
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene-based phenformin carriers for cancer cell treatment: a comparative study between oxidized and pegylated pristine graphene in human cells and zebrafish†
Abdelnour
Alhourani
 *a,
Jan-Lukas
Førde
bc,
Mojdeh
Nasrollahzadeh
a,
Lutz Andreas
Eichacker
a,
Lars
Herfindal
b and
Hanne Røland
Hagland
*a
*a,
Jan-Lukas
Førde
bc,
Mojdeh
Nasrollahzadeh
a,
Lutz Andreas
Eichacker
a,
Lars
Herfindal
b and
Hanne Røland
Hagland
*a
aDepartment of Chemistry, Biosciences and Environmental Engineering, University of Stavanger, Stavanger, Norway. E-mail: hanne.r.hagland@uis.no
bCentre for Pharmacy, Department of Clinical Science, University of Bergen, Bergen, Norway
cDepartment of Internal Medicine, Haukeland University Hospital, Bergen, Norway
First published on 9th February 2022
Abstract
Graphene is an attractive choice for the development of an effective drug carrier in cancer treatment due to its high adsorption area and pH-responsive drug affinity. In combination with the highly potent metabolic drug phenformin, increased doses could be efficiently delivered to cancer cells. This study compares the use of graphene oxide (GO) and polyethylene glycol stabilized (PEGylated) pristine graphene nanosheets (PGNSs) for drug delivery applications with phenformin. The cytotoxicity and mitotoxicity of the graphene-based systems were assessed in human cells and zebrafish larvae. Targeted drug release from GO and PGNSs was evaluated at different pH levels known to arise in proliferating tumor microenvironments. PGNSs were less cytotoxic and mitotoxic than GO, and showed an increased release of phenformin at lower pH in cells, compared to GO. In addition, the systemic phenformin effect was mitigated in zebrafish larvae when bound to GO and PGNSs compared to free phenformin, as measured by flavin metabolic lifetime imaging. These results pave the way for improved phenformin-based cancer therapy using graphene nano-sheets, where PGNSs were superior to GO.
Introduction
Despite recent advances in targeted therapies, resistance to chemotherapy is a primary reason for cancer relapse.1,2 Therapeutics that are effective against chemo-resistant cancer cells, such as biguanides, have been extensively studied in cancer therapy to reduce cancer relapse.3,4 Metformin, a dimethyl biguanide derivative, and a type 2 diabetes drug,2,4 has been suggested as a potential adjuvant cancer treatment agent after meta-analysis studies of diabetic patients showed lower cancer incidence compared to diabetic cancer patients that did not use metformin.2In vitro experiments have demonstrated its anti-proliferative effect in various cancer cell lines.5,6 A phenethyl biguanide, known as phenformin, is a more potent derivative,7–9 but its use was discontinued after incidences of lactic acidosis in diabetic patients.9 However, its potential re-use as a metabolic anti-cancer drug has been argued to offset its side effects,10,11 and is currently being evaluated in a clinical trial (NCT03026517).Biguanide derivatives act by targeting the mitochondrial electron transport chain (ETC), limiting the cell's ability to utilize oxidative phosphorylation for energy production,2,11–13 thus, depleting the tricarboxylic acid (TCA) cycle intermediates, which are critical for tumor growth.7,14 ETC complexes are regulated by multiple co-enzymes, such as flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN).15,16 The redox state and subsequent fluorescence of FAD and FMN are tied to the electron flow through ETC during normal ATP production,17–19 making the fluorescence of these co-enzymes sensitive to ETC disruption.20–22 Interestingly, inhibition of complex I of the ETC is associated with a metabolic shift towards alternative energy pathways, such as glycolysis, leading to an increased response to chemotherapy.2,11–13 Therefore, biguanide has the potential to revert chemotherapy resistance,11,13 enhancing treatment outcomes.3,23
A challenge in utilizing metabolic anti-cancer drugs is their inability to target malignant cells innately, showing no preference towards proliferating cells, contrary to most chemotherapeutic agents, which target actively proliferating cells, malignant and non-malignant, giving rise to their common side effects.24 Targeted nano-sized drug carriers can thus be used to increase the concentration of metabolic agents at the tumor site by exploiting the dysfunctional vasculature often found in fast-growing tumors, causing an enhanced permeability and retention (EPR) effect.25–28 Another feature of solid tumors is a harsh surrounding microenvironment, often with areas of lower pH due to increased hypoxia.29–31
The utilization of graphene in drug delivery has been an attractive area for investigation since its discovery.30,32–34 Compared to many other nano-sized drug delivery platforms, graphene has many advantages, such as high absorption area35,36 to efficiently load higher amounts of the drug, and pH responsiveness.37–39
The oxidized form of graphene, graphene oxide (GO), is the most studied form of graphene in drug carrier applications due to its affordable production and ability to disperse in aqueous solutions.29 However, previous biotoxicity studies showed that GO induced the generation of reactive oxygen species (ROS), leading to cytotoxicity in several cellular models40–43 and in zebrafish.44–48 The extent of cytotoxicity has been shown to correlate with the oxygen content of GO in human umbilical vein endothelial cells (HUVECs).49 Additionally, residual impurities from the oxidation process during the production of GO50–52 could also be a source of toxicity.
Contrary to GO, pristine graphene has been less studied as a nano-sized drug carrier due to its relatively complicated production process.53 Furthermore, its hydrophobicity54 leads to low stability in aqueous solutions. Recently, higher yield production processes of pristine graphene have emerged,55 including a plasma-enhanced CVD (PE-CVD) production method, resulting in low-defect graphene sheets with low oxygen content56,57 and less chemical impurities that would make it a more promising drug carrier candidate compared to GO.
Drug delivery of phenformin using a nano-sized delivery system has been previously demonstrated using micelles.8 However, we just recently demonstrated that phenformin can be loaded and released in a pH-responsive manner using graphene as a nano-dug carrier.39 Here, we wanted to test the applicability of polyethylene glycol stabilized (pegylated) pristine graphene sheets (PGNSs) compared to GO as a potential drug carrier of the metabolic drug phenformin in biological systems. In our assessment, we used cell lines of different origins and growth properties to test the effect, considering that the graphene-based drug carriers may be administered intravenously. In addition, we evaluated the zebrafish tolerance towards free phenformin compared to loading on GO and PGNSs by tracking the fluorescence lifetime of FAD and FMN.
Results and discussion
Particle stability and phenformin retention capacity of GO and PGNSs
To enhance compatibility with aqueous solutions, covalent attachment of amine-terminated polyethylene glycol (PEG) onto PE-CVD graphene was performed as described in our previous work39 to produce pegylated graphene nano-sheets (PGNSs) which were further assessed for loading and binding capacity for phenformin as described in detail previously. We compared the produced PGNSs to commercially available graphene oxide (GO).To determine the retention ratio of phenformin on GO and PGNSs, we used increasing concentrations of the drug carriers and tracked the diffusion of phenformin through an impermeable membrane over 96 hours. We found that both GO and PGNSs were able to retain on average a weight/weight percentage (wt%) of 10% phenformin/graphene over the measurement time (Fig. S1†).
The stability of GO and PGNSs was assessed in a human cell culture medium by measuring changes in the size and zeta potential. Dynamic light scattering measurements (DLS) of 100 μg mL−1 suspensions of GO and PGNSs in Dulbecco's modified essential medium (DMEM) with 10% fetal bovine serum (FBS) were performed 1 hour after preparation and again after five days.
GO had higher polydispersity in DMEM compared to PGNSs, as two distinct size distributions were observed for GO on the day of preparation (day zero) (Fig. 1a and Table S1†), whereas the intensity derived size of PGNSs maintained a stable distribution over the five-day period under the same conditions (Fig. 1b and Table S1†). The larger size-intensity derived histograms seen on day five for GO indicated the formation of agglomerates.
 | ||
| Fig. 1 Stability of GO and PGNSs in complete cell culture media. Intensity-weighted size histograms of GO (a) and PGNSs (b) were obtained via dynamic light scattering (DLS) at 37 °C with a five day interval. Normalized data are displayed as histogram averages (N = 3). | ||
While DLS is not able to report accurate size distributions of non-spherical materials, the measured trends would indicate a change in size over time. Contrary to GO, there was no indication of agglomeration of PGNSs at either of the time points measured. The cause of GO agglomeration could be the higher ionic strength of the culture media than that of DiH2O. Ions can disturb the repulsive stabilization mechanism in GO, while the sterically stabilized PGNSs remain unaffected.39,58–60 The instability of GO in physiological solutions has previously been shown to be caused by the “charge screening effect”,61 thus resulting in agglomeration. This agglomeration can be mitigated to some extent using FBS which was shown to lower GO cytotoxicity in cell models.40
GO is more cytotoxic than PGNSs in adherent and suspension cell models
The biocompatibility of a drug carrier is a critical aspect of its safe use in vivo to minimize the side effects of loaded drugs effectively. The cytotoxicity of GO and PGNSs was therefore tested in three cell models representing different tissue types that would be exposed to the graphene drug carrier if used in vivo. MOLM-13 is a non-adherent myeloid cell model representing white blood cells; HUVECs are endothelial cells, and the colorectal cancer (CRC) cell line SW948 represents cancer cells originating from adenocarcinoma. The viability of these cell lines was assessed after 48 hours of exposure to GO and PGNSs (Fig. 2).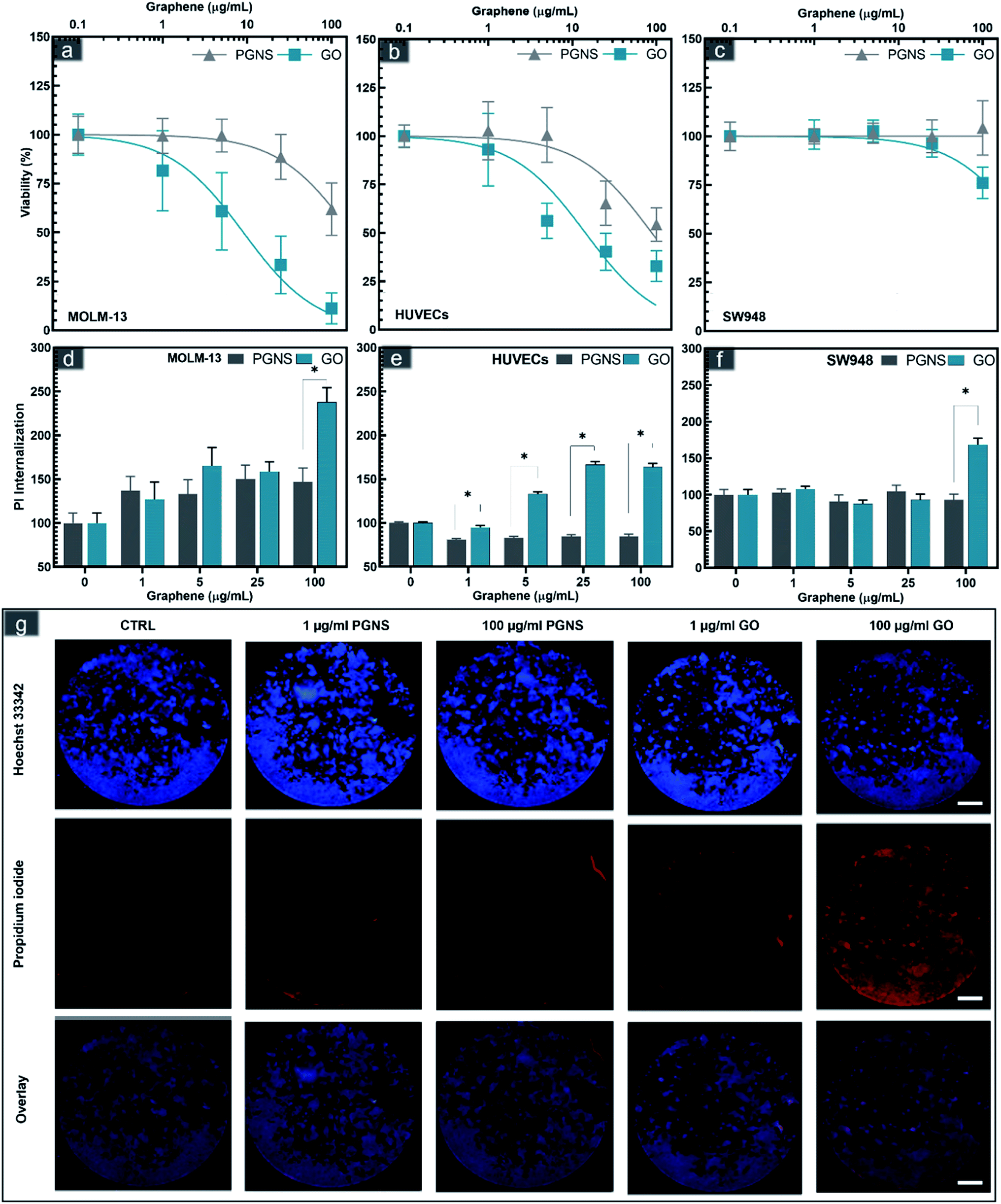 | ||
| Fig. 2 Cytotoxicity of PGNSs (grey) and GO (blue) in MOLM-13 cells (a and d), HUVECs (b and e), and SW948 cells (c and f) using CCK8 viability assay expressed as mean ± SEM; N = 8 for HUVECs and N = 9 for SW948 and MOLM-13 (a–c); 95% CI ranges of IC50 values are displayed as insets within for each cell line. Membrane internalization of propidium iodide (Pi) normalized by Hoechst counterstaining is shown as bars representing means normalized to control ± SEM, N = 5 (d–f). Statistically significant changes between the same concentrations of PGNS and GO using a Student's t-test where significant differences are denoted as * for P < 0.05. Widefield fluorescence imaging of the whole well Pi and Hoechst stained SW948 cells after 48 hours of GO and PGNS exposure (g). | ||
Cytotoxic effects were observed in all three cell lines, and the myeloid cell model, MOLM-13 was the most sensitive. GO induced significant cytotoxicity compared to control at concentrations as low as 5 μg mL−1 in MOLM-13 cells and the cytotoxicity increased in a concentration-dependent manner (Fig. 2a). PGNS induced less toxicity in MOLM-13 cells, but at the highest concentration of 100 μg mL−1, we found a significant decrease in viability compared to control.
GO was also more cytotoxic than PGNS towards HUVECs. The viability of HUVECs (Fig. 3b) was below 50% at the higher GO doses, whereas the viability did not decrease below 50% in PGNS treated cells even at a concentration of 100 μg mL−1. The least affected cells were of the CRC model SW948, where viability was only reduced to 75% at 100 μg mL−1 of GO, whereas there was no decrease in viability after PGNS exposure compared to control (Fig. 2c). The results show that GO induced higher toxicity in all cell models (Fig. 2a–c) at all concentrations compared to PGNS. Furthermore, PGNS did not cause significant toxicity in treatment concentrations as high as 25 μg mL−1, compared to control, where lower concentrations showed no discernable differences from control. Finally, to exclude the possibility of endotoxin contamination in the GO or PGNS solutions used to influence the viability measurements, we performed a limulus amebocyte lysate (LAL) based endotoxin assay on both GO and PGNS filtrates. Endotoxin levels were found to be well below the acceptable concentration (0.5 EU per mL) in both filtrates. Thus the decrease in viability we observed was not caused by the presence of endotoxins in the GO and PGNS filtrates.
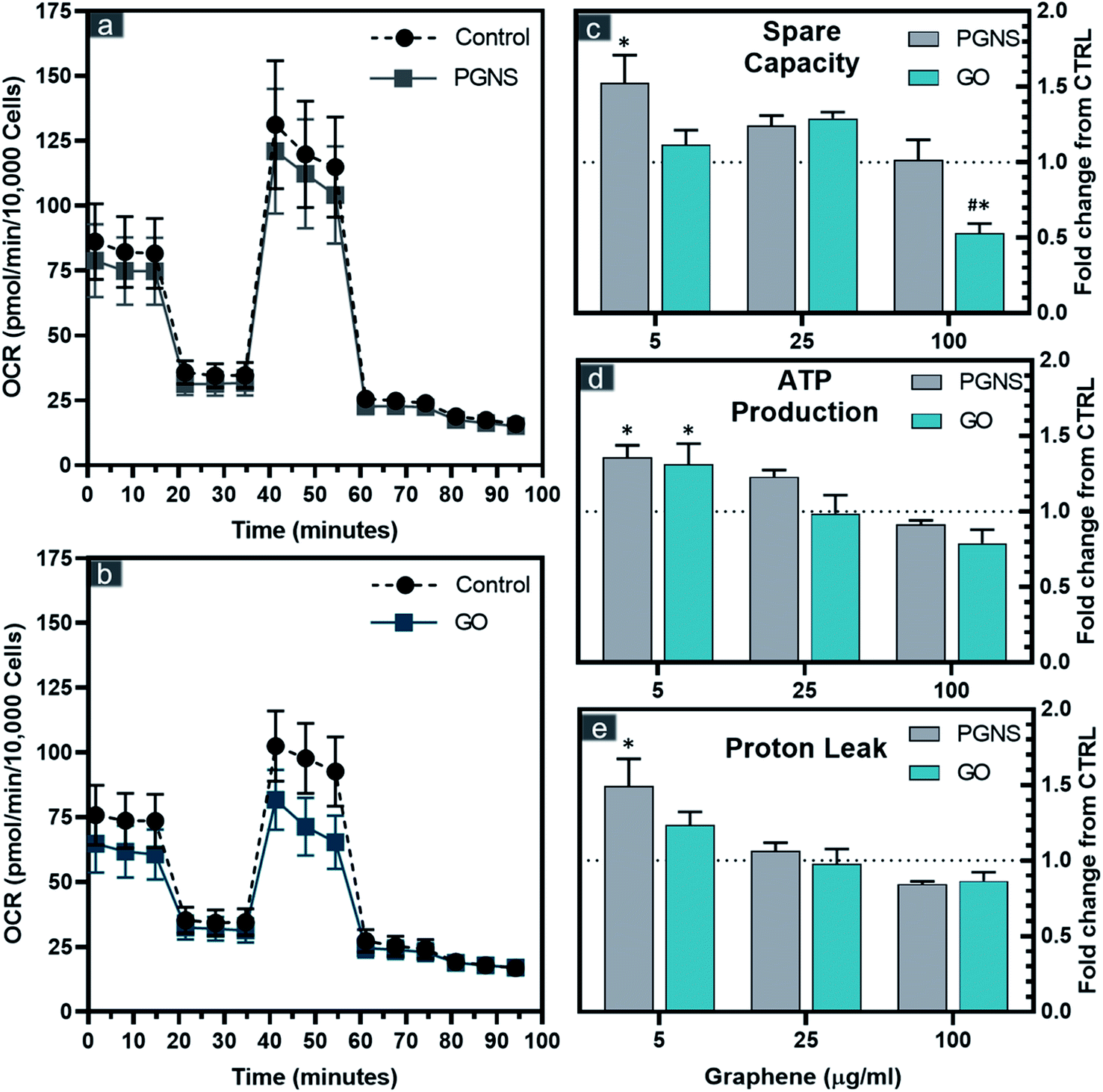 | ||
| Fig. 3 Bioenergetic parameters of SW948 cells after 48 hours of exposure to GO and PGNSs. Oxygen consumption rates (OCR) of SW948 after 100 μg mL−1 exposure to GO (a) and PGNSs (b) are shown with the sequential injection ETC inhibitors of oligomycin, CCCP, rotenone, and antimycin A. Calculated spare capacity (c), ATP production (d), and proton leak (e) after exposure to GO and PGNSs are displayed as fold-changes from control ± SEM, N = 7 for 5 and 25 μg mL−1 concentrations and N = 9 for 100 μg mL−1 concentrations from 3 independent experiments. The dotted line represents no change from control. Statistical significance (P < 0.05) with respect to graphene-free CTRL using a two-way ANOVA test is denoted as * and between GO and PGNSs for the same concentrations is denoted as #. | ||
The increase in cytotoxicity was accompanied by increased propidium iodide (PI) internalization, a measurement of cell membrane damage (Fig. 2d–g and S2†). Previous studies have suggested that graphene sheets may pierce the cell membrane,43,62,63 possibly causing membrane disintegration, which is supported by our findings. The higher toxicity in MOLM-13, compared to the other two adherent cell models, especially in the case of GO, could be due to their increased endocytotic capacity leading to an increase in reactive oxygen species as has been described previously for hematological cells.64 In contrast, the significantly lower toxicity seen in MOLM-13 cells by PGNS could be due to the polyethylene glycol (PEG) functionalization as well as the lower oxygen content, both of which are shown to reduce endocytosis.65 The covalent PEGylation method used in PGNSs relies on the amidification of the edge dominant carboxyl groups,57,66 which could result in a decreased uptake in cells.65
The data converge on oxidative stress, mediated by increased oxygen content, is the primary mechanism for graphene-mediated cytotoxicity.41,49,67,68 PEGylation has been shown to increase the in vivo biocompatibility of GO in mice, associated with normal hematology markers.69 In contrast, non-PEGylated GO has been shown to trigger hemolysis when mixed with blood samples.43 Thus, the toxicity in adherent cell models also shows that PEGylation and lower oxygen content offset the toxicity response towards higher concentrations of graphene, which could be pivotal for the tolerance to the drug carrier.
Mitochondrial metabolic effects of GO and PGNSs on colon cancer cells
The correlation found between reduced viability and membrane integrity in GO-treated cells could potentially be caused by ROS, whereas the PGNS treatment did not result in loss of viability to the same extent. To test if there was a possible mitochondrial involvement in the treatment response, we performed an extracellular flux analysis probing the different electron chain complexes after 48 hours of GO and PGNS exposure (Fig. 3).PGNS treatment at 5 μg mL−1 induced a significant change in mitochondrial maximal respiration compared to control, whereby spare capacity increased to 1.5-fold that of control (Fig. 3c). Although a slight increase in spare respiratory capacity was seen at the lowest concentration of GO, this was not statistically significant with respect to control. A significant decrease in spare capacity was induced upon the highest concentration treatment of GO at 100 μg mL−1, where the same concentration of PGNSs induced no change (Fig. 3c). There was a minor change in the coupling efficiency after GO and PGNS treatment as ATP production was only affected at the highest dose of treatment (Fig. 3d). The increase in spare capacity could be due to an increased electron transport chain (ETC) uncoupling from the TCA cycle, consequently achieving higher proton leak levels measured without a reduction in ATP production (Fig. 3d and e). The proton leak was particularly increased after 5 μg mL−1 PGNS exposure up to 1.5-fold that of the control, indicating that low concentrations of PGNSs affect the mitochondria of SW948, without compromising viability (Fig. 1c). The overall elevated bioenergetic parameters observed after the exposure to 5 and 25 μg mL−1 concentrations of graphene indicate a mitochondrial stress response.
The mitotoxicity of graphene has been previously shown to prompt a dose-dependent depolarization of the membrane potential (MMP) by GO42 and pristine graphene,70 both leading to increased intracellular ROS.42,70 These effects are claimed to be a direct consequence of the inhibition of ETC complexes by graphene.71 The mitotoxicity of graphene has been previously shown to correlate with increased oxygen levels, where the GO mediated decrease in mitochondrial activity was 12-fold that of non-oxidized graphene.67 This corresponds with our data showing that PGNSs are better tolerated by mitochondria than GO.
Phenformin delivery using GO and PGNSs in cancer cells
To assess the effectiveness of graphene loaded phenformin in vitro, viability of SW948 cancer cells was measured after 24 and 48 hours of incubation with increasing concentrations of phenformin (2–250 μM) both in the free form and loaded to GO and PGNSs at 10 wt%.The viability of SW948 cells in response to GO or PGNS loaded phenformin was equal to that of the cells exposed to the free drug after 48 hour exposure (Fig. 4). However, upon 24 hour exposure, there was a differential response to the different treatments. These results indicate that phenformin uptake in cells is unhindered by loading the drug onto graphene, at least over longer exposure times such as 48 hours. The cytotoxicity results were further confirmed using a modified LDH assay (Fig. S3†) over 48 hours.
 | ||
| Fig. 4 The cytotoxicity of phenformin-loaded GO and PGNS on SW948 cancer cells. Cellular viability of SW948 cells after 24 and 48 hours of treatment with free phenformin (2–250 μM) compared with the same molar concentrations of phenformin loaded GO and PGNSs at 10 wt% measured by CCK8 viability assay. The viability is presented as an averaged ratio from control ± SEM, and N = 7 from 3 independent experiments. | ||
Phenformin may be actively transported into cells via organic cation transporters,72 although the exact mechanism of membrane transport is not yet clear.73 As we found a differential response after 24 hour exposure in cancer cells, we wanted to test whether acute short exposure of cells to free phenformin treatment would induce a clearer effect compared to GO and PGNS retained drug (Fig. 5). Here, we treated cells for 2 hours using a high phenformin concentration (200 μM) before removing the drug and replenished with a fresh cell culture medium and performed a viability assay after another 24 hours. Furthermore, we simulated the drop in pH that is normally seen after 24 hour drug exposure in cells by testing the drug response at two different pH levels of 6 and 7.4 before returning the cells to the original 7.4 pH medium for the 24 hour period.
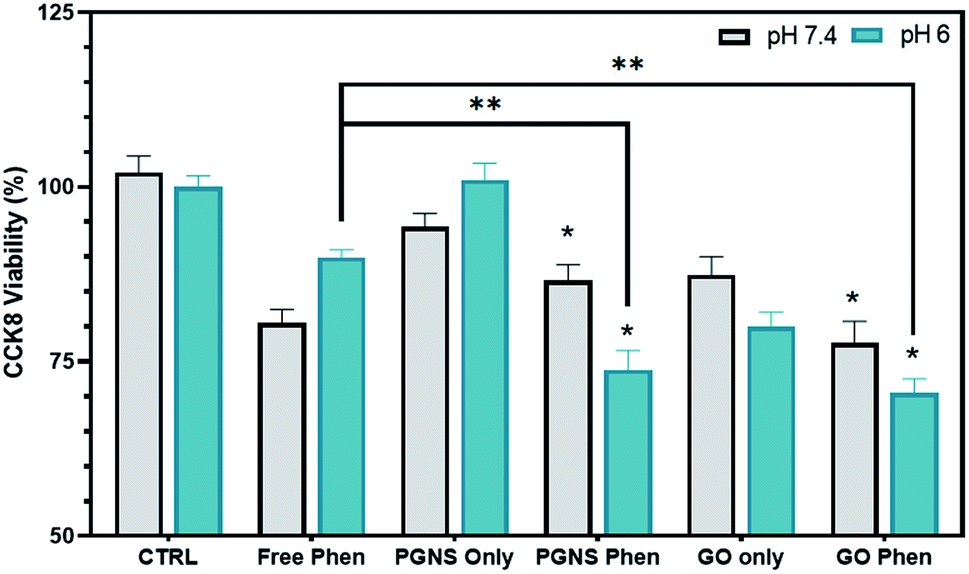 | ||
| Fig. 5 pH effect on the release of phenformin from GO and PGNSs. Cytotoxicity after a prompt 2 hour exposure to 10 wt% free and bound phenformin at pH 7.4 and pH 6 in SW948 cells (N = 9 ± SEM, P < 0.05 significance via a two-way ANOVA is denoted as * for a change from the non-phenformin CTRL and ** for a significant change from free phenformin at the same pH). | ||
At pH 7.4, no significant difference in toxicity was observed between the free phenformin drug and PGNS and GO loaded phenformin exposure (Fig. 5). However, GO only exposure did induce a significant decline in viability (20%) compared to control that was not seen in the PGNS treated cells (Fig. 5). Interestingly, the viability at pH 6 was significantly decreased after PGNS- and GO-loaded phenformin delivery compared to the free drug, at 74% and 71% versus 90% (P < 0.05), respectively. Furthermore, at pH 6, there was a 20% reduction in viability by GO treatment alone, which was not seen in PGNSs. The increased cytotoxic effect of graphene-loaded phenformin at pH 6 compared to that at pH 7 (Fig. 5) is of interest as it could reserve a higher proportion of the drug to be released in tumor microenvironments where pH is lower than physiological levels. In GO-loaded phenformin, the increased toxicity could be a result of both phenformin and GO, as GO on its own induced toxicity (Fig. 5). While toxicity in cancer cells is considered desirable, possible GO toxicity in other cells as measured in normal HUVECs (Fig. 3b) would be less desirable for a nanocarrier. Interestingly, the phenformin loaded PGNSs showed increased toxicity at pH 6 compared to the free drug at the same pH. This is highly desirable as a pH-responsive drug release would protect normal cells growing at pH 7.4, but more importantly target cancer cells growing in more hostile conditions of lower pH.74,75 This pH-responsive effect was not significant in GO, giving PGNSs a clear advantage as the more promising nano-drug carrier.
Maintaining a continuous drug exposure in cancer treatment is important, and often requires repeated dosage over time. As phenformin is known to act on mitochondria,7,76 we wanted to measure the effect of phenformin adsorption on PGNSs and GO in real-time as a function of cellular changes in oxygen consumption rates (OCR). This was accomplished by measuring the OCR levels in SW948 cells after a two-phase exposure regimen of free and PGNS- or GO-loaded phenformin. We found that PGNS- and GO-loaded phenformin caused a decrease in oxygen consumption over time compared to the response seen during the free drug exposure (Fig. 6). Although cells exposed to the PGNS- and GO-loaded phenformin had a delayed decline in the oxygen consumption compared to the free drug, this effect was not statistically significant, indicating that the drug was reaching its target.
 | ||
| Fig. 6 Oxygen consumption rates (OCR) over 250 min of SW948 after the acute injection of free phenformin, PGNS–phenformin, or GO–phenformin towards an end concentration of 600 μM per well distributed over 2 injections after 15 and 100 min (N = 4 ± SEM, from 2 independent experiments). | ||
Metabolic FLIM of phenformin treated zebrafish larvae
To test whether the retained drug effect seen in in vitro cell models could be further assessed in a more complex in vivo model system, we used the transparent zebrafish larvae (Danio rerio) “Casper”.77 Specifically, we wanted to assess if the phenformin drug bound to GO and PGNSs would reduce effects compared to free phenformin by measuring changes in the fluorescence lifetime of mitochondrial flavins. Phenformin has previously been reported to cause yolk sac edema in zebrafish larvae after aquatic exposure.78 Furthermore, it was found to accumulate in the liver, causing toxicity after prolonged aquatic exposure in adult zebrafish.79Phenformin was injected as a free or graphene-bound drug into the yolk sac of zebrafish larvae and monitored for 24 hours after injection (Fig. 7). No mortality was recorded among the injected zebrafish at 10 wt%, indicating that the selected phenformin concentration did not exert acute toxic effects within 24 hours of treatment. However, higher concentrations of free phenformin induced dose dependent mortality (Fig. S4†).
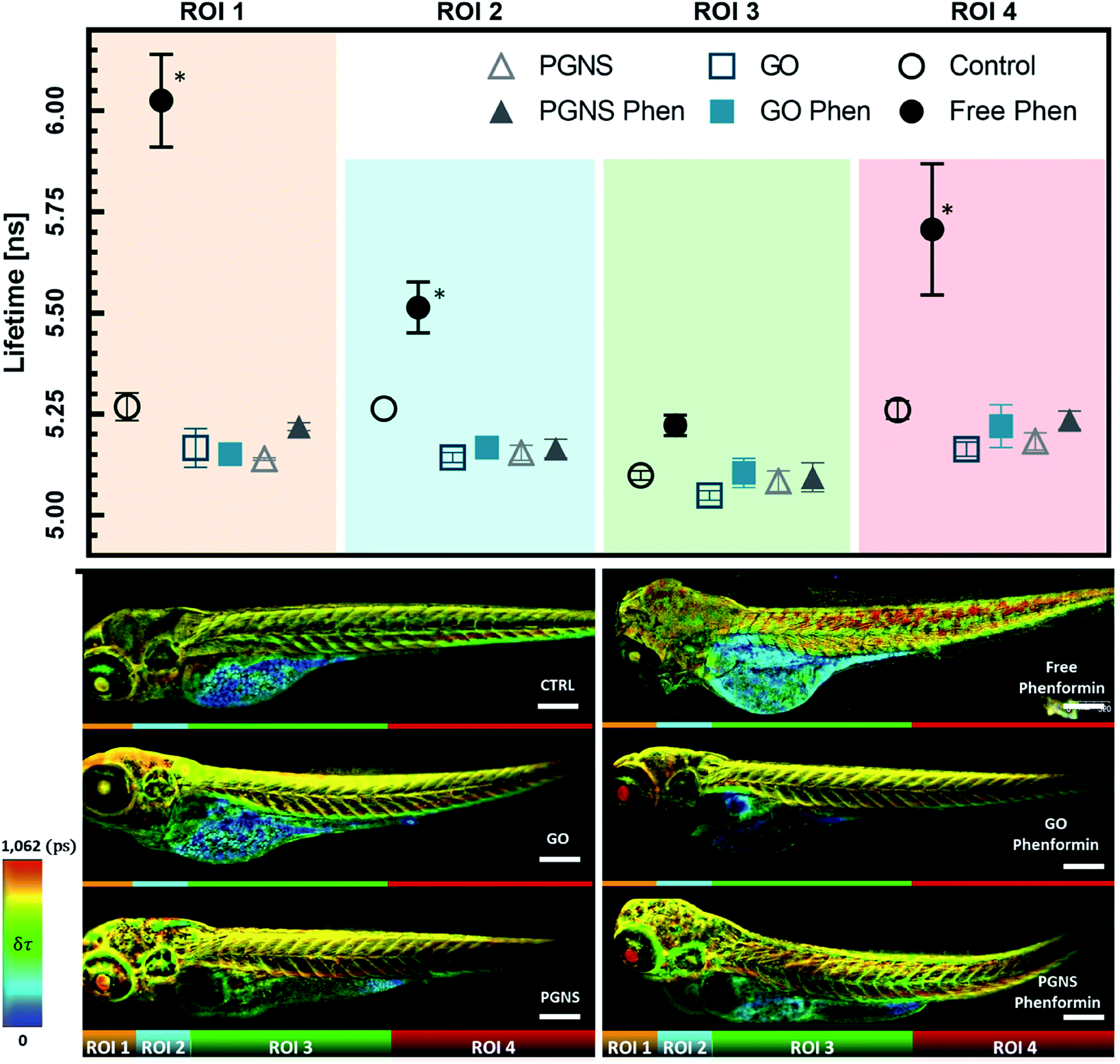 | ||
| Fig. 7 Label-free metabolic fluorescence lifetime, gated for FAD and FMN in 4 dpf zebrafish larvae segmented into four main regions of interest. Time-gated (>10 ns lived photons, after a 470 nm excitation pulse) amplitude weighted fluorescence lifetime of free and graphene-loaded phenformin; values are represented as mean ± SEM and N = 3; significant changes from CTRL via a two-way ANOVA (P < 0.05) are denoted as * (top). Representative Z-maximum projection of zebrafish images for each condition overlayed with a rainbow pseudo-colored overall lifetime mask denoting lifetime change (bottom). | ||
Fluorescence lifetime imaging (FLIM) was used to track endogenous fluorescence of mitochondrial flavins in zebrafish larvae. The fluorescence spectra of FAD and FMN overlap, leading to indistinguishable emission.80 However, their recorded fluorescence lifetime (τ) varies significantly, with FMN fluorescence being almost twice as long-lived as FAD and exhibiting a 10-fold higher intensity.17,81,82 The τ of both FAD and FMN is much longer-lived than that of other reported endogenously fluorescing molecules in zebrafish (Table S2†), and therefore can be conveniently time-gated for improved selectivity. A shift in the fluorescence lifetime of these co-factors indicates a disruptive ETC flow which would be an expected response after phenformin treatment.
We found that zebrafish larvae, treated with the free phenformin drug, had a longer fluorescence lifetime (τ) than control (Fig. 7), which could be due to an increase of τ from FAD in response to decreasing intracellular pH,83 a subsequent effect of high lactate levels after increased glycolysis.76 Additionally, this could be due to a higher fluorescence contribution of FMN, a redox-active group noncovalently bound to complex I.15 FMN would normally be reduced by NADH to FMNH2 during electron transport before oxidation restores its affinity to complex I of the ETC.84 As phenformin is known to inhibit complex I,76 FMNH2 could become dissociated,84–86 unable to bind its oxidized form to complex I,22,85 which can contribute to longer τ due to its fluorescent state.17 Therefore, a prolonged τ based on the FAD and FMN endogenous fluorescence would be indicative of phenformin mitochondrial effects on complex I, whereby a reduced τ in the GO and PGNS bound phenformin treated fish compared to the free drug would indicate a retained toxicity of phenformin.
To see if the changes in τ of FAD and FMN correspond to certain areas of the zebrafish larvae, the acquired FLIM images were segmented into four regions of interest (ROIs), segmented by straight lines to avoid biases when analyzing the different zebrafish larvae, where ROI 1 included the eye and parts of the central nervous system (CNS), ROI 2 included an area with the parts of the liver, heart, and CNS, ROI 3 mainly encompasses skeletal muscles together with the yolk sac, GI tract, and swim bladder, whereas ROI 4 mainly included the skeletal muscle from the tail area. The τ measurements were consistent across the control larvae, whereas there was a significant shift towards longer τ observed in ROIs 1, 2, and 4 after free phenformin drug injection, indicating a systemic effect of the drug exposure. Interestingly, this shift was not detected for the larvae injected with the phenformin drug loaded onto PGNSs or GO (Fig. 7), most likely due to its prolonged retention on graphene.
Conclusions
The delivery of phenformin for cancer treatment using graphene-based drug carriers has not previously been investigated. We found that GO and PGNSs by themselves had a differential cell response, where GO treatment induced a reduction in viability that was not seen when treating with PGNSs in three different cell models. The toxicity response from GO could be due to plasma membrane damage as it was accompanied by an increase in PI internalization, making PGNSs the better candidate for targeted drug delivery, avoiding possible toxic side effects on peripheral tissues. Moreover, the phenformin drug effect when loaded onto GO and PGNSs was significantly increased at pH 6 compared to free phenformin at the same pH when tested in cancer cells. Interestingly, the phenformin drug effects were retained completely when coupled to GO or PGNSs in a more advanced in vivo zebrafish model, even after 24 hours post-injection.This suggests that both GO and PGNSs were effectively delaying the drug effects of phenformin under normal “physiological pH” conditions in contrast to free phenformin, making them attractive candidates to study further for targeted drug delivery. Furthermore, the pH-responsive effect seen in our cell model system will be important to further explore using in vivo models with a tumor load for assessing targeted drug delivery in the future.
Experimental section
Preparation of PGNSs and GO
10 mg of PECVD graphene flakes (as obtained from CealTech AS) were dispersed in MilliQ water and sonicated in a glass vial for 10 minutes. To increase the carboxyl groups on the surface, the dispersion was adjusted to basic conditions and stirred at 55c for 4 hours. Afterward, the pH was brought back to 5.5 with HCl. Excess salts were removed by washing with water using 100 kDa MWCO concentrators. 50 mg of monofunctional 2k mPEG–amine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 wt%) was added and sonicated with the graphene for 5 min, and then cross-linking was performed using 1.5 mg of EDC under sonication for 30 min. The dispersion was left under constant agitation overnight. The excess EDC and PEG were removed through dialysis. After two days of dialysis, the PEG–graphene was repeatedly sonicated and washed using concentrator tubes until the filtrate had no clear peaks via UV-vis spectroscopy. The final concentration was estimated using UV-vis spectroscopy.
:5 wt%) was added and sonicated with the graphene for 5 min, and then cross-linking was performed using 1.5 mg of EDC under sonication for 30 min. The dispersion was left under constant agitation overnight. The excess EDC and PEG were removed through dialysis. After two days of dialysis, the PEG–graphene was repeatedly sonicated and washed using concentrator tubes until the filtrate had no clear peaks via UV-vis spectroscopy. The final concentration was estimated using UV-vis spectroscopy.
Phenformin retention time on GO and PGNSs (HPLC)
13.34 μg of phenformin was added to GO and PGNS dispersions containing 12.5, 25, and 50 μg of graphene in duplicates of 1 mL each, and then sonicated for 10 min and stirred gently overnight. The mixtures were then separated from a measurement compartment containing 300 μL of DiH2O by using a 20k MWCO regenerated cellulose membrane. 200 μL from the measurement compartment was taken at 6, 24, 48 and 96 hours and substituted with an equal volume of DiH2O. The samples were measured for free phenformin using high-performance liquid chromatography (Hitachi High Technologies, Tokyo, Japan) at 233 nm over a phenyl–hexyl, 2.7 μm column using a gradient of DiH2O and acetonitrile as the mobile phase.Stability in cell culture media
PGNSs and GO were dispersed in DMEM cell culture media at a concentration of 100 μg mL−1 by sonicating for 10 min and then left for one hour to equilibrate; 1 mL samples of each preparation were then analyzed using dynamic light scattering (DLS) for hydrodynamic radius and zeta potential using a Zetasizer Nano ZSP (Malvern Panalytical, Malvern, United Kingdom). The measurements were repeated again after five days to compare the stability over time.Assessment of endotoxins in GO and PGNSs
1 mL of GO and PGNSs at a 100 μg mL−1 concentration were filtered using 100000 MWCO Vivaspin columns. The endotoxin concentrations were then measured in the filtrate using a Pierce™ LAL Chromogenic Endotoxin Quantitation Kit according to the manufacturer's instructions.
Cell culture
Epithelial colorectal cancer cells SW948 from ETCC, hematopoietic acute myeloid leukemia cells MOLM-13 from the German Collection of Microorganisms and Cell Culture (DSMZ, Braunschweig, Germany), and human umbilical vein endothelial cells (HUVECs) from Lonza were maintained in 5 mM glucose complete DMEM (Corning, New York, USA), complete RPMI (Corning, New York, USA), and Endothelial Cell Growth Medium-2 (EGM-2) Lonza, Basel, Switzerland) culture media, respectively. Complete DMEM and RPMI contained 2 mM L-glutamine (Corning, New York, USA), 10% fetal bovine serum (FBS) (BioWest, Nuaillé, France), penicillin (100 U mL−1) and streptomycin (100 μg mL−1) (Merck Millipore Corporation, Burlington, USA). EGM-2 was supplemented with FBS 10.00 mL, hydrocortisone 0.20 mL, hFGF-B 2.00 mL, VEGF 0.50 mL, R3-IGF-1 0.50 mL, ascorbic acid 0.50 mL, hEGF 0.50 mL, GA-1000 0.50 mL, and heparin 0.50 mL. The cells were cultured and maintained under humidified conditions in a 5% CO2 incubator at 37 °C and passaged at approximately 80% confluency using 0.5% trypsin–EDTA for SW948 and MOLM-13 and 0.05% trypsin–EDTA for HUVECs. HUVECs were also maintained under passage 8 throughout this work.Cellular viability and membrane integrity assays
Cells were cultured at 15k cells per well in a 96 well cell culture plate and then incubated with 1, 5, 25, and 100 μg mL−1 of GO or PGNSs for 48 h. Adherent cells were left to attach overnight prior to graphene exposure.Seahorse mitostress test
The mitochondrial oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of SW948 cells were measured using a Seahorse XFp flux analyzer (Agilent Technologies, Santa Clara, USA). 15k cells were seeded in multiple XFp cell cultures and left overnight to attach prior to GO and PGNS exposure at 5, 25, and 100 μg mL−1 for 48 hours. Each run contained a triplicate of graphene-free control cells to use as a baseline for the measurement. Prior to measurement, wells were washed with PBS to remove the graphene and re-cultured in an unbuffered serum-free, phenol red-free DMEM (D5030, Sigma-Aldrich, St. Louis, USA). The DMEM medium was adjusted to pH 7.4 and supplemented with 2 mM L-glutamine (Corning, New York, USA) and 5 mM glucose (Sigma-Aldrich, St. Louis, USA), and the cells were left for one hour to equilibrate at 37 °C in a CO2-free incubator. After three 15 min cycles of basal OCR measurements, sequential in-well injections of oligomycin (3 μM), CCCP (0.25 μM), rotenone (1 μM), and antimycin A (1 μM) were performed. Each injection was also followed by three mixing and measuring cycles of the OCR and ECAR. After finishing the measurements, each plate was washed with PBS, and a 4% paraformaldehyde fixation agent was added to each well with the addition of 10 μg mL−1 Hoechst. The plates were then imaged using a Leica TCS SP8 confocal microscope (Leica Microsystems, Mannheim, Germany), and the number of cells was counted using the LAS X 2D analysis workflow.The OCR after acute phenformin exposure
The cells were seeded at 15k in multiple XFp cell culture plates and left overnight to attach prior to conducting the experiment. The following day, medium was removed and cells were washed with PBS before addition of unbuffered serum-free, phenol red-free DMEM (D5030, Sigma-Aldrich, St. Louis, USA). The DMEM medium was adjusted to pH 7.4 and supplemented with 2 mM L-glutamine (Corning, New York, USA) and 5 mM glucose (Sigma-Aldrich, St. Louis, USA), and the cells were left for one hour to equilibrate at 37 °C in a CO2-free incubator. The measurements included three basal oxygen readings (5 minutes apart) before addition of 100 μM free phenformin or phenformin loaded to GO or PGNSs to initiate an acute response. The decrease in oxygen consumption was monitored over 75 minutes before increasing the total concentration to 600 μM phenformin, with sampling of data every 20 minutes until completion of the experiment (250 minutes).Phenformin 2 hour exposure assay viability and cell integrity
Cells were cultured at 15k per well in a 96 cell culture plate and incubated with serial dilutions (2–250 μM) of free phenformin, PGNS–phenformin, and GO–phenformin at 10 wt%. After the duration of the exposure, cells were washed with PBS and incubated for 2 hours with complete cell media with added CCK8 before measuring viability. For the 2 hours of short-term exposure to the treatments listed above, the cells were rinsed and re-incubated in complete cell media for another 22 hours, whereby the last 2 hours included the CCK8 reagent, before measuring the absorbance as detailed above.Modified LDH assay
The same experimental parameters were used as in the viability assay, whereby 48 hour graphene exposure was performed before the cells were washed using PBS, before incubation with a LDH reagent and a lysis solution for 30 min. The reaction was stopped after 30 min using acetic acid, and LDH absorbance was measured at 650 nm using a SpectraMax Paradigm plate reader (Molecular Devices, San Jose, USA).FLIM analysis of zebrafish models
Zebrafish larvae were injected four days after fertilization (4df) with free phenformin or 10% wt graphene-loaded drug phenformin (∼1.5 ng per zebrafish) in their yolk sacs while the control was injected with equal volumes of DiH2O. A concentration of 7.1 nMol of phenformin (∼1.5 ng per zebrafish) was selected based on previous pilot experiments (data not shown) to not cause mortality or extreme toxicity. 24 hours post-injection, the fish were fixed in 2% paraformaldehyde and imaged using a Leica TCS SP8 falcon microscope. The fluorescence lifetime was recorded upon 470 nm pulsed laser excitation using a time-correlated single-photon counting instrumentation setup. The fluorescence lifetime decays were analyzed using a tail-fit model with three lifetime components for photons arriving between 3.5 and 45 ns and with two time components for time-gated analysis for photons arriving between 10 and 45 ns. The average lifetime of all components was then weighted by amplitude to calculate the fluorescence lifetime. All X2 values were close to 1, with no fit residuals observed.Statistical analysis
Statistical comparisons were made using GraphPad PRISM (version 8, GraphPad Software, Inc., USA) software with a two-way ANOVA to determine significant differences between several treatment groups. Post hoc corrections for multiple comparisons were applied according to recommendations by GraphPad for each experimental data set (Dunnett). A student's unpaired t-test was employed when only two groups were compared. The number of biological replicates (N) is given in the figures and legends, together with what was considered a statistically significant P-value and the standard error of the mean (SEM).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially funded by grant from the Western Norway Health Authorities to (grant no.: F-12533).References
- C. Denise, P. Paoli, M. Calvani, M. L. Taddei, E. Giannoni, S. Kopetz, S. M. A. Kazmi, M. M. Pia, P. Pettazzoni, E. Sacco, A. Caselli, M. Vanoni, M. Landriscina, P. Cirri and P. Chiarugi, 5-Fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits, Oncotarget, 2015, 6, 41706–41721 CrossRef PubMed.
- B. Viollet, B. Guigas, N. Sanz Garcia, J. Leclerc, M. Foretz and F. Andreelli, Cellular and molecular mechanisms of metformin: an overview, Clin. Sci., 2012, 122, 253–270 CrossRef CAS PubMed.
- X. Sui, Y. Xu, X. Wang, W. Han, H. Pan and M. Xiao, Metformin: A Novel but Controversial Drug in Cancer Prevention and Treatment, Mol. Pharmaceutics, 2015, 12, 3783–3791 CrossRef CAS PubMed.
- P. Sancho, E. Burgos-Ramos, A. Tavera, T. Bou Kheir, P. Jagust, M. Schoenhals, D. Barneda, K. Sellers, R. Campos-Olivas, O. Graña, C. R. Viera, M. Yuneva, B. Sainz and C. Heeschen, MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells, Cell Metab., 2015, 22, 590–605 CrossRef CAS PubMed.
- A. H. Alhourani, T. R. Tidwell, A. A. Bokil, G. V. Røsland, K. J. Tronstad, K. Søreide and H. R. Hagland, Metformin treatment response is dependent on glucose growth conditions and metabolic phenotype in colorectal cancer cells, Sci. Rep., 2021, 11, 10487 CrossRef CAS PubMed.
- W. K. Miskimins, H. J. Ahn, J. Y. Kim, S. Ryu, Y.-S. Jung and J. Y. Choi, Synergistic anti-cancer effect of phenformin and oxamate, PLoS One, 2014, 9, e85576 CrossRef PubMed.
- A. Janzer, N. J. German, K. N. Gonzalez-Herrera, J. M. Asara, M. C. Haigis and K. Struhl, Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10574–10579 CrossRef CAS PubMed.
- S. Krishnamurthy, V. W. L. Ng, S. Gao, M.-H. Tan and Y. Y. Yang, Phenformin-loaded polymeric micelles for targeting both cancer cells and cancer stem cells in vitro and in vivo, Biomaterials, 2014, 35, 9177–9186 CrossRef CAS PubMed.
- C. R. Chong and B. A. Chabner, Mysterious metformin, Oncologist, 2009, 14, 1178–1181 CrossRef CAS PubMed.
- L. M. Berstein, Modern approach to metabolic rehabilitation of cancer patients, Future Oncol., 2010, 6, 1313–1323 CrossRef CAS PubMed.
- P. S. Kharkar, Cancer Stem Cell (CSC) Inhibitors in Oncology-A Promise for a Better Therapeutic Outcome: State of the Art and Future Perspectives, J. Med. Chem., 2020, 63, 15279–15307 CrossRef CAS PubMed.
- J. L. Counihan, E. A. Grossman and D. K. Nomura, Cancer Metabolism: Current Understanding and Therapies, Chem. Rev., 2018, 118, 6893–6923 CrossRef CAS PubMed.
- B. Zhao, J. Luo, T. Yu, L. Zhou, H. Lv and P. Shang, Anticancer mechanisms of metformin: A review of the current evidence, Life Sci., 2020, 254, 117717 CrossRef CAS PubMed.
- T. Griss, E. E. Vincent, R. Egnatchik, J. Chen, E. H. Ma, B. Faubert, B. Viollet, R. J. DeBerardinis and R. G. Jones, Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis, PLoS Biol., 2015, 13, e1002309 CrossRef PubMed.
- C. D. Barker, T. Reda and J. Hirst, The flavoprotein subcomplex of complex I (NADH:ubiquinone oxidoreductase) from bovine heart mitochondria: insights into the mechanisms of NADH oxidation and NAD+ reduction from protein film voltammetry, Biochemistry, 2007, 46, 3454–3464 CrossRef CAS PubMed.
- T. Udhayabanu, A. Manole, M. Rajeshwari, P. Varalakshmi, H. Houlden and B. Ashokkumar, Riboflavin Responsive Mitochondrial Dysfunction in Neurodegenerative Diseases, J. Clin. Med., 2017, 6(52) CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- M. Y. Berezin and S. Achilefu, Fluorescence lifetime measurements and biological imaging, Chem. Rev., 2010, 110, 2641–2684 CrossRef CAS PubMed.
- L. P. Zaino, D. A. Grismer, D. Han, G. M. Crouch and P. W. Bohn, Single occupancy spectroelectrochemistry of freely diffusing flavin mononucleotide in zero-dimensional nanophotonic structures, Faraday Discuss., 2015, 184, 101–115 RSC.
- S. Kalinina, C. Freymueller, N. Naskar, B. von Einem, K. Reess, R. Sroka and A. Rueck, Bioenergetic Alterations of Metabolic Redox Coenzymes as NADH, FAD and FMN by Means of Fluorescence Lifetime Imaging Techniques, Int. J. Mol. Sci., 2021, 22, 5952 CrossRef CAS PubMed.
- A. C. Rueck, P. Schäfer, B. von Einem and S. Kalinina, in Multiphoton Microscopy in the Biomedical Sciences XX, ed. A. Periasamy, P. T. So and K. König, SPIE, 2020, p. 21 Search PubMed.
- A. Stepanova, S. Sosunov, Z. Niatsetskaya, C. Konrad, A. A. Starkov, G. Manfredi, I. Wittig, V. Ten and A. Galkin, Redox-Dependent Loss of Flavin by Mitochondrial Complex I in Brain Ischemia/Reperfusion Injury, Antioxid. Redox Signaling, 2019, 31, 608–622 CrossRef CAS PubMed.
- M. J. Cantoria, H. Patel, L. G. Boros and E. J. Meuillet, in Pancreatic Cancer – Insights into Molecular Mechanisms and Novel Approaches to Early Detection and Treatment, ed. K. D. McCall, InTech, 2014 Search PubMed.
- A. Luengo, D. Y. Gui and M. G. Vander Heiden, Targeting Metabolism for Cancer Therapy, Cell Chem. Biol., 2017, 24, 1161–1180 CrossRef CAS PubMed.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Progress and challenges towards targeted delivery of cancer therapeutics, Nat. Commun., 2018, 9, 1410 CrossRef PubMed.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Active targeting schemes for nanoparticle systems in cancer therapeutics, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS PubMed.
- M. De, P. S. Ghosh and V. M. Rotello, Applications of Nanoparticles in Biology, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- M. Björnmalm, K. J. Thurecht, M. Michael, A. M. Scott and F. Caruso, Bridging Bio-Nano Science and Cancer Nanomedicine, ACS Nano, 2017, 11, 9594–9613 CrossRef PubMed.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. Huang, Graphene Oxide Sheets at Interfaces, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano-Graphene Oxide for Cellular Imaging and Drug Delivery, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- V. C. Sanchez, A. Jachak, R. H. Hurt and A. B. Kane, Biological Interactions of Graphene-Family Nanomaterials, Chem. Res. Toxicol., 2012, 25, 15–34 Search PubMed.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, PEGylated nanographene oxide for delivery of water-insoluble cancer drugs, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- T. H. Tran, H. T. Nguyen, T. T. Pham, J. Y. Choi, H.-G. Choi, C. S. Yong and J. O. Kim, Development of a Graphene Oxide Nanocarrier for Dual-Drug Chemo-phototherapy to Overcome Drug Resistance in Cancer, ACS Appl. Mater. Interfaces, 2015, 7, 28647–28655 CrossRef CAS PubMed.
- X. Yang, X. Zhang, Z. Liu, Y. Ma, Y. Huang and Y. Chen, High-Efficiency Loading and Controlled Release of Doxorubicin Hydrochloride on Graphene Oxide, J. Phys. Chem. C, 2008, 112, 17554–17558 CrossRef CAS.
- K. Yang, L. Feng and Z. Liu, The advancing uses of nano-graphene in drug delivery, Expert Opin. Drug Delivery, 2015, 12, 601–612 CrossRef CAS PubMed.
- L. Feng, L. Wu and X. Qu, New Horizons for Diagnostics and Therapeutic Applications of Graphene and Graphene Oxide, Adv. Mater., 2013, 25, 168–186 CrossRef CAS PubMed.
- Y. T. Fong, C.-H. Chen and J.-P. Chen, Intratumoral Delivery of Doxorubicin on Folate-Conjugated Graphene Oxide by In-Situ Forming Thermo-Sensitive Hydrogel for Breast Cancer Therapy, Nanomaterials, 2017, 7, 388 CrossRef PubMed.
- D. Depan, J. Shah and R. Misra, Controlled release of drug from folate-decorated and graphene mediated drug delivery system: Synthesis, loading efficiency, and drug release response, Mater. Sci. Eng., C, 2011, 31, 1305–1312 CrossRef CAS.
- A. Alhourani, J.-L. Førde, L. A. Eichacker, L. Herfindal and H. R. Hagland, Improved pH-Responsive Release of Phenformin from Low-Defect Graphene Compared to Graphene Oxide, ACS Omega, 2021, 6(38), 24619–24629 CrossRef CAS PubMed.
- S. Vranic, A. F. Rodrigues, M. Buggio, L. Newman, M. R. H. White, D. G. Spiller, C. Bussy and K. Kostarelos, Live Imaging of Label-Free Graphene Oxide Reveals Critical Factors Causing Oxidative-Stress-Mediated Cellular Responses, ACS Nano, 2018, 12, 1373–1389 CrossRef CAS PubMed.
- Y. Chang, S.-T. Yang, J.-H. Liu, E. Dong, Y. Wang, A. Cao, Y. Liu and H. Wang, In vitro toxicity evaluation of graphene oxide on A549 cells, Toxicol. Lett., 2011, 200, 201–210 CrossRef CAS PubMed.
- T. Lammel, P. Boisseaux, M.-L. Fernández-Cruz and J. M. Navas, Internalization and cytotoxicity of graphene oxide and carboxyl graphene nanoplatelets in the human hepatocellular carcinoma cell line Hep G2, Part. Fibre Toxicol., 2013, 10, 27 CrossRef CAS PubMed.
- K.-H. Liao, Y.-S. Lin, C. W. Macosko and C. L. Haynes, Cytotoxicity of Graphene Oxide and Graphene in Human Erythrocytes and Skin Fibroblasts, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CrossRef CAS PubMed.
- X. T. Liu, X. Y. Mu, X. L. Wu, L. X. Meng, W. B. Guan, Y. Q. Ma, H. Sun, C. J. Wang and X. F. Li, Toxicity of multi-walled carbon nanotubes, graphene oxide, and reduced graphene oxide to zebrafish embryos, Biomed. Environ. Sci., 2014, 27, 676–683 CAS.
- M. Chen, J. Yin, Y. Liang, S. Yuan, F. Wang, M. Song and H. Wang, Oxidative stress and immunotoxicity induced by graphene oxide in zebrafish, Aquat. Toxicol., 2016, 174, 54–60 CrossRef CAS PubMed.
- C. Ren, X. Hu, X. Li and Q. Zhou, Ultra-trace graphene oxide in a water environment triggers Parkinson's disease-like symptoms and metabolic disturbance in zebrafish larvae, Biomaterials, 2016, 93, 83–94 CrossRef CAS PubMed.
- Z. Chen, C. Yu, I. A. Khan, Y. Tang, S. Liu and M. Yang, Toxic effects of different-sized graphene oxide particles on zebrafish embryonic development, Ecotoxicol. Environ. Saf., 2020, 197, 110608 CrossRef CAS PubMed.
- G. Xiong, Y. Deng, X. Liao, J. Zhang, B. Cheng, Z. Cao and H. Lu, Graphene oxide nanoparticles induce hepatic dysfunction through the regulation of innate immune signaling in zebrafish (Danio rerio), Nanotoxicology, 2020, 14, 667–682 CrossRef CAS PubMed.
- S. Das, S. Singh, V. Singh, D. Joung, J. M. Dowding, D. Reid, J. Anderson, L. Zhai, S. I. Khondaker, W. T. Self and S. Seal, Oxygenated Functional Group Density on Graphene Oxide: Its Effect on Cell Toxicity, Part. Part. Syst. Charact., 2013, 30, 148–157 CrossRef CAS.
- J. N. Coleman, Liquid exfoliation of defect-free graphene, Acc. Chem. Res., 2013, 46, 14–22 CrossRef CAS PubMed.
- M. P. Del Lavin-Lopez, A. Romero, J. Garrido, L. Sanchez-Silva and J. L. Valverde, Influence of Different Improved Hummers Method Modifications on the Characteristics of Graphite Oxide in Order to Make a More Easily Scalable Method, Ind. Eng. Chem. Res., 2016, 55, 12836–12847 CrossRef.
- H. Yue, W. Wei, Z. Yue, B. Wang, N. Luo, Y. Gao, D. Ma, G. Ma and Z. Su, The role of the lateral dimension of graphene oxide in the regulation of cellular responses, Biomaterials, 2012, 33, 4013–4021 CrossRef CAS PubMed.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Graphene-based materials: synthesis, characterization, properties, and applications, Small, 2011, 7, 1876–1902 CrossRef CAS PubMed.
- H. Tao, Y. Zhang, Y. Gao, Z. Sun, C. Yan and J. Texter, Scalable exfoliation and dispersion of two-dimensional materials an update, Phys. Chem. Chem. Phys., 2017, 19, 921–960 RSC.
- J. Y. Lim, N. M. Mubarak, E. C. Abdullah, S. Nizamuddin, M. Khalid and Inamuddin, Recent trends in the synthesis of graphene and graphene oxide based nanomaterials for removal of heavy metals — A review, J. Ind. Eng. Chem., 2018, 66, 29–44 CrossRef CAS.
- D. A. Boyd, W.-H. Lin, C.-C. Hsu, M. L. Teague, C.-C. Chen, Y.-Y. Lo, W.-Y. Chan, W.-B. Su, T.-C. Cheng, C.-S. Chang, C.-I. Wu and N.-C. Yeh, Single-step deposition of high-mobility graphene at reduced temperatures, Nat. Commun., 2015, 6, 6620 CrossRef CAS PubMed.
- W. Tian, W. Li, W. Yu and X. Liu, A Review on Lattice Defects in Graphene: Types, Generation, Effects and Regulation, Micromachines, 2017, 8, 163 CrossRef.
- A. Angelopoulou, E. Voulgari, E. K. Diamanti, D. Gournis and K. Avgoustakis, Graphene oxide stabilized by PLA-PEG copolymers for the controlled delivery of paclitaxel, Eur. J. Pharm. Biopharm., 2015, 93, 18–26 CrossRef CAS PubMed.
- M. M. Gudarzi, Colloidal Stability of Graphene Oxide: Aggregation in Two Dimensions, Langmuir, 2016, 32, 5058–5068 CrossRef CAS PubMed.
- I. Chowdhury, M. C. Duch, N. D. Mansukhani, M. C. Hersam and D. Bouchard, Colloidal properties and stability of graphene oxide nanomaterials in the aquatic environment, Environ. Sci. Technol., 2013, 47, 6288–6296 CrossRef CAS PubMed.
- K. Yang, L. Feng, X. Shi and Z. Liu, Nano-graphene in biomedicine: theranostic applications, Chem. Soc. Rev., 2013, 42, 530–547 RSC.
- Y. Li, H. Yuan, A. von dem Bussche, M. Creighton, R. H. Hurt, A. B. Kane and H. Gao, Graphene microsheets enter cells through spontaneous membrane penetration at edge asperities and corner sites, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12295–12300 CrossRef CAS PubMed.
- Y. Li, Q. Wu, Y. Zhao, Y. Bai, P. Chen, T. Xia and D. Wang, Response of microRNAs to in vitro treatment with graphene oxide, ACS Nano, 2014, 8, 2100–2110 CrossRef CAS PubMed.
- A. Schinwald, F. A. Murphy, A. Jones, W. MacNee and K. Donaldson, Graphene-based nanoplatelets: a new risk to the respiratory system as a consequence of their unusual aerodynamic properties, ACS Nano, 2012, 6, 736–746 CrossRef CAS PubMed.
- M. Vila, M. T. Portolés, P. A. A. P. Marques, M. J. Feito, M. C. Matesanz, C. Ramírez-Santillán, G. Gonçalves, S. M. A. Cruz, A. Nieto and M. Vallet-Regi, Cell uptake survey of pegylated nanographene oxide, Nanotechnology, 2012, 23, 465103 CrossRef CAS PubMed.
- K. Yang, L. Feng, H. Hong, W. Cai and Z. Liu, Preparation and functionalization of graphene nanocomposites for biomedical applications, Nat. Protoc., 2013, 8, 2392–2403 CrossRef CAS PubMed.
- M. Pelin, L. Fusco, V. León, C. Martín, A. Criado, S. Sosa, E. Vázquez, A. Tubaro and M. Prato, Differential Cytotoxic Effects of Graphene and Graphene Oxide on Skin Keratinocytes, Sci. Rep., 2017, 40572 CrossRef CAS PubMed.
- S. Liu, T. H. Zeng, M. Hofmann, E. Burcombe, J. Wei, R. Jiang, J. Kong and Y. Chen, Antibacterial activity of graphite, graphite oxide, graphene oxide, and reduced graphene oxide: membrane and oxidative stress, ACS Nano, 2011, 6971–6980 CrossRef CAS PubMed.
- K. Yang, J. Wan, S. Zhang, Y. Zhang, S.-T. Lee and Z. Liu, In vivo pharmacokinetics, long-term biodistribution, and toxicology of PEGylated graphene in mice, ACS Nano, 2011, 5, 516–522 CrossRef CAS PubMed.
- Y. Li, Y. Liu, Y. Fu, T. Wei, L. Le Guyader, G. Gao, R.-S. Liu, Y.-Z. Chang and C. Chen, The triggering of apoptosis in macrophages by pristine graphene through the MAPK and TGF-beta signaling pathways, Biomaterials, 2012, 33, 402–411 CrossRef CAS PubMed.
- H. Zhou, B. Zhang, J. Zheng, M. Yu, T. Zhou, K. Zhao, Y. Jia, X. Gao, C. Chen and T. Wei, The inhibition of migration and invasion of cancer cells by graphene via the impairment of mitochondrial respiration, Biomaterials, 2014, 35, 1597–1607 CrossRef CAS PubMed.
- Y. Sogame, A. Kitamura, M. Yabuki and S. Komuro, A comparison of uptake of metformin and phenformin mediated by hOCT1 in human hepatocytes, Biopharm. Drug Dispos., 2009, 30, 476–484 CrossRef CAS PubMed.
- Y. Shitara, N. Nakamichi, M. Norioka, H. Shima, Y. Kato and T. Horie, Role of organic cation/carnitine transporter 1 in uptake of phenformin and inhibitory effect on complex I respiration in mitochondria, Toxicol. Sci., 2013, 132, 32–42 CrossRef CAS PubMed.
- H. Liu, J. Yao, H. Guo, X. Cai, Y. Jiang, M. Lin, X. Jiang, W. Leung and C. Xu, Tumor Microenvironment-Responsive Nanomaterials as Targeted Delivery Carriers for Photodynamic Anticancer Therapy, Front. Chem., 2020, 8, 758 CrossRef CAS PubMed.
- H. Lu, R. A. Forbes and A. Verma, Hypoxia-inducible Factor 1 Activation by Aerobic Glycolysis Implicates the Warburg Effect in Carcinogenesis, J. Biol. Chem., 2002, 277, i–ii CrossRef.
- L. Di Magno, S. Manni, F. Di Pastena, S. Coni, A. Macone, S. Cairoli, M. Sambucci, P. Infante, M. Moretti, M. Petroni, C. Nicoletti, C. Capalbo, E. de Smaele, L. Di Marcotullio, G. Giannini, L. Battistini, B. M. Goffredo, E. Iorio, E. Agostinelli, M. Maroder and G. Canettieri, Phenformin Inhibits Hedgehog-Dependent Tumor Growth through a Complex I-Independent Redox/Corepressor Module, Cell Rep., 2020, 30, 1735–1752 CrossRef CAS PubMed.
- R. M. White, A. Sessa, C. Burke, T. Bowman, J. LeBlanc, C. Ceol, C. Bourque, M. Dovey, W. Goessling, C. E. Burns and L. I. Zon, Transparent adult zebrafish as a tool for in vivo transplantation analysis, Cell Stem Cell, 2008, 2, 183–189 CrossRef CAS PubMed.
- S. Ali, J. Aalders and M. K. Richardson, Teratological effects of a panel of sixty water-soluble toxicants on zebrafish development, Zebrafish, 2014, 11, 129–141 CrossRef CAS PubMed.
- W. Lin, Y. Yan, S. Ping, P. Li, D. Li, J. Hu, W. Liu, X. Wen and Y. Ren, Metformin-Induced Epigenetic Toxicity in Zebrafish: Experimental and Molecular Dynamics Simulation Studies, J. Environ. Sci. Technol., 2021, 55, 1672–1681 CrossRef CAS PubMed.
- C. Kang, H.-L. Wu, M.-L. Xu, X.-F. Yan, Y.-J. Liu and R.-Q. Yu, Simultaneously quantifying intracellular FAD and FMN using a novel strategy of intrinsic fluorescence four-way calibration, Talanta, 2019, 197, 105–112 CrossRef CAS PubMed.
- A. J. Visser and F. Müller, in Vitamins and Coenzymes Part E, Elsevier, 1980, pp. 373–385 Search PubMed.
- P. Wahl, J. C. Auchet, A. J. Visser and F. Müller, Time resolved fluorescence of flavin adenine dinucleotide, FEBS Lett., 1974, 44, 67–70 CrossRef CAS.
- M. S. Islam, M. Honma, T. Nakabayashi, M. Kinjo and N. Ohta, pH dependence of the fluorescence lifetime of FAD in solution and in cells, Int. J. Mol. Sci., 2013, 14, 1952–1963 CrossRef CAS PubMed.
- I. S. Gostimskaya, V. G. Grivennikova, G. Cecchini and A. D. Vinogradov, Reversible dissociation of flavin mononucleotide from the mammalian membrane-bound NADH:ubiquinone oxidoreductase (complex I), FEBS Lett., 2007, 581, 5803–5806 CrossRef CAS PubMed.
- P. J. Holt, R. G. Efremov, E. Nakamaru-Ogiso and L. A. Sazanov, Reversible FMN dissociation from Escherichia coli respiratory complex I, Biochim. Biophys. Acta, 2016, 1857, 1777–1785 CrossRef CAS PubMed.
- A. Kahl, A. Stepanova, C. Konrad, C. Anderson, G. Manfredi, P. Zhou, C. Iadecola and A. Galkin, Critical Role of Flavin and Glutathione in Complex I-Mediated Bioenergetic Failure in Brain Ischemia/Reperfusion Injury, Stroke, 2018, 49, 1223–1231 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00778e |
| This journal is © The Royal Society of Chemistry 2022 |