
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neural network embeddings based similarity search method for atomistic systems†
Yilin
Yang‡
,
Mingjie
Liu‡
and
John R.
Kitchin
 *
*
Department of Chemical Engineering, Carnegie Mellon University, 5000 Forbes Ave., Pittsburgh, PA 15213, USA. E-mail: jkitchin@andrew.cmu.edu
First published on 8th August 2022
Abstract
With the popularity of machine learning growing in the field of catalysis there are increasing numbers of catalyst databases becoming available. These databases provide us with the opportunity to search for catalysts with desired properties, which could lead to the discovery of new catalysts. However, while there are search methods for molecules based on similarity metrics, for solid-state catalyst systems there is not yet a straightforward search method. In this work, we propose a neural network embeddings based similarity search method that is applicable for both molecules and solid-state catalyst systems. We illustrate how the search method works and show search examples for the QM9, Materials Project (MP) and Open Catalyst 2020 (OC20) databases. We show that the configurations found present similarity in terms of geometry, composition, energy and in the electronic density of states. These results imply the neural network embeddings have encoded effective information that could be used to retrieve molecules and materials with similar properties.
1 Introduction
Data is the central part of almost all machine learning applications. With the increasing capacity to generate and store more data, efficient methods to retrieve target data of interest has become much more in-demand. In the chemistry field, the sizes of the datasets have grown dramatically in the past decade. For example, the Materials Project has more than 140 thousand inorganic compounds, 530 thousand nanoporous materials, and their properties.1 PubChem includes more than 100 million compounds.2 Open Catalyst 2020 (OC20) provides DFT calculations of more than 130 million adsorption structures.3 Given the huge sizes of existing datasets and potentially larger datasets in the future, we need fast methods to explore and search in these datasets. While a lot of progress has been made for searching molecules, the ability of searching catalyst systems is still lacking.Usually, researchers may want to search for molecules or materials with similar properties in applications like discovering new drugs or cheaper materials.4–6 Many similarity search methods have been developed for this purpose.7,8 In general, a similarity search approach consists of three essential components: a molecular representation method, a quantitative metric to measure the similarity of two molecules, and a search algorithm. The search process usually starts with one or more query molecules (e.g., configurations that have desired properties). Then the representation method converts them into numerical representations which could be used to calculate the pair-wise similarities. After that, the search algorithm retrieves candidates on the basis of the similarity measurement. The retrieved molecules are ranked by their similarities to the query molecule(s) in descending order. Significant efforts have been spent on designing fingerprints to represent molecules. For example, SMIfp (SMILES fingerprint) converts a molecule into a 34-dimension scalar fingerprint.9 Each element of the fingerprint counts the occurrences of 34 symbols in SMILES, where SMILES (Simplified Molecular Input Line Entry System) is a chemical language and information system used to represent different atoms and bonds with ASCII characters.10,11
The substructure-based fingerprint is also a popular choice to represent molecules. Each item of the fingerprint encodes whether or not a substructure is present in a molecule. Typical examples include the Molecular ACCess System (MACCS) and the Barnard Chemical Information Ltd. (BCI) fingerprint.12,13 MACCS uses 166 structural fragments as the keys while BCI contains 1052 substructures. These fingerprints rely on a pre-built library of substructures as the keys, which limits their applications only for molecules which have well-defined bondings. Also, they cannot be used for chemical systems represented with the periodic boundary condition (PBC). However, catalyst systems, such as complex adsorbates on solid surface, are usually represented with PBC and there is no well-defined bonding between atoms. Therefore, these methods cannot be used. To cope with the PBC problem, several molecular representation methods have been proposed such as the Atom-Centered Symmetry Functions (ACSF),14 and Smooth Overlap of Atomic Positions (SOAP).15 However, these methods have very poor scaling regarding the number of elements in the database. Hence, a method that deals with PBC and that has good scaling regarding the number of elements is needed.
In the past decade, the development of deep learning methods has changed the way we can represent data like text and images. Deep learning models like the convolutional neural network (CNN) and recurrent neural network (RNN) have been widely applied in computer vision and language processing tasks.16–19 For most of the deep learning models, the last layer of the deep neural network represents the input data as a numerical vector which contains rich information about the data. This vector representation is also called an embedding. Since the neural network output usually depends linearly on the embedding, we can also regard the embedding as a nonlinear dimensional transform of the input into a space where the output is linear. More importantly, this numerical vector is a fingerprint of the input that may be useful in search. The promising performance of the deep learning models in various tasks implies the embedding must represent the data in a reasonable way. Therefore, these neural network embeddings have been applied in many information retrieval systems involving images and text.20–22
For molecular data, several graph neural network (GNN) models have been proposed to learn the embeddings to represent the atomic configurations, such as the CGCNN and the GemNet model.23,24 The atomic embeddings contain information including the element type of the central atom, positions, and elemental information of the neighboring atoms. When applied in specific tasks (e.g., energy and force prediction), it is reasonable to think that neural networks could be trained to generate atomic embeddings in a space where the specific property (e.g., atomic energy) is linearly related to the embedding vectors. Therefore, the distance between the embeddings in this space could serve as a similarity measure of a specific property.
In addition to the molecular representation part, the other important component in the molecular similarity search is the search algorithm. Exhaustive search for similar vectors to a high-dimensional query vector in a large database is both time- and resource-consuming. Therefore, many approximate nearest neighbor (ANN) search methods have been proposed to find approximate results with much less time and fewer resources.25 The ANN search methods can be classified as hashing-based, quantization-based, tree-based, or graph-based methods according to the techniques used to accelerate the search process.26 Typical examples include locality-sensitive hashing, SPTAG, and ScaNN.27–29 There are also packages released to implement these ANN search algorithms such that more research work can be benefited from these ANN search methods. For example, Facebook AI Similarity Search (FAISS) is a library containing implementations for several ANN search algorithms.30
In this work, we show a method based on neural network embeddings to search for similar structures. We demonstrate that the method can be applied to any atomistic system including organic molecules, bulk materials, and adsorption systems. When combined with ANN search methods, neural network embeddings can be used to retrieve similar atomic structures efficiently in large databases. We also show that the similarity is related to the specific property which is used to train the neural network models. Therefore, this method has the potential to search for similar molecular structures in a property-oriented way.
2 Methods
2.1 Searching similar molecules via neural network embeddings
In this section, we introduce the overall framework to search for similar molecules in a database using neural network embeddings. This framework is shown in Fig. 1. The whole workflow can be divided into two stages: preparation and query. In the preparation stage, we use a database of molecules to train a neural network model on some property. Then we use the trained model to calculate the embeddings of the atoms in the database. These atom embeddings are processed into a specific data structure for future searching by an approximate nearest neighbor (ANN) search algorithm. In the query stage, given a query molecule, we use the same trained neural network model to get the embeddings for the atoms in the molecule. Then we retrieve neighboring atom embeddings using an ANN search method and return the corresponding molecules as the results for the query event. If the query wants to find similar atomic environments, then the atoms corresponding to the embeddings are directly returned. In this work we used GemNet to generate the atomic embeddings.24 The FAISS package was used for the ANN search algorithm.30 More details of each step will be discussed in the following sections. | ||
| Fig. 1 The schematic of the search method. In the building phase, a GemNet is trained with the database. Then, GemNet is used to calculate the atomic embedding for each atom in the database. With all the atomic embeddings, the FAISS package is used to build an index for the atomic embeddings so that similar atomic embeddings can be efficiently queried. In the searching phase, the atomic embeddings of atoms in the target local structure will be calculated with the trained GemNet. The top k similar atomic embeddings for each target embedding can be queried using the built index. The tables for each target embedding will be merged with a heuristic to get the top n systems that contain the similar local structure. | ||
2.2 GemNet to generate atom embeddings
To query similar chemical systems from a database, it is important for each chemical system to have a characteristic fingerprint, so the similarity between different systems can be evaluated. In this work, the fingerprint for each chemical system is a collection of atomic fingerprints (atomic embeddings) obtained from a modified version of the GemNet-dT. The overall architecture of the modified GemNet-dT is shown in Fig. 2. The modification is in the output atomic embeddings. Instead of using the output atomic embeddings of all interaction blocks to predict the atomic energy, the modified version only uses the output atomic embedding from the last interaction block. The drawback of the modification is that the atomic embeddings will be less detailed. While the output atomic embedding from the last interaction block contains all information of the center atom and its local environments, the output atomic embeddings from previous interaction blocks have more detailed information of the center atom and its neighboring atoms in smaller neighborhoods. However, the modification is necessary for two main reasons. First, including those output embeddings will make the dimensionality of the atomic embeddings too high and every atomic embedding could seem to be far from each other due to the curse of dimensionality.31 Second, the high dimensionality will make the computational cost higher. The detailed architecture of each block of the modified GemNet-dT is the same as the original GemNet-dT. A detailed analysis of the GemNet architecture is out of the scope of this work but it can be found in the original paper.24 The GemNet code we used is adapted from the Open Catalyst Project models codebase.3 The hyperparameters used are shown in the ESI.†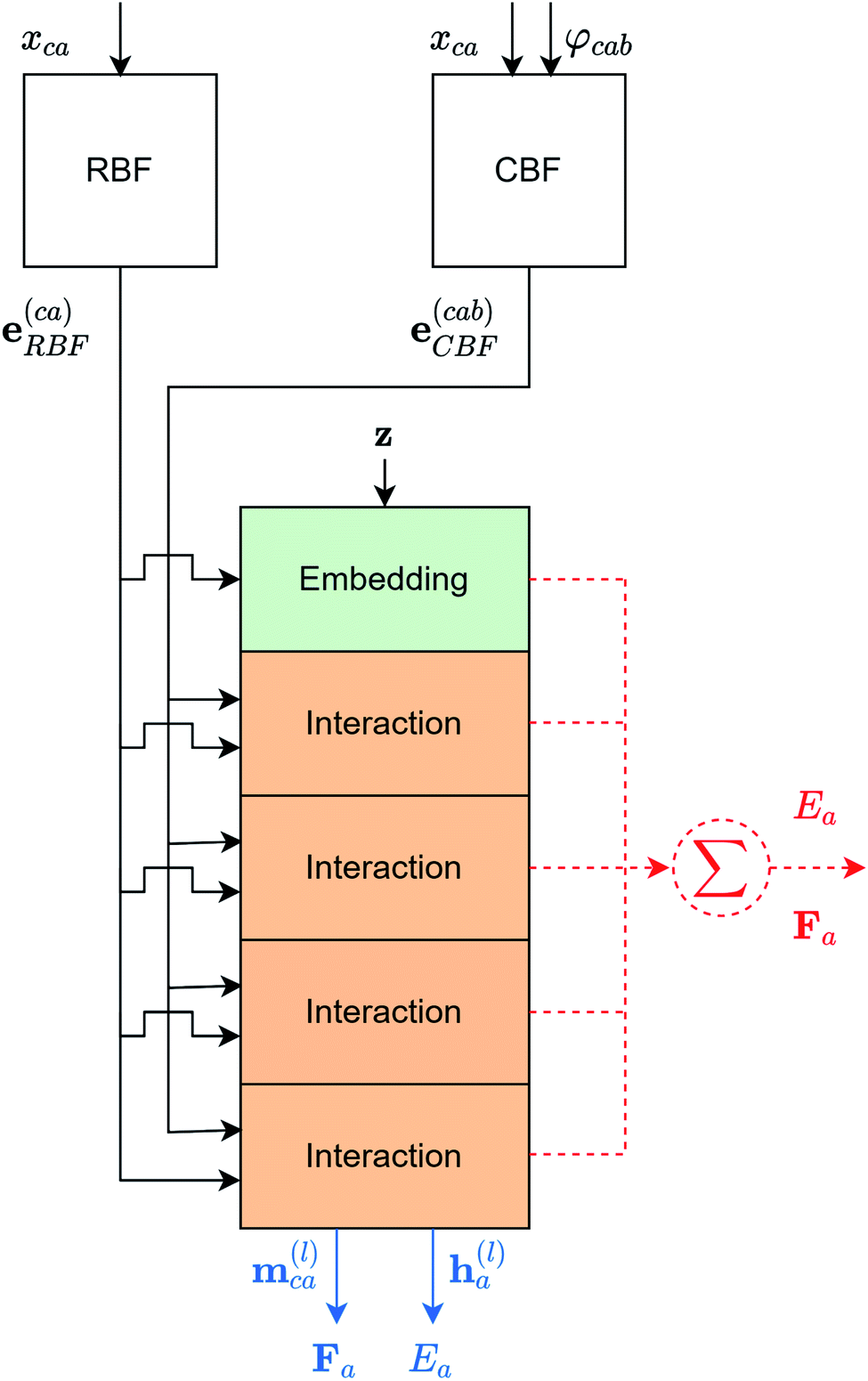 | ||
| Fig. 2 The overall architecture of the modified GemNet-dT. The red dotted parts are from the original GemNet-dT and the blue parts are from the modified GemNet-dT. xca is the distance between atom a and atom c. φcab is the angle ∠cab. RBF and CBF are the spherical Fourier–Bessel bases with polynomial envelopes developed by Klicpera et al.24 Therefore, e(ca)RBF and e(cab)CBF are the distances and angles expanded into those bases. z is the element information of all atoms in the chemical system. m(l)ca is the edge embedding between atom c and atom a. Edge embeddings for all arbitrary atom c regarding atom a, will be used to predict the atomic forces of atom a. h(l)a is the atomic embedding of atom a. It will be used to predict the atomic energy of atom a and it is the atomic fingerprint used in this work. (mlca is the edge embedding between atom c and atom a. While it is not used in this work, it can be used to predict the contribution to the atomic forces of atom a from atom c). | ||
To obtain the GemNet atomic embedding, we trained the GemNet on the energies of each entry in the database. Once the GemNet is trained, the output atomic embedding, h(l)a, which is used to predict the atomic energy Ea, will be the atomic embedding used to describe the local environment of the atom in the chemical system.
2.3 Approximate nearest neighbor search
Searching for the exact k closest results to a query vector is computationally expensive for large databases and high-dimensional data. Therefore, we used ANN search as the search engine to obtain approximately neighboring results in our work. Specifically, we used the FAISS library to implement the ANN search part.30 Its IndexIVFPQ mode is used in our work. The bases of IndexIVFPQ are an inverted file system and product quantization.32,33 The inverted file system is built by applying k-means clustering on a database of vectors to form a set of centroids. These centroids allow rapid access to a small fraction of nearby vectors for a query vector, which avoids exhaustive comparisons against each vector in a database. Then, the search efficiency is further enhanced by using product quantization on the residual query vector (subtracting the corresponding centroid from the query vector). The essential idea of product quantization is dividing a vector into small subvectors, applying k-means clustering on these subvectors, and using the corresponding centroids of the subvectors to represent the original vector. Recording the centroid index uses less memory compared to saving the whole real vector. The computational complexity of IndexIVFPQ for querying k nearest neighbors is about O(n + k![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logkloglogn) assuming only search for one partition of vectors,33 where n is the number of vectors in that partition. The parameters of the FAISS IndexIVFPQ method used in our work are attached in the ESI.† FAISS supports similarity metrics like L2 distance and the inner product. L2 distance was used in our work.
logkloglogn) assuming only search for one partition of vectors,33 where n is the number of vectors in that partition. The parameters of the FAISS IndexIVFPQ method used in our work are attached in the ESI.† FAISS supports similarity metrics like L2 distance and the inner product. L2 distance was used in our work.
In this work, the similarity search is operated on two aspects, at the atomic level and the molecular level. At the atomic level, we can directly use ANN search on the atom embeddings based on the Euclidean distance. However, for the nearest molecules search, we need to convert the similarity of atom embeddings into the similarity of the molecules. This process is shown in Algorithm 1. Basically, for each atom in a molecule, we search for k approximate nearest atom embeddings in a database. These atom embeddings are considered matched for the query atom. The corresponding molecules containing these matched atoms are added to a candidate set. After looping over all atoms in the query molecule, we rank the molecules in the candidate set according to the number of matched atoms in descending order (sum of Euclidean distances between the matched atom embeddings is used to break ties). Top n molecules are the n approximate nearest neighbors. Large k prefers the candidates that are globally similar to the query molecule while small k favors the molecules containing local environments with high similarity to the query molecule. We used k around 10 times n in our work.

2.4 Datasets
We demonstrate the ANN search on three datasets across organic molecules, bulk materials and surfaces. For the organic molecules, we applied our search method on the QM9 dataset.34,35 QM9 contains properties of 134k small organic molecules with elements of C, H, O, N, and F. In terms of the bulk materials, we adopted the Materials Project dataset which includes more than 126k bulk crystals.1 The QM9 and Materials Project databases were obtained from the SchNetPack package.36 For the surface systems, we used the IS2RS subset from the newly released OC20 dataset,3 which contains about 460k relaxed adsorption configurations. For each of the above dataset, we train a GemNet model on their potential energy data to learn the atom embeddings. After that, the atom embeddings were used in the search tasks.3 Results
In this section, we demonstrate the GemNet embedding-based ANN search results for different molecular systems: small organic molecules, metallic bulk materials, and metallic surfaces with adsorbates.3.1 ANN search for organic molecules
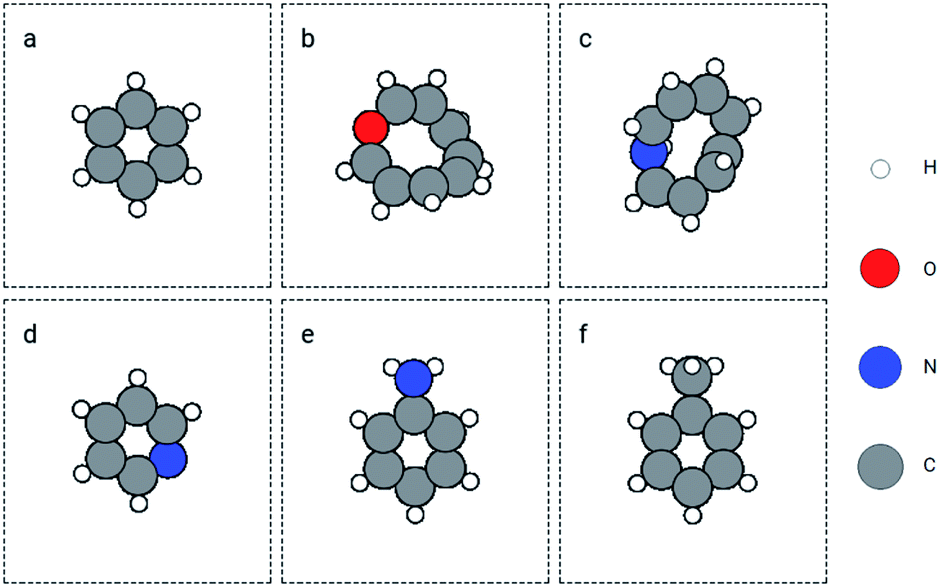
The first case of ANN search is for small organic molecules, which was performed in the QM9 dataset. The whole dataset was split into the training and validation sets randomly with a ratio of 0.8:0.2. A GemNet model was built on the training set. The energy mean absolute error (MAE) on the training and validation sets was 4.57 eV and 4.97 eV separately. Noticeably, the errors are much larger than the benchmark results on QM9.36 This is because we trained the GemNet on the raw energy instead of the scaled energy which has already accounted for the contributions from different elements. The benefit of using the raw energy is that the GemNet embedding will be able to better learn elemental information from the data. We then used this model to obtain the embeddings for atoms in the QM9 dataset and search for similar molecules in this database. We chose several molecules and functional groups as the queries to search for similar (sub)structures. The examples include molecules of benzene and toluene, as well as groups of hydroxyl, amino, and imino. Here, we only discuss the results for benzene and a joint search of amino and hydroxyl groups. Results for other examples can be found in the ESI.† For benzene, we used the GemNet embedding of each atom as the query vector to search for similar atomic environments. Then we sorted the candidate molecules based on their number of matched atoms and the sum of the L2 distances as mentioned in Section 2.3. The found molecules are shown in Fig. 3. The top left molecule is the query benzene while the Fig. 3b to f are the nearest 5 molecules. They all contain a 6-atom ring structure with some small difference against the query benzene. Basically, the 6 atoms in the ring are all carbon. Except in Fig. 3c and f, one carbon atom is replaced by a nitrogen atom. There are also some extra groups on the rings like hydroxyl and amino groups. But generally, these searched molecules are similar to benzene in terms of elemental and geometric features.
 | ||
| Fig. 3 Retrieved similar molecules (top 5) for benzene. Figure (a) is benzene used as the query molecule. Figures (b) to (f) show the nearest molecules in the QM9 dataset. | ||
Because QM9 is a molecule database, methods developed for drug discovery can also be used. Therefore, to understand the search results from our method, we compared them with the results obtained using a typical molecular similarity search method, which served as a baseline for the search results. For the molecular similarity search, we used the Morgan Fingerprint with a diameter of four,37,38 and the similarity metric used was the Tanimoto similarity.39 The Morgan fingerprint is a bit vector that essentially tells whether there are certain local structure (e.g. aromaticity, double bond, hydroxyl group, etc.) in the molecule. The Tanimoto similarity is calculated by eqn (1).
 | (1) |
Therefore, if two molecules contain a lot of the same local structures, the Tanimoto similarity will be high and the molecules will be considered similar.
The search results of the Morgan fingerprints and the Tanimoto coefficient are shown in Fig. 4. The main difference from the GemNet result is in Fig. 4b and c, where larger rings are retrieved in the search results. These two molecules are less similar to benzene from the aspect of atom numbers and bond angles in the ring structure. According to the top 5 nearest molecules, GemNet embedding retrieves more similar molecules than Morgan fingerprint. This shows that the kind of vector fingerprint used in the search is important.
 | ||
| Fig. 4 Searched similar molecules (top 5) using Morgan fingerprint with Tanimoto coefficient as a distance measure. Figure (a) is the query molecule. Figures (b) to (f) show the nearest molecules in the QM9 dataset. | ||
In addition to the qualitative evaluation of the similarity by visual comparison over the elemental and geometric features, we also analyze the similarity among the molecules by investigating their relevance in the energetic embedding space. We built Gaussian process regression (GPR) models using the found molecules as the training set. The hypothesis is that training on similar molecules will result in a more accurate model more quickly for a query than training on random molecules.
We used the FLARE package as the implementation of the GPR models.40 The hyperparameters for the GPR model are provided in the ESI.† During the training process, we iteratively added the found molecules one by one into the training set and updated the GP model. Then we used the GPR model to predict the energy of benzene and compared the prediction with the true label. The results of the GPR models are shown in Fig. 5. We included the results from the training set searched using GemNet embeddings and Morgan fingerprints, as well as a set of random molecules from the QM9 dataset as the baseline. In Fig. 5, we can see that as we add more configurations into the training set, the prediction error and standard deviation are generally decreasing. However, using molecules found in different ways, the GPR models have different performances. The GPR model trained on the molecules retrieved by GemNet embeddings has the smallest prediction error (0.04 eV) and standard deviation (0.02 eV). The GPR model from Morgan fingerprints has a larger error (3.64 eV) and standard deviation (17.14 eV). The GPR model from the random molecules has the largest error and prediction uncertainty, which is 5.76 eV and 43.44 eV respectively with up to 15 configurations. These results imply that GemNet embedding has a representation of the atomic environments that is more relevant to the energetic property of the molecules. This is not surprising since the GemNet model was trained on the energy data of the molecules and the atomic energy prediction is a linear regression on the atom embeddings.
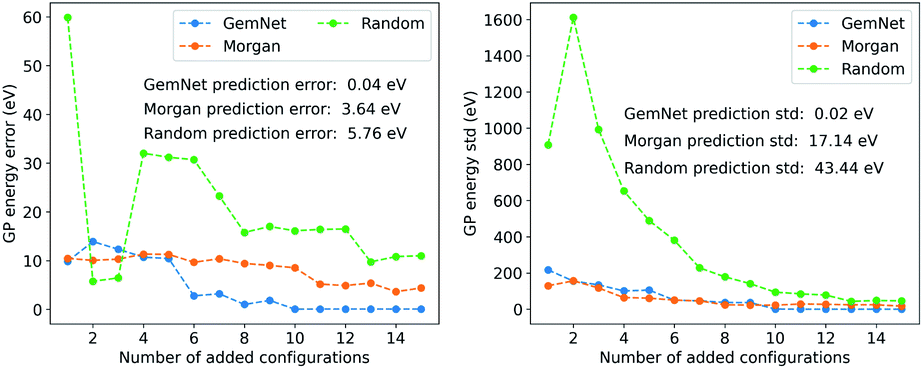 | ||
| Fig. 5 Prediction performance of GPR models trained on molecules searched using GemNet embeddings and Morgan fingerprints, as well as a set of molecules randomly sampled from the QM9 dataset. The left figure shows the prediction error while the right figure shows the standard deviation (std) of the GPR prediction. The number of added configuration means the number of molecules added into the training set to build the GP regression model. The annotations in the figures are the mininum prediction error and standard deviation for the models trained on different number of configurations. | ||
In addition to the search for a whole molecule, we can also use the GemNet embeddings to search for substructures. Here, we demonstrate an example of searching for a molecule containing similar substructures to the hydroxyl group of the butanol and the amino group of the glycine. The search procedure is similar to the method for benzene but has an additional step to join the search results from hydroxyl and amino groups, which is similar to an and operator on two sets. Fig. 6 shows the query substructures and the searched molecule. The atoms in the query and matched substructures are marked with crosses. Both the hydroxyl and amino groups are retrieved in the resulting molecule. In addition, the retrieved hydroxyl and amino groups are somehow similar to the queries. For the hydroxyl groups, they are both at the end of a three-carbon chain for the query and searched molecules. For the amino groups, they are at the end of a two-carbon chain and there is a –OH group at the other end.
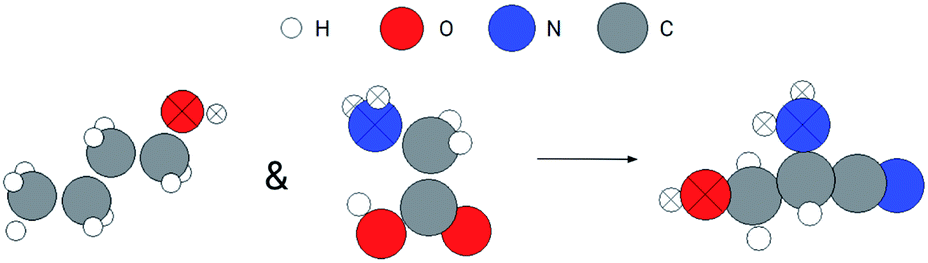 | ||
| Fig. 6 Joint search result for hydroxyl and amino groups. The query substructures are marked as crossed at the left of the arrow. The retrieved molecule is at the right side with matched atoms also marked as crossed. | ||
3.2 ANN search for bulk local environments
We next applied the ANN search method on metallic bulk systems with the Materials Project dataset.1 A critical difference in bulk environments from molecular systems is that bulk systems are typically described in unit cells with periodic boundary conditions. Consequently, the fingerprints must account for this.Similar to the QM9 case, the whole Materials Project database was split into the training and validation sets randomly with a ratio of 0.8:0.2. A GemNet model was trained on the training set. The energy MAE on the training and validation set was 0.62 eV and 1.42 eV respectively. There is an apparent gap between the accuracy of the GemNet model on the training and validation set. We attribute this gap to the configuration extrapolation in the validation set. At the point we stop the training, there was no increase of the MAE on the validation set along with the training steps, which implied the model was not located in the overfitting region. We then used the trained model to search for similar atomic environments in the Materials Project training dataset.
As an example query, we search for an oxygen atom in a Al2Cu3O6 bulk cell. The query and found atoms are shown in Fig. 7. The distances of the searched atoms to the query atom and their ranks are shown in Fig. 7. The query oxygen atom is atom 9 in Fig. 7a, which is closely neighboring to a copper atom (atom 3). There is also an aluminum atom (atom 0) near the query oxygen atom. These three atoms form an angle around 135° with the aluminum and copper atoms at two ends and the oxygen atom at the vertex. There is also another oxygen atom (atom 5) at the opposite position to the query oxygen atom across the copper atom. These geometric features also appear in the searched atoms in Fig. 7b (atoms 10, 13, 15, 17) and Fig. 7c (atoms 6, 8). Periodic boundary conditions should be considered when examining the geometric similarity for atom 10 and atom 13 in Fig. 7b. In Fig. 7d, atom 17 is also the found atom and it is ranked as 7th in all atomic environments although its neighboring environment looks not so similar to the query atom. This is because there are no more similar atoms like the previous ones in the remaining pool.
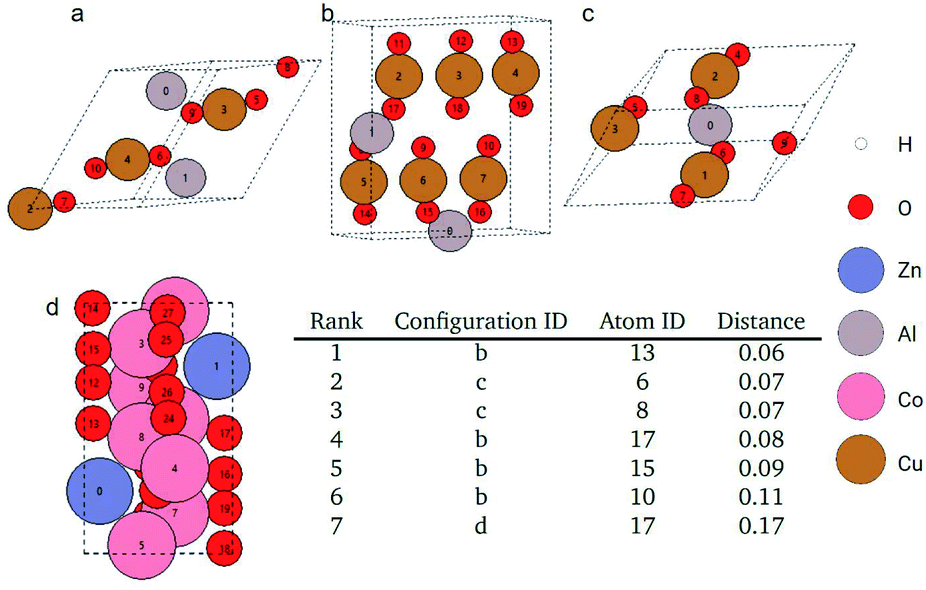 | ||
| Fig. 7 Top 7 nearest atoms to the query oxygen atom in the Materials Project dataset. Atom 9 in figure (a) is the query atom. Atom 10, 13, 15, and 17 in figure (b), atom 6, 8 in figure (c), and atom 17 in figure (d) are the searched atoms. The table in the figure shows the Euclidean distances between the GemNet embeddings of the searched atoms (excluding the query atom itself) and the query atom. | ||
As shown in Fig. 7, the Euclidean distance of the atom embeddings for the searched atoms in Fig. 7b and c to the query oxygen atom ranges between 0.06–0.11. This distance jumps to 0.17 for atom 17 in Fig. 7d. The distance of the atom embeddings also implies that atom 17 of Fig. 7d is not so similar to the query oxygen atom from the view of the GemNet model. It is also worth noting that during the searching process, we did not explicitly restrict the searching pool to be oxygen atoms. This atomic identity feature was already encoded into the GemNet atomic embeddings, and this is why the retrieved atoms are all oxygen atoms in Fig. 7 although with different local environments. For more examples, please refer to the ESI.†
3.3 ANN search for surfaces
In this section, we move on to a more complicated system: metallic surfaces with adsorbates. Relaxed configurations in the OC20 dataset were used in this case. There are more than 460k configurations in the training set and about 24k configurations in the validation set. A GemNet model was trained on the training set with the energy MAEs of 0.82 eV and 0.92 eV on the training and validation sets respectively. Atom embeddings were generated by this GemNet model to be searched during the query events. We illustrate the application of the GemNet embeddings to search for similar adsorption configurations via two examples.The first query example is an oxygen atom adsorbed on a tilted hollow site consisting of two Pd atoms and one Ag atom. The local configurations are shown in Fig. 8a. We seek examples from the database that are similar to this query. During the searching, we did not explicitly provide information about the element types of the central and surrounding atoms. Only the GemNet embeddings were used to measure the similarities. According to the search result in Fig. 8, (a zoomed-in view can be found in the ESI†) this elemental information as well as the geometric information of the adsorption site has already been encoded into the GemNet embeddings. On the one hand, the retrieved atoms are all oxygen atoms. On the other hand, the adsorption sites are all hollow sites with two Pd atoms.
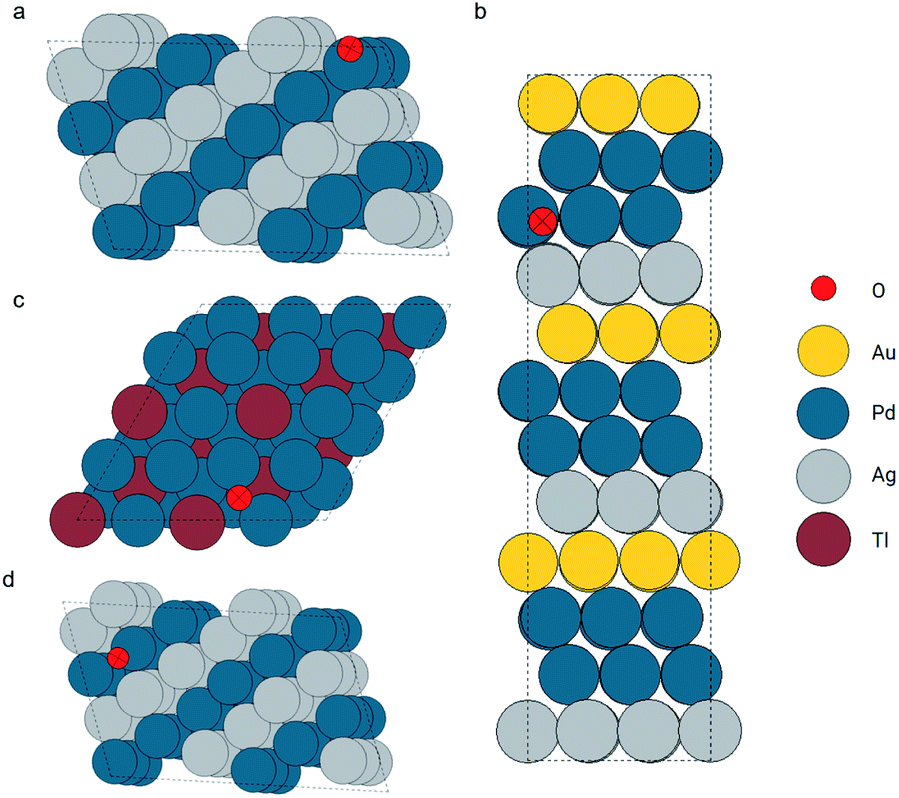 | ||
| Fig. 8 Configurations of the query and found atoms (marked as crossed). Configuration (a) is the query oxygen and configurations (b) to (d) are the retrieved atoms. | ||
In addition to these apparent similar geometric features, we also present the similarity between the query atom and the searched atoms via the density of states projected to these atoms (ADOS). The ADOS data was calculated by the Vienna Ab initio Simulation Package (VASP).41 The ADOS data is shown in Fig. 9. For the searched atoms, their ADOS curves almost overlap with the query oxygen atom. Their cosine similarities are all above 0.6 (1.0 would be identical ADOS). As a comparison, we show the ADOS data of four randomly selected oxygen atoms (see detailed configurations in the ESI†) in the OC20 dataset in Fig. 10. These random atoms have different ADOS from the query atom and their cosine similarities are generally smaller than the searched ones. This example shows that the GemNet embeddings are able to search for elementally and geometrically similar local environments for a single atom adsorbed on metallic surfaces. These similarities also lead to the similarity in the density of states (DOS). This example also implies a potential application of searching for similar local structures using the projected DOS with vector search methods, since similar DOS suggests similar elemental and geometric environments, as well as potentially similar chemical properties. Storing DOS data when building a database with some extra resources would be beneficial to this kind of application in the future.
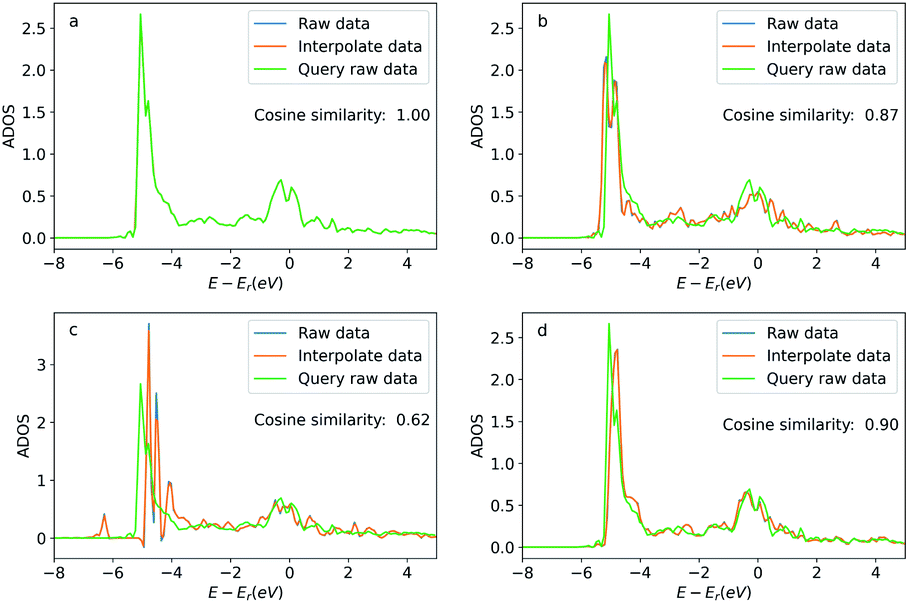 | ||
| Fig. 9 Density of states projected onto the p-orbital of the query and searched oxygen atoms. Figures (a) to (d) correspond the configurations (a) to (d) in Fig. 8. The blue curve is the original DOS energy and density data. The orange line is the linearly interpreted data from the original DOS data to make the energy stamps to be the same across the configurations. Cosine similarity was calculated using the interpreted data. | ||
 | ||
| Fig. 10 Density of states projected onto the p-orbital of four randomly selected oxygen atoms. The blue curve is the original DOS energy and density data. The orange line is the linearly interpreted data from the original DOS data to make the energy stamps to be the same across the configurations. Cosine similarity was calculated using the interpreted data. | ||
Next, we demonstrate that our method not only works for simple atoms like oxygen, but also for larger adsorbates like acetylene. In the OC20 dataset, we search for similar atoms with embeddings similar to that of the two carbon atoms in the query acetylene. We did not include the searching for similar atom embeddings to the hydrogen atoms since the carbon atom is the main feature of acetylene. Ignoring hydrogen atoms is also adopted in other molecular fingerprints like the SMILES.10 The query and searched configurations are shown in Fig. 11. The query object is an acetylene molecule adsorbed on a hollow site formed by three Cu atoms. The retrieved adsorption configurations are similar to the query one. The first point is that the found adsorbates are all acetylene without explicitly setting the search pool to be acetylene molecules. The second point is that the adsorption sites of the top two results (Fig. 11b and c) are hollow sites with three Cu atoms which are the same as the query one. This is not so clear in Fig. 11b, more details of the local structure can be found in the ESI.†
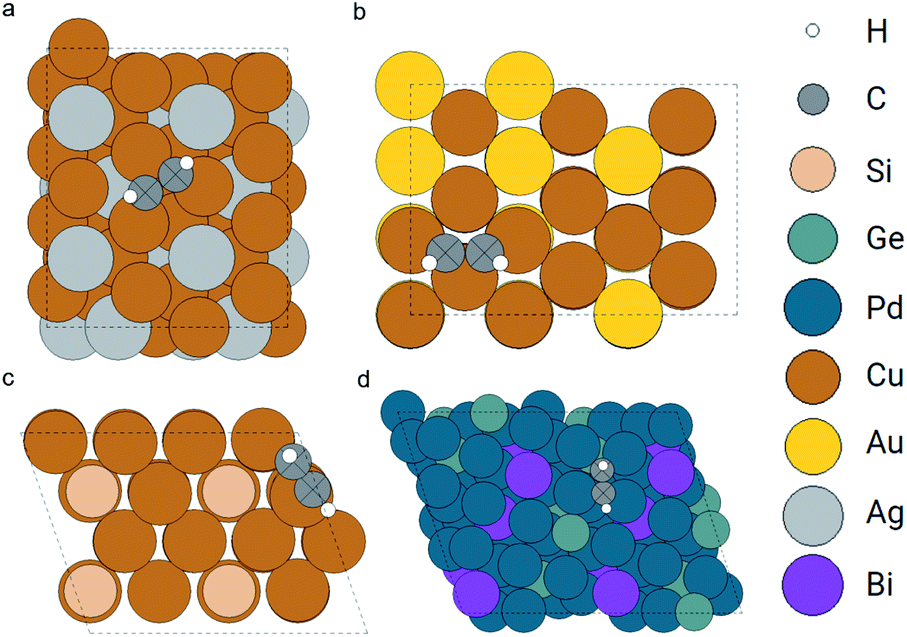 | ||
| Fig. 11 Configurations of the query (config. a) and top 3 retrieved acetylene adsorption configurations (config. b to d). The query and matched carbon atoms are marked as crossed. | ||
Similar to the oxygen case, we also compare the ADOS of the query and searched configurations. Fig. 12 shows the ADOS of a selected carbon atom of acetylene molecule in these systems. We can see the ADOS of the searched configurations are similar to the query one, and their cosine similarities are all above 0.65 which is much higher than that of four randomly selected configurations shown in Fig. 13. The similarities in terms of the adsorbates, adsorption sites, and DOS between the query and searched configurations suggest that our method also works well for adsorption systems with large adsorbates.
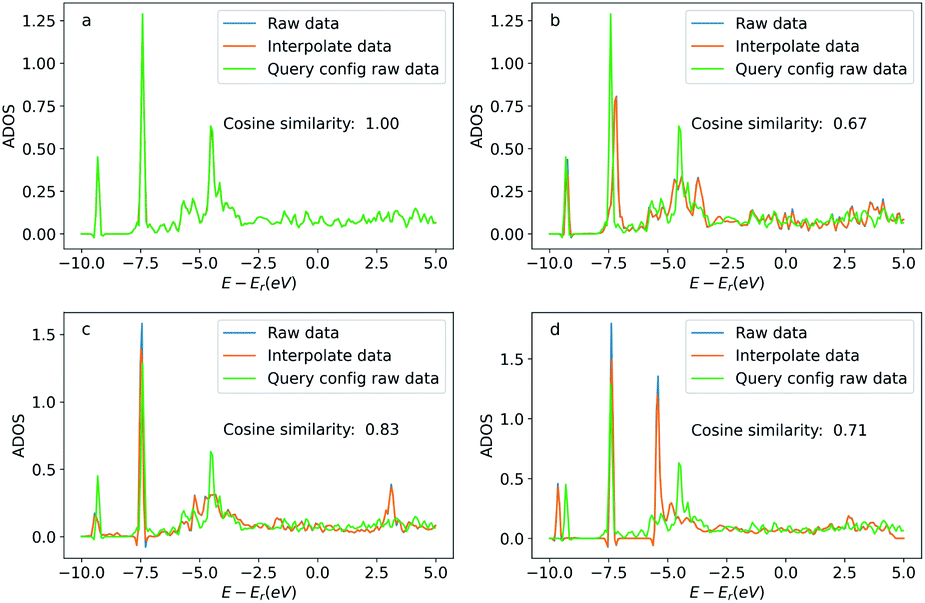 | ||
| Fig. 12 Density of states projected onto the p-orbital of the selected query and searched carbon atoms. Figures (a) to (d) correspond the configurations (a) to (d) in Fig. 11. The blue curve is the original DOS energy and density data. The orange line is the linearly interpreted data from the original DOS data to make the energy stamps to be the same across the configurations. Cosine similarity was calculated using the interpreted data. | ||
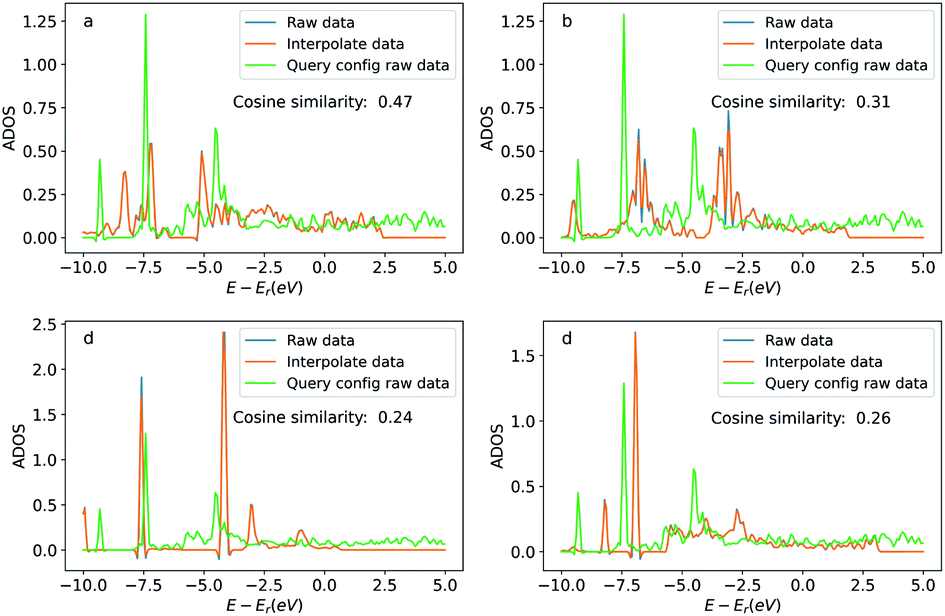 | ||
| Fig. 13 Density of states projected onto the p-orbital of the carbon atoms in four randomly selected acetylene adsorption configurations. The blue curve is the original DOS energy and density data. The orange line is the linearly interpreted data from the original DOS data to make the energy stamps to be the same across the configurations. Cosine similarity was calculated using the interpreted data. | ||
The results from the OC20 examples demonstrate that the method is able to find atoms in similar chemical environments illustrated by their similar ADOS. This could be very useful for catalyst design. One potential direction would be to find cheaper alternative catalyst materials which could maintain similar chemical environments for the adsorbates.
4 Conclusion
In this work, we showed how to use neural network embedding-based approximate nearest neighbor search framework to search for similar chemical (sub)structures in large chemical databases. We discussed two components of this framework: the neural network embedding and the approximate nearest neighbor search. The former enables us to represent local atomic configurations precisely. The latter provides us with a fast and cheap way to search for neighboring real vectors in a large database. In our work, we used GemNet and FAISS as the neural network model and the ANN search implementation. However, the usage of this framework is not limited to these two examples. Any molecular descriptors and other deep learning models can be used to generate the representing vectors for atoms or molecules. Factors such as computational scaling, expressiveness, and computational cost need to be considered when we make a choice. Similarly any vector search method can be used as the search engine. A cheap, fast, and user-friendly package would be favorable, such as FAISS.We illustrated the idea with examples across organic molecules, bulk systems, and solid surfaces with adsorbates, and showed the ability of this framework to find similar configurations in different databases. We presented the similarities from different aspects: elemental types, geometric features, energetic relevance, and the electronic density of states. These examples also reflect the generalizability of this framework for different types of atomistic systems.
Data availability
Data and processing scripts for this paper, including IPython notebooks, and the indexes are available at https://kilthub.cmu.edu/ at https://doi.org/10.1184/R1/19968323.Author contributions
Yilin Yang: investigation (equal); writing – original draft (equal). Mingjie Liu: investigation (equal); writing – original draft (equal). John R. Kitchin: conceptualization (equal); supervision (equal); writing – review and editing (equal).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported under NSF DMREF Award CBET-1921946. We would like to thank Professor Zachary W. Ulissi for generously providing the computational resources for constructing the search algorithm, and Muhammed Shuaibi for providing helps on using the Open Catalyst Project models codebase.References
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2020, 49, D1388–D1395 CrossRef PubMed.
- L. Chanussot, A. Das, S. Goyal, T. Lavril, M. Shuaibi, M. Riviere, K. Tran, J. Heras-Domingo, C. Ho, W. Hu, A. Palizhati, A. Sriram, B. Wood, J. Yoon, D. Parikh, C. L. Zitnick and Z. Ulissi, ACS Catal., 2021, 11, 6059–6072 CrossRef CAS.
- K. Moffat, V. J. Gillet, M. Whittle, G. Bravi and A. R. Leach, J. Chem. Inf. Model., 2008, 48, 719–729 CrossRef CAS PubMed.
- D. Stumpfe and J. Bajorath, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2011, 1, 260–282 CrossRef CAS.
- A. Cereto-Massagué, M. J. Ojeda, C. Valls, M. Mulero, S. Garcia-Vallvé and G. Pujadas, Methods, 2015, 71, 58–63 CrossRef PubMed.
- M. A. Skinnider, C. A. Dejong, B. C. Franczak, P. D. McNicholas and N. A. Magarvey, J. Cheminf., 2017, 9, 46 Search PubMed.
- O. Laufkötter, T. Miyao and J. Bajorath, ACS Omega, 2019, 4, 15304–15311 CrossRef PubMed.
- J. Schwartz, M. Awale and J.-L. Reymond, J. Chem. Inf. Model., 2013, 53, 1979–1989 CrossRef CAS PubMed.
- D. Weininger, J. Chem. Inf. Model., 1988, 28, 31–36 CrossRef CAS.
- D. Weininger, A. Weininger and J. L. Weininger, J. Chem. Inf. Comput. Sci., 1989, 29, 97–101 CrossRef CAS.
- J. M. Barnard and G. M. Downs, J. Chem. Inf. Comput. Sci., 1997, 37, 141–142 CrossRef CAS.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280 CrossRef CAS PubMed.
- J. Behler, J. Chem. Phys., 2011, 134, 074106 CrossRef PubMed.
- A. P. Bartók, R. Kondor and G. Csányi, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 184115 CrossRef.
- K. Fukushima, Biol. Cybern., 1980, 36, 193–202 CrossRef CAS PubMed.
- S. Hochreiter and J. Schmidhuber, Neural Comput., 1997, 9, 1735–1780 CrossRef CAS PubMed.
- K. Fukushima, Neural Networks, 2013, 37, 103–119 CrossRef PubMed.
- A. Sherstinsky, Phys. D, 2020, 404, 132306 CrossRef.
- A. Irtaza, M. A. Jaffar, E. Aleisa and T.-S. Choi, Multimed. Tools. Appl., 2013, 72, 1911–1931 CrossRef.
- A. B. Yandex and V. Lempitsky, 2015 IEEE International Conference on Computer Vision (ICCV), 2015 Search PubMed.
- H. Palangi, L. Deng, Y. Shen, J. Gao, X. He, J. Chen, X. Song and R. Ward, IEEE/ACM Transactions on Audio, Speech, and Language Processing, 2016, vol. 24, pp. 694–707 Search PubMed.
- T. Xie and J. C. Grossman, Phys. Rev. Lett., 2018, 120, 145301 CrossRef CAS PubMed.
- J. Klicpera, F. Becker and S. Günnemann, Gemnet: Universal Directional Graph Neural Networks for Molecules, 2021, https://arxiv.org/abs/2106.08903v8 Search PubMed.
- A. Andoni, P. Indyk and I. Razenshteyn, Approximate Nearest Neighbor Search in High Dimensions, 2018, http://arxiv.org/abs/1806.09823v1 Search PubMed.
- M. Wang, X. Xu, Q. Yue and Y. Wang, Proceedings of the VLDB Endowment, 2021, vol. 14, pp. 1964–1978 Search PubMed.
- Q. Huang, J. Feng, Y. Zhang, Q. Fang and W. Ng, Proceedings of the VLDB Endowment, 2015, vol. 9, pp. 1–12 Search PubMed.
- Q. Chen, H. Wang, M. Li, G. Ren, S. Li, J. Zhu, J. Li, C. Liu, L. Zhang and J. Wang, SPTAG: a library for fast approximate nearest neighbor search, 2018 Search PubMed.
- R. Guo, P. Sun, E. Lindgren, Q. Geng, D. Simcha, F. Chern and S. Kumar, Accelerating Large-Scale Inference With Anisotropic Vector Quantization, 2019, https://arxiv.org/abs/1908.10396v5 Search PubMed.
- J. Johnson, M. Douze and H. Jegou, IEEE Transactions on Big Data, 2021, 7, 535–547 Search PubMed.
- R. B. Marimont and M. B. Shapiro, IMA Journal of Applied Mathematics, 1979, 24, 59–70 CrossRef.
- J. Sivic and A. Zisserman, Proceedings Ninth IEEE International Conference on Computer Vision, 2003, pp. 1470–1477 Search PubMed.
- H. Jégou, M. Douze and C. Schmid, IEEE Transactions on Pattern Analysis and Machine Intelligence, 2011, 33, 117–128 Search PubMed.
- L. Ruddigkeit, R. van Deursen, L. C. Blum and J.-L. Reymond, J. Chem. Inf. Model., 2012, 52, 2864–2875 CrossRef CAS PubMed.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Scientific Data, 2014, 1, 140022 CrossRef CAS PubMed.
- K. T. Schütt, P. Kessel, M. Gastegger, K. A. Nicoli, A. Tkatchenko and K.-R. Müller, J. Chem. Theory Comput., 2018, 15, 448–455 CrossRef PubMed.
- H. L. Morgan, J. Chem. Doc., 1965, 5, 107–113 CrossRef CAS.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed.
- D. Bajusz, A. Rácz and K. Héberger, J. Cheminf., 2015, 7, 20 Search PubMed.
- J. Vandermause, S. B. Torrisi, S. Batzner, Y. Xie, L. Sun, A. M. Kolpak and B. Kozinsky, npj Comput. Mater., 2020, 6, 20 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2dd00055e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |