. Jang, S. H. Choi and J. S. Lee, Nanoscale, 2015, 7, 19144–19151 RSC.
- F. L. Formal, M. Grätzel and K. Sivula, Adv. Funct. Mater., 2010, 20, 1099–1107 CrossRef.
- J. Xiao, H. Huang, Q. Huang, L. Zhao, X. Li, X. Hou, H. Chen and Y. Li, J. Catal., 2017, 350, 48–55 CrossRef CAS.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef CAS PubMed.
- F. Cardon and W. P. Gomes, J. Phys. D: Appl. Phys., 1978, 11, L63–L67 CrossRef CAS.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 122, 6549–6552 CrossRef.
- H. Dotan, K. Sivula, M. Grätzel, A. Rothschild and S. C. Warren, Energy Environ. Sci., 2011, 4, 958–964 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2na00029f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |