
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MOFUN: a Python package for molecular find and replace
Paul
Boone
 a and
Christopher E.
Wilmer
*abc
a and
Christopher E.
Wilmer
*abc
aDepartment of Chemical and Petroleum Engineering, University of Pittsburgh, 3700 O'Hara Street, Pittsburgh, Pennsylvania 15261, USA. E-mail: wilmer@pitt.edu
bDepartment of Electrical and Computer Engineering, University of Pittsburgh, 3700 O'Hara Street, Pittsburgh, Pennsylvania 15261, USA
cClinical and Translational Science Institute, University of Pittsburgh, 3700 O'Hara Street, Pittsburgh, Pennsylvania 15261, USA
First published on 23rd August 2022
Abstract
MOFUN is an open-source Python package that can find and replace molecular substructures in a larger, potentially periodic, system. In the context of molecular simulations, find and replace is a useful operation for adding/swapping functional groups, adding/removing solvent molecules or defect sites, and many other helpful system perturbations. MOFUN can also be used to alter force field terms on certain atoms while leaving the geometry/composition otherwise unchanged. The package is easily automated, which is particularly helpful for preparing input files for large-scale screenings. The package is freely available on GitHub at https://github.com/WilmerLab/mofun.
Introduction
MOFUN is a general purpose, open-source Python package for searching an arbitrary molecular structure for a pattern and replacing any instances of it with a replacement pattern – i.e., find and replace for molecular systems. We built MOFUN, initially, to support our own investigations of metal–organic frameworks (MOFs), which are a class of porous materials composed of linkers and metal centers which self-assemble into periodic crystalline structures.1 In the context of MOFs, MOFUN was used for (1) modifying the linkers of MOFs with various functional groups, (2) adding defects to MOFs, and (3) parameterizing MOFs with flexible force field terms. Although these operations can be carried out by editing the underlying files in a text editor or by adding and removing atoms individually in a visual editor such as Avogadro,2 this is time-consuming, error-prone and impractical when scaling to greater numbers of structures or structures containing more atoms. We wrote MOFUN to be an automated solution to these problems that can operate on periodic structures with over 100k atoms.Prior to the development of MOFUN, numerous packages have been developed that can perform ligand replacement or substituent replacement on smaller non-periodic molecules,3–5 for preparing structures for quantum calculations. Pattern replacement can also be performed on non-periodic molecules represented as human-readable SMILES strings.6 Parts of a molecule can be searched for using a SMARTS7 pattern or even simpler (but much less precisely) by using a regex-based text find and replace on the SMILES string itself.
Before MOFUN, Wilmer8 developed a molecular search-and-replace program called FunctionalizeThis! designed for crystalline structures such as MOFs. FunctionalizeThis! did not support finding and replacing bonds and other force field terms, which limited its use particularly when attempting to generate structures that can be used with flexible force fields. More recently, a free and open-source Julia package named PoreMatMod.jl was reported by Henle et al.9 Like MOFUN, the highly versatile package by Henle et al. can be used for automating crystal structure modifications and was at least partly motivated to facilitate research on hypothetical MOFs. While there is significant overlap in functionality between MOFUN and PoreMatMod.jl, there are also a few key differences. Whereas MOFUN searches for patterns via comparisons of distances between atoms, PoreMatMod.jl analyzes the molecular graph defined by how a structure's atoms are bonded. Both approaches can handle many common use cases but sometimes one approach is more suitable than the other, in terms of what kinds of patterns can be searched for. For example, when using distances between atoms, it is possible to search for patterns that are not bonded, such as a molecule physisorbed to a binding site or matching defects between two different layers of stacked graphene. In contrast, molecular graph searches are much better suited to searching for substructures in a conformation-invariant manner, such as when looking for hydrocarbon chains that can assume varied configurations in space while their molecular graphs stay the same. While PoreMatMod.jl can be applied to many of the same problems as MOFUN, like FunctionalizeThis! it does not support finding and replacing bonds or higher-order force field terms.
In addition to simpler use cases, MOFUN was built to support find and replace of substructures that are fully parameterized to use with flexible force fields for molecular dynamics (MD) simulations (in particular, for LAMMPS10). By releasing MOFUN as open-source and announcing it here, we hope that other researchers will also benefit from this general-purpose package and can use it to accelerate and expand their research.
MOFUN is available on GitHub at https://github.com/WilmerLab/mofun under the open-source MIT license. The version described in this paper is version 1.0.1 (https://doi.org/10.5281/zenodo.6950355). MOFUN can be installed from the GitHub source code, or from PyPI using pip.
MOFUN: algorithm details
In this section, we will discuss the implementation of both the find and the replace parts of the algorithm. We use the word structure here to mean a list of atoms with elements and positions along with optional periodic boundaries that we want to exhaustively examine for instances of a search pattern, which is also a list of atoms with elements and positions. When we find an instance of the search pattern in the structure (a list of atoms in the structure whose relative positions are the same as the relative positions in the search pattern), we call this a match. Every match found can be replaced with a replacement pattern, which is another list of atoms with elements and positions. While we use the word structure here because we predominantly use MOFUN on periodic crystal lattice structures, the software works equally well on molecules or any other aperiodic grouping of atoms.To find all instances of a search pattern in a structure, we first calculate the distances between all pairs of atoms in the search pattern. For there to be a match of the search pattern in the structure, there must be a list of atoms in the structure that share both the same distances (within a tolerance), and the same atom elements. Let rp be an N-length list of all positions in the search pattern. Let rs be the list of all atom positions in the structure plus each atom's periodic images from immediately adjacent unit cells. Let rm be an N-length list containing N positions from rs that we will examine as a trial match in the structure. We will refer to specific positions in both rp and rm as rp,i and rm,i, where i ∈ [1, …, N] refers to the ith element in the list.
The trial match rm is a good match if three conditions are met. The first condition is that the distances (or Euclidian norm denoted by ‖…‖) between all pairs of atoms in the trial pattern must match their corresponding pairs in the search pattern within a specified tolerance δ.
| ∀i, j ∈ [1, …, N], |‖rp,i − rp,j‖ − ‖rm,i − rm,j‖| < δ |
If the distance between any pair of atoms differs from its proposed matching pair by an amount greater than the tolerance, the trial match is not a match. The tolerance can be set higher or lower for cases when a looser or tighter match is appropriate.
The second condition is that the atom elements for the pattern must be the same as the atom elements of the trial match. If we let Ep,i be the ith element of the pattern and Em,i be the ith element for the trial match, then:
| ∀i ∈ [1, …, N], Ep,i = Em,i |
The third condition is that there must exist rotation and translation operations such that when they are applied to the search pattern, the atom positions of the search pattern match the atom positions in the trial match. This condition is necessary to handle cases of symmetry and chirality in the search pattern. For each trial match, we calculate the translation and rotation operations necessary to transform the search pattern to the location of the trial match and then we exclude any matches where the atom positions of the transformed search pattern are not the same as those in the trial match.
To rotate the search pattern into place, we select three points in the search pattern, two to define a direction axis, and the third to use as an orientation point. MOFUN will pick the two atoms in the search pattern that are farthest from each other to define the direction axis and it will pick the atom that is farthest from the infinite line defined by the direction axis to be the orientation point. If the pattern only contains two atoms, or all the atoms are colinear, then the orientation point can be ignored. Rotating the search pattern to align with the match pattern requires two rotation transformations that are implemented using quaternions: (1) we rotate the search pattern so that the two atoms of the direction axis are pointed in the same direction as those same atoms in the match pattern, and (2) we then rotate the search pattern around the directional axis so that the orientation point is in the correct direction. Since all the atoms should now be offset by the location of the match in space, we can translate the search pattern by the difference in position between any corresponding pair of atoms between the search pattern and the match pattern. This alignment procedure is fast as it is precisely defined and doesn't require an optimization, but for structures where the match positions do not closely match the search pattern (i.e. finding a pattern requires a tolerance δ), the resulting replacement atoms may be aligned sub-optimally proportional to the required tolerance.
To demonstrate the problem posed by symmetry, let us consider searching for the CH3 group at the ends of an octane molecule. We can define the CH3 search pattern by arbitrarily labeling one hydrogen as ‘A’, and then labeling the other hydrogens ‘B’, and ‘C’ clockwise as shown in Fig. 1A. The atom pairs (A, B) and (A, C) are equally far apart so we arbitrarily choose (A, B) to be the directional axis and C to be the orientation point. If we search an octane for this pattern, we will find six possible matches for each actual CH3 in the structure (or 12 matches total), one for each possible ordering of the hydrogens: ABC, ACB, BAC, BCA, CAB, CBA. If we look at the matches that start with the ‘A’ hydrogen – ABC, ACB – there is a clockwise ordering of atoms and a counter-clockwise ordering of atoms (see Fig. 1B). If the atoms in the match pattern are ordered clockwise like the search pattern and if we align the directional axis atoms (A, B) in the search pattern to the same atoms in the trial match and rotate the orientation point C into place (see Fig. 1C) then all three hydrogens and the carbon will be in the correct locations (see Fig. 1D). However, if the match pattern was numbered counter-clockwise (opposite numbering to the search pattern), and if we follow the same process to align (A, B) and rotate C into place, then the carbon will be in the wrong position even though all three hydrogens are correctly located. For this example, three of the six possible trial matches have the same counter-clockwise ordering as the search pattern and are good matches, and three trial matches have clockwise ordering and are not matches. Similarly, a chiral search pattern in a structure will match either enantiomorph since the distances between all atoms are the same regardless of which enantiomorph is found, but only an enantiomorph which matches the chirality of the search pattern will be able to be rotated into position of the match pattern.
 | ||
| Fig. 1 (A) A search pattern for the CH3 group in octane with clockwise ordering of hydrogens, (B) trial matches with clockwise and counter-clockwise ordering of hydrogens, (C) the directional axis points AB of the search pattern are aligned with the trial match and the orientation point C is rotated into place for both matches, and (D) the result after alignment for when the structure ordering matches the search pattern ordering (all atoms match) and when the ordering is opposite (the carbon is out of place). | ||
Thus, the third condition – that the atom positions of a rotated and translated search pattern must be the same as the atom positions of the trial match – removes the bad matches caused by symmetric and chiral search patterns. Once we have all the possible good matches, if we still have multiple matches for the same group of atoms caused by symmetry, then we randomly pick one of the good matches.
At this stage, inserting the replacement atoms is now straightforward. The replacement pattern is defined on the same coordinate system as the search pattern, so to insert the replacement pattern into the structure at the right position and orientation, we take the transformations we calculated above to transform the search pattern into place and apply them to the replacement pattern. We insert the replacement pattern atoms and topology into the structure, delete the matched atoms and existing topology and our find and replace is complete.
MOFUN: optimization and performance
Here we describe some subtleties to the implementation of MOFUN that were necessary to optimize its performance.First, depending on the length of the search pattern, we do not search all the atoms in each neighboring unit cell. Searching every atom in a replicated 3 × 3 × 3 expanded unit cell could be prohibitively inefficient for larger structures, so we limit the set of atoms searched to only those within a distance d of one of the boundaries of the original unit cell, where d is the length of the search pattern (see Fig. 2C).
 | ||
| Fig. 2 Overview of algorithm as applied to (A) an idealized MOF structure with blue metal center and (B) a four atom red and grey linker; (C) the unit cell is expanded to include neighboring atoms, (D) starting atoms that match the first element in the search pattern are identified, (E) for each starting atom, nearby atoms are isolated, (F) distances between all nearby atoms are precalculated, and (G) nearby atoms are matched for element and pair distances. | ||
Second, we do not generate all trial matches at once as this would lead to running out of memory for all but the smallest systems; instead, we build up trial matches of the search pattern one atom at a time. The first atom in the search pattern is matched by finding all atoms in the structure that share the same element as the first atom in the search pattern (see Fig. 2D). For each of these starting matching structure atoms, we create a list of nearby structure atoms – those atoms that are within a box x ± d, y ± d, z ± d about the matched atom (see Fig. 2E) – and precalculate the distances between every pair of atoms in this list (see Fig. 2F). The calculation for identifying nearby structure atoms is implemented as a filter on a presorted NumPy11 array of the coordinates and is therefore executed as highly-optimized compiled C code. The distances between all nearby atoms are precalculated as a group using the SciPy12 library, which is again executed as highly optimized C code. To match the second atom, we find all nearby structure atoms that match the element of the second search atom and select only the atoms where the distance from the second atom to the first atom matches that of the search pattern. Continuing through the search pattern one atom at a time, we match all nearby atoms based on their elements, and select only those atoms where the distances between the atom and all prior atoms match the distances in the search pattern. At any stage, if there are no viable matches, then we can abort looking for matches using this starting element. If we reach the last atom in the search pattern and have built up one or more complete matches (see Fig. 2G), then these are added to the list of successful matches. Another advantage of generating matches in this manner is that the algorithm can easily be made to operate in parallel, where each starting structure atom is run on a separate core. We have not found it necessary to implement this yet due to MOFUN's current performance being more-than sufficient for our needs but parallelization is available to us if it becomes necessary.
The performance of the optimized algorithm is O(N2), where N is the number of atoms in the system, and the memory usage is O(M2), where M is the maximum number of atoms within a distance d of any other atom. Practically, this means that system size and CPU speed will limit the kinds of systems that can be run. Our early naïve implementations took over 10 minutes to search for all linkers in a 2 × 2 × 2 replicated UiO-66 unit cell – 3496 atoms, 192 linkers – and outright failed for systems bigger than that due to running out of memory. With the optimized algorithm, we can search for and replace all linkers in an 8 × 8 × 8 replicated UiO-66 unit cell – 221![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 184 atoms and 12288 linkers – in less than 6 minutes on a single core of an Apple MacBook Pro M1 Pro laptop (see Fig. 3). This size system greatly exceeds our lab's current needs of approximately a 4 × 4 × 4 system with 40k atoms for use in thermal conductivity calculations; however, we did run a larger find and replace on a 20 × 20 × 20 system – 3.5M atoms and 192k linkers – and it completed in 15 hours. Additional optimizations could be implemented if there is a need for a find and replace operation at that scale.
184 atoms and 12288 linkers – in less than 6 minutes on a single core of an Apple MacBook Pro M1 Pro laptop (see Fig. 3). This size system greatly exceeds our lab's current needs of approximately a 4 × 4 × 4 system with 40k atoms for use in thermal conductivity calculations; however, we did run a larger find and replace on a 20 × 20 × 20 system – 3.5M atoms and 192k linkers – and it completed in 15 hours. Additional optimizations could be implemented if there is a need for a find and replace operation at that scale.
 | ||
| Fig. 3 Time required to find or find + replace all linkers in a UiO-66 unit cell replicated to 2 × 2 × 2, 4 × 4 × 4, 5 × 5 × 5, 6 × 6 × 6, 7 × 7 × 7, and 8 × 8 × 8. | ||
MOFUN: usage details
Structures, search patterns, and replacement patterns can be defined directly in Python, or loaded in from a CML file (for typical output from Avogadro2), a P1 CIF file, a LAMMPS10 data file, or from any file format supported by the ASE13 package, such as XYZ, PDB, RES, etc. Structures and patterns must include coordinates and elements and can optionally contain periodic boundaries, charges and other metadata. Structures can be defined with either a cubic or triclinic unit cell. MOFUN supports reading and writing LAMMPS data files directly, including the LAMMPS pair, bond, angle, dihedral, and improper styles and all coefficients necessary for defining a flexible force field. When using a parameterized LAMMPS data file as a replacement pattern, MOFUN can insert the appropriate force field terms for all interactions into the resulting structure file. When using a CIF file format, all extra columns attached to an atom collection as well as bond, angle, and torsion geometry collections are supported when doing a replace. MOFUN also supports optionally replacing only a fraction of the search pattern matches found in a structure. A replacement fraction can be defined so that only a given% of matched search patterns will be replaced. One can also run a find operation without replacing anything; in this case, in addition to returning what atoms were matched, the quaternions necessary to rotate the search pattern into place are also returned. All features can be used via the Python interface, and a command line tool is also included that handles many common use cases.One of the more advanced features of MOFUN is that it can manage structure topology and force field parameters for flexible force fields and apply them correctly when inserting a parameterized replacement pattern into a structure. This supports the insertion of two-body pair and bond potentials, three-body angle potentials, and four-body dihedral and improper potentials, as defined by LAMMPS. For each defined potential in the replacement pattern, the atoms that make up the potential and the potential type are inserted into structure alongside the replacement pattern atom positions and types. If the structure already has defined topology, then any topology associated with the search pattern is deleted along with its atoms prior to insertion of the replacement pattern, with one very important exception: if any of the atoms are shared between both the search pattern and the replace pattern (i.e. if the atoms share the same element and position), then any force field potentials defined on these atoms are overridden, rather than all existing terms being deleted and replaced by what's in the replacement pattern. This is necessary to handle parameterizing a structure like the one shown below in example 3, or when using find and replace to override the force-field terms in part of a structure while leaving the structure intact. If the replacement pattern defines the potential parameters (i.e. via a “* coeffs” section in the LAMMPS data file), then the potential parameters will also be carried forward into the resulting structure. While this only supports LAMMPS data files (and direct code in Python) at the moment, this is primarily because there is no standardized file format that is commonly-used to define periodic molecular structures along with full topological data and force-field parameters. For LAMMPS users such as ourselves, writing to a LAMMPS data file is extremely convenient as we can immediately simulate systems after find and replace; for users of other simulation packages, the LAMMPS data file should contain sufficient information to be converted into the file formats required by other simulation packages.
Examples
We have chosen three examples to demonstrate MOFUN's capabilities: (1) functionalizing the MOF UiO-66, (2) adding defects to UiO-66, and (3) fully parameterizing UiO-66 across periodic boundary conditions starting with an unparameterized UiO-66 structure and parameterized metal center and linker fragments. These examples (and others) are also available online with all supporting files and there is expanded guidance in the software's documentation. Since MOFUN is living software, the syntax shown below may change and/or additional features may be added in the future. When in doubt, please refer to the online documentation. For each example, we describe the process we used to prepare files and run the find/replace; we include this level of detail so the example properly illustrates what the task involves, but a user does not have to follow this process exactly.Example 1: functionalizing linkers in UiO-66
The first example is how to use find and replace to functionalize a structure. We will take the MOF UiO-66 (Fig. 4A) and functionalize all linkers with hydroxyl groups (Fig. 4B). We will need a structure file for UiO-66 and files for a standard UiO-66 linker and a linker functionalized with the hydroxyl. To create the UiO-66 linker file, we used Vesta14 to pick one linker in the structure, deleted all other atoms, then exported to a file format that Avogadro2 can read. We opened the file in Avogadro and saved as CML. The replacement pattern needs to lie in the same coordinate system as the search pattern. The easiest way to do this is to start with the search pattern and simply not move any of the atoms unless you intend to move them with the replacement operation. We took the search pattern CML, replaced one of the hydrogens on the linker with an oxygen atom, and added the attached hydrogen to make the hydroxyl. We used Avogadro's “Fix Selected Atoms” feature to prevent all the atoms from moving except for the newly added ones, then ran optimize structure to let the OH group find a more appropriate position. If you do not fix all the atoms except for the hydroxyls, many of the atoms will move when you optimize and the atoms of your replacement pattern will not correspond to the same atoms in the search pattern. Once all the files are prepared, you can run MOFUN either using the Python interface or the command line interface. For Python:
 | ||
| Fig. 4 (A) Search pattern and search structure (a “slice” of UiO-66), (B) example 1 resulting structure after find and replace using a replacement pattern with an added OH functional group, and (C) example 2 resulting structure after find and replace using a replacement pattern of a defective linker (two capping formate groups), as applied to 25% of the linkers. | ||
When we are functionalizing a MOF using find and replace, we are typically replacing a pattern that has fewer atoms with a pattern that has more, and the larger the functional group is in the replacement pattern, the more likely that functional groups from different linkers will overlap. This may not be a problem, for example, when adding hydroxyl groups to the linker in UiO-66, but if one were to add more bulky functional groups overlap would likely occur. The replacement operation inserts the functional group exactly as specified, and the resulting structure may need to be relaxed using molecular dynamics for the functional group to find a more reasonable configuration. When we are adding bulky functional groups to a structure, we create a replacement pattern where the functional groups are tightly placed near the linker, as much parallel to the linker as possible, to limit any overlapping with functional groups on other linkers. While this tight configuration may be high in energy, since we then relax the structure using a flexible force field, the functional groups can relax into a lower energy configuration.
Example 2: adding defects to UiO-66
While we often assume a MOF is perfectly formed when we evaluate it computationally, it is well known that synthesized MOFs have a variety of defects, typically missing linkers or missing metal centers or both.15–17 Missing linker defect rates of 5–20% is normal, depending on what MOF is being synthesized and the experimental synthesis method used. With MOFUN, we can search for a linker and replace it with a defect site, typically just a pair of capping groups – such as two formates – on the metal centers the linker was formerly connected to. Since MOFUN supports replacing a specified fraction of all instances of a pattern found in a structure, we can create structures with varying defect densities.For this example, we will introduce defects into UiO-66 by randomly removing 25% of the linkers from the structure. We will first replicate the structure to a 2 × 2 × 2 so it fulfills minimum image conventions, which needs to be done before adding defects so that the defects aren't repeated in the structure. We can reuse the structure and search pattern files from example 1, but we will need to create a replacement pattern from the search pattern where the biphenyl ring is removed and replaced with formate caps where the linker would attach to the metal center (see Fig. 4C). This replacement pattern can be created in a similar manner to that described in example 1.

Example 3: parameterizing UiO-66 with flexible force field terms
For some simulations of gas adsorption in MOFs, it is common to assume the positions of the structure's atoms are fixed and only the adsorbate gases move.18 However, we do not always want to assume this (e.g. for thermal conductivity calculations,19 which require the atoms to move for heat to transfer), and many flexible force-fields have been developed that enable structures to flex and move.20–25 Despite these force-fields already existing, it can be challenging and time consuming to apply these force-fields to new structures: one needs to define atom types for every atom in the system, all topology required by the force field, and all force field parameters across the entire system, which may be tens of thousands of atoms and topology terms. This is a significant amount of work to do manually. There have been attempts to automate this,26 but it is hard to automate this process effectively and still allow for the parameter assignment process to be easily modified so that a researcher can validate and fix any parameter assignment issues when the automated system doesn't assign reasonable parameters. Even if one starts with a fully and correctly parameterized structure, expanding the structure to a larger number of unit cells can also be non-trivial, because bonds that cross periodic boundaries need to be “remapped” across the new periodic boundaries of the larger, expanded unit cell in order to run a LAMMPS simulation.One tactic to overcome these challenges for structures that can be deconstructed into distinct parts is to assign force-field parameters to the constituent parts of the structure and then use find and replace to apply the force field to the entire structure. In this example, we apply this technique to MOFs: we assign parameters to a MOF's metal center and linker and then replace all unparameterized metal centers and linkers in the full structure with their corresponding parameterized versions.
The patterns for the parameterized linker and the parameterized metal center will need to overlap; every desired force-field term will need to be included fully in at least one of the patterns so some atoms and force-field terms will be defined in both files. For 2-body terms, only the atom that connects the metal center to the linker needs to be shared between the patterns. For 3-body (angle) or 4-body (dihedral/improper) terms there will need to be two or three atoms of overlap, respectively (see Fig. 5). Linker and metal center files can be prepared similarly to example 1 above and parameterized manually, or possibly in an automated manner using the rough UFF parameterizer included in MOFUN (information on the UFF parameterizer is beyond the scope of this paper, but can be found in the documentation online), or with other packages.26 For Python:

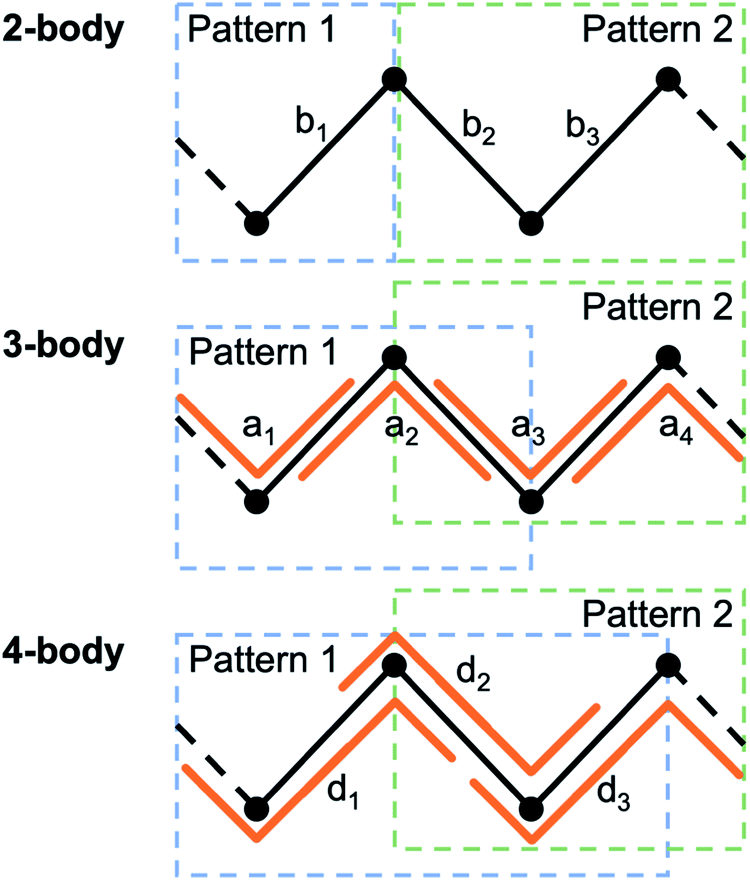 | ||
| Fig. 5 When parameterizing a structure using two parameterized patterns, for all two-body bond terms b1, …, bn to be replicated, the patterns must share an atom, for all three-body angle terms a1, …, an to be replicated the patterns must share two atoms, and for all four-body dihedral terms d1, …, dn to be replicated the patterns must share three atoms. | ||
Conclusion
MOFUN is an open-source Python package for generalized molecular find and replace. In our own lab, this is enabling us to quickly screen MOFs with various functional groups at different defect percentages, and easily apply force field parameters to structures. MOFUN is a great tool for automation, but there are some limitations. Since MOFUN identifies patterns using relative positions, a search pattern may not match all expected instances of the pattern in the structure if the positions vary in the structure, for example for longer alkane chains. While MOFUN fully supports force-fields defined in LAMMPS, there is no inherent format support for other molecular packages, except for outputting CIF files containing topology and force-field parameters. When doing a replacement operation, MOFUN places the replacement atoms exactly as specified and does not check if this placement overlaps with other atoms in the system, so using MOFUN requires the researcher to setup the find and replace operation in a reasonable manner and potentially relax the system after the replace operation. At present, MOFUN is primarily optimized for smaller systems (<40k atoms), though still works (albeit, more slowly) for larger systems. By making this code available to other labs, we hope that this will enable other labs to perform more ambitious screening and simulation studies.Data availability
The code for MOFUN can be found at https://github.com/WilmerLab/mofun. The version of the code employed for this study is version v1.0.1 (https://doi.org/10.5281/zenodo.6950355).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
P. B. and C. E. W. gratefully acknowledge support from the National Science Foundation (NSF award OAC-1931436) and the U.S Department of Energy NETL (UCFER_5-UPitt-S1-22). This research was also supported in part by the University of Pittsburgh Center for Research Computing through the resources provided.Notes and references
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Design and Synthesis of an Exceptionally Stable and Highly Porous Metal-Organic Framework, Nature, 1999, 402(6759), 276–279, DOI:10.1038/46248.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform, J. Cheminf., 2012, 4(1), 17, DOI:10.1186/1758-2946-4-17.
- J.-G. Sobez and M. Reiher, Molassembler: Molecular Graph Construction, Modification, and Conformer Generation for Inorganic and Organic Molecules, J. Chem. Inf. Model., 2020, 60(8), 3884–3900, DOI:10.1021/acs.jcim.0c00503.
- E. I. Ioannidis, T. Z. H. Gani and H. J. Kulik, MolSimplify: A Toolkit for Automating Discovery in Inorganic Chemistry, J. Comput. Chem., 2016, 37(22), 2106–2117, DOI:10.1002/jcc.24437.
- V. M. Ingman, A. J. Schaefer, L. R. Andreola and S. E. Wheeler, QChASM: Quantum Chemistry Automation and Structure Manipulation, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11(4), e1510, DOI:10.1002/wcms.1510.
- D. Weininger, SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36, DOI:10.1021/ci00057a005.
- Daylight Theory: SMARTS – A Language for Describing Molecular Patterns, https://www.daylight.com/dayhtml/doc/theory/theory.smarts.html, accessed, 2022-07-31 Search PubMed.
- Y.-S. Bae, J. Liu, C. E. Wilmer, H. Sun, A. N. Dickey, M. B. Kim, A. I. Benin, R. R. Willis, D. Barpaga, M. D. LeVan and R. Q. Snurr, The Effect of Pyridine Modification of Ni–DOBDC on CO2 Capture under Humid Conditions, Chem. Commun., 2014, 50(25), 3296–3298, 10.1039/C3CC44954H.
- E. A. Henle, N. Gantzler, P. K. Thallapally, X. Z. Fern and C. M. Simon, PoreMatMod.Jl: Julia Package for in Silico Postsynthetic Modification of Crystal Structure Models, J. Chem. Inf. Model., 2022, 62(3), 423–432, DOI:10.1021/acs.jcim.1c01219.
- S. Plimpton, Fast Parallel Algorithms for Short-Range Molecular Dynamics, J. Comput. Phys., 1995, 117(1), 1–19 CrossRef CAS.
- C. R. Harris, K. J. Millman, S. J. van der Walt, R. Gommers, P. Virtanen, D. Cournapeau, E. Wieser, J. Taylor, S. Berg, N. J. Smith, R. Kern, M. Picus, S. Hoyer, M. H. van Kerkwijk, M. Brett, A. Haldane, J. F. del Río, M. Wiebe, P. Peterson, P. Gérard-Marchant, K. Sheppard, T. Reddy, W. Weckesser, H. Abbasi, C. Gohlke and T. E. Oliphant, Array Programming with NumPy, Nature, 2020, 585(7825), 357–362, DOI:10.1038/s41586-020-2649-2.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, İ. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa and P. van Mulbregt, SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python, Nat. Methods, 2020, 17(3), 261–272, DOI:10.1038/s41592-019-0686-2.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, The Atomic Simulation Environment—a Python Library for Working with Atoms, J. Phys.: Condens. Matter, 2017, 29(27), 273002, DOI:10.1088/1361-648X/aa680e.
- K. Momma and F. Izumi, VESTA: A Three-Dimensional Visualization System for Electronic and Structural Analysis, J. Appl. Crystallogr., 2008, 41(3), 653–658, DOI:10.1107/S0021889808012016.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, Unusual and Highly Tunable Missing-Linker Defects in Zirconium Metal–Organic Framework UiO-66 and Their Important Effects on Gas Adsorption, J. Am. Chem. Soc., 2013, 135(28), 10525–10532, DOI:10.1021/ja404514r.
- O. V. Gutov, M. G. Hevia, E. C. Escudero-Adán and A. Shafir, Metal–Organic Framework (MOF) Defects under Control: Insights into the Missing Linker Sites and Their Implication in the Reactivity of Zirconium-Based Frameworks, Inorg. Chem., 2015, 54(17), 8396–8400, DOI:10.1021/acs.inorgchem.5b01053.
- N. Al-Janabi, X. Fan and F. R. Siperstein, Assessment of MOF's Quality: Quantifying Defect Content in Crystalline Porous Materials, J. Phys. Chem. Lett., 2016, 7(8), 1490–1494, DOI:10.1021/acs.jpclett.6b00297.
- J. Rouquerol, F. Rouquerol, P. Llewellyn, G. Maurin and K. S. W. Sing, Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, Academic Press, Amsterdam, 2nd edn, 2013 Search PubMed.
- K. B. Sezginel, P. A. Asinger, H. Babaei and C. E. Wilmer, Thermal Transport in Interpenetrated Metal–Organic Frameworks, Chem. Mater., 2018, 30(7), 2281–2286, DOI:10.1021/acs.chemmater.7b05015.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, DREIDING: A Generic Force Field for Molecular Simulations, J. Phys. Chem., 1990, 94(26), 8897–8909, DOI:10.1021/j100389a010.
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations, J. Am. Chem. Soc., 1992, 114(25), 10024–10035 CrossRef.
- H. Sun, COMPASS: An Ab Initio Force-Field Optimized for Condensed-Phase Applications Overview with Details on Alkane and Benzene Compounds, J. Phys. Chem. B, 1998, 102(38), 7338–7364 CrossRef CAS.
- M. A. Addicoat, N. Vankova, I. F. Akter and T. Heine, Extension of the Universal Force Field to Metal–Organic Frameworks, J. Chem. Theory Comput., 2014, 10(2), 880–891, DOI:10.1021/ct400952t.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules, J. Am. Chem. Soc., 1995, 117(19), 5179–5197, DOI:10.1021/ja00124a002.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins, J. Phys. Chem. B, 1998, 102(18), 3586–3616, DOI:10.1021/jp973084f.
- P. G. Boyd, S. M. Moosavi, M. Witman and B. Smit, Force-Field Prediction of Materials Properties in Metal-Organic Frameworks, J. Phys. Chem. Lett., 2017, 8(2), 357–363, DOI:10.1021/acs.jpclett.6b02532.
| This journal is © The Royal Society of Chemistry 2022 |