
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceData mining crystallization kinetics†
Diego A.
Maldonado
 ,
Antony
Vassileiou
,
Blair
Johnston
,
Alastair J.
Florence
and
Cameron J.
Brown
*
,
Antony
Vassileiou
,
Blair
Johnston
,
Alastair J.
Florence
and
Cameron J.
Brown
*
EPSRC Future Manufacturing Research Hub for Continuous Manufacturing and Advanced Crystallisation (CMAC), University of Strathclyde, Technology and Innovation Centre, 99 George Street, Glasgow G1 1RD, UK. E-mail: cameron.brown.100(at)strath.ac.uk
First published on 1st August 2022
Abstract
The population balance model is a valuable modelling tool which facilitates the optimization and understanding of crystallization processes. However, in order to use this tool, it is necessary to have previous knowledge of the crystallization kinetics, specifically crystal growth and nucleation. The majority of approaches to achieve proper estimations of kinetic parameters require experimental data. Over time, a vast amount of literature on the estimation of kinetic parameters and population balances has been published. Considering the availability of data, in this work a database was built with information on solute, solvent, kinetic expression, parameters, crystallization method and seeding. Correlations were assessed and cluster structures identified by hierarchical cluster analysis. The final database contains 336 datapoints of kinetic parameters from 185 different sources. The data were analysed using kinetic parameters of the most common expressions. Subsequently, clusters were identified for each kinetic model. With these clusters, classification random forest models were made using solute descriptors, seeding, solvent, and crystallization methods as classifiers. Random forest models had an overall classification accuracy higher than 70% whereby they were useful for providing rough estimates of kinetic parameters, although these methods have some limitations.
1. Introduction
Year by year the challenges that the pharmaceutical sector has to face do not cease to increase. Regulatory requirements, patients' needs, and market competition are becoming more challenging, which has led the industry to rethink the model of business and seek alternatives to improve its productivity. Historically, the business model has been based on the discovery of new molecules and patent protection to a certain extent. However, the cost of developing new drugs increases with time and the patent expiry time remains the same.1,2 In addition, the pharmaceutical industry has been characterized by problems of innovation, flexibility, and efficacy in its processes, which increase costs and hinder the response to customers' demands as required.2 As a result, the industry is seeking to optimize resources and improve its procedures to satisfy its needs and produce better medicines.In this way, various initiatives have been introduced in the industry in the last few decades. They include the use of process analytical technology (PAT), the concept of quality by design (QbD), and the development of continuous pharmaceutical manufacturing (CPM), which has come along with technological and scientific advances.3,4 Consequently, many methodologies that optimize resources and create more efficient processes have been adopted. In particular, modelling techniques are of great interest given their ability to predict and provide information in an efficient manner.1,5
Modelling techniques aim to depict a material property or a process through a mathematical expression which can be founded on either a physical or empirical relationship.1,5,6 These representations enable the simulation of a process and assess different scenarios in which a condition or property changes.1,3 Likewise, modelling techniques facilitate the evaluation and analysis of the effect of factors on process performance or product quality.4 In light of these potential usages, the advantages that these models offer are numerous; an adequate model may enable the number of experiments necessary to obtain certain information to be reduced,3 or it may help with quality improvement as modelling provides a valuable insight into the design of a process, which would allow conditions to be selected or specifications to be established systematically with a scientific base.4 As a result, these tools have been used more frequently in recent years.
For crystallization, a critical unit operation in the control and delivery of APIs with desired specifications, the most common form of modelling is through a population balance model (PBM), typically combined with momentum, mass and energy balances.7 The main attraction of a PBM is the ability to predict the crystal size distribution (CSD). To fully resolve a PBM, expressions representing various crystallization phenomena, such as growth, primary nucleation, secondary nucleation, breakage, and agglomeration, are required. For each phenomenon several expressions are available, ranging from mechanistic to semi-empirical.7 Therefore, the selection of the most appropriate kinetic expression and the determination of the respective parameters are crucial in order to obtain accurate predictions. Currently, these activities require the collection of data through an experimental approach, with subsequent application of optimization algorithms that enable proper estimations. Nonetheless, there exists a vast amount of literature tackling PBM and the calculation of kinetic parameters, considering numerous factors such as solute, solvent, operational conditions, etc.
Theoretically, crystallization sub-processes are strongly affected by interactions between the solute and solvent and process conditions. In this regard, it could be observed that some kinetic parameters include terms that describe directly any property related to the solute and solvent, e.g. surface tension and molar volume. In the same way, it might be expected that kinetic parameters employed in nucleation and growth models, which do not have an explicit relation with the physical or chemical properties of the components involved, follow a distribution or correlate with some variables associated with the solute, solvent or process. Finding these relations could potentially be helpful to provide a reasonable range of values within kinetic parameters or an approximate estimation of these which may be used in PBM.
This work aims to (1) build a database containing information on kinetic parameters of primary nucleation and crystal growth of different crystallization processes, including solute, solvent, crystallization technique, seeding, and kinetic expression, and (2) establish the feasibility of a model that enables estimation of kinetic parameters of growth and primary nucleation by analysis for patterns and correlations with some molecular and process descriptors.
2. Theory
In the modelling of crystallization, supersaturation plays a major role as the driving force that makes this process occur. Supersaturation is a condition where there is an excess of solute dissolved in a particular solvent with respect to its solubility. Usually, supersaturation can be expressed in terms of absolute supersaturation (ΔC = C − C*), relative supersaturation (S = C/C*) or degree of supersaturation (σ = S − 1), where C and C* are solute concentration and solubility, respectively.8 Due to its importance in crystallization, supersaturation is included in the modelling of crystallization subprocesses such as primary nucleation and growth which are described below.Nucleation involves the generation of small crystals or nuclei that will serve as a template for growth. Nucleation can be primary and secondary.9 In the latter, nuclei are formed by breakage or attrition of existing crystals.9 On the other hand, crystals are formed from a clear solution in primary nucleation. In turn, primary nucleation can be homogeneous and heterogeneous depending on the influence of impurities or other substances in the solution.9 The modelling of primary nucleation can be derived from classical nucleation theory (CNT), or empirical equations which can be seen in Table 1.
| Model | Equation | Comments |
|---|---|---|
| Heterogeneous nucleation (CNT) |
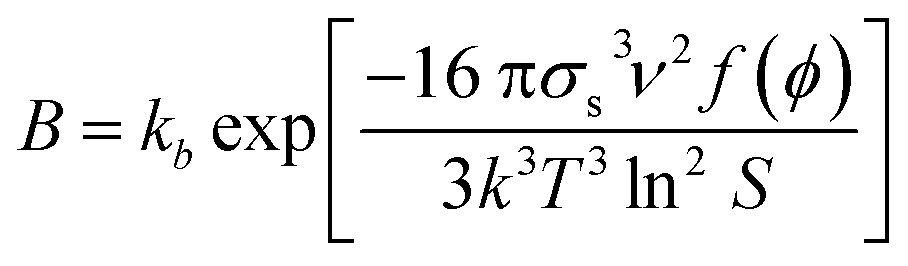
|
k b : nucleation rate pre-exponential constant, k: Boltzmann constant, σs: interfacial surface, S: relative supersaturation, T: temperature, ν: molecular volume, f(ϕ): factor for the effect of impurities or other substances |
| Homogeneous nucleation (CNT) |
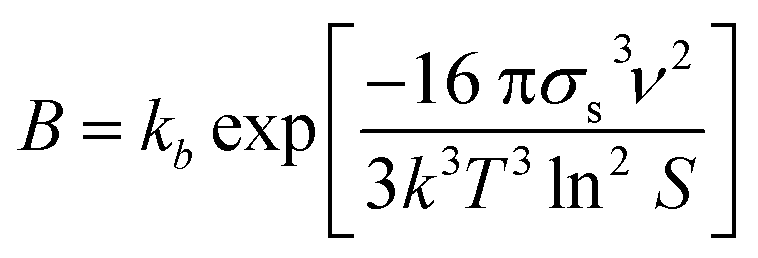
|
|
| Empirical expression | B = kbσb | k b : nucleation rate pre-exponential constant, σ: degree of supersaturation |
Generated nuclei and existing crystals undergo growth over time. In this process, there is a mass transfer that can happen by a convective transport and diffusion of solute molecules towards the surface.8 Then, the solvent is displaced from solute units and crystal union sites, and the solute integrates into the available sites. The growth rate is limited by the slowest step that can be mass transfer or surface integration.8 Thus, expressions that describe the growth rate at every step have been proposed. For instance, in the case of surface integration, models such as rough growth, birth & spread, and spiral growth are found.10 Some of the most used expressions for growth rate are shown in Table 2.
| Model | Equation | Comments |
|---|---|---|
| Size independent growth | G = kgσg | k g : growth rate pre-exponential constant |
| Temperature dependent |
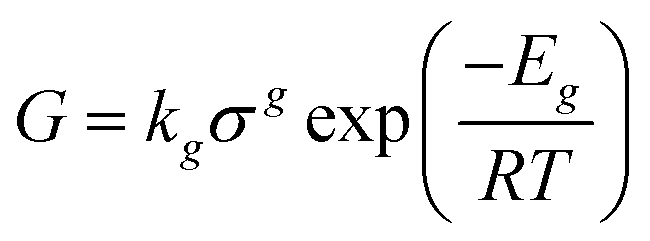
|
E g : activation energy, T: temperature, R: ideal gas constant |
| Burton–Cabrera–Frank |
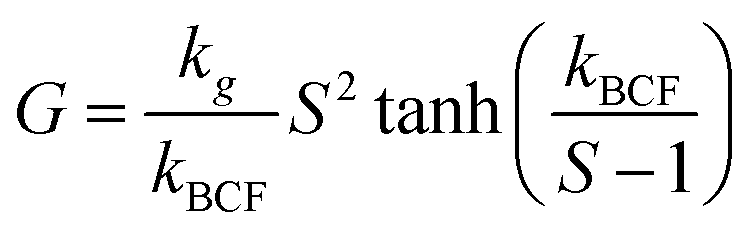
|
S: relative supersaturation, kBCF: Burton–Cabrera–Frank constant |
3. Methods
3.1. Data collection
Initially, a sample frame of potential articles containing the information of interest was built by web-scraping search results from different scientific databases. This procedure was conducted as described by Kwartler.11 To obtain these results, several search strategies were implemented in the following databases: ScienceDirect, ACS Publications, AIChE, and Scientific Research. The combinations of keywords, inclusion and exclusion criteria are detailed in the ESI.† All the searches were performed between June 4 and 6, 2019. The searches were limited to research articles in English – avoiding, for instance, reviews or book chapters – as the main objective was to obtain experimental data. Subsequently, the information on title, journal, and authors was extracted from the respective websites. The data was next stored and pre-processed. Pre-processing consisted of text cleaning, duplicate removal and filtering. Text cleaning involved stripping extra white spaces and fixing corrupted characters to then remove duplicates, which resulted in a list of 1938 articles. All these tasks were carried out using the R statistical program version 3.5.1 and Microsoft Excel (2016).This list was later filtered by journal and title. Firstly, all the results were published in a total of 125 journals where around 85% of these papers corresponded to solely 15 journals. Therefore, journals with the number of results lower than 16 were discarded since the remaining 15% did not reach this number of papers. To verify that important data was not omitted, articles in the discarded journals went through a non-exhaustive review and most of the search results turned out to contain non-relevant information. Thus, with the remaining articles, a word frequency analysis of the titles was carried out. Further information on text mining and frequency analysis can be found in Kwartler.11 Words with a frequency higher than 3 and identified as non-relevant can be seen in the ESI.† The article titles containing these words were excluded to finally obtain a list of 1187 articles.
The remaining articles were then reviewed manually and data were collected. During the review, various documents were found to have incomplete information or to have taken data from another source; therefore, more results were discarded. Likewise, articles that initially were not included in the list were added by considering the source stated in the reviewed papers. The criteria used to select the articles in this stage are illustrated in Fig. 1. Information on description, name, data type and comments was recorded and can be seen in the ESI.†
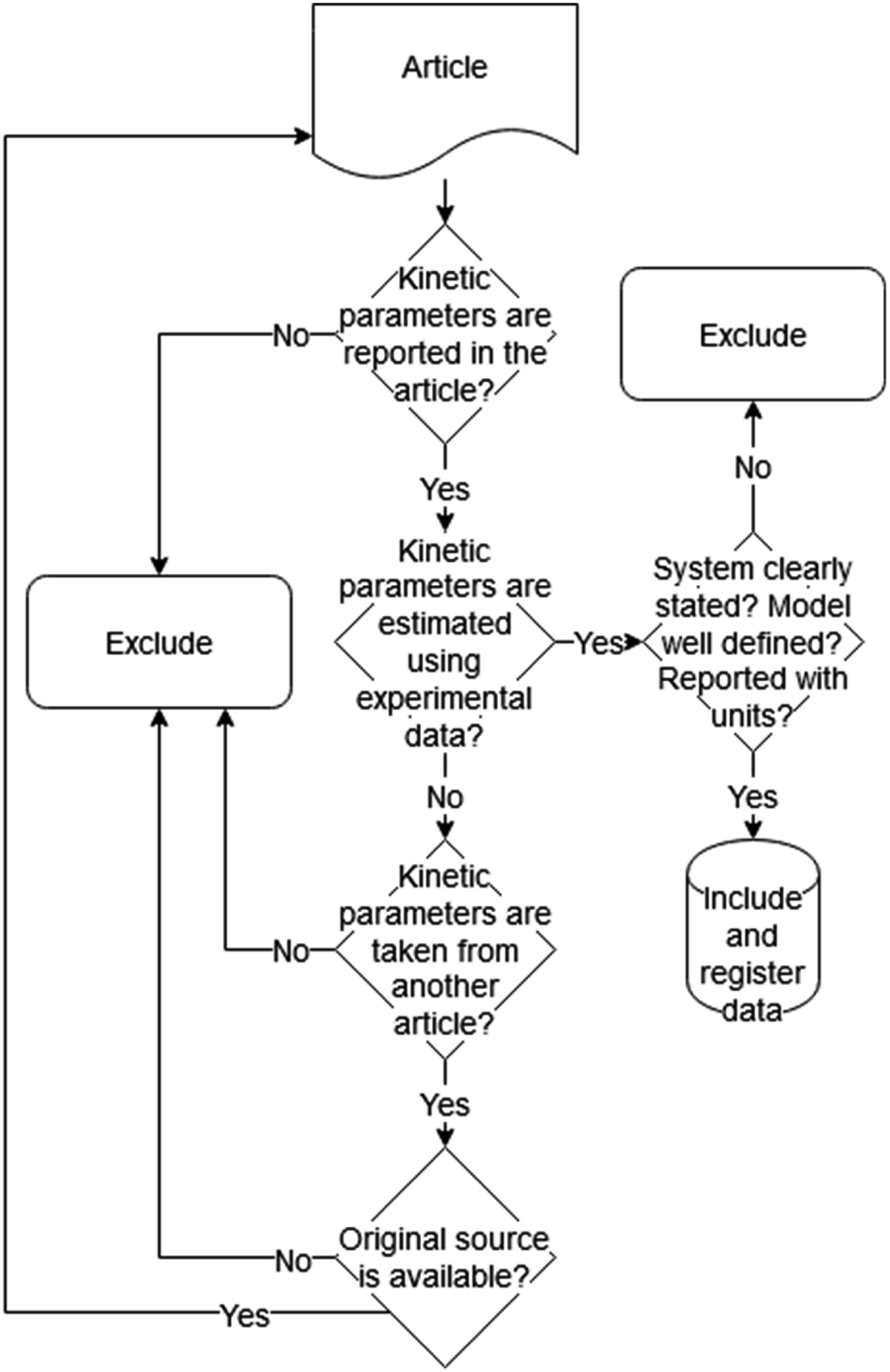 | ||
| Fig. 1 Exclusion/inclusion criteria for the final list of articles. | ||
3.2. Data analysis
Before conducting the analysis, the collected data went through several cleaning steps. Firstly, the units of kg and kb were converted into international system units (SI). Most units of kg were either m s−1 or m s−1(g per g solvent)−1, while kb was mostly in # m−3 s−1. However, the units of kb and kg depended on the factors considered in the kinetic models. Therefore, it was not possible to transform all the units into the same unit and ensure all the data were comparable in this aspect. Additionally, there were also a few articles in which an equation was given to calculate the constants and other articles which estimated the bidimensional growth rate. These cases were recorded but not considered during the analysis. Another adjustment was the scale where logarithm transformation was applied to kg and kb, given the order of magnitude that these constants presented. On the other hand, the kinetic equation nomenclature was harmonized since several models could be considered equivalent but were expressed in different terms according to the author. Finally, to evaluate whether or not it is feasible to build a model to estimate kinetic parameters, associations between these and molecular descriptors were assessed. Analyses and visualizations were carried out using the R statistical environment software version 3.5.1.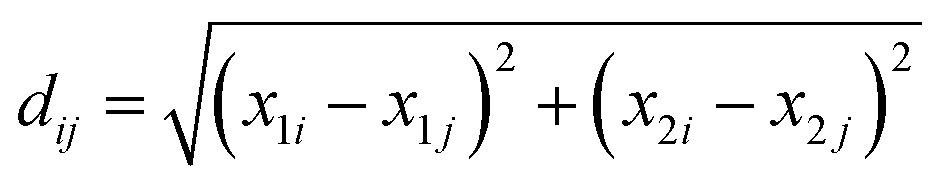 | (1) |
Thus, a model of classification was first built and the model parameters were tuned. The groups were created by cluster analysis and the classifiers, or potential predictors, corresponded to the molecular descriptors, solvent, method, and seeding. Subsequently, the importance of the predictors was estimated as the mean decrease in accuracy (MDA). RF implementation was performed as detailed elsewhere.15 The top 15 most important variables were analysed in detail. To conclude, the selected classifiers were analysed in detail with respect to kinetic constants to assess how they are related.
4. Results and discussion
4.1. Data description
The database contains 336 datapoints of kinetic parameters obtained from 185 articles, of which 21 were not included in the initial sample frame, which means around 1 in 10 revised articles had relevant information. Most of the excluded papers contained incomplete data – for example, the solute identity was stated generically or not provided – or consisted of reviews wherein the primary focus was on the mathematical or theoretical aspects of crystallization kinetics. Thus, if this approach is to be used in future work, the search strategies ought to be refined to reduce the content which was unrelated and increase search efficacy by including additional keywords, limiting the search to certain journals or considering other filters.In the recorded data, 297 corresponded to growth rate and 145 related to primary nucleation rate. The data are distributed over 87 solutes and 27 solvents. In particular, solutes are mostly of low molecular weight (<500 Da) and diverse chemical structure, being 25 inorganic and 62 organic molecules. Another important aspect to highlight is the large predominance of data related to crystallization in aqueous systems. As stated previously, there was a total of 27 solvents where 12 corresponded to aqueous–organic mixtures that, along with water, represented 72.6% of the collected data. Moreover, when the antisolvent technique was applied, water was frequently used as an antisolvent (74.5%), which reinforced the aqueous system preponderance. As a consequence, the analysis of this study concerning the effect of solvent on kinetic parameters may be limited due to scarce information on other solvents apart from water. A breakdown of the information related to solute, solvent, method, seeding, and kinetic expressions can be seen in Table 3.
| Solute | |
| Paracetamol | 8.93% |
| Glutamic acid | 6.85% |
| Felodipine | 3.87% |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| Solvent | |
| Water | 65.2% |
| Ethanol | 9.8% |
| Methanol | 8.3% |
|
|
| Method | |
| Cooling | 62.2% |
| Precipitation | 18.2% |
| Antisolvent | 15.2% |
| Evaporative | 1.2% |
| Combinations | 3.2% |
|
|
| Seeding | |
| Seeded | 50.0% |
| Unseeded | 47.6% |
| Combination of seeded and unseeded | 2.4% |
|
|
| Growth rate expression | |
| G = kgΔCg | 31.6% |
| G = kg(S − 1)g | 25.3% |
| G = kg(S − 1)ge(−Eg/RT) | 12.1% |
|
|
| Nucleation rate expression | |
| B = kbΔCb | 42.8% |
| B = kbe(−B/ln2S) | 19.3% |
| B = kb(S − 1)be(−Eb/RT) | 5.5% |
Regarding kinetic equations, the expressions used to model growth rate were more diverse than the primary nucleation rate. In total, 38 different expressions for growth and 22 different expressions for nucleation rate were found. However, the majority of the crystal growth expressions were derived from the first two shown in Table 3. In these cases, the models included multiplicative terms related to stirring rate, crystal size, or temperature adjustment by Arrhenius, the last being the most frequent. More complex equations like the birth & spread model were also found, but they were isolated cases. For nucleation rate, while there were various ways of modelling, a clear tendency to use the empirical nucleation rate and, to a lesser extent, equations derived from CNT was observed. As can be seen, the power-law models are predominant in both crystal growth and primary nucleation modelling. During the revision, a specific reason to use one or another expression was not found. However, the power-law expressions have long been used in crystallization kinetics modelling since experimental data generally fit well to these equations.16
Fig. 2 illustrates the sampling distribution of different kinetic parameters. It can be observed that the most frequent values were in the order of 108 and 10−6, in international units, for nucleation and growth rate constants, respectively. Likewise, the most common estimations of b and g corresponded approximately to 2.0 and 1.0. All the distributions were right-skewed to a certain extent. However, this behaviour was more notable for the exponents. In this particular case, it was more frequent to find low values of b and g. This fact was emphasized by seeing that 50% of the data were contained within the intervals between 1.0 and 2.0 for g, and between 1.5 and 5.9 for b, which may be considered relatively narrow compared to all the possible values. Returning to kinetic constants, logkb values lower than 0 or higher than 30 were not common since they only represented around 13% of the data, while the majority of logkg values were lower than 0 at about 75%. Nonetheless, although similar distributions for kinetic parameter values can be seen when separated by kinetic models, some differences between models were observed.
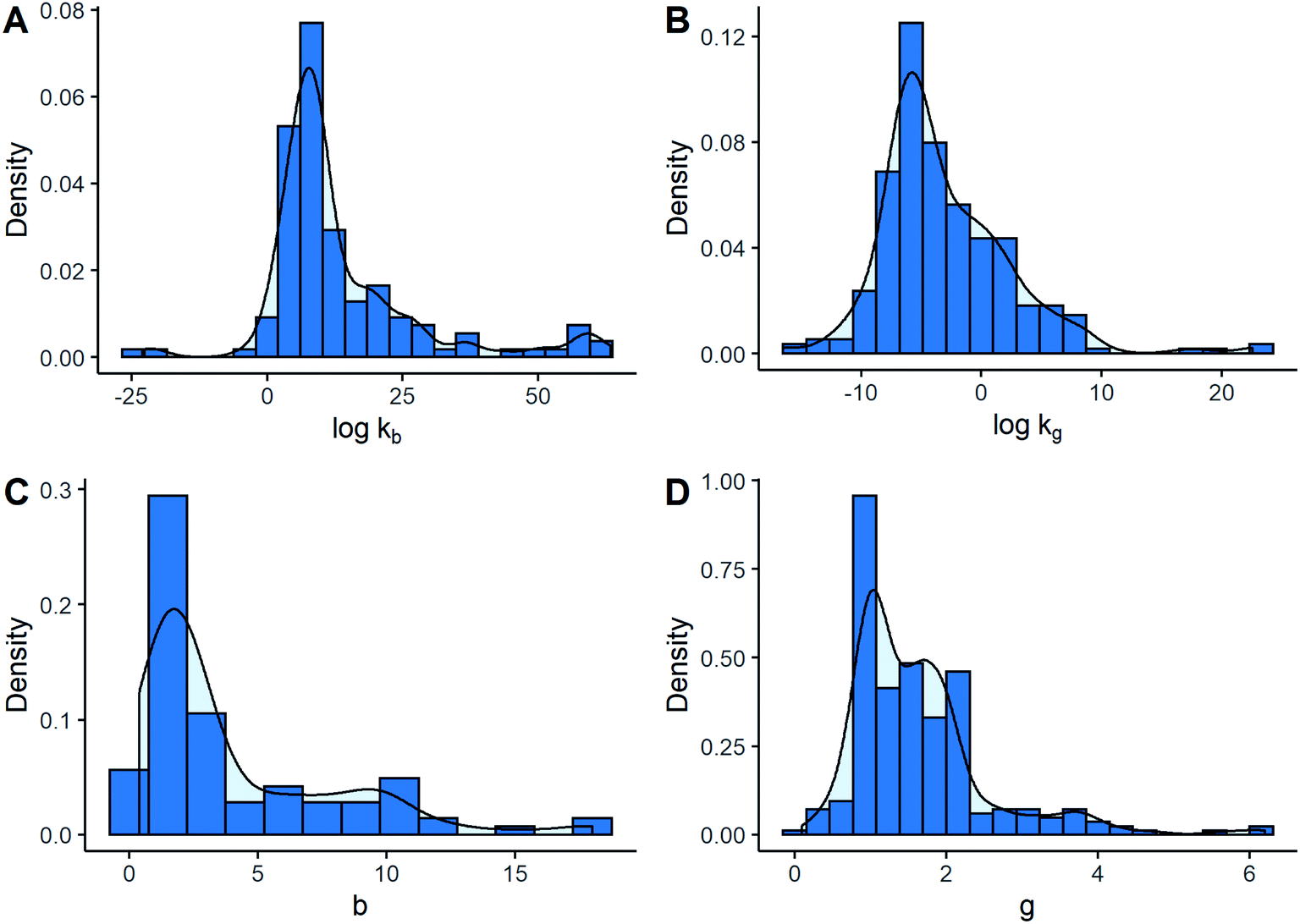 | ||
| Fig. 2 Histograms of kinetic parameters: (A) primary nucleation rate constants; (B) growth rate constants; (C) exponential term associated with supersaturation in primary nucleation rate; (D) exponential term associated with supersaturation in growth rate. | ||
The comparison of the most common kinetic models is displayed in Fig. 3. All the distributions were right-skewed and had a similar shape compared to those discussed previously. By contrasting cumulative distributions, it was possible to notice that kg values were lower when growth was a function of supersaturation ratio instead of absolute supersaturation (Mann–Whitney U = 1945.5, p-value < 0.05). This difference was around two orders of magnitude. On the other hand, there seems to have been no significant difference in g between growth models (Mann–Whitney U = 2828.5, p-value = 0.301). In the same way, when kb values from the empirical model were contrasted with the CNT model, a high level of coincidence was observed, by which it could be said that the available evidence does not allow detection of significant differences (Mann–Whitney U = 821, p-value = 0.774). Thus, the only constant significantly affected by the model was kg.
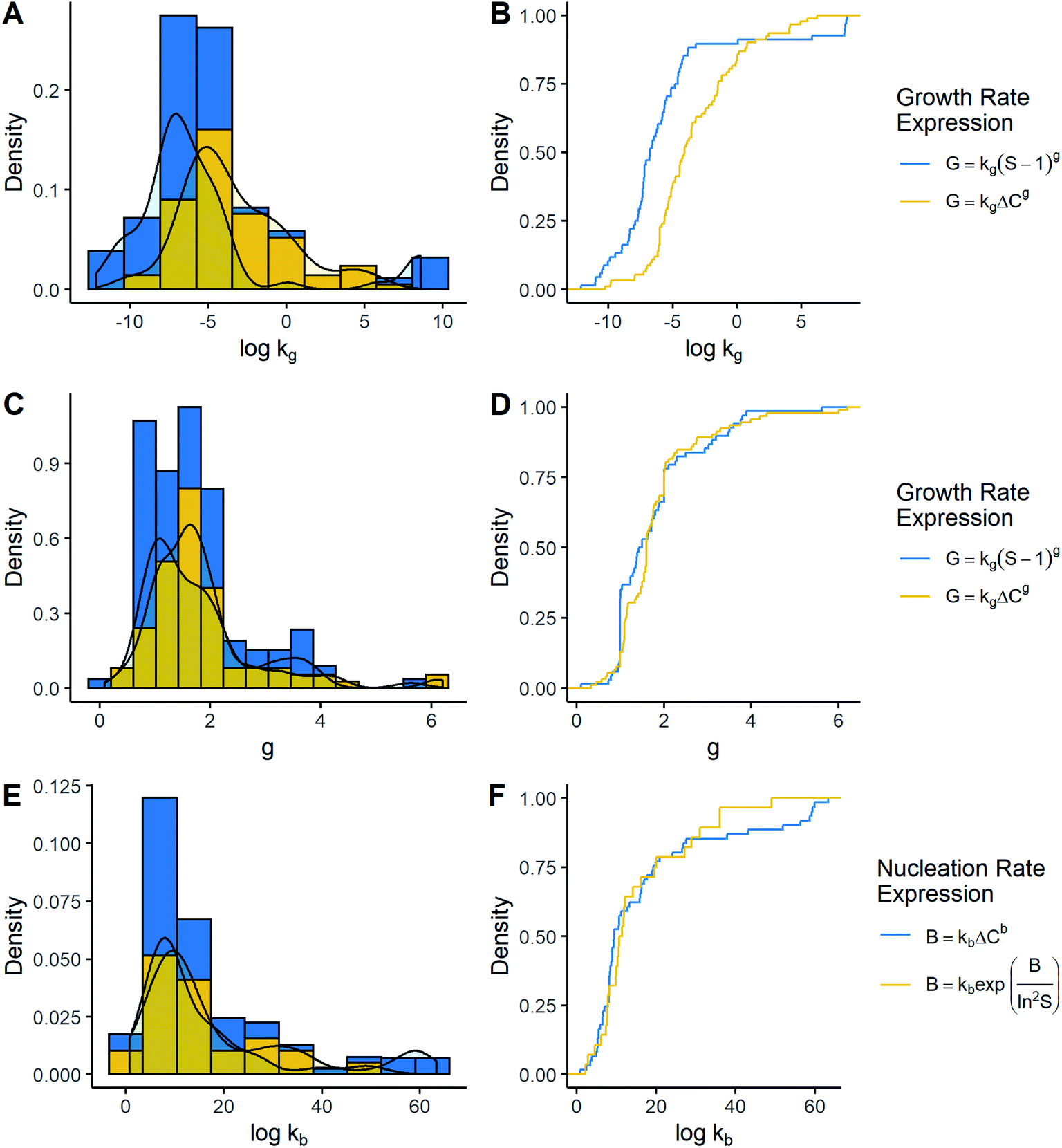 | ||
| Fig. 3 Histograms and empirical cumulative distribution of the kinetic parameter by kinetic expression. (A) Histogram of logkg and (B) cumulative logkg for empirical growth rate expressions using the supersaturation ratio and absolute supersaturation. (C) Histogram of g and (D) cumulative g for empirical growth rate expressions using the supersaturation ratio and absolute supersaturation. (E) Histogram of logkb and (F) cumulative logkb for empirical nucleation rate and CNT. | ||
As for crystal growth, g depends – among other factors – on the growth mechanism which in turn depends on the supersaturation degree.10 It has been reported that g generally is between 1.0 and 2.0, which coincides with the results found in this work, although many datapoints were outside this range.10,16 Additionally, g does not seem to be affected by the way supersaturation is expressed. However, kg showed different values caused by the kinetic model. In line with this, these differences in the magnitude of kg are expected. Having as a reference the models G = kg(S − 1)g and G = kgΔCg, it could be said that k{ΔC}g = k{S}g/C*g, which explains the difference. Finally, the tendency shows that differences may be between 2 and 3 orders of magnitude, where the values of k{ΔC}g and k{S}g are around 10−4.11 m s−1(g per g solvent)−1 and 10−6.77 m s−1, in that respective order. On the other hand, reference values of kg were not found for either model. However, according to the literature, growth rates may be in the order of 10−7 m s−1 and 10−9–10−8 m s−1 at supersaturation (S − 1) of 0.01 and 10 to 100, respectively.9,10,17 Assuming g = 1 due to being the most common, kg might take values in the order of 10−11 to 10−5 m s−1 for the model using the supersaturation ratio. Consequently, it can be noted that most of the recorded data are within the interval previously described, indicating a certain agreement with what would be expected.
Concerning primary nucleation, neither reference ranges of kb or b were found for the power-law empirical model. Thus, the pre-exponential terms in CNT were compared to rate constant kb. In terms of magnitude, no large differences were observed in the models. Therefore, this suggests that the expected values and interpretation of both constants might be similar. In the CNT model, the pre-exponential term is expected to be around 1030 #m3 s−1 or 1010–1020 # m3 s−1, depending on whether nucleation is homogeneous or heterogeneous.10 As a result, it can be seen that a big portion of the constants fitted in either CNT or the power-law model is within these intervals, indicating a certain level of concordance compared to previous revisions.
To conclude this part, a database of kinetic parameters was built and, considering all the points exposed for growth and primary nucleation, it can be said that there are no major deviations between the collected data and the information available in other studies. This fact provides a certain level of reliability in the data. Additionally, since the source of data is varied in terms of methods and solutes, it is possible to establish approximate intervals in which some kinetic parameters would be expected to belong. However, in this scenario, some constraints are the limited variety of solvents and that most data are concentrated in a few models, due to which the studied kinetic parameters in the next sections were limited to the most common models and there might be a bias toward aqueous systems.
4.2. Association between kinetic parameters and descriptors
kg and g. In the same way, the variables correlated with g did not match between models, and for logkg, some overlap such as b_max1len, PEOE_VSA+4, and vsurf_DW13 which were lower in G = kgΔCg. Generally speaking, a similar behaviour was seen in nucleation rate constants compared to growth models. logkb and b also showed mainly weak to moderate correlations. Nonetheless, logkb, in contrast to the other model kinetic parameters, had a strong correlation (|r| > 0.7) with two descriptors a_nCl (number of chlorine atoms) and vsurf_DW12 (contact distance between lowest hydrophilic energies) with values of around 0.78 for both descriptors. However, by analysing these correlations thoroughly, some extreme values were observed which might have caused an overestimation of these relationships. To conclude, overall, strong correlations between solute descriptors and kinetic parameters could not be identified, except for logkb, which suggested that linear relationships between the assessed solute properties and the kinetic parameters are poor. These results indicate that these molecular descriptors may not be appropriate predictors or classifiers using linear models, by which, to discard definitely these variables, non-linear associations should be assessed.
kg associated with MEK and ACN were significantly higher than the rest of the solvents and these values exceed 0.0. On the other hand, the rate order g was similar among the distinct solvents, being lower than 2.0. In line with this, ACN values were the lowest with respect to the other solvents. In relation to the model G = kg(S − 1)g, the values of logkg and g were comparable to the majority of solvents, solely seeing a large difference of logkg in aqueous mixtures and g in EtOH. The results of kb and b for each solvent are shown in Fig. 4. Note that while MEK and the aqueous mixtures had the highest values of b, they presented the lowest values of logkb. In contrast, even though EA also possessed a high b, its logkb was comparable to that of water and MeOH. As for MeOH, the data were very scattered for both logkb and b, thereby hindering the determination of a difference with respect to the other solvents. Thus, for primary nucleation as well as growth models, it was difficult to find significant variations of kinetic parameters with relation to the solvent given the majority of solvents showed the tendency to be around the same range and the number of datapoints and solutes per solvent was rather unbalanced. Nonetheless, there are two cases to highlight: MEK in growth and MeOH in nucleation. It has been documented that numerous solvent properties such as viscosity, polarity, and chemical nature can affect crystal growth as well as primary nucleation processes.10,18 Thus, significative differences among solvents were expected to be observed. However, despite the fact that there were some solvents of different nature, kinetic parameters were rather similar. By observing the particular cases of MEK in growth and MeOH in nucleation, it can be seen that these two have a wide scattering of their parameters compared to water, which is the most frequently employed solvent. MeOH data comprised two solutes – paracetamol and felodipine – crystallized by precipitation and antisolvent methods; every system exhibited substantial differences in its nucleation parameters. On the other hand, MEK had a wide dispersion of g which is explained by changes in cooling rate in co-crystallization of agomelatine–citric acid. Considering water, there were many more possible combinations of methods, solutes and process conditions but such scattering was not exhibited. These facts indicate there might be interactions between solvent and several other factors, such as process conditions, and the solvent effect may not be evaluated in isolation. In future studies, a better approach might be to analyse interactions with other factors or use solvent descriptors like viscosity, in order to identify potential associations in a clearer way.
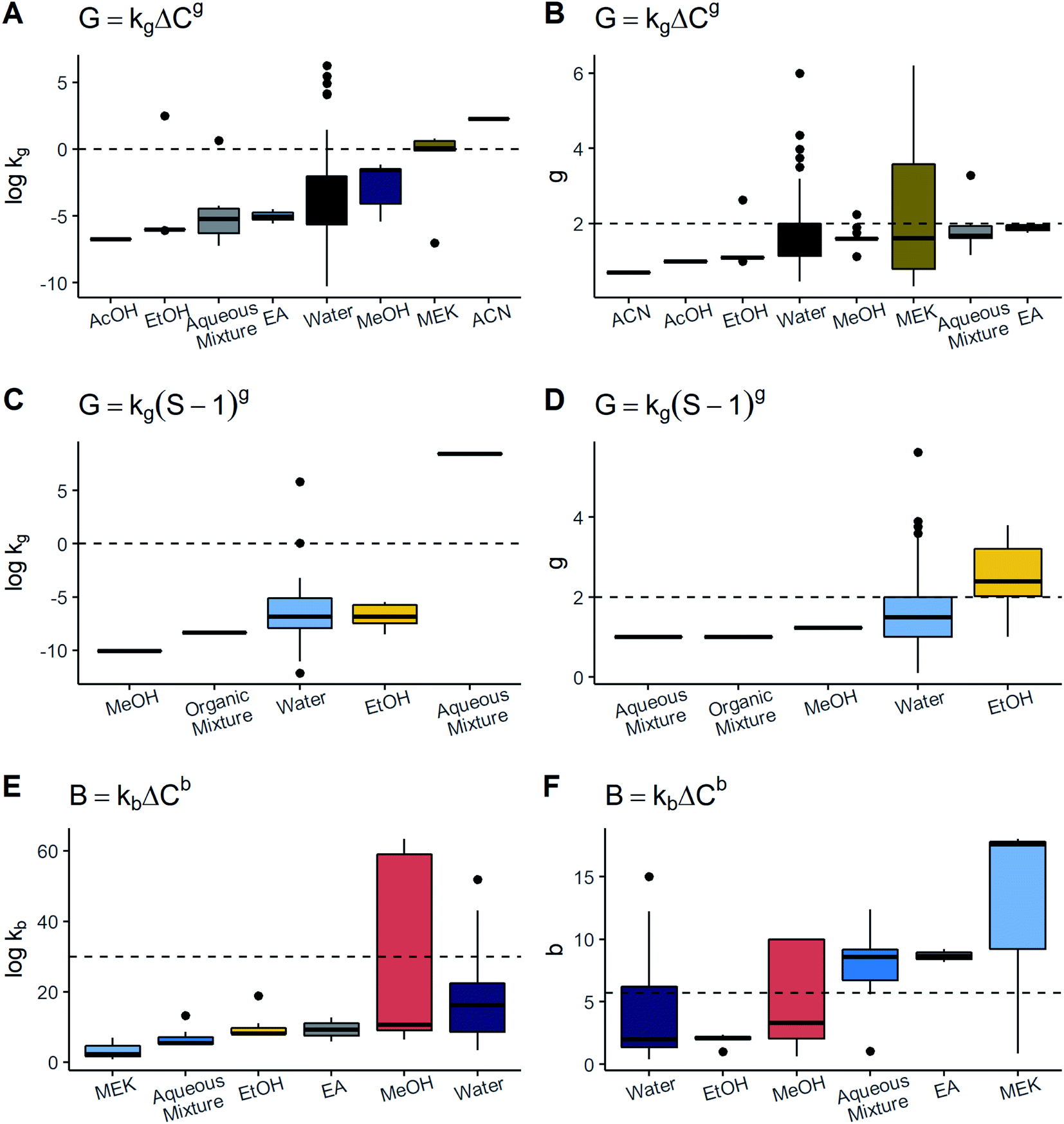 | ||
| Fig. 4 Association between kinetic parameters and solvents. Boxplots for (A) logkg in the growth rate expression with absolute supersaturation vs. solvent, (B) g in the growth rate expression with absolute supersaturation vs. solvent, (C) logkg in the growth rate expression with supersaturation ratio vs. solvent, (D) g in the growth rate expression with supersaturation ratio vs. solvent, (E) logkb in the nucleation rate expression vs. solvent, and (F) b in the nucleation rate expression vs. solvent. AcOH, acetic acid; EtOH, ethanol; EA, ethyl acetate; MeOH, methanol; MEK, methyl ethyl ketone; Aqueous mixture, mixture of water + an organic solvent; Organic mixture, mixture of several organic solvents. From left to right, solvents are placed in ascending order of medians. | ||
kg and g in the model G = kgΔCg followed by evaporative and antisolvent crystallization. In cooling crystallization, it was observed that the data exhibited the highest scattering in both parameters due to which, despite having the highest values of both kinetic parameters, these were not notably different to the other techniques. These results contrasted with the model G = kg(S − 1)g since the same patterns were not seen. In this model, for example, precipitation and cooling had the lowest values of logkg and g. The values of g in both models tended to be high or moderately higher than 1.0 for precipitation and antisolvent methods. These techniques are characterised by reaching a very high level of supersaturation.9,10,17 Under these conditions, g is generally higher than 2.0 given the low solubility in the system.10 Thus, the results are consistent. Conversely, kg does not exhibit the same behaviour, suggesting that kg may not necessarily show a pattern related to the technique. As for cooling crystallization, the dispersion is generally wider than the other techniques. A reason might be that cooling crystallization was the most frequent and more variations of process conditions can be found. Thus, all of these changes may lead to a larger variance in growth constants.
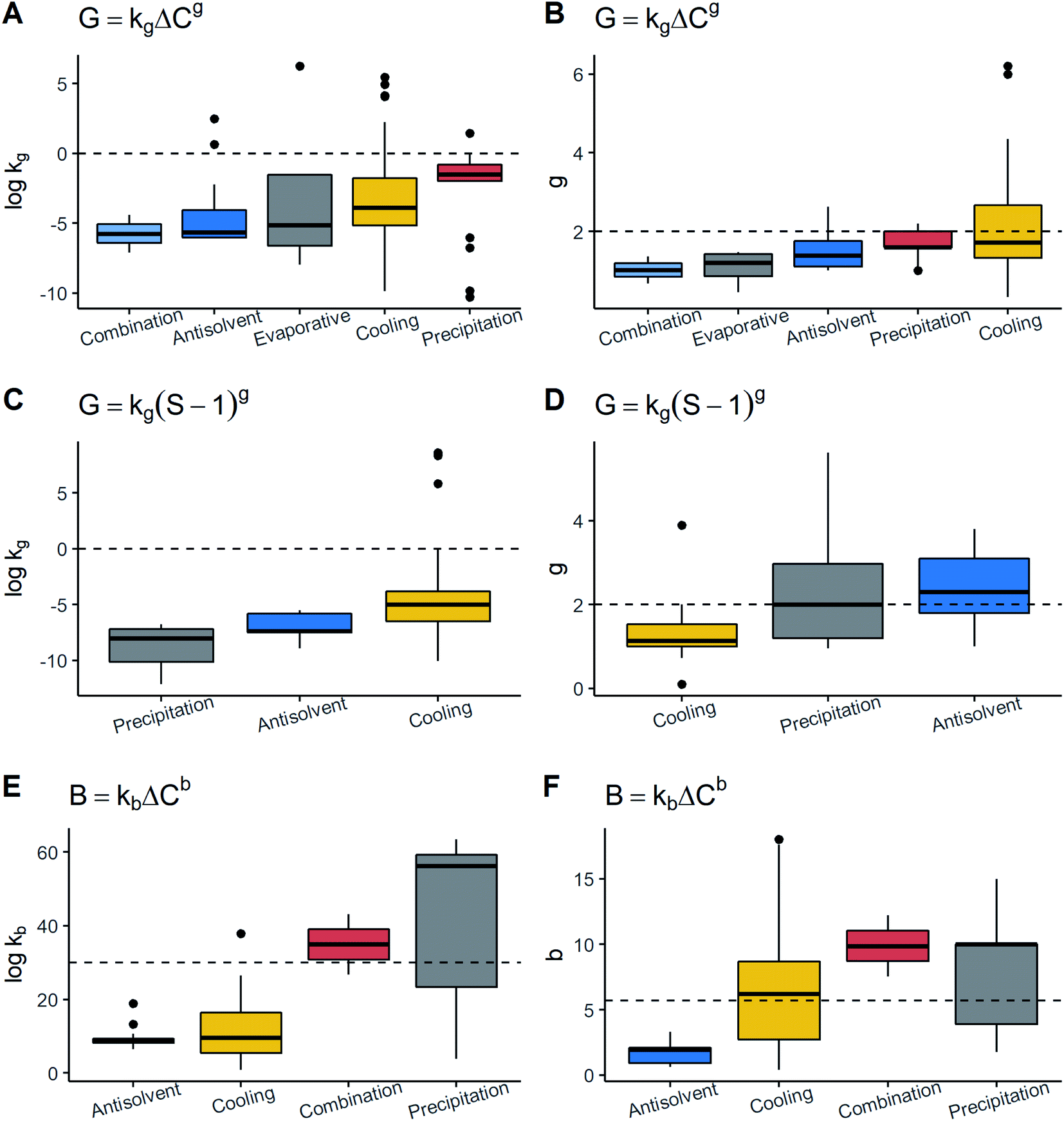 | ||
| Fig. 5 Association between kinetic parameters and crystallization methods. Boxplots for (A) logkg in the growth rate expression with absolute supersaturation vs. method, (B) g in the growth rate expression with absolute supersaturation vs. method, (C) logkg in the growth rate expression with supersaturation ratio vs. method, (D) g in the growth rate expression with supersaturation ratio vs. method, (E) logkb in the nucleation rate expression vs. method, and (F) b in the nucleation rate expression vs. method. From left to right, techniques are placed in ascending order of medians. | ||
Nucleation rate data showed that logkb and b have the same pattern i.e., a technique with high b has high logkb. Although the scattering was the highest, precipitation exhibited the largest kb and b followed by cooling crystallization. It could also be observed that the majority of methods displayed values of b higher than 5.9. In opposition, the antisolvent technique shows the lowest values for both nucleation parameters. The precipitation and antisolvent methods are characterised by large nucleation rates.10 In this way, their parameters are expected to show the same tendency. This trend was seen for precipitation but not for the antisolvent method. A possible reason is that the solutes crystallized by the antisolvent method show a moderate solubility in the solvent–antisolvent system.10 The results are portrayed in Fig. 5. Finally, the data indicate that there may be patterns such as in the case of precipitation where, especially for primary nucleation, higher values of all the parameters compared to the others were observed.
kg in the G = kg(S − 1)g model exhibited the opposite trend, where seeded processes have greater values of kg. Thus, kinetic parameters were different depending on seeding but this difference did not appear substantial overall, due to which this parameter may not be useful for characterising growth rate parameters.
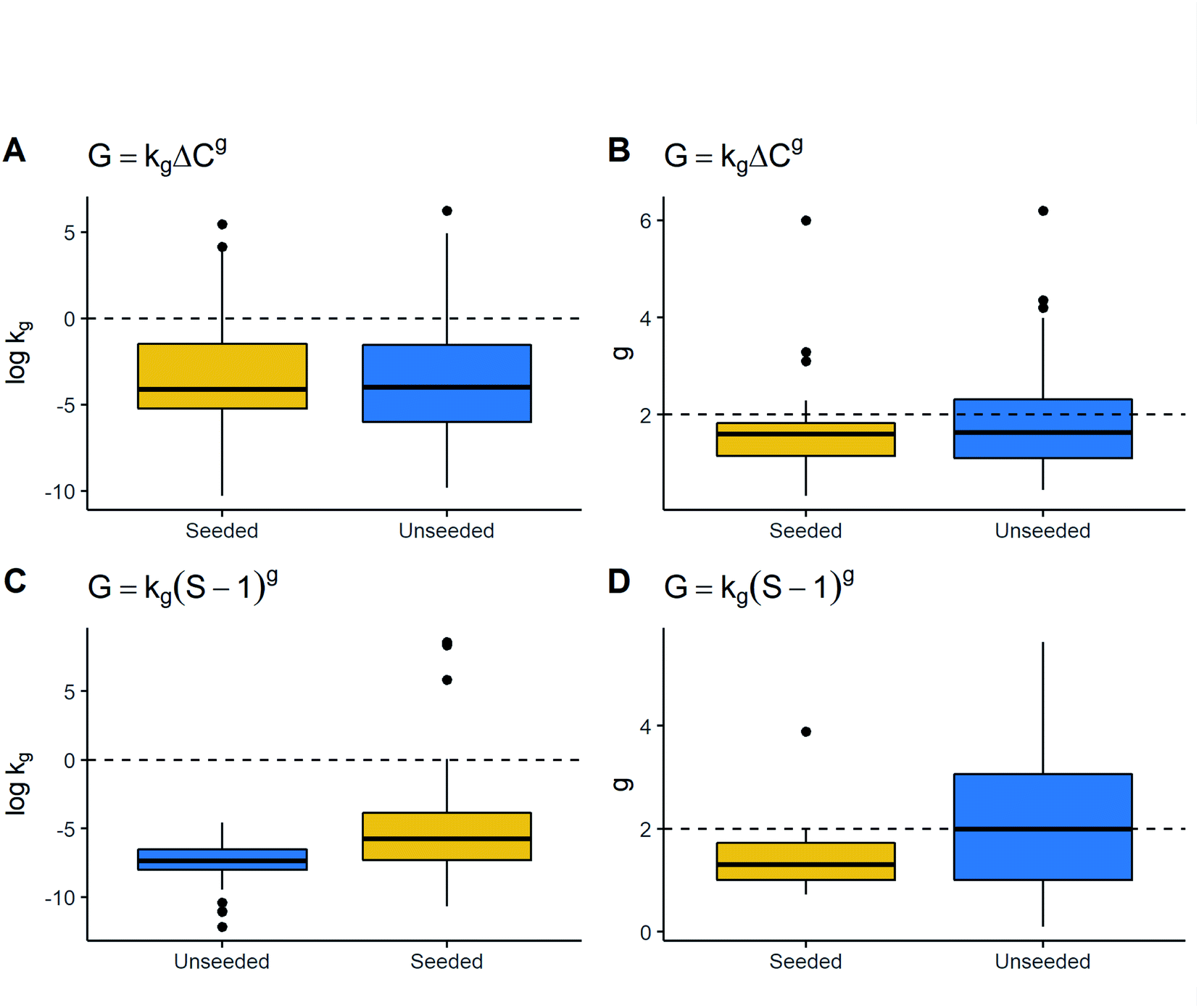 | ||
| Fig. 6 Association growth kinetic parameters and seeding. Boxplots for (A) logkg in the growth rate expression with absolute supersaturation vs. seeding, (B) g in the growth rate expression with absolute supersaturation vs. seeding, (C) logkg in the growth rate expression with supersaturation ratio vs. seeding, and (D) g in the growth rate expression with supersaturation ratio vs. seeding. From left to right, seed and unseeded processes are placed in ascending order of medians. | ||
4.3. Cluster analysis
In response to previous results where no clear associations could be established between certain properties and kinetic parameters, AHC was performed on kinetic parameters. The objective was to identify whether the data could be grouped into homogeneous clusters based on the kinetic parameters, and then, through a complementary methodology, to find characteristics that enable classification of a solute crystallised under certain conditions in a group and provide a rough estimation for its kinetic parameters. Thus, AHC was carried out over the next models G = kgΔCg (G1), G = kg(S − 1)g (G2), and B = kbΔCb (B1).Initially, the optimal number of clusters was 3 in the model G1, while it was 2 for the others based on the maximum silhouette index (see the ESI†). Nonetheless, in the models G2 and B1, 2 clusters did not provide a good differentiation between groups in relation to the rate constant and the supersaturation rate order together. Therefore, the chosen number of clusters for these cases was the second optimal number according to the index. Thus, the final number of clusters of 3, 3, and 5 was chosen for models G1, G2, and B1, respectively.
The results for model G1 are shown in Fig. 7 and summary statistics of the cluster are provided in the ESI.† Clusters 1, 2, and 3 had median values of g 3.50, 1.60, and 1.57, respectively. The clusters also showed logkg of −3.19, −0.40, and −5.55. As can be seen, all the clusters had different values for their kinetic parameters. However, the difference of logkg and g of cluster 1 compared to the others is more remarkable, in particular, g parameter, which is, in turn, larger than the average. This suggests that the growth rate behaviour in cluster 1 is more sensitive to changes in supersaturation than in the other clusters. On the other hand, when comparing clusters 2 and 3, these showed a similar distribution of g values where their main difference is due to logkg. Thus, the growth rate behaviour of clusters 2 and 3 is more dependent on kg. Thus, observations in cluster 2 will have higher growth rates compared to cluster 3 at a similar supersaturation as their kg values tend to be higher.
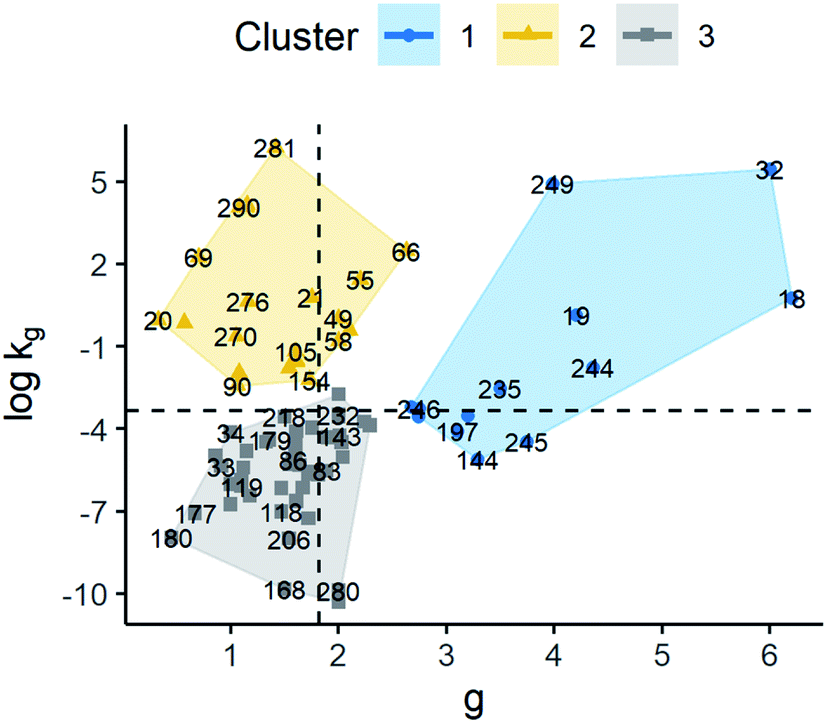 | ||
| Fig. 7 Scatter plot of standardised logkg and g for the model G = kgΔCg (G1). The labels represent the identification number of the observations. Cluster observations are distributed as follows: cluster 1 (n = 13), cluster 2 (n = 27), and cluster 3 (n = 52). Dashed lines represent the average values of g and logkg. | ||
3 clusters were also identified based on kinetic parameters of model G2. The median values of g for clusters 1, 2 and 3 were 1.43, 3.62 and 1.05, respectively. As for logkg, median values of −6.83, −7.19, and 8.42 were obtained for groups 1, 2 and 3. Although clusters 1 and 2 showed close values of logkg, the g values of cluster 2 are larger. As a result, cluster 2 exhibits an analogous behaviour to cluster 1 in model G1, where supersaturation seems to have greater importance compared to the other clusters. By comparing clusters 1 and 3, the opposite is observed where the main difference is due to kg, given g values are similar, showing that kg has a greater weight in growth rate determination. Thus, observations in cluster 1 have a slower growth rate compared to cluster 3 at the same supersaturation. Finally, it is worth noting that cluster 3 has a lower dispersion in the data compared to the other clusters. This can be related to the small number of observations in this cluster. These results can be observed in Fig. 8 and the ESI.†
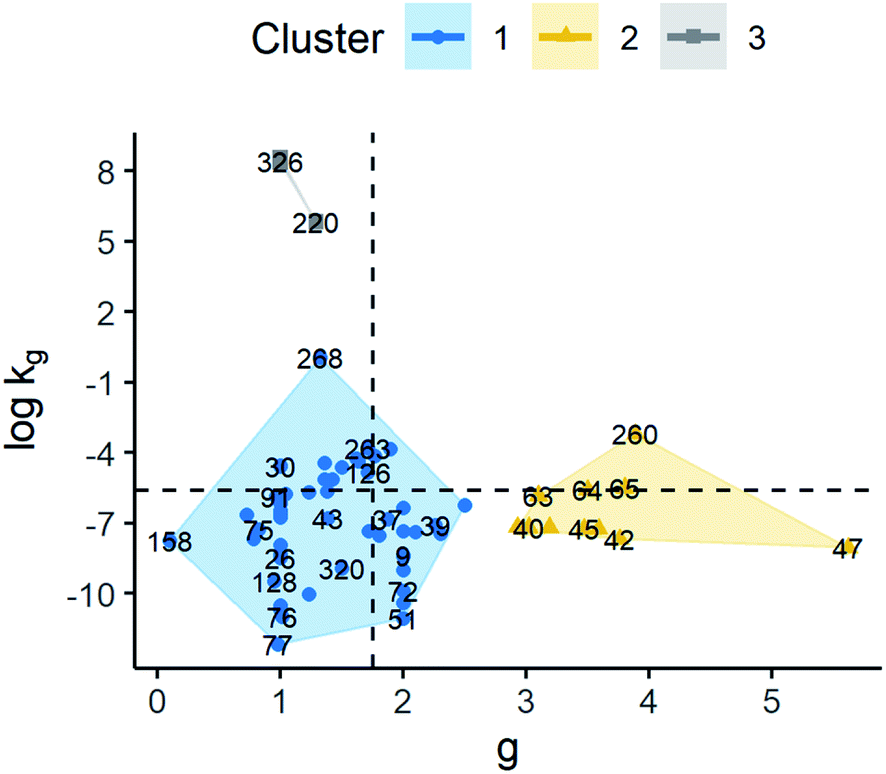 | ||
| Fig. 8 Scatter plot of standardised logkg and g for the model G = kg(S − 1)g (G2). The labels represent the identification number of the observations. Cluster observations are distributed as follows: cluster 1 (n = 51), cluster 2 (n = 11), and cluster 3 (n = 6). Dashed lines represent the average values of g and logkg. | ||
Regarding primary nucleation, 5 clusters were identified. The scatter plot and summary statistics can be found in Fig. 9 and the ESI,† respectively. Although all the groups presented different means for all the kinetic parameters, they still had some values that could overlap. Note that cluster 3 was composed of 2 observations only which belonged to the same solute. These observations corresponded to an experiment related to co-crystallization of agomelatine/citric acid. Given the characteristics of the solutes, this group was not included in the later analysis since molecular descriptors were not appropriate.
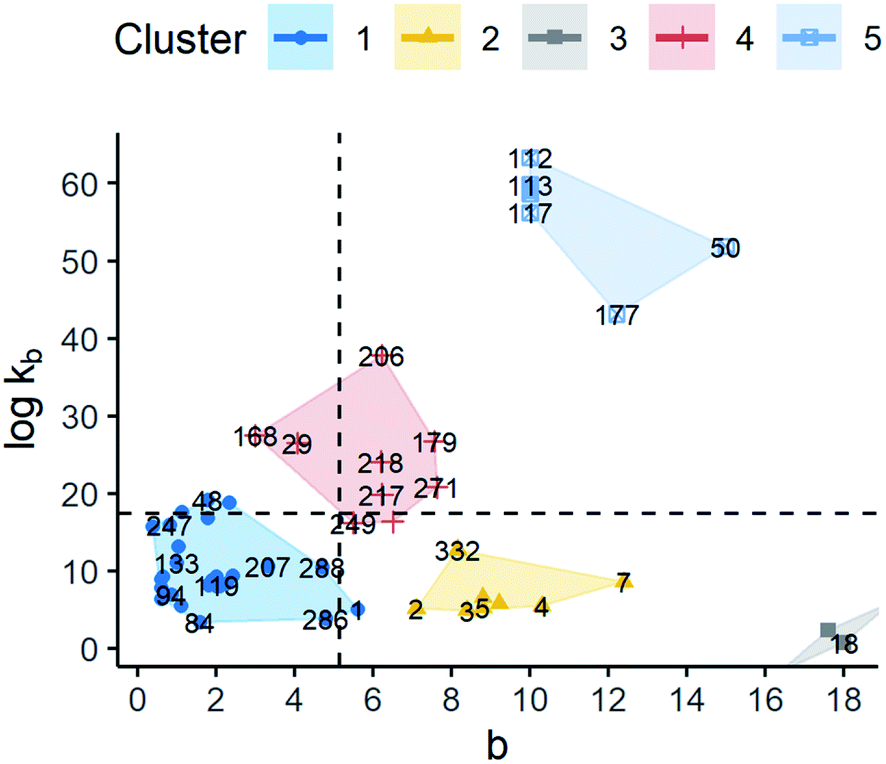 | ||
| Fig. 9 Scatter plot of standardised logkb and b for the model B = kbΔCb (B1). The labels represent the identification number of the observations. Cluster observations are distributed as follows: cluster 1 (n = 34), cluster 2 (n = 8), cluster 3 (n = 2), cluster 4 (n = 9), and cluster 5 (n = 8). Dashed lines represent the average values of b and logkb. | ||
By taking cluster 1 as a reference since it has the greater number of observations, two types of relative behaviours can be seen as a function of kinetic parameters. The first behaviour is observed in cluster 2 with respect to cluster 1. All of these clusters had values of logkb below the average showing a big difference in the b-parameter with median values of 1.92 and 9.15, respectively. Large values of b make the nucleation rate more sensitive to changes in supersaturation with respect to cluster 1. In addition, the median values of logkb were 8.89 and 5.69. As discussed in the descriptive analysis, logkb in the empirical model seems to have a high concordance with the pre-exponential term in CNT. Thus, the main form of nucleation in these clusters might be assumed to be heterogeneous as logkb is lower than 20.10
The second type of behaviour was seen by comparing clusters 4 and 5 to cluster 1. In this scenario, there seems to be a relationship between b and logkb where high values of b and high values of logkb are observed. In these clusters, contrary to clusters 2 and 3, logkb is above the average, even being higher than 30. Thus, homogeneous nucleation is expected to be dominant in many observations that belong to clusters 4 and 5, but mainly the latter.
Finally, the data were segmented into different groups for each model. Cluster analysis provides the relative behaviour of growth and nucleation rate as a function of their kinetics parameters, establishing how dependent rate is on supersaturation and rate constant. In addition, as in nucleation, clusters might be associated with a particular nucleation form. Similarly, every cluster showed characteristic values in terms of its parameters. Thus, if a molecule could be classified in a specific cluster, information on its relative behaviour and a range of its kinetic parameters might be obtained. In the next section, this idea is explored by using a Random Forest (RF) as a method to classify chemical entities in a cluster and assess the relationships between clusters and molecular descriptors, solvent, methods and seeding.
4.4. Importance of descriptors
RF possesses the ability to deal with non-linear relationships and redundant information, and assign importance to the classifiers, which is useful in the selection of variables and search of patterns. With this in mind, RF models were built for the kinetic expressions G1, G2, and B1 with the following parameters: number of trees (ntree) = 15000, number of variables per tree (mtry) = 10, and set.seed = 50. In these models, 15000 decision trees were built, where each used 10 variables randomly selected from molecular descriptors, technique, and solvent. Then, each decision tree assigns a new sample to a cluster based on its input variables, and the final classification is defined by the majority of votes. To measure the importance of a variable in the model, the mean decrease in accuracy (MDA) is assessed. The procedure to evaluate MDA for a variable consists of permutating its values, retraining the model and determining the change in the accuracy with respect to the original model. Thus, if a particular variable is decisive for classifying a sample in a cluster, a significant reduction in the accuracy is expected due to the permutation. This process is done for each input. The metrics and results for the trained model are discussed below.
The out-of-bag (OOB) and class prediction errors are listed below in Table 4. The high errors within groups were generally associated with the smallest size class. Additionally, the predictability was evaluated via leave-one-out cross-validation. The overall classification accuracy was 74.11%, 85.45%, and 83.05% for the models G1, G2, and B1, respectively. Previous studies dealing with the application of RF in the crystallization phenomenon showed a level of accuracy of around 70%.19 Therefore, the proposed models can be considered acceptable in this aspect.
| G = kgΔCg (G1) | G = kg(S − 1)g (G2) | B = kbΔCb (B1) | |
|---|---|---|---|
| OOB error (%) | 25.88 | 14.55 | 16.95 |
|
|||
| Class error (%) | |||
| Cluster 1 | 36.36 | 10.26 | 2.94 |
| Cluster 2 | 30.43 | 30.00 | 0.00 |
| Cluster 3 | 21.56 | 16.67 | — |
| Cluster 4 | — | — | 77.78 |
| Cluster 5 | — | — | 25.00 |
Fig. 10 shows the top 15 of the most important variables for RF classification. All the models included solvent, method, seeding and 110 molecular descriptors as classifiers. For all three models, among the most common and important classifiers were found mostly descriptors related to partial charges (PEOE), topological indices such as BCUT and GCUT, and volume-surface-shape indices (vsurf). Variables such as seeding and solvent were not as relevant as the other descriptors. Instead, the crystallization technique (method) was among the top 15 only in the primary nucleation rate model. Fig. 10 also shows that after the first one or two ranked variables, MDA is reduced slowly which suggests that there are no large differences in the importance after the first one. Thus, this might indicate that the contribution of the majority of variables to the model predictability is similar. As a result, there are no outstanding variables but most of them contribute equally.
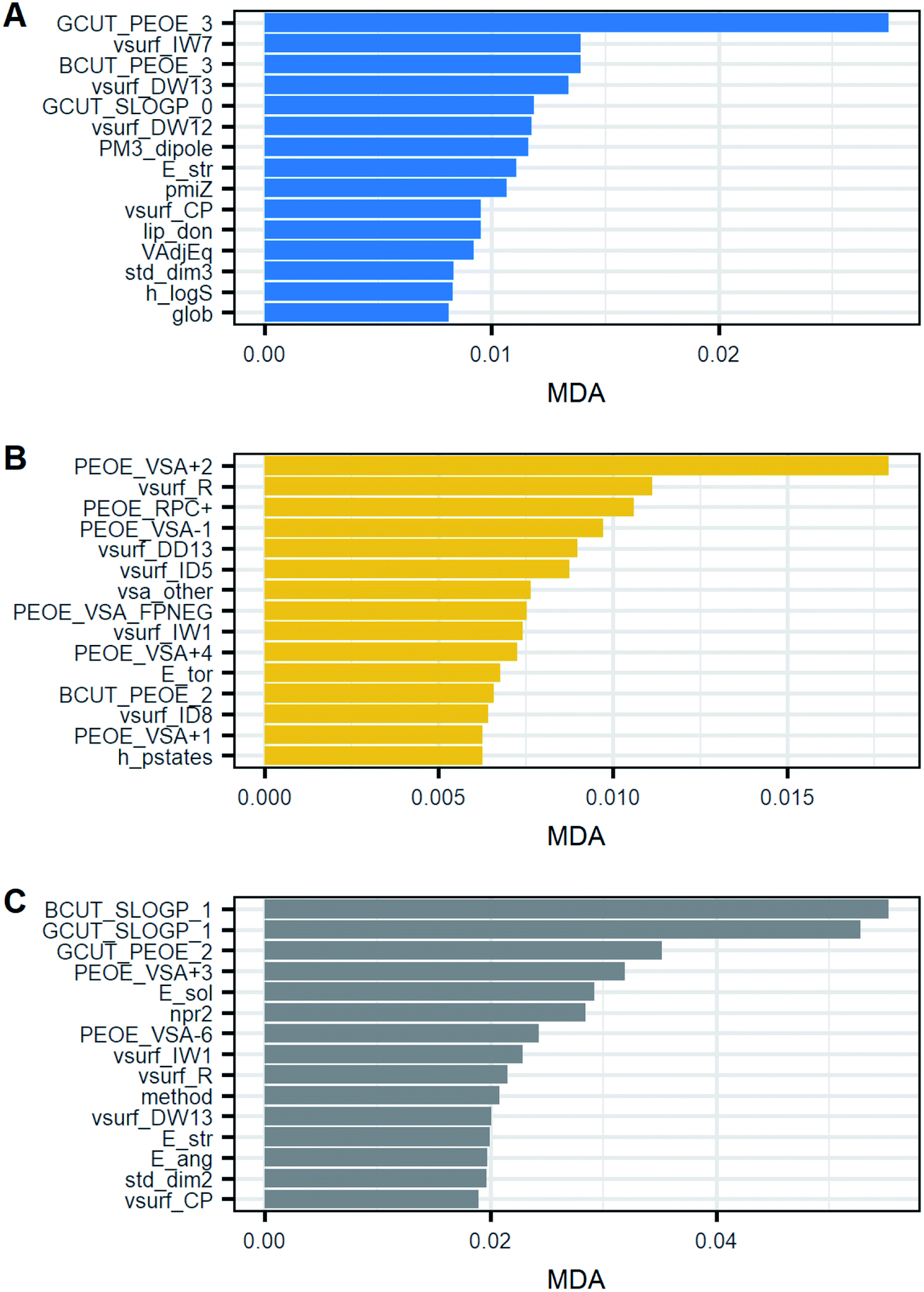 | ||
| Fig. 10 Top 15 of the most important classifiers based on the mean decrease in accuracy (MDA). (A) Model G1, G = kgΔCg; (B) model G2, G = kg(S − 1)g; (C) model B1, B = kbΔCg. | ||
By observing Table 5, it is possible to notice that the 3 most important variables were different with respect to mean throughout all clusters. As a result, these classifiers can be potentially useful for distinguishing one group from another. However, some clusters had a high standard deviation and so a high scattering. Therefore, the observations of those clusters may overlap. Thus, the most important descriptors may not be enough to provide accurate discrimination between groups. This can be seen for instance in the descriptor GCUT_PEOE_3 of model G1. Cluster 1 had a lower value than the others but the descriptor in clusters 2 and 3 was rather similar, around 2.1. Thus, only the best descriptor can identify cluster 1 from the rest in this case. Furthermore, cluster 1 has a wide scattering with respect to its average, which means some observations of this group might overlap with the others, thereby being confused. In light of the mentioned limitations of the descriptors, the high scattering within clusters may provide an explanation for why the MDA is rather similar and low in the models given the descriptors may separate a cluster from another but not all the clusters. Consequently, this suggests that a variable in isolation cannot explain the variability between clusters and the best model requires many variables.
| Cluster | Descriptors | ||
|---|---|---|---|
| Model G1 | GCUT_PEOE_3 | vsurf_IW7 | BCUT_PEOE_3 |
| 1 | 1.56 (0.66) | 1.95 (2.23) | 1.80 (0.65) |
| 2 | 2,12 (0.50) | 1.26 (2.12) | 2.24 (0.37) |
| 3 | 2.17 (0.46) | 3.53 (2.13) | 2.44 (0.34) |
|
|||
| Model G2 | PEOE_VSA+2 | vsurf_R | PEOE_RPC+ |
| 1 | 6.65 (11.13) | 1.61 (0.16) | 0.51 (0.36) |
| 2 | 20.23 (16.04) | 1.38 (0.15) | 0.22 (0.19) |
| 3 | 24.72 (12.11) | 1.23 (0.02) | 0.11 (0.04) |
|
|||
| Model B1 | BCUT_SLOGP_1 | GCUT_SLOGP_1 | GCUT_PEOE_2 |
| 1 | −0.68 (0.43) | −0.53 (0.48) | 0.14 (0.14) |
| 2 | −0.57 (0.06) | −0.43 (0.10) | 0.09 (0.00) |
| 4 | −0.63 (0.52) | −0.59 (0.54) | 0.28 (0.31) |
| 5 | −0.21 (0.06) | −0.28 (0.08) | −0.05 (0.04) |
By comparing the most important descriptor in the proposed models to those in previous studies on crystallization and solubility, several coincidences can be found. Specifically, MOE descriptors such as BCUT, GCUT and partial charge (PEOE) have been found to be useful for predicting solubility and crystallisability,19,20 which matches with the findings in this work to a certain extent. From a conceptual point of view, BCUT and GCUT descriptors are topological indices which are calculated based on molecular graphs.21 This group of indices has been related to chemical features like branching, size and cyclicity which in turn are related to molecular flexibility and rigidity.22 These properties have been found to influence crystallization tendency and kinetics.23 In this way, descriptors that measure properties like molecular flexibility are expected to be relevant in crystallization models. Similarly, partial charge is important since it affects the solute–solvent and solute–solute interactions.16,24 These descriptors were primarily relevant in the model G2 and model B1. The difference between models G1 and G2 may be given by the definition of the rate constant in which, as mentioned in previous sections, k{ΔC}g = k{S}g/C*g. As can be seen, kg in model G1 is more solubility-dependent whereby differences in important descriptors can arise, even though both models describe the same process. Lastly, vsurf descriptors comprise indices that characterise surface properties which include hydrophobic and hydrophilic interactions, shape, etc.25 This group of indices is calculated considering molecular conformation which makes them different from partial charge descriptors, for example.25 These types of interactions are also important in nucleation and crystal growth.10 Thus, descriptors that represent interactions between the solute and solvent or solute and solute may be of help to describe crystallization kinetics.
To highlight, seeding, solvent, and methods were not important for growth models, and only the crystallization technique had some relevance in the primary nucleation model. These results were expected since no associations between kinetic parameters and these variables were observed, except between the crystallization technique and nucleation parameters, as discussed in previous sections. By revising the results of model B1, a clearer association between the crystallization technique and nucleation parameters can be observed given there is a dominant method in every cluster as follows: cluster 1: 64.7% antisolvent, cluster 2: 100% cooling, cluster 4: 77.8% cooling and cluster 5: 87.5% precipitation. This might suggest that every cluster may also be associated with a determined crystallization method. Nonetheless, this result did not include evaporative crystallization as there were not data of primary nucleation under this condition. In the end, this indicates that RF models were able to discriminate irrelevant variables and select the most important ones in the corresponding model.
To summarise, RF classification models with acceptable, >70%, accuracy were built. These models may yield very rough estimates of kinetic parameters for the models G = kgΔCg, G = kg(S − 1)g, and B = kbΔCb, by providing mostly information on certain molecular descriptors and the crystallization technique. Among the main limitations of these models, it can be found that most training data were limited to water. Although solvent was not important, a possible reason is that there was no sufficient variety of solvents to capture the variability and have an appropriate measurement of its effect, whereby it would be recommended to incorporate more solvents and study solvent molecular descriptors. Another constraint was the sample size per cluster. It would have been desirable to have a larger sample with a greater number of solutes to produce better groups and obtain more accurate models. A final limitation was concerning molecular descriptors. Specifically, 3D descriptors such as vsurft are dependent on the molecule conformation. For this work, the optimal conformation was not selected, so in future studies, this might be considered to obtain more accurate values.
5. Conclusions
A database containing 336 datapoints of crystal growth and primary nucleation kinetic parameters in different solvents under several conditions of seeding and crystallization technique for more than 90 solutes was built. The collected data were compared to expected ranges from the literature and shown to be consistent, thereby being useful for developing other analyses.The most common kinetic models were G = kgΔCg, G = kg(S − 1)g, and B = kbΔCb. No strong linear correlations were found between the molecular descriptors and kinetic parameters of these expressions. Similarly, clear associations of kinetic parameters with seeding or solvent were not observed. On the other hand, while the crystallization technique did not display any tendency in regards to growth parameters, a notable association was seen with primary nucleation parameters where all the kinetic constants are high in reactive crystallization.
A cluster structure was identified and the observations were assigned to a group by using hierarchical cluster analysis over the kinetic parameter of the most common expressions. Through random forest models, new molecules can be classified into a cluster, which is related to its kinetic parameters, using as inputs molecular descriptors and indicating the crystallization technique with an accuracy higher than 70%. Three random forest models were obtained for each kinetic model. The most important variables for classification were topological (BCUT and GCUT), partial charge (PEOE), and vsurf descriptors showing a certain association with kinetic parameters. In addition, the crystallization technique was relevant to classify observation in primary nucleation, which confirms its relationship with nucleation parameters.
These models may be employed to yield a rough estimate of kinetic parameters of crystal growth and primary nucleation. However, the models are mostly constrained to aqueous systems. Thus, it was possible to establish that developing a model to predict kinetic constants is feasible. Future studies in this field should focus on providing more accurate estimations. In this scenario, considering the following factors might be useful:
1 Increase the number of solutes for each model.
2 Increase the number and nature of solvents.
3. Model solvent molecular descriptor.
4. Select an optimal conformation to calculate solute molecular descriptors.
To aid in points 1 and 2, the authors welcome contributions from researchers to expand the database. Original and updated versions of the database will remain freely available from the University of Strathclyde KnowledgeBase at https://doi.org/10.15129/8f47a175-3ac7-4791-a310-82e6652bd9f5.
Data availability statement
All data underpinning this publication are openly available from the University of Strathclyde KnowledgeBase at https://doi.org/10.15129/8f47a175-3ac7-4791-a310-82e6652bd9f5:• All the data collected with and without pre-processing, observations whose kinetic parameters were a function of solvent or antisolvent concentration, observations whose growth was measured as volume, data adjusted according to what was explained in the article (dataset_raw.csv and dataset_preprocessed.csv).
• Molecular descriptors employed in random forests of the compounds in the database (moe_descriptors.csv).
• Code employed to perform cluster analysis and random forests in R (script_v2.html).
Author contributions
Diego A. Maldonado – methodology, software, formal analysis, investigation, writing – original draft, review & editing. Antony Vassileiou – methodology, software, resources, supervision. Blair Johnston – supervision. Alastair J. Florence – supervision. Cameron J. Brown – conceptualization, writing – review & editing, supervision.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to thank EPSRC and the Future Continuous Manufacturing and Advanced Crystallisation Research Hub (Grant Ref: EP/P006965/1) for funding this work.References
- J. Rantanen and J. Khinast, The Future of Pharmaceutical Manufacturing Sciences, J. Pharm. Sci., 2015, 104(11), 3612–3638, DOI:10.1002/jps.24594PubMed.
- S. L. Lee, T. F. O'Connor, X. Yang, C. N. Cruz, S. Chatterjee, R. D. Madurawe, C. M. V. Moore, L. X. Yu and J. Woodcock, Modernizing Pharmaceutical Manufacturing: from Batch to Continuous Production, J. Pharm. Innov., 2015, 10(3), 191–199, DOI:10.1007/s12247-015-9215-8.
- K. V. Gernaey, A. E. Cervera-Padrell and J. M. Woodley, A perspective on PSE in pharmaceutical process development and innovation, Comput. Chem. Eng., 2012, 42, 15–29, DOI:10.1016/j.compchemeng.2012.02.022.
- A. Rogers and M. Ierapetritou, Challenges and opportunities in modeling pharmaceutical manufacturing processes, Comput. Chem. Eng., 2015, 81, 32–39, DOI:10.1016/j.compchemeng.2015.03.018.
- P. Pandey, R. Bharadwaj and X. Chen, 1 – Modeling of drug product manufacturing processes in the pharmaceutical industry, in Predictive Modeling of Pharmaceutical Unit Operations, ed. Pandey, P. and Bharadwaj, R., Woodhead Publishing, 2017, pp. 1–13 Search PubMed.
- D. M. Kremer and B. C. Hancock, Process Simulation in the Pharmaceutical Industry: A Review of Some Basic Physical Models, J. Pharm. Sci., 2006, 95(3), 517–529, DOI:10.1002/jps.20583.
- H. M. Omar and S. Rohani, Crystal Population Balance Formulation and Solution Methods: A Review, Cryst. Growth Des., 2017, 17(7), 4028–4041, DOI:10.1021/acs.cgd.7b00645.
- N. Yazdanpanah and Z. K. Nagy, The Handbook of Continuous Crystallization, Royal Society of Chemistry, 2020 Search PubMed.
- P. Rudolph, Handbook of Crystal Growth: Bulk Crystal Growth, Elsevier Science, 2014 Search PubMed.
- A. Lewis, M. Seckler, H. Kramer and G. van Rosmalen, Industrial Crystallization: Fundamentals and Applications, Cambridge University Press, 2015 Search PubMed.
- T. Kwartler, Text Mining in Practice with R, Wiley, 2017 Search PubMed.
- R. A. Johnson and D. W. Wichern, Applied Multivariate Statistical Analysis (Classic Version), Pearson, 2018 Search PubMed.
- A. Kassambara Practical Guide to Cluster Analysis in R: Unsupervised Machine Learning; STHDA, 2017 Search PubMed.
- T. Hastie; R. Tibshirani and J. FriedmanThe Elements of Statistical Learning: Data Mining, Inference, and Prediction; Springer, New York, 2009, 2nd edn Search PubMed.
- G. Williams, Data Mining with Rattle and R: The Art of Excavating Data for Knowledge Discovery; Springer, New York, 2011 Search PubMed.
- A. S. Myerson; D. Erdemir and A. Y. Lee, Handbook of Industrial Crystallization; Cambridge University Press, 2019 Search PubMed.
- T. Nishinaga, Handbook of Crystal Growth: Fundamentals, Elsevier Science, 2014 Search PubMed.
- W. Du, Q. Yin, J. Gong, Y. Bao, X. Zhang, X. Sun, S. Ding, C. Xie, M. Zhang and H. Hao, Effects of Solvent on Polymorph Formation and Nucleation of Prasugrel Hydrochloride, Cryst. Growth Des., 2014, 14(9), 4519–4525, DOI:10.1021/cg5006067.
- R. M. Bhardwaj, A. Johnston, B. F. Johnston and A. J. Florence, A random forest model for predicting the crystallisability of organic molecules, CrystEngComm, 2015, 17(23), 4272–4275, 10.1039/C4CE02403F.
- D. S. Palmer, N. M. O'Boyle, R. C. Glen and J. B. O. Mitchell, Random Forest Models To Predict Aqueous Solubility, J. Chem. Inf. Model., 2007, 47(1), 150–158, DOI:10.1021/ci060164k.
- K. Roy; S. Kar and R. N. Das, Chapter 2 – Chemical Information and Descriptors. in Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment, ed. Roy, K., Kar, S. and Das, R. N., Academic Press, 2015, pp. 47–80 Search PubMed.
- Q.-N. Hu, Y.-Z. Liang, H. Yin, X.-L. Peng and K.-T. Fang, Structural Interpretation of the Topological Index. 2. The Molecular Connectivity Index, the Kappa Index, and the Atom-type E-State Index, J. Chem. Inf. Comput. Sci., 2004, 44(4), 1193–1201, DOI:10.1021/ci049973z.
- (a) J. Bai, H. Fang, Y. Zhang and Z. Wang, Studies on crystallization kinetics of bimodal long chain branched polylactides, CrystEngComm, 2014, 16(12), 2452–2461, 10.1039/C3CE42319K; (b) L. Yu, S. M. Reutzel-Edens and C. A. Mitchell, Crystallization and Polymorphism of Conformationally Flexible Molecules: Problems, Patterns, and Strategies, Org. Process Res. Dev., 2000, 4(5), 396–402, DOI:10.1021/op000028v.
- M. Kowacz, M. Prieto and A. Putnis, Kinetics of crystal nucleation in ionic solutions: Electrostatics and hydration forces, Geochim. Cosmochim. Acta, 2010, 74(2), 469–481, DOI:10.1016/j.gca.2009.10.028.
- G. Cruciani; R. Mannhold; H. Kubinyi and G. Folkers, Molecular Interaction Fields: Applications in Drug Discovery and ADME Prediction, Wiley, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2dd00033d |
| This journal is © The Royal Society of Chemistry 2022 |